

Abstract
Background. Neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD) is a Mendelian disorder arising from biallelic SLC25A13 mutations, and SLC25A13 genetic analysis was indispensable for its definite diagnosis. However, conventional SLC25A13 analysis could not detect all mutations, especially obscure large insertions/deletions. This paper aimed to explore the obscure SLC25A13 mutation in an NICCD infant. Methods. Genomic DNA was extracted to screen for 4 high-frequency SLC25A13 mutations, and then all 18 exons and their flanking sequences were analyzed by Sanger sequencing. Subsequently, cDNA cloning, SNP analyses, and semiquantitative PCR were performed to identify the obscure mutation. Results. A maternally inherited mutation IVS16ins3kb was screened out, and then cDNA cloning unveiled paternally inherited alternative splicing variants (ASVs) featuring exon 5 skipping. Ultimately, a large deletion c.329-1687_c.468+3865del5692bp, which has never been described in any other references, was identified via intensive study on the genomic DNA around exon 5 of SLC25A13 gene. Conclusions. An NICCD patient was definitely diagnosed as a compound heterozygote of IVS16ins3kb and c.329-1687_c.468+3865del5692bp. The large deletion enriched the SLC25A13 mutation spectrum, and its identification supported the concept that cDNA cloning analysis, along with other molecular tools such as semiquantitative PCR, could provide valuable clues, facilitating the identification of obscure SLC25A13 deletions.
1. Introduction
Human citrin deficiency is an autosomal recessive disease due to dysfunction of citrin, the liver-type calcium-stimulated aspartate-glutamate carrier isoform 2 (AGC2) encoded by the SLC25A13 gene [1, 2]. Up to now, three age-dependent clinical phenotypes had been reported for this disorder, that is, adult-onset citrullinemia type 2 (CTLN2) in adolescents and adults, neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD) in neonates or infants, and failure to thrive and dyslipidemia caused by citrin deficiency (FTTDCD) in older children [3–8]. Although most reported patients were Asian [9–14], citrin deficiency has been recognized as a worldwide panethnic disorder nowadays [15–19].
Due to the lack of well-recognized clinical/biochemical diagnostic criteria for NICCD, SLC25A13 genetic analysis has been regarded as reliable tool for the definite diagnosis of such patients. However, routine SLC25A13 genetic analyses, such as polymerase chain reaction (PCR), long and accurate-PCR (LA-PCR), PCR-restriction fragment length polymorphism (RFLP), and Sanger sequencing, could not detect all SLC25A13 mutations, especially large deletions or insertions of an obscure nature. It has been estimated that approximately 15% of compound heterozygotes or homozygotes carrying SLC25A13 mutations in both alleles could not be definitely diagnosed just by the above conventional approaches [20]. In such cases, other molecular tools, although usually labor-intensive and cost-expensive, were needed to identify the obscure mutations.
In this study, a large SLC25A13 deletion was identified via sophisticated molecular analyses using peripheral blood lymphocytes (PBLs) in an NICCD patient, who responded well to a lactose-free and medium-chain triglycerides- (MCTs-) enriched formula. We herein reported the clinical and molecular findings.
2. Subjects and Methods
2.1. Subjects and Ethics
The research subjects in this study were a male patient suspected to have NICCD and his parents as well. With the informed consent from the parents and the ethical approval by the medical ethical committee of our hospital, we performed intensive clinical and genetic study on this family.
2.2. Conventional DNA Analysis
The DNA was extracted from the peripheral venous blood following the genomic DNA extraction kit (Omega, USA) instructions. Four high-frequency SLC25A13 mutations, c.851_854del, c.1638_1660dup, IVS6+5G>A, and IVS16ins3kb, were screened by PCR, LA-PCR, and PCR-RFLP procedures, and then Sanger sequencing of all the 18 exons and their flanking sequences was undertaken, using direct sequencing of DNA fragments amplified by genomic DNA-PCR to identify novel mutation in the gene SLC25A13 [21].
2.3. Reverse Transcriptional PCR (RT-PCR) and Nested PCR
PBLs were separated using lymphocyte separation medium (LSM, MP) from 2-3 milliliters of ethylene diamine tetraacetic acid- (EDTA-) anticoagulant peripheral venous blood [20]. PBLs were cracked with ribonucleic acid (RNA) trizol reagent (The Life Technologies) and total RNA was extracted using a variety of organic solvents, as described previously [12, 22–25]. The cDNAs were synthesized from total RNA by Moloney murine leukemia virus (MMLV) reverse transcriptase (TaKaRa). Nested PCR was used to amplify SLC25A13 open reading frame (ORF), and the primers and PCR temperature profile were described in detail as in previous references [21, 22]. The two primer pairs in Nested PCR were RAS2 and RACEA1 in the first PCR and RAS3 and Ex18R in the second one, with expected PCR products of 3107 bp and 2191 bp in size, respectively. Both of them contained the citrin-coding sequence (CDS).
2.4. Molecular Cloning and Alternative Splicing Variants (ASVs) Analyses
The Nested PCR products of paternal origin were purified by using a gel extraction kit (Omega) and then connected with the sequence of the pMD 18-T vector (TaKaRa) and transformed into DH5α Escherichia coli competent cells, as in our previous publications [12, 22, 24, 25]. Following that, the transformed cells were cultured in the shaking table for 4 h and then coated on plates after centrifugation on 1000 rpm for 5 min. After cultivation in 37°C for 16 hours, the positive clones which had the targeted bands after PCR amplification were selected and sequenced. The ASV sequences were analyzed by using the ContigExpress and DNAMAN software, and their designation was according to the nomenclature guidelines [26, 27].
2.5. Further Location and Identification of the Obscure Mutation
According to the ASV structures, to further locate the DNA span around exon 5 that might contain the obscure mutation, semiquantitative PCR was performed in a total volume of 50 μL, containing 5 μL of 10x Buffer (Mg2+ plus) (TaKaRa), 4 μL of dNTP (10 mM), 37.75 μL of sterilized distilled water, 0.25 μL of Taq (TaKaRa), 2 μL of the forward and reverse primers together, and 1 μL of DNA template. Then, all tubes were placed into a thermal cycler and the parameters were set in 94°C for 5 minutes followed by 28 to 30 cycles of 94°C for 30 seconds, 60°C for 40 seconds, 72°C for 40 seconds, and a final extension step at 72°C for 10 minutes. The two primer pairs were IVS5S5 and IVS5A6 in Set 1 and IVS 1NF and IVS 2NB in Set 2, whose sequences and locations were displayed in Figure 1, respectively.
Figure 1.

Semiquantitative PCR in positioning analysis of the novel large deletion. Figure 1① was a representative electrophoresis of the semiquantitative PCR products. Compared with the mother (M1), the patient (P1) and the father (F1) had less signal intensity of the PCR products when using primer Set 1, while this is not the case when using primer Set 2; note that the signal intensity of the PCR products in the patient (P2) and the father (F2) was similar to that in the mother (M2). Figure 1② depicted the positions of the primer Sets 1 and 2. The primer sequences in Set 1 were 5′-GAGCTTCTTAGAAACCACCATGTGG-3′ (IVS5S5) and 5′-TCCAATGAGG AAGAAGACTACAGGAAG-3′ (IVS5A6), while in Set 2, 5′-TTTATGCACTGGGGCAACATG-3′ (IVS 1NF) and 5′-TGCCGGGCTGACACTTTGG-3′ (IVS 2NB), respectively. The results suggested that the patient (P) and the father (F) might harbor an obscure large deletion around the primer Set 1 but not Set 2.
Based on the findings above, a PCR approach using the primers IVS4S3 and J5.6KbDelR1 (Set 3) was carried out to identify the obscure mutation. The reaction mixture was a total volume of 50 μL, containing 5 μL of 10x LA Buffer (Mg2+ plus) (TaKaRa), 8 μL of dNTP (10 mM), 30.3 μL of sterilized distilled water, 0.2 μL of Taq, 0.5 μL of LA Taq (TaKaRa), 4 μL of the forward and reverse primers together, and 2 μL of DNA template. The temperature profile was 95°C for 5 minutes followed by 40 cycles of 94°C for 40 seconds, 55°C for 40 seconds, 68°C for 10 minutes, and a final extension step at 68°C for 10 minutes.
2.6. Statistical Analysis
The proportions of the ASVs with exon 5 skipping in all ASVs from the patient and 8 healthy volunteers were statistically assessed using SPSS (Version 13.0) chi-square test, and a P value of less than 0.05 was considered statistically significant.
3. Results
3.1. Clinical Findings
A male infant at the age of 2 months was referred to the local hospital with the chief complaint of jaundiced skin over 10 days. Physical examination at referral revealed a mildly pale face and hemorrhagic spots scattered on the skin of whole body. The lungs were clear on auscultation. No abnormal sound or murmur was heard on heart auscultation. An enlarged liver, 5.0 cm below the right costal margin, was palpated. No pathological reflexes could be found on nervous system examination. Slightly visible pitting edema could be found in both lower extremities.
Biochemical test demonstrated elevated aspartate aminotransferase (AST), gamma-glutamyl transpeptidase (GGT), total bilirubin (Tbil), direct bilirubin (Dbil), indirect bilirubin (Ibil), and total bile acids (TBA), which indicated intrahepatic cholestasis. The albumin level was decreased while that of alpha-feto protein (AFP) elevated markedly (Table 1). Ultrasonography revealed hepatomegaly, ascites, and abdominal bloating, along with a patent oval foramen of the heart. The urinary gas chromatography mass spectrometry (GC-MS) analysis discovered large quantity of hexanedioic acid and 4-hydroxyphenyl lactate (4HPL), and on tandem mass spectrometry (MS-MS) analysis, elevated levels of citrulline and arginine were detected.
Table 1.
Biochemical changes over time in the NICCD infant.
| Biochemical indices | 2.0M | 2.3M | 2.4M | 2.5M | 2.6M | 2.8M | 3.0M | 3.7Ma | 4.8M | 8.8M | 17.7M |
|---|---|---|---|---|---|---|---|---|---|---|---|
| ALT (5–40 U/L) | 46.00 | 29.80 | 40.39 | 34.48 | 33.00 | 30.00 | 19.23 | 20.00 | 174.00 | 63.00 | 27.00 |
| AST (5–40 U/L) | 83.00 | 127.50 | 116.92 | 73.47 | 72.00 | 62.00 | 45.02 | 37.00 | 172.00 | 54.00 | 30.00 |
| GGT (8–50 U/L) | — | — | 131.13 | 139.63 | 183.00 | 185.00 | 475.33 | 211.00 | 109.00 | 54.00 | 26.00 |
| ALP (20–500 U/L) | — | — | 246.65 | 205.63 | 259.00 | 201.00 | 269.91 | 385.00 | 418.00 | 243.00 | 251.00 |
| TP (60.0–83.0 g/L) | 33.90 | 66.20 | 67.53 | 66.32 | 62.80 | 63.70 | 65.39 | 54.40 | 61.70 | 66.00 | 62.80 |
| Alb (35.0–55.0 g/L) | 21.30 | 36.94 | 39.66 | 39.23 | 35.00 | 37.60 | 40.87 | 37.50 | 41.80 | 45.80 | 43.80 |
| Glb (20.0–30.0 g/L) | — | 29.26 | 27.87 | 27.09 | 27.80 | 26.10 | 24.52 | 16.90 | 19.90 | 20.20 | 19.00 |
| Tbil (2–19 μmol/L) | 178.60 | 163.09 | 172.99 | 92.56 | 56.50 | 60.90 | 63.61 | 38.40 | 44.70 | 3.20 | 6.20 |
| Dbil (0–6 μmol/L) | 70.70 | 66.76 | 79.86 | 48.95 | 35.10 | 31.90 | 35.04 | 31.00 | 36.30 | 1.00 | 2.00 |
| Ibil (2.56–20.9 μmol/L) | 107.9 | 96.33 | 93.13 | 43.61 | 21.40 | 29.00 | 28.57 | 7.40 | 8.40 | 2.20 | 4.20 |
| TBA (0–10 μmol/L) | — | 33.86 | 39.35 | 54.43 | — | 73.50 | 44.45 | 173.60 | 169.7 | 11.30 | 8.80 |
| AFP(0–12 ng/mL) | — | — | — | — | — | — | — | 10709.20 | 887.20 | — | 1.90 |
ALT, alanine aminotransferase; AST, aspartate aminotransferase; GGT, γ-glutamyl transpeptidase; ALP, alkaline phosphatase; TP, total protein; Alb, albumin; Glb, globulin; Tbil, total bilirubin; Dbil, direct bilirubin; Ibil, indirect bilirubin; TBA, total bile acid; AFP, alpha-feto protein. M, months of age. —, not tested. aWhen the breast milk feeding was stopped while a galactose-free and MCT-enriched therapeutic formula was introduced.
NICCD was thus suspected based on the above clinical and biochemical findings, and according to the pediatrician's advice, breast feeding was stopped and a lactose-free and MCT-enriched therapeutic formula was introduced immediately. In the subsequent follow-up, the clinical manifestations were alleviated gradually. At his age of 3.7 months, the infant was referred to our hospital to confirm the NICCD diagnosis by SLC25A13 gene analyses.
3.2. Screening and Sequencing Results
Screening of the 4 high-frequency mutations proved that the patient harbored a maternally inherited mutation IVS16ins3kb, but another mutation was not detected. Even after Sanger sequencing of all the 18 exons and their flanking sequences, the SLC25A13 mutation of paternal origin remained obscure. Therefore, cDNA cloning analysis was undertaken to facilitate the identification of the obscure mutation.
3.3. Findings of cDNA Cloning Analysis
As the maternal mutation IVS16ins3kb led to production of an aberrant mRNA molecule with a new exon 17, losing the normal exons 17 and 18 [21], only the ASVs transcribed from the paternal SLC25A13 allele were amplified by the Nested PCR approach using the two reverse primers both within the normal exon 18 of SLC25A13 gene. In other words, the 26 ASVs in Table 2 all resulted from the paternally inherited SLC25A13 allele.
Table 2.
The SLC25A13 ASVs detected by cDNA analysis in the NICCD patient.
| Clones | Alternative splicing variants (ASVs) | Annotations |
|---|---|---|
| C0282-1 | r.213_468del; r.1194A>G | Exons 4 and 5 skipping |
| C0282-2 | r.70_468del; r.1194A>G | Exons 3, 4, and 5 skipping |
| C0282-3 | r.70_468del; r.1194A>G | Exons 3, 4, and 5 skipping |
| C0282-4 | r.213_468del; 1311_1312ins1311+102_1312+176; r.1194A>G | Exons 4 and 5 skipping |
| C0282-5 | r.213_468del; r.616_848del; r.1194A>G | Exons 4, 5, 7, and 8 skipping |
| C0282-6 | r.70_468del; r.1194A>G | Exons 3, 4, and 5 skipping |
| C0282-7 | r.329_468del; r.1194A>G | Exon 5 skipping |
| C0282-8 | r.213_468del; r.755_848del; r.1194A>G | Exons 4, 5, and 8 skipping |
| C0282-9 | r.329_468del; r.1194A>G | Exon 5 skipping |
| C0282-10 | r.70_468del; r.1194A>G | Exons 3, 4, and 5 skipping |
| C0282-11 | r.70_468del; r.1194A>G | Exons 3, 4, and 5 skipping |
| C0282-12 | r.213_468del; 1311_1312ins1311+102_1312+176; r.1194A>G | Exons 4 and 5 skipping |
| C0282-13 | r.213_468del; r.993_1018del; r.1194A>G | Exons 4 and 5 skipping |
| C0282-14 | r.213_468del; r.1194A>G | Exons 4 and 5 skipping |
| C0282-15 | r.213_468del; r.1194A>G | Exons 4 and 5 skipping |
| C0282-16 | r.70_468del; r.1194A>G | Exons 3, 4, and 5 skipping |
| C0282-17 | r.213_468del; r.755_848del; r.1194A>G | Exons 4, 5, and 8 skipping |
| C0282-18 | r.213_468del; r.1194A>G | Exons 4 and 5 skipping |
| C0282-19 | r.70_468del; r.1194A>G | Exons 3, 4, and 5 skipping |
| C0282-20 | r.213_468del; 1311_1312ins1311+102_1312+176; r.1194A>G | Exons 4 and 5 skipping |
| C0282-21 | r.213_468del; r.1194A>G | Exons 4 and 5 skipping |
| C0282-22 | r.213_468del; r.755_848del; r.1194A>G | Exons 4, 5, and 8 skipping |
| C0282-23 | r.213_468del; r.755_848del; r.1194A>G | Exons 4, 5, and 8 skipping |
| C0282-24 | r.213_468del; r.1194A>G | Exons 4 and 5 skipping |
| C0282-25 | r.213_468del; r.1194A>G | Exons 4 and 5 skipping |
| C0282-26 | r.213_468del; 1311_1312ins1311+102_1312+176; r.1194A>G | Exons 4 and 5 skipping |
Similar to our previous findings [22], these ASVs demonstrated remarkable structural heterogeneity. However, all these ASVs featured exon 5 skipping (r.329_468del), as shown in Table 2. When compared with the ASVs in the 8 healthy volunteers which had been reported previously [24], exon 5 skipping was a unique structural feature of the ASVs in this patient (χ 2 = 129.2 and P < 0.01 as in Table 3), strongly suggesting the existence of a large insertion or deletion around exon 5 at the DNA level.
Table 3.
SLC25A13 ASVs harboring r.329_468del in the patient C0282 and 8 healthy volunteers.
| Subjects | SLC25A13 ASVs | χ 2 | P | |
|---|---|---|---|---|
| With r.329_468 skipping | Without r.329_468 skipping | |||
| C0282 | 26 (100%) | 0 (0.0%) | 129.2 | <0.01 |
| Volunteers | 1 (0.9%) | 115 (99.1%) | ||
Correction for continuity was performed for χ 2 calculation in this table.
3.4. Positioning and Sequencing Analysis of the Obscure Mutation
Based on the findings above, a diversity of primers was designed and PCR amplification conducted to analyze a variety of SNPs within the DNA fragment around exon 5 in all 3 family members. Although no clues suggestive of the obscure mutation could be detected by these analyses, semiquantitative PCR had positive findings—when amplified using the primer Set 1 near exon 5, the DNA samples of the patient (P1) and the father (F1) had less PCR products in comparison to that of the mother (M1), but this is not the case when using the primer Set 2 covering the entire exon 2 (Figure 1), indicating that the patient and the father might have a large deletion or insertion involving the positions of the primer Set 1. According to this result, LA-PCR amplification using the primer Set 3 (Figure 2) yielded an unexpected band of 632 bp in size in the patient and father, besides the expected 6324 bp product as in the mother, and subsequent Sanger sequencing of the unexpected product uncovered a large deletion c.329-1687_c.468+3865del5692bp.
Figure 2.
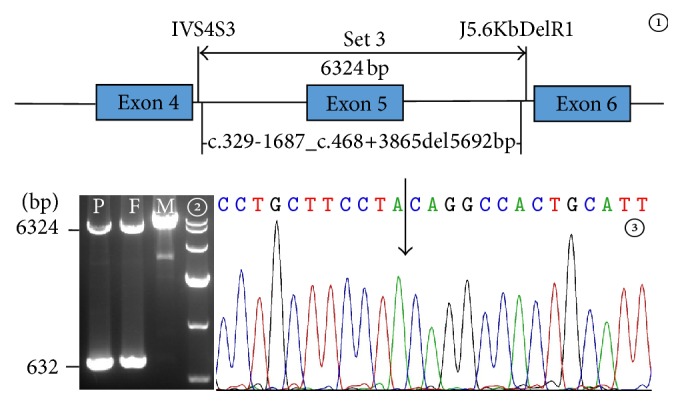
The large deletion mutation in SLC25A13 gene of the infant and his father. Figure 2① depicted the positions of the primer Set 3 and the novel large deletion. The primer sequences were 5′-AAGATTGTTGTTTATGGTGAGAC-3′ for IVS4S3 and 5′-ATGGTTTGCCCGACATGAGTAATC-3′ for J5.6KbDelR1, respectively. In Figure 2②, LA-PCR with the primer Set 3 revealed that the patient (P) and his father (F), but not the mother (M), had an unexpected band of 632 bp in size besides the normal product of 6324 bp. Figure 2③ was a segmental sequencing result of the unexpected PCR product. The arrow indicated the breakpoint arising from the large deletion.
4. Discussion
Although a maternally originated SLC25A13 mutation IVS16ins3kb had been discovered as a high-frequency mutation in this study, conventional genetic analyses, such as PCR/LA-PCR, PCR-RFLP, and Sanger sequencing, could not unveil the paternally inherited mutation in the infant with typical clinical and biochemical features of intrahepatic cholestasis, making the definite diagnosis of NICCD a challenge. It was by using cDNA cloning along with SNP analysis and semiquantitative PCR in all family members that c.329-1687_c.468+3865del5692bp, a large deletion, was finally identified, which has never been described in any other references. Although time- and cost-consuming, the identification of this novel deletion confirmed the feasibility of SLC25A13 cDNA cloning analysis using PBLs as a molecular tool facilitating the identification of large deletions and provided reliable evidence for the definite diagnosis of NICCD in the patient along with the mutation IVS16ins3kb of maternal origin. This was important not only for the proband himself but also for future genetic counseling and antenatal diagnosis in the family.
This 5692 bp deletion resulted in exon 5 skipping at mRNA level, as described in Table 2, theoretically yielding a truncated citrin protein of 127 amino acids—the exon 5 skipping gave rise to a frameshift at codon 110, continued to translate 17 amino acids, and then encountered a terminator codon at the position of 127, eventually producing an aberrant citrin molecule p.E110fs127X. Interestingly, this truncated citrin molecule was the same as the one arising from the large insertion IVS4ins6kb which had been reported very recently by our group [24]. The aberrant mRNA with exon 5 skipping could be a target for nonsense-mediated mRNA decay (NMD). Moreover, according to the molecular structural feature of mitochondrial AGCs (aspartate/glutamate carriers) [28], such a truncated citrin molecule predictively lost the entire mitochondrial carrier domain and had no AGC2 function to export aspartate and import glutamate together with a proton into the mitochondrial matrix, thus causing a series of metabolic disturbances and finally leading to the formation of the laboratory and clinical presentations in the patient.
The lactose-free and MCT-enriched formulas have been reported to be clinically and biochemically effective for NICCD patients in increasing clinical cases [12, 25, 29–31]. The clinical process in this study further supported this concept. Overall, the increased cytosolic NADH/NAD+ ratio in hepatocytes has been well-recognized as a key biochemical alteration for citrin deficiency [6]. Breast milk or common formula contained high carbohydrate in the form of lactose, a well-known disaccharide that was easily digested by lactase on the intestinal mucosa into glucose and galactose, both of which were then rapidly absorbed into the blood flow. In hepatocytes, galactose was conversed via Leloir pathway into glucose [32], the metabolism of which produced NADH, and thus increased the cytosolic NADH/NAD+ ratio in the liver and caused liver damage in patients with citrin deficiency [6, 31]. Moreover, secondary galactosemia due to citrin deficiency might be involved in the NICCD pathogenesis—the increased cytosolic NADH/NAD+ ratio in citrin-deficient hepatocytes might inhibit the activity of uridine diphosphate- (UDP-) galactose-4-epimerase [33], leading to accumulation of a large quantity of galactitol and galactonate [34], and galactitol has been suggested as one of the substrates causing jaundice, hepatosplenomegaly, hepatocellular insufficiency, and cataracts [30, 35, 36].
Energy shortage in the liver caused by an impairment of glycolysis due to an increased NADH/NAD+ ratio has been proposed as an important pathophysiology in citrin deficiency [31, 37]. Since the galactose kinase reaction in Leloir pathway was energy-consuming [32], the galactose metabolism in hepatocytes inevitably exacerbated such a pathophysiology. On the other hand, the absorption of MCTs was not bile acid-dependent, which might reduce the burden of the liver to synthesize and excrete bile salt into the gut [30]. Of particular note, MCTs can be better absorbed and transported via the portal vein into the liver and were mainly metabolized to acetyl-CoA along with FADH2 and NADH, which could supply more such substrates to hepatic cells as energy sources [31]. Taking all these factors together, it was not surprising for the NICCD infant in this study to respond well to the lactose-free and MCT-enriched therapeutic formula.
In summary, by sophisticated molecular analysis using PBLs, this study definitely diagnosed an NICCD patient who was a compound heterozygote of the IVS16ins3kb mutation and a novel deletion c.329-1687_c.468+3865del5692bp in the SLC25A13 gene. The large deletion constituted a novel component in the SLC25A13 mutation spectrum, and its identification lent further support to the concept that SLC25A13 cDNA cloning analysis using PBLs, along with other molecular tools such as semiquantitative PCR, could provide valuable clues, facilitating the identification of unknown large deletions.
Acknowledgments
The authors would like to express their deep appreciation to all research subjects for their kind cooperation. The research was financially supported by the Cultivation Foundation for Scientific Research (nos. 2014208 and A2014101) approved by the First Affiliated Hospital, Jinan University, the Medical Scientific Research Foundation of Guangdong Province (A2014385), the Fundamental Research Funds for the Central Universities (no. 21615470), and the projects supported by the National Natural Science Foundation of China (nos. 81270957 and 81570793).
Abbreviations
- NICCD:
Neonatal intrahepatic cholestasis caused by citrin deficiency
- SLC25A13:
Solute carrier family 25, member 13
- DNA:
Deoxyribonucleic acid
- cDNA:
Complementary deoxyribonucleic acid
- SNP:
Single nucleotide polymorphism
- PCR:
Polymerase chain reaction
- ASVs:
Alternative splicing variants
- CTLN2:
Adult-onset citrullinemia type 2
- FTTDCD:
Failure to thrive and dyslipidemia caused by citrin deficiency
- LA-PCR:
Long and accurate-polymerase chain reaction
- PCR-RFLP:
Polymerase chain reaction-restriction fragment length polymorphism
- PBLs:
Peripheral blood lymphocytes
- MCTs:
Medium-chain triglycerides
- RT-PCR:
Reverse transcriptional polymerase chain reaction
- LSM:
Lymphocyte separation medium
- EDTA:
Ethylene diamine tetraacetic acid
- RNA:
Ribonucleic acid
- MMLV:
Moloney murine leukemia virus
- ORF:
Open reading frame
- CDS:
Coding sequence
- AST:
Aspartate aminotransferase
- GGT:
Gamma-glutamyl transpeptidase
- Tbil:
Total bilirubin
- Dbil:
Direct bilirubin
- Ibil:
Indirect bilirubin
- TBA:
Total bile acids
- AFP:
Alpha-feto protein
- GC-MS:
Gas chromatography mass spectrometry
- 4HPL:
4-Hydroxyphenyl lactate
- MS-MS:
Tandem mass spectrometry
- NMD:
Nonsense-mediated mRNA decay
- AGC:
Aspartate/glutamate carrier
- NADH:
Reduced form of nicotinamide adenine dinucleotide
- NAD+:
Nicotinamide adenine dinucleotide
- UDP-galactose-4-epimerase:
Uridine diphosphate-galactose-4-epimerase.
Competing Interests
The authors declare that they have no competing interests.
References
- 1.Kobayashi K., Sinasac D. S., Iijima M., et al. The gene mutated in adult-onset type II citrullinaemia encodes a putative mitochondrial carrier protein. Nature Genetics. 1999;22(2):159–163. doi: 10.1038/9667. [DOI] [PubMed] [Google Scholar]
- 2.Palmieri L., Pardo B., Lasorsa F. M., et al. Citrin and aralar1 are Ca2+-stimulated aspartate/glutamate transporters in mitochondria. The EMBO Journal. 2001;20(18):5060–5069. doi: 10.1093/emboj/20.18.5060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ohura T., Kobayashi K., Tazawa Y., et al. Neonatal presentation of adult-onset type II citrullinemia. Human Genetics. 2001;108(2):87–90. doi: 10.1007/s004390000448. [DOI] [PubMed] [Google Scholar]
- 4.Tazawa Y., Kobayashi K., Ohura T., et al. Infantile cholestatic jaundice associated with adult-onset type II citrullinemia. The Journal of Pediatrics. 2001;138(5):735–740. doi: 10.1067/mpd.2001.113264. [DOI] [PubMed] [Google Scholar]
- 5.Tomomasa T., Kobayashi K., Kaneko H., et al. Possible clinical and histologic manifestations of adult-onset type II citrullinemia in early infancy. The Journal of Pediatrics. 2001;138(5):741–743. doi: 10.1067/mpd.2001.113361. [DOI] [PubMed] [Google Scholar]
- 6.Saheki T., Kobayashi K. Mitochondrial aspartate glutamate carrier (citrin) deficiency as the cause of adult-onset type II citrullinemia (CTLN2) and idiopathic neonatal hepatitis (NICCD) Journal of Human Genetics. 2002;47(7):333–341. doi: 10.1007/s100380200046. [DOI] [PubMed] [Google Scholar]
- 7.Song Y.-Z., Deng M., Chen F.-P., et al. Genotypic and phenotypic features of citrin deficiency: five-year experience in a Chinese pediatric center. International Journal of Molecular Medicine. 2011;28(1):33–40. doi: 10.3892/ijmm.2011.653. [DOI] [PubMed] [Google Scholar]
- 8.Kobayashi K., Saheki T., Song Y. Z. Citrin deficiency. In: Pagon R. A., Adam M. P., Ardinger H. H., et al., editors. GeneReviews®. Seattle, Wash, USA: University of Washington; 1993–2015. [Google Scholar]
- 9.Thong M. K., Boey C. C. M., Sheng J. S., Ushikai M., Kobayashi K. Neonatal intrahepatic cholestasis caused by citrin deficiency in two Malaysian siblings: outcome at one year of life. Singapore Medical Journal. 2010;51(1):e12–e14. [PubMed] [Google Scholar]
- 10.Lee B. H., Jin H. Y., Kim G.-H., Choi J.-H., Yoo H.-W. Nonalcoholic fatty liver disease in 2 siblings with adult-onset type II citrullinemia. Journal of Pediatric Gastroenterology and Nutrition. 2010;50(6):682–685. doi: 10.1097/MPG.0b013e3181d67fbf. [DOI] [PubMed] [Google Scholar]
- 11.Kikuchi A., Arai-Ichinoi N., Sakamoto O., et al. Simple and rapid genetic testing for citrin deficiency by screening 11 prevalent mutations in SLC 25A13. Molecular Genetics and Metabolism. 2012;105(4):553–558. doi: 10.1016/j.ymgme.2011.12.024. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Z.-H., Lin W.-X., Deng M., et al. Clinical, molecular and functional investigation on an infant with neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD) PLoS ONE. 2014;9(2) doi: 10.1371/journal.pone.0089267.e89267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang M.-H., Gong J.-Y., Wang J.-S. Citrin deficiency presenting as acute liver failure in an eight-month-old infant. World Journal of Gastroenterology. 2015;21(23):7331–7334. doi: 10.3748/wjg.v21.i23.7331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bijarnia-Mahay S., Häberle J., Rüfenacht V., Shigematsu Y., Saxena R., Verma I. Citrin deficiency: a treatable cause of acute psychosis in adults. Neurology India. 2015;63(2):220–222. doi: 10.4103/0028-3886.156285. [DOI] [PubMed] [Google Scholar]
- 15.Dimmock D., Kobayashi K., Iijima M., et al. Citrin deficiency: a novel cause of failure to thrive that responds to a high-protein, low-carbohydrate diet. Pediatrics. 2007;119(3):e773–e777. doi: 10.1542/peds.2006-1950. [DOI] [PubMed] [Google Scholar]
- 16.Hutchin T., Preece M. A., Hendriksz C., et al. Neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD) as a cause of liver disease in infants in the UK. Journal of Inherited Metabolic Disease. 2009;32(1, supplement):S151–S155. doi: 10.1007/s10545-009-1116-x. [DOI] [PubMed] [Google Scholar]
- 17.Fiermonte G., Parisi G., Martinelli D., et al. A new Caucasian case of neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD): a clinical, molecular, and functional study. Molecular Genetics and Metabolism. 2011;104(4):501–506. doi: 10.1016/j.ymgme.2011.08.022. [DOI] [PubMed] [Google Scholar]
- 18.Vitoria I., Dalmau J., Ribes C., et al. Citrin deficiency in a Romanian child living in Spain highlights the worldwide distribution of this defect and illustrates the value of nutritional therapy. Molecular Genetics and Metabolism. 2013;110(1-2):181–183. doi: 10.1016/j.ymgme.2013.06.011. [DOI] [PubMed] [Google Scholar]
- 19.Avdjieva-Tzavella D. M., Ivanova M. B., Todorov T. P., et al. First bulgarian case of citrin deficiency caused by one novel and one recurrent mutation in the SLC25A13 gene. Genetic Counseling. 2014;25(3):271–276. [PubMed] [Google Scholar]
- 20.Tokuhara D., Iijima M., Tamamori A., et al. Novel diagnostic approach to citrin deficiency: analysis of citrin protein in lymphocytes. Molecular Genetics and Metabolism. 2007;90(1):30–36. doi: 10.1016/j.ymgme.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 21.Tabata A., Sheng J.-S., Ushikai M., et al. Identification of 13 novel mutations including a retrotransposal insertion in SLC25A13 gene and frequency of 30 mutations found in patients with citrin deficiency. Journal of Human Genetics. 2008;53(6):534–545. doi: 10.1007/s10038-008-0282-2. [DOI] [PubMed] [Google Scholar]
- 22.Zhang Z.-H., Lin W.-X., Deng M., Zhao X.-J., Song Y.-Z. Molecular analysis of SLC25A13 gene in human peripheral blood lymphocytes: marked transcript diversity, and the feasibility of cDNA cloning as a diagnostic tool for citrin deficiency. Gene. 2012;511(2):227–234. doi: 10.1016/j.gene.2012.09.049. [DOI] [PubMed] [Google Scholar]
- 23.Lin W.-X., Zhang Z.-H., Deng M., Cai X.-R., Song Y.-Z. Multiple ovarian antral follicles in a preterm infant with neonatal intrahepatic cholestasis caused by citrin deficiency: a clinical, genetic and transcriptional analysis. Gene. 2012;505(2):269–275. doi: 10.1016/j.gene.2012.06.012. [DOI] [PubMed] [Google Scholar]
- 24.Song Y.-Z., Zhang Z.-H., Lin W.-X., et al. SLC25A13 gene analysis in citrin deficiency: sixteen novel mutations in East Asian patients, and the mutation distribution in a large pediatric cohort in China. PLoS ONE. 2013;8(9) doi: 10.1371/journal.pone.0074544.e74544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zeng H.-S., Zhao S.-T., Deng M., et al. Inspissated bile syndrome in an infant with citrin deficiency and congenital anomalies of the biliary tract and esophagus: identification and pathogenicity analysis of a novel SLC25A13 mutation with incomplete penetrance. International Journal of Molecular Medicine. 2014;34(5):1241–1248. doi: 10.3892/ijmm.2014.1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.den Dunnen J. T., Antonarakis S. E. Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Human Mutation. 2000;15(1):7–12. doi: 10.1002/(sici)1098-1004(200001)15:1<7::aid-humu4>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 27.den Dunnen J. T., Antonarakis E. Nomenclature for the description of human sequence variations. Human Genetics. 2001;109(1):121–124. doi: 10.1007/s004390100505. [DOI] [PubMed] [Google Scholar]
- 28.Thangaratnarajah C., Ruprecht J. J., Kunji E. R. S. Calcium-induced conformational changes of the regulatory domain of human mitochondrial aspartate/glutamate carriers. Nature Communications. 2014;5, article 5491 doi: 10.1038/ncomms6491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ohura T., Kobayashi K., Tazawa Y., et al. Clinical pictures of 75 patients with neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD) Journal of Inherited Metabolic Disease. 2007;30(2):139–144. doi: 10.1007/s10545-007-0506-1. [DOI] [PubMed] [Google Scholar]
- 30.Song Y. Z., Wen F., Chen F. P., Kobayashi K., Saheki T. Neonatal intrahepatic cholestasis caused by citrin deficiency: efficacy of therapeutic formulas and update of clinical outcomes. Japanese Journal for Inherited Metabolic Diseases. 2010;26(1):57–69. [Google Scholar]
- 31.Hayasaka K., Numakura C., Toyota K., Kimura T. SSIEM. Treatment with lactose (galactose)-restricted and medium-chain triglyceride-supplemented formula for neonatal intrahepatic cholestasis caused by citrin deficiency. (JIMD Reports).JIMD Reports—Case and Research Reports, 2011/2. 2012;2:37–44. doi: 10.1007/8904_2011_42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frey P. A. The Leloir pathway: a mechanistic imperative for three enzymes to change the stereochemical configuration of a single carbon in galactose. The FASEB Journal. 1996;10(4):461–470. [PubMed] [Google Scholar]
- 33.Saheki T., Kobayashi K., Iijima M., et al. Pathogenesis and pathophysiology of citrin (a mitochondrial aspartate glutamate carrier) deficiency. Metabolic Brain Disease. 2002;17(4):335–346. doi: 10.1023/a:1021961919148. [DOI] [PubMed] [Google Scholar]
- 34.Song Y.-Z., Li B.-X., Chen F.-P., et al. Neonatal intrahepatic cholestasis caused by citrin deficiency: clinical and laboratory investigation of 13 subjects in mainland of China. Digestive and Liver Disease. 2009;41(9):683–689. doi: 10.1016/j.dld.2008.11.014. [DOI] [PubMed] [Google Scholar]
- 35.Ning C., Reynolds R., Chen J., et al. Galactose metabolism by the mouse with galactose-1-phosphate uridyltransferase deficiency. Pediatric Research. 2000;48(2):211–217. doi: 10.1203/00006450-200008000-00015. [DOI] [PubMed] [Google Scholar]
- 36.Bosch A. M. Classical galactosaemia revisited. Journal of Inherited Metabolic Disease. 2006;29(4):516–525. doi: 10.1007/s10545-006-0382-0. [DOI] [PubMed] [Google Scholar]
- 37.Hayasaka K., Numakura C., Watanabe H. Treatment and pathomechanism of citrin deficiency. Brain and Nerve. 2015;67(6):739–747. doi: 10.11477/mf.1416200211. [DOI] [PubMed] [Google Scholar]