

Abstract
Sickle cell disease (SCD) is the most common cause of stroke in childhood and results primarily from a mismatch of cerebral oxygen supply and demand rather than arterial obstruction. However, resting cerebral blood flow (CBF) has not been examined in the general African American population, in whom obesity, hypertension, cerebrovascular disease, and diminished cerebrovascular reserve capacity are common. To better understand the underlying physiological substrate upon which SCD is superimposed, we measured CBF in 32 young (age 28 ± 10 yr), asymptomatic African American subjects with and without sickle cell trait (n = 14). To characterize the effects of chronic anemia, in isolation of sickle hemoglobin we also studied a cohort of 13 subjects with thalassemia major (n = 10), dyserythropoetic anemia (n = 1), or spherocytosis (n = 2). Blood was analyzed for complete blood count, hemoglobin electrophoresis, cell free hemoglobin, and lactate dehydrogenase. Multivariate regression analysis showed that oxygen content was the strongest predictor of CBF (r2 = 0.33, P < 0.001). CBF declined rapidly in the second and third decades of life, but this drop was explained by reductions in cerebral gray matter. However, age effects persisted after correction for brain composition, possibly representing microvascular impairment. CBF was independent of viscosity, hemoglobin S%, and body mass index. Hyperoxia resulted in reduced CBF by 12.6% (P = 0.0002), and CBF changes were proportional to baseline oxygen content (r2 = 0.16, P = 0.02). These data suggest that these hemoglobin subtypes do not alter the normal CBF regulation of the balance of oxygen supply and demand.
Keywords: African Americans, anemia, cerebral blood flow, hyperoxia, stroke
sickle cell disease (scd) is the most common cause of stroke in the pediatric population. In patients with SCD, abnormal red cell rheology, hemolysis, anemia, oxygen-dissociation curve changes, and endothelial dysfunction impair oxygen delivery, whereas rapid changes in the maturing brain affect oxygen consumption. More than 95% of patients with SCD are also African American, in whom factors such as obesity and hypertension are known cerebrovascular risk factors. In fact, African Americans develop cerebrovascular disease at twice the rate of white Americans (15) and have significantly diminished cerebrovascular reserve capacity even in apparently healthy young adults (18), and more than 1.5% of all children born in the United States and 7.3% of African Americans have sickle cell trait (SCT); despite these observations, resting cerebral blood flow (CBF) in African Americans has not been systemically examined.
Therefore, the primary goal of this study was to determine the predictors of CBF in asymptomatic young African American subjects with and without SCT to establish reference values for patients with SCD. To thoroughly characterize the effect of anemia on CBF independent of sickle hemoglobin it was necessary to recruit subjects with thalassemia major, dyserythropoetic anemia, and spherocytosis. This was necessitated because of a lack of appropriate African American subjects with severe, chronic anemia (Hb <10) and without either sickle hemoglobin or other vasculopathy in the age group required. Our overarching hypotheses were that 1) the presence of SCT (one sickle cell gene) would not affect CBF, and 2) blood oxygen content would be the largest determinant of CBF in our study population, overwhelming any effect of blood viscosity, blood pressure, hemolysis, and body mass index.
MATERIALS AND METHODS
This study was approved by the Children's Hospital Los Angeles Committee on Clinical Investigations (CCI 11-00083) and informed consent was obtained from all patients. Two ethnically distinct cohorts were recruited. The first cohort consisted of African Americans older than 12 yr of age (n = 32). Exclusion criteria included prior history of neurologic insult; developmental delay; seizures; learning disability; or serious, chronic illness requiring daily medications. The second cohort consisted of patients with chronic anemia, excluding sickle cell anemia, who were otherwise healthy and age-matched to the first cohort. The etiology of anemia consisted of thalassemia major (n = 10), congenital dyserythropoetic anemia (n = 1), and spherocytosis (n = 2). Exclusion criteria included prior neurologic insult, developmental delay, seizures, learning disability, or major medical problems in addition to chronic anemia.
Patients underwent measurement of vital signs and phlebotomy. Complete blood count, reticulocyte total, quantitative hemoglobin electrophoresis, and lactate dehydrogenase (LDH) and cell free hemoglobin levels were analyzed in the clinical laboratory. A 10-ml aliquot of each sample was heparanized, equilibrated with humidified room air using a hollow tube gas exchanger (Living Systems Instrumentation, Burlington, VT), and analyzed for viscosity at a controlled temperature of 37° (Rheolog Health Vector, Pennsauken, NJ).
MRI was performed using an eight-element phased-array head coil on a 3T Philips Achieva. A large field-of-view magnetic resonance angiogram was reformatted into coronal and sagittal images to better localize the phase-contrast imaging plane approximately 1 cm above the carotid bifurcation and orthogonal to both carotids. Vertebral arteries exhibited obliquity of up to 15 degrees in ∼10% of the patients. Image parameters for the phase-contrast examination were as follows: repetition time, 12.3 ms; echo time, 7.5 ms; field of view, 260 mm; thickness, 5 mm; signal averages, 10; acquisition matrix, 204 × 201; reconstruction matrix, 448 × 448; bandwidth, 244 Hz/pixel; and velocity encoding gradient, 200 cm/s.
Initially, the within-session stability of phase-contrast measurements was unknown. As a result, phase-contrast measurements were interleaved (without relocalizing) with the other imaging experiments.
After baseline anatomic and phase-contrast imaging, patients were fitted with a custom rebreathing apparatus with a 4-liter inspiratory reservoir to provide low-resistance tidal breathing. The reservoir was refreshed with 10-15 l/min of inflowing gas, which was typically 21% oxygen/79% nitrogen. Patients breathed through a SCUBA mouthpiece coupled via a three-way valve to the breathing reservoir. Nose plugs blocked nasal inflow. Phase-contrast imaging was subsequently repeated with patients breathing a mixture of 21% oxygen and 79% nitrogen. The patients then underwent two blood oxygen level-dependent (BOLD) imaging experiments (not reported in this manuscript). During the first BOLD experiment, five breaths of pure nitrogen were administered to transiently reduce patient oxygen saturations to 80–85%. During the second BOLD experiment, 100% oxygen was administered for 4 min to increase blood oxygen content. Phase-contrast measurements were repeated before and after both oxygen challenges. Arterial blood oxygen content was estimated using the following equation: O2 content (ml O2/dl blood) = 1.34 × [Hb] × O2sat% + 0.003 × Po2, where O2sat% represents the hemoglobin saturation estimated by pulse oximetry (7550VO; Nonin, Plymouth, MN) and Po2 was the estimated partial pressure of arterial oxygen. Arterial blood gases were not measured. Po2 was assumed to be 100 torr for room air ventilation and 400 torr for 100% oxygen nonrebreather ventilation (34).
Phase-contrast images were analyzed offline. Stationary tissue pixels were identified in the complex difference images by simple thresholding using a mean plus two standard deviations from a remote, nonvascular region. Phase differences of stationary pixels were fit to a second-order two-dimensional polynomial and used to remove the background phase from vessels. Vessel boundaries were identified semiautomatically from the complex difference image processed using a Canny edge-detector. We then performed a single-pixel dilation of vessel boundaries and retained pixels whose complex difference was greater than the stationary tissue threshold. Once vessel boundaries were defined, the CBF was calculated voxel-wise using the following equation: CBF (ml blood/min) = ∑ni = 1Ai × Vi, where there are n voxels inside the vessel, and for each voxel (i) the area (Ai) is a defined by MRI parameters, and Vi is the velocity measured by the phase-contrast MRI. Processing was performed in MATLAB (The MathWorks, Natwick, MA).
Total CBF was corrected for changes in brain size and composition by measuring gray and white matter volumes and assuming a brain density of 1.05 g/ml. Three-dimensional T1-weighted images were processed using BrainSuite (www.brainsuite.org) to skull-strip and classify tissues, manual corrections were made to the cortex and gray-white boundaries, and the brainstem was cut at the bottom of the cerebellum. Surface-volume registration was performed using the BrainSuite SVReg15a tool to calculate average cortical thickness and cortical surface area.
Vital signs, laboratory data, brain volumes, and CBF were imported into JMP10.0 software (SAS, Cary, NC) for analysis. Two sample comparisons were performed using the Student's t-test. Univariate regression was first performed to identify candidate contributors to CBF. Because CBF is proportional to the ratio of oxygen consumption divided by oxygen content, CBF and all its predictors were log-transformed before being entered in the regressions. All variables having a univariate value of P < 0.10 were included as candidates for the stepwise multivariate regression, and retained in the final model if their value was P < 0.05.
RESULTS
Table 1 summarizes the demographics of the African American (nonanemic) and non-African American (anemic) cohorts. Statistically significant differences between the two groups are highlighted with an asterisk. The differences in blood parameters, body mass index, and blood pressure are intuitive consequences of the chronic anemia in the non-African American cohort. Fourteen patients in the African American cohort had SCT with a mean hemoglobin S of 39%. Three of the non-African American patients had detectable hemoglobin E but most of their circulating hemoglobin was normal AA hemoglobin from chronic transfusion therapy. The two patients with spherocytosis exhibited polycythemia (Hb 17.5), although the other 11 non-African American patients had hemoglobin levels of 10.2 ± 0.7; patients with spherocytosis were retained in the regression analyses because they provided additional dynamic range to assess the effect of hemoglobin on CBF.
Table 1.
Baseline patient demographics
| Parameter | African American | Non-African American | Range | Normal Range |
|---|---|---|---|---|
| Age, yr | 28.7 ± 10.5 | 22.5 ± 5.2† | 12.3–50.5 | |
| Sex | 9 M, 23 W | 7 M, 6 W† | ||
| Racial background | African 32 | Chinese, 4 | ||
| Thai, 1 | ||||
| Pakistani, 2 | ||||
| Filipino, 2 | ||||
| Hispanic, 3 | ||||
| Indian, 1 | ||||
| Body surface area, m2 | 1.82 ± 0.24 | 1.74 ± 0.19 | 1.40–2.37 | |
| Body mass index, kg/m2 | 26.3 ± 7.0 | 23.8 ± 2.8† | 16.8–45.1 | |
| Systolic blood pressure, torr | 119 ± 13 | 111 ± 12† | 90–152 | |
| Diastolic blood pressure, torr | 70 ± 10 | 63 ± 9† | 47–89 | |
| Heart rate, bpm | 74 ± 18 | 78 ± 11 | 56–122 | |
| Respiratory rate, bpm | 18 ± 2 | 22 ± 12 | 16–61 | |
| Temperature, °C | 36.8 ± 0.2 | 36.7 ± 0.1 | 36.4–37.1 | |
| Oxygen saturation, % | 99.2 ± 1.1 | 99.1 ± 1 | 95–100 | |
| White blood cell, 103/μl | 5.5 ± 1.5 | 7.0 ± 1.8† | 3.1–11 | 5.0–13.0 |
| Absolute neutrophil count, 103/μl | 3.1 ± 1.3 | 4.3 ± 1.5† | 1.0–8.5 | |
| Hemoglobin, g/dl | 13.6 ± 1.3 | 11.3 ± 2.8† | 9.2–17.5 | 12.0–15.5 |
| Hematocrit, % | 39.9 ± 3.5 | 32.6 ± 7.1†† | 27.3–47.6 | 36.0–50.0 |
| Mean corpuscular hemoglobin concentration, % | 34.0 ± 1.0 | 34.5 ± 1.3 | 32.3–37.6 | 30.0–36.0 |
| Platelets, 103/μl | 249 ± 63 | 246 ± 122 | 120–537 | 150–450 |
| Red cell distribution width, % | 13.2 ± 1.2 | 17 ± 4.1† | 14.4–24.8 | 11.5–14.5 |
| Reticulocytes, % | 1.4 ± 0.6 | 1.3 ± 1.9 | 0.1–7 | 0.5–1.5 |
| Lactate dehydrogenase, U/l | 529 ± 92 | 427 ± 162† | 253–909 | 313–618 |
| Cell free hemoglobin, mg/dl | 5.2 ± 3.8 | 21.8 ± 24.5† | 0.5–44.5 | 0–10 |
| Hemoglobin electrophoresis | AA 18 | AA 10 | ||
| AS 14 | AE 3 | |||
| Hemoglobin A in patients with AE hemoglobin, % | 84.1 ± 6.0 | 78.1–90.0 | ||
| Hemoglobin S in patients with AS hemoglobin, % | 39.0 ± 3.2 | 33.2–42.5 | ||
| Total brain mass, g | 1,176 ± 106 | 1,171 ± 104 | 1,003–1,380 | |
| Gray matter mass, g | 633 ± 68 | 652 ± 51 | 535–786 | |
| White matter mass, g | 542 ± 56 | 519 ± 58 | 409–686 | |
| Average cortical thickness, mm | 3.91 ± 0.13 | 4.02 ± 0.11† | 3.61–4.19 | |
| Average cortical surface area, cm2 | 995 ± 97 | 898 ± 234 | 825–1,186 |
AE, hemoglobin E trait; AS, hemoglobin S trait; bpm, beats per minute;
Values are means ± SD.
Statistically significant differences at the 0.05 level.
A representative phase-contrast acquisition with vessel boundaries and time course of CBF measurements is shown in Figure 1. Within-session stability of the CBF measurements was assessed in 21 patients (17 with normal hemoglobin and 4 with anemia). An average of 4.7 ± 1.1 (range, 2–7) CBF measurements were performed across 30.3 ± 9.5 (range 4–50) min. The coefficient of variation was 4.6% ± 3.0% (range, 0.6–12.4) with a median of 3.5%.
Fig. 1.

A: phase-contrast imaging plane referenced to a coronal angiogram. B: magnitude image. C: phase image depicting the voxels selected by the vessel-selection algorithm. D: representative time course from our youngest control subject (12.6 yr of age).
Not surprisingly, the strongest predictor of CBF normalized to brain weight was oxygen content (r2 = 0.33, P < 0.001, Fig. 2). After removing the contribution from oxygen content, the dominant predictors of CBF are summarized in Table 2. Age was a negative correlate of CBF. Systolic and diastolic blood pressures were also negatively associated with CBF. CBF was equally correlated with gray matter volume and cortical surface area, and less strongly associated with total brain volume. Notably, CBF was independent of white matter volume and cortical thickness.
Fig. 2.
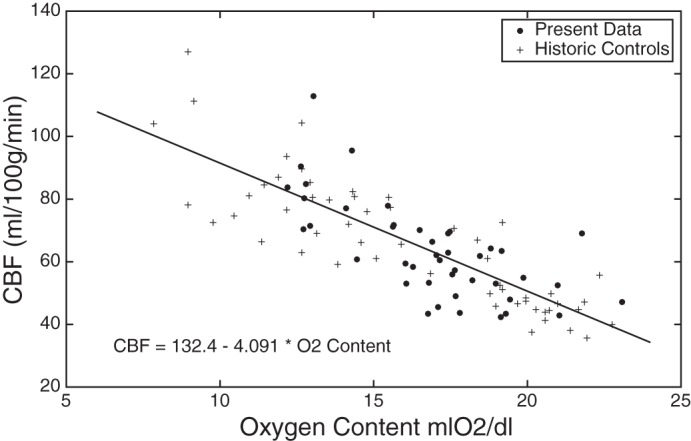
Plot of cerebral blood flow (CBF) normalized to per 100 g of total brain weight vs. oxygen content. Large dots indicate our experimental data. Plus signs (+) represent controls described by Brown et al. (8).
Table 2.
Significant predictors of first residual of cerebral blood flow and O2 content
| Parameter | r2, Direction | P |
|---|---|---|
| Age | 0.20, ↓ | <0.001 |
| Total brain volume | 0.12, ↑ | 0.008 |
| Gray matter volume | 0.14, ↑ | 0.003 |
| Cortical surface area | 0.14, ↑ | 0.003 |
| Cortical thickness | 0.02, ∼ | 0.28 |
| White matter volume | 0.02, ∼ | 0.23 |
| Systolic blood pressure | 0.1, ↓ | 0.01 |
| Diastolic blood pressure | 0.12, ↓ | 0.007 |
| Lactate dehydrogenase | 0.17, ↓ | 0.01 |
All variables were log-transformed in the analysis.
The observed age-related reduction in CBF during adolescence and early adulthood could be explained by normal brain maturation. There was a dramatic decrease in gray matter volume as a function of patient age up to 25 yr of age (Fig. 3, open circles), paralleling the changes in CBF corrected for oxygen content (Fig. 3, diamonds). Because gray matter consumes more energy than white matter, we entered gray matter volume into the multivariate model before entering age. Age remained a powerful independent predictor of CBF, but no effect was observed until after 30 years of age. After inclusion of oxygen content, gray matter volume, and age, CBF was independent of other patient demographics, vital signs, blood viscosity, and all clinical laboratory variables except LDH. The multivariate model for CBF log-transformed both CBF and all the following parameters. The resulting model used log-transformed parameter values as follows: log oxygen content (ml O2·100 g−1·min−1), −0.542 (r2 = 0.35, P < 0.001); log total gray matter (mm3), 0.337 (r2 = +0.16, P < 0.001); log age (years), −0.245 (r2 = +0.08, P = 0.008), and log LDH, −0.322 (r2 = +0.07, P = 0.007). The total model r2 was 0.67.
Fig. 3.
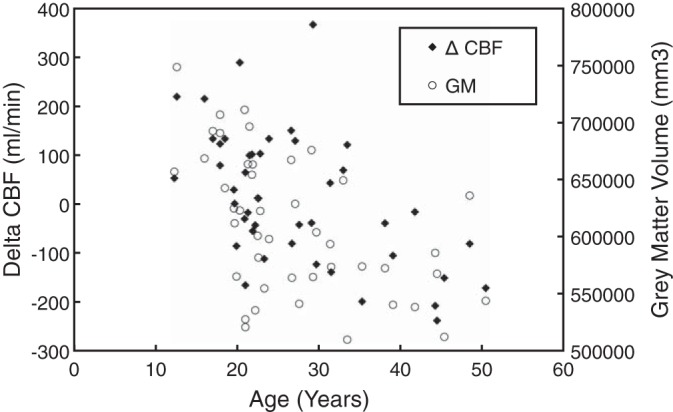
First residual of CBF, after removing the contribution from oxygen content plotted as a function of age (solid diamonds). Gray matter volume as a function of age (open circles).
Following baseline CBF assessments, patients were given the breathing apparatus, and CBF was remeasured while they breathed room air. Breathing through the circuit resulted in a CBF decrease from 752 to 695 ml/min (standard error 15 ml/min, P < 0.001). After the brief exposure to 100% nitrogen and subsequent 5-min recovery, CBF was unchanged (P = 0.85). Following the 5-min exposure to 100% oxygen, CBF was measured again (Fig. 4). CBF declined 12.6% (P < 0.001) and response magnitude was inversely proportional to hemoglobin level (Fig. 4, right; r2 = 0.19, P = 0.01). The CBF reduction with 100% oxygen administration was exactly balanced by the improved oxygen content provided by the dissolved oxygen, such that oxygen delivery was unchanged by exogenous oxygen administration.
Fig. 4.

Left: plot of CBF and cerebral oxygen delivery when breathing room air and 100% oxygen through nonbreathing ventilatory circuit. CBF decreased 7.6% when patients breathed oxygen, roughly balancing the projected increase in oxygen carrying capacity. As a result, cerebral oxygen delivery is completely neutral to inhaled oxygen. Right: percent reduction in CBF with 100% oxygen treatment as a function of hemoglobin level.
DISCUSSION
Our data are the first in humans to systematically explore the relative importance of oxygen content, brain weight and composition, age, and viscosity in determining CBF in a predominantly African American population. These data were in excellent agreement with a large historical cohort of Caucasians (8) and reinforce the hypothesis that CBF is controlled to maintain a stable oxygen delivery to the brain.
The inverse relationship between CBF and oxygen content at baseline was expected, but the reduction in CBF with 100% oxygen has not been universally observed (8, 9, 25, 28, 32, 35, 36). Some authors attribute the decreased CBF to reductions in cerebral metabolic rate (35), others dispute this (36) and attribute the response to the stimulatory effect of oxygen on ventilation (9, 25, 35). However, the larger CBF decrease observed in patients with anemia cannot be attributed to hyperventilation. Instead, our observations suggest that the brain is adjusting CBF to maintain oxygen delivery, and the dissolved oxygen fraction is a much larger fraction of total oxygen carrying capacity in patients with anemia, producing a greater effect on CBF. This study cannot identify how local oxygen demand in the brain is transduced and regulated. Partial pressure of oxygen has been proposed as the regulatory stimulus (7, 13, 24). We speculate that if this is true, the oxygen-sensing apparatus must be sufficiently distal in the microvascular distribution network that dissolved oxygen fraction is reflective of supply-demand balance.
The second striking observation was that the sharp reduction in CBF we observed in the second and third decades of life was explained by the reduction in gray matter volume and surface area that occurs during normal brain maturation (6, 12, 33). The independence of CBF from white matter volume underscores the metabolic dominance of gray matter (3, 16). The practice of normalizing total CBF to total brain volume may not be appropriate in children and young adults in whom significant gray matter-to-white matter ratio changes are expected.
Interestingly, gray matter volume and cortical surface area (CSA) were equally important in determining CBF, but CBF was independent of cortical thickness. Gray matter volume is dependent on both CSA and cortical thickness, but these parameters account for different morphological traits of the neocortex. The radial unit hypothesis explains that CSA is affected by the number of neuronal columns, whereas cortical thickness is determined by the number of neurons in each column (29). Expansion of CSA increases the number of columns, thereby increasing the complexity of neuronal networks (17), and hence increasing energy consumption. Determinants of cortical thickness on the other hand are not as straightforward. Cortical thickening is a normal developmental process, cortical thinning is typically the result of myelination and synaptic pruning (20), and later in adulthood brain atrophy due to normal aging and disease further thins the cortex.
Systolic and diastolic blood pressures were negatively associated with CBF after controlling for oxygen content. However, systolic and diastolic blood pressures both rise sharply through childhood and adolescence, so we believe that blood pressure associations were merely surrogates for patient age. After accounting for age, blood pressure disappeared as a predictor of CBF.
After correcting for changes in brain size and composition, further age-related CBF decline remained apparent in the fourth and fifth decades of life, which could not be accounted for by cortical atrophy. Similar findings have been described in positron emission tomography, computed tomographic perfusion, magnetic resonance arterial spin labeling, and phase-contrast MRI studies (2, 19, 19a, 27, 31). Recent work by Lu and colleagues (22) suggests that CBF reductions with age are not the result of declining cerebral metabolic rate, but instead they reflect microvascular disease.
This is particularly interesting given the perplexing relationship between CBF, LDH, and absolute neutrophil count (ANC). We could find no precedent for these associations in the literature. LDH can reflect intravascular hemolysis in patients with SCD, but neither reticulocyte count nor cell free hemoglobin (which are better hemolytic markers) correlated with CBF. We speculate that the LDH may serve as a nonspecific marker of endothelial damage. ANC could represent a surrogate of systemic inflammation; we did not measure high-sensitivity C-reactive protein, which would have been a better surrogate for vascular inflammation. Regardless, the CBF associations with LDH and ANC should be independently repeated in a larger cohort before too much weight is placed on these observations.
A number of negative findings were noteworthy. After controlling for hemoglobin and gray matter volume, sex differences in CBF disappeared. CBF was also independent of hemoglobin electrophoresis results, despite the known effect of SCT on the hemoglobin dissociation curve (4). Blood viscosity had no detected effect on CBF after correction for differences in oxygen carrying capacity. This does not imply that blood viscosity is unimportant in mediating changes in CBF in response to stimuli (8, 30), but that it plays no apparent role in establishing the “set point” for baseline CBF (7). CBF was also independent on BMI over a fairly broad range. This is interesting given accumulating evidence that obesity directly promotes vascular inflammation via adipose-based synthesis of cytokines and accelerates cerebral microvascular disease (21). However, to observe this, an older cohort of individuals with higher BMI may have been required.
Our study had limitations. Our patients with chronic anemia had Asian or Middle Eastern ancestry, whereas the primary control cohort was of African descent. This was necessitated by design because we wanted to study the effect of anemia alone without the confounding influences of SCD (e.g., vaso-occlusion, intravascular hemolysis, and stroke, which usually occur in patients with SCD). We measured CBF in a single axial plane rather than four acquisitions optimized to vessel angle as advocated by some because the temporal variation in any one vessel is higher than the variability in their sum. We estimated pressure of arterial oxygen rather than directly measuring it via arterial blood gases. Although this would have little effect on our regression analyses, patient-specific differences in alveolar-arterial oxygen gradient could affect results of the 100% oxygen challenge. Our study would have benefited from continuous measurements of CO2. We speculate that the reduction in CBF that occurred when subjects transitioned from breathing room air freely to breathing room air through a breathing apparatus was caused by mild hyperventilation. However, without an MRI-compatible transcutaneous CO2 measuring device, we cannot confirm this hypothesis.
In summary, we demonstrated that oxygen-demand principles dominate resting CBF and its response to inhaling 100% oxygen. Increased CBF in patients with chronic anemia is sufficient to preserve brain oxygen delivery. Marked changes in CBF observed in the second and third decades of life were entirely explained by normal brain maturation (33). After correction for oxygen content and gray matter volume, no effect of sex, sickle cell hemoglobin, or blood viscosity was evident. Age-related decline in CBF was still observed starting around 30 yr of age. Our results in this predominately African American cohort provide key normative data for studying impaired oxygen delivery in patients with sickle cell anemia.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grant 1U01-HL-117718-01 and Minority Supplement to Grant 1U01-HL-117718-01, and by the National Center for Research through the Clinical Translational Science Institute at Children's Hospital Los Angeles. Philips Healthcare provided support for protocol development and applications engineering on a support-in-kind basis.
DISCLOSURES
J. Wood is a consultant for AMAG Pharmaceuticals, ISIS Pharmaceuticals, ApoPharma, Pfizer, Celgene, WorldCare Clinical, and BiomedInformatics. He also receives support-in-kind from Philips Healthcare. T. Coates is a consultant for Acceleron, ApoPharma, Celgene, ISIS Pharma, and Novartis. None of the others authors have commercial affiliations.
AUTHOR CONTRIBUTIONS
M.T.B., A.M.B., and J.C.W. conception and design of research; M.T.B., A.M.B., and J.C.W. performed experiments; M.T.B., A.M.B., S.C., A.J.N., L.V., T.D.C., and J.C.W. analyzed data; M.T.B., A.M.B., and J.C.W. interpreted results of experiments; M.T.B., A.M.B., and J.C.W. prepared figures; M.T.B., A.M.B., S.C., A.J.N., L.V., T.D.C., and J.C.W. drafted manuscript; M.T.B., A.M.B., S.C., A.J.N., L.V., T.D.C., and J.C.W. edited and revised manuscript; M.T.B., A.M.B., S.C., A.J.N., L.V., T.D.C., and J.C.W. approved final version of manuscript.
REFERENCES
- 1.Adams RJ, McKie VC, Hsu L, Files B, Vichinsky E, Pegelow C, Abboud M, Gallagher D, Kutlar A, Nichols FT, Bonds DR, Brambilla D. Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial Doppler ultrasonography. N Engl J Med 339: 5–11, 1998. [DOI] [PubMed] [Google Scholar]
- 2.Ances BM, Liang CL, Leontiev O, Perthen JE, Fleisher AS, Lansing AE, Buxton RB. Effects of aging on cerebral blood flow, oxygen metabolism, and blood oxygenation level dependent responses to visual stimulation. Hum Brain Mapp 30: 1120–1132, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Attwell D, Laughlin SB. An energy budget for signaling in the grey matter of the brain. J Cereb Blood Flow Metab 21: 1133–1145, 2001. [DOI] [PubMed] [Google Scholar]
- 4.Becklake MR, Griffiths SB, McGregor M, Goldman HI, Schreve JP. Oxygen dissociation curves in sickle cell anemia and in subjects with the sickle cell trait. J Clin Invest 34: 751–755, 1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bernaudin F, Verlhac S, Arnaud C, Kamdem A, Chevret S, Hau I, Coic L, Leveille E, Lemarchand E, Lesprit E, Abadie I, Medejel N, Madhi F, Lemerle S, Biscardi S, Bardakdjian J, Galacteros F, Torres M, Kuentz M, Ferry C, Socie G, Reinert P, Delacourt C. Impact of early transcranial Doppler screening and intensive therapy on cerebral vasculopathy outcome in a newborn sickle cell anemia cohort. Blood 117: 1130–1140, 2011. [DOI] [PubMed] [Google Scholar]
- 6.Borzage M, Bluml S, Seri I. Equations to describe brain size across the continuum of human lifespan. Brain Struct Funct 219: 141–150, 2012. [DOI] [PubMed] [Google Scholar]
- 7.Brown MM, Marshall J. Regulation of cerebral blood-flow in response to changes in blood-viscosity. Lancet 1: 604–609, 1985. [DOI] [PubMed] [Google Scholar]
- 8.Brown MM, Wade JP, Marshall J. Fundamental importance of arterial oxygen content in the regulation of cerebral blood flow in man. Brain 108, Pt 1: 81–93, 1985. [DOI] [PubMed] [Google Scholar]
- 9.Bulte DP, Chiarelli PA, Wise RG, Jezzard P. Cerebral perfusion response to hyperoxia. J Cereb Blood Flow Metab 27: 69–75, 2006. [DOI] [PubMed] [Google Scholar]
- 10.DeBaun MR, Armstrong FD, McKinstry RC, Ware RE. Silent cerebral infarcts: a review on a prevalent and progressive cause of neurologic injury in sickle cell anemia. Blood 119: 4587–4596, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DeBaun MR, Sarnaik SA, Rodeghier MJ, Minniti CP, Howard TH, Iyer RV, Inusa B, Telfer PT, Kirby-Allen M, Quinn CT, Bernaudin F, Airewele G, Woods GM, Panepinto JA, Fuh B, Kwiatkowski JK, King AA, Rhodes MM, Thompson AA, Heiny ME, Redding-Lallinger RC, Kirkham FJ, Sabio H, Gonzalez CE, Saccente SL, Kalinyak KA, Strouse JJ, Fixler JM, Gordon MO, Miller JP, Noetzel MJ, Ichord RN, Casella JF. Associated risk factors for silent cerebral infarcts in sickle cell anemia: low baseline hemoglobin, sex, and relative high systolic blood pressure. Blood 119: 3684–3690, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dekaban A, Sadowsky D. Changes in brain weights during the span of human life: relation of brain weights to body heights and body weights. Ann Neurol 4: 345–356, 1978. [DOI] [PubMed] [Google Scholar]
- 13.Demchenko IT. Regulation of the brain's vascular responses to oxygen. Circ Res 91: 1031–1037, 2002. [DOI] [PubMed] [Google Scholar]
- 14.Dowling MM, Quinn CT, Rogers ZR, Buchanan GR. Acute silent cerebral infarction in children with sickle cell anemia. Ped Blood Cancer 54: 461–464, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gorelick PB. Cerebrovascular disease in African Americans. Stroke 29: 2656–2664, 1998. [DOI] [PubMed] [Google Scholar]
- 16.Harris JJ, Attwell D. The energetics of CNS white matter. J Neurosci 32: 356–371, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hofman MA. Design principles of the human brain: an evolutionary perspective. Evolution of the primate brain: from neuron to behavior. London: Elsevier, 2012. [DOI] [PubMed] [Google Scholar]
- 18.Hurr C, Kim K, Harrison ML, Brothers RM. Attenuated cerebral vasodilatory capacity in response to hypercapnia in college-aged African Americans. Exp Physiol 100: 35–43, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kety SS. Human cerebral blood flow and oxygen consumption as related to aging. Res Publ Assoc Res Nerv Ment Dis 35: 31–45, 1956. [PubMed] [Google Scholar]
- 19a.Kety SS, Schmidt CF. The determination of cerebral blood flow in man by the use of nitrous oxide in low concentrations. Am J Physiol 143: 53–66, 1945. [Google Scholar]
- 20.Lenroot R, Giedd J. Brain development in children and adolescents: insights from anatomical magnetic resonance imaging. Neurosci Biobehav Rev 30: 718–729, 2006. [DOI] [PubMed] [Google Scholar]
- 21.Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation 105: 1135–1143, 2002. [DOI] [PubMed] [Google Scholar]
- 22.Lu H, Xu F, Rodrigue KM, Kennedy KM, Cheng Y, Flicker B, Hebrank AC, Uh J, Park DC. Alterations in cerebral metabolic rate and blood supply across the adult lifespan. Cereb Cortex 21: 1426–1434, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lynch JK. Cerebrovascular disorders in children. Curr Neurol Neurosci Rep 4: 129–138, 2004. [DOI] [PubMed] [Google Scholar]
- 24.Morikawa T, Kajimura M, Nakamura T, Hishiki T, Nakanishi T, Yukutake Y, Nagahata Y, Ishikawa M, Hattori K, Takenouchi T, Takahashi T, Ishii I, Matsubara K, Kabe Y, Uchiyama S, Nagata E, Gadalla MM, Snyder SH, Suematsu M. Hypoxic regulation of the cerebral microcirculation is mediated by a carbon monoxide-sensitive hydrogen sulfide pathway. Proc Natl Acad Sci USA 109: 1293–1298, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nishimura N, Iwasaki KI, Ogawa Y, Shibata S. Oxygen administration, cerebral blood flow velocity, and dynamic cerebral autoregulation. Aviat Space Environ Med 78: 1121–1127, 2007. [DOI] [PubMed] [Google Scholar]
- 26.Ohene-Frempong K, Weiner SJ, Sleeper LA, Miller ST, Embury S, Moohr JW, Wethers DL, Pegelow CH, Gill FM. Cerebrovascular accidents in sickle cell disease: rates and risk factors. Blood 91: 288–294, 1998. [PubMed] [Google Scholar]
- 27.Pantano P, Baron JC, Lebrun-Grandié P, Duquesnoy N, Bousser MG, Comar D. Regional cerebral blood flow and oxygen consumption in human aging. Stroke 15: 635–641, 1984. [DOI] [PubMed] [Google Scholar]
- 28.Prohovnik I, Hurlet-Jensen A, Adams R, De Vivo D, Pavlakis SG. Hemodynamic etiology of elevated flow velocity and stroke in sickle-cell disease. J Cereb Blood Flow Metab 29: 803–810, 2009. [DOI] [PubMed] [Google Scholar]
- 29.Rakic P. A small step for the cell, a giant leap for mankind: a hypothesis of neocortical expansion during evolution. Trends Neurosci 18: 383–388, 1995. [DOI] [PubMed] [Google Scholar]
- 30.Reilly P, Bullock R. Brain Metabolism, and Cerebral Blood Flow. In: Head injury: pathophysiology and management of severe closed injury. New York: Chapman & Hall, p. 90–98. [Google Scholar]
- 31.Shaw TG, Mortel KF, Meyer JS, Rogers RL, Hardenberg J, Cutaia MM. Cerebral blood flow changes in benign aging and cerebrovascular disease. Neurology 34: 855–862, 1984. [DOI] [PubMed] [Google Scholar]
- 32.Sicard KM, Duong TQ. Effects of hypoxia, hyperoxia, and hypercapnia on baseline and stimulus-evoked BOLD, CBF, and CMRO2 in spontaneously breathing animals. Neuroimage 35: 850–858, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sowell ER, Peterson BS, Thompson PM, Welcome SE, Henkenius AL, Toga AW. Mapping cortical change across the human life span. Nat Neurosci 6: 309–315, 2003. [DOI] [PubMed] [Google Scholar]
- 34.Weibel ER. The pathway for oxygen: structure and function in the mammalian respiratory system. Cambridge, MA: Harvard University Press, 1984. [Google Scholar]
- 35.Xu F, Liu P, Pascual JM, Xiao G, Lu H. Effect of hypoxia and hyperoxia on cerebral blood flow, blood oxygenation, and oxidative metabolism. J Cereb Blood Flow Metab 32: 1909–1918, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zaharchuk G, Martin AJ, Dillon WP. Noninvasive imaging of quantitative cerebral blood flow changes during 100% oxygen inhalation using arterial spin-labeling MR imaging. Am J Neuroradiol 29: 663–667, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]