

Abstract
Transforming growth factor-β1 (TGF-β1) is established to be involved in the pathogenesis of diabetic nephropathy. The diabetic milieu enhances oxidative stress and induces the expression of TGF-β1. TGF-β1 promotes cell hypertrophy and extracellular matrix accumulation in the mesangium, which decreases glomerular filtration rate and leads to chronic renal failure. Recently, TGF-β1 has been demonstrated to regulate urinary albumin excretion by both increasing glomerular permeability and decreasing reabsorption in the proximal tubules. TGF-β1 also increases urinary excretion of water, electrolytes and glucose by suppressing tubular reabsorption in both normal and diabetic conditions. Although TGF-β1 exerts hypertrophic and fibrogenic effects in diabetic nephropathy, whether suppression of the function of TGF-β1 can be an option to prevent or treat the complication is still controversial. This is partly because adrenal production of mineralocorticoids could be augmented by the suppression of TGF-β1. However, differentiating the molecular mechanisms for glomerulosclerosis from those for the suppression of the effects of mineralocorticoids by TGF-β1 may assist in developing novel therapeutic strategies for diabetic nephropathy. In this review, we discuss recent findings on the role of TGF-β1 in diabetic nephropathy.
Keywords: nephrin, megalin, podocyte, proximal tubule, sodium-glucose cotransporter
transforming growth factor-β1 (TGF-β1) is a multifunctional cytokine that controls numerous biological processes including immunity (92), differentiation (29), tumor suppression (2), tumor metastasis (107), senescence (68), migration (17), wound healing (105), apoptosis (108), cell division (2), adipogenesis (76), and osteogenesis (27). In addition, the renal expression of TGF-β1 mRNA and protein are increased in patients with diabetes mellitus (110), and it enhances the synthesis and cross-linking of extracellular matrix (ECM) (3, 81).
In the US population, the most frequent cause of chronic renal failure is diabetes mellitus (∼44%). No more than 20∼40% of diabetic subjects, however, develop nephropathy despite having similar blood glucose levels, suggesting the presence of genetic predisposition to the complication. The T869C polymorphism in the human TGF-β1 gene, leading to the L10P variant of the coding protein, is associated with an increased risk of diabetic nephropathy (71, 77, 109), but how the functional change in TGF-β1 protein increases the incidence of diabetic nephropathy remains elusive.
In incipient diabetic nephropathy, the glomerular filtration rate (GFR) increases by 25∼50% (glomerular hyperfiltration) (1) by the reduction of tubuloglomerular feedback, which is caused by the increase in sodium/glucose reabsorption and hence the reduced sodium delivery in the macula densa (101). However, in the late stage of diabetic nephropathy, the GFR eventually declines as the number of functional nephrons decreases, which leads to the insufficiency of renal excretory function. The decline in GFR is associated with the expansion of mesangial area, which is caused by cell proliferation and accumulation of ECM.
Although the increase in urinary excretion of albumin is considered to be the early indicator of diabetic nephropathy, the mechanisms whereby hyperalbuminuria occurs in diabetic subjects are not fully understood. However, impairment of barrier function at the slit diaphragm between podocytes and the decrease in reabsorption of filtered albumin by proximal tubules have recently been identified as pivotal in the development of diabetic albuminuria.
Mice having the heterozygous Akita diabetogenic mutation expressing ∼10, 50, 100, 200, and 300% normal Tgfb1 mRNA levels have recently been generated. In these mice, the severity of glomerulosclerosis and albuminuria is enhanced as the expression of TGF-β1 is increased, despite blood pressure being negatively correlated with TGF-β1 expression (31). It is noteworthy that the diabetic mice with 10% normal TGF-β1 expression exhibit near-normal glomerular histology, GFR, and urinary albumin excretion, despite the presence of primary aldosteronism and hypertension. Additionally, the markedly increased urinary excretion of water, electrolytes, and glucose in Akita type 1 diabetes was diminished to levels comparable to those in nondiabetic wild-type mice by the genetic insufficiency of TGF-β1.
These previous findings suggest that TGF-β1 plays a pathophysiological role not only in promoting glomerulosclerosis, interstitial fibrosis, and the decline in GFR but also in increasing urinary excretion of albumin, water, electrolytes, and glucose in diabetes. In the current review, we will discuss the mechanisms whereby TGF-β1 causes renal morphological and functional changes in diabetes mellitus. As such, therapeutic strategies targeting TGF-β signaling may be developed to effectively treat chronic excretory insufficiency in patients with diabetes.
TGF-β1 Facilitates Accumulation of ECM in Diabetic Nephropathy
Previous studies show that neutralizing anti-TGF-β antibodies prevented glomerulosclerosis and interstitial fibrosis, and reduced expression of ECM genes including fibronectin and type IV collagen in mice with type 1 and type 2 diabetes (90, 116), suggesting that TGF-β signaling plays a critical role in ECM accumulation in diabetic nephropathy. Both canonical and alternative signaling of TGF-β1 have been suggested to be involved in the development of diabetic nephropathy (15, 19).
Since glucose availability is impaired in diabetes, the metabolic shift occurs from glycolysis toward oxidative phosphorylation by using more fatty acids as an energy source (Fig. 1). As a result, the mitochondrial electron transport chain increases superoxide production and stimulates three major pathways of hyperglycemic damage (activation of the polyol pathway, advanced glycation end products generation, and protein kinase C activation) (72). Furthermore, high glucose induces reactive oxygen species (ROS) via NAD(P)H oxidase, mitochondrial electron transport chain, and protein kinase C (56).
Fig. 1.
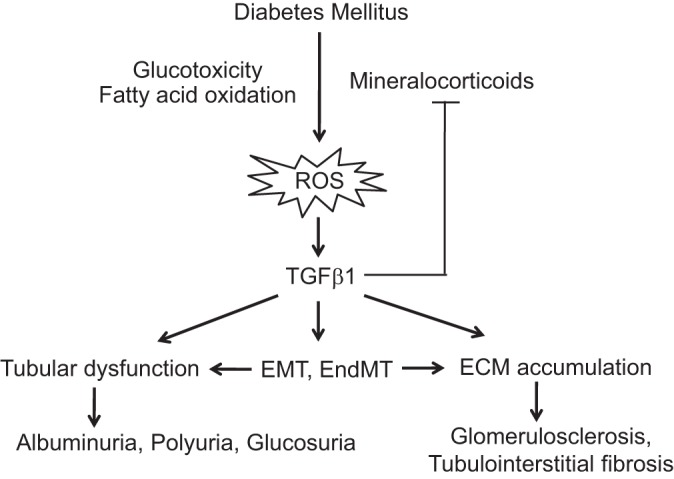
Diagram depicting the proposed effects of transforming growth factor-β1 (TGF-β1) on the features of diabetic nephropathy. ROS, reactive oxygen species; EMT, epithelial-mesenchymal transition; EndMT, endothelial-mesenchymal transition.
Hydrogen peroxide upregulates TGF-β1 and its downstream ECM-related genes, including integrin-linked kinase, fibronectin, and collagen types I, III, and IV (25, 48). In the reverse direction, pharmacological inhibition of different ROS sources including NAD(P)H oxidase and mitochondrial respiratory chain decreases the transcription of TGF-β1 via reduced activity of activated protein-1 (26), indicating that ROS-induced enhancement of the transcription of TGF-β1 may at least in part account for the increase in TGF-β1 expression in diabetes.
Previous studies have demonstrated that TGF-β1 stimulates the transcription of the components of ECM, including collagen, fibronectin, and laminin (5, 41). It has been demonstrated that TGF-β1 also increases the expression of lysyl oxidase, which forms cross-links between collagen and elastin fibers that stabilizes their structure (3). TGF-β1 also stimulates the transcription of procollagen lysyl hydroxylase 2, which hydroxylates lysyl residues of collagen telopeptides and is essential for collagen cross-linking (24). In addition, TGF-β1 augments the expression of plasminogen activator inhibitor-1 (39) and tissue inhibitor of metalloproteinases-1 (100), both of which inhibit the activity of ECM-degradating matrix metalloproteinases.
The effect of TGF-β1 on the expression of matrix metalloproteinase 9 (MMP9) is controversial. TGF-β1 increases the expression of MMP9 in cultured cells and in isolated, perfused kidneys (13, 82), whereas transgenic overexpression of TGF-β1 decreases MMP9 expression in mice (100, 112). The exact reason for this discrepancy is unclear, but the suppressive effect of TGF-β1 on mineralocorticoid production may be related. Previous studies have demonstrated that aldosterone increases the transcription of MMP9 via phosphoinositide 3-kinase (PI3K), p38 MAPK, ERK (23), and oxidized Ca2+/calmodulin-dependent protein kinase II (oxCaMKII) (32), suggesting that TGF-β1 decreases tissue MMP9 expression via suppressing circulating aldosterone produced in adrenocortical cells.
Recently, TGF-β type 1 receptor antagonists and aldosterone have also been found to increase the expression of a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS)-1, which also degrades ECM (16, 54).
Thus TGF-β1 facilitates ECM accumulation by increasing production and stabilization of ECM and by suppressing its degradation, which plays a causative role in developing glomerulosclerosis and interstitial fibrosis in diabetic nephropathy.
TGF-β1 Facilitates Dedifferentiation of Renal Cells
Previous studies suggest that TGF-β1 induces epithelial-mesenchymal transition/transdifferentiation (EMT), a dedifferentiation process by which an epithelial cell is transformed into a myofibroblast, a type of mesenchymal stem cell (96). In the kidney, tubular cells are normally lining the tubular basement membrane and have a highly polarized structure for efficient reabsorption of solutes and proteins from urinary space into peritubular capillaries, but through the EMT process they lose cell polarity and gain migratory and fibrogenic properties of myofibroblasts (35). Indeed, TGF-β1 has been demonstrated to induce EMT in proximal tubules (115), collecting duct cells (43), glomerular podocytes (33, 88), and glomerular parietal epithelial cells (93).
It has been demonstrated that hypoxia-inducible factor 1α (HIF-1α) enhances EMT in murine proximal tubular epithelial cells (34). Since the effect of hypoxia on EMT is only partially inhibited by anti-TGF-β antibodies, TGF-β signaling is unlikely to be the only mechanism to induce EMT (34). Prolyl hydroxylase domain-containing proteins (PHDs) hydroxylate HIF-1α. Hydroxylated HIF-1α binds to von Hippel-Lindau tumor suppressor protein and is subject to polyubiquitination and degradation by proteasomes (44). TGF-β1 has been shown to decrease both mRNA and protein levels of PHD2 and hence stabilize HIF-1α protein (66). Overexpression of the HIF-1α PHD2 transgene, which enhances degradation of HIF-1α, completely prevents TGF-β1-induced changes in the expression of HIF-1α and marker genes for EMT, suggesting that HIF-1α mediates TGF-β1-induced EMT (30).
It has also been shown that TGF-β1 activates the Jagged/Notch signaling pathway and that activated Notch triggers EMT (74). These results suggest that TGF-β1-induced EMT is also mediated at least in part by Jagged/Notch signaling.
Previous studies suggest that endothelial-mesenchymal transition/transdifferentiation (EndMT), a process by which endothelial cells are transformed into myofibroblasts, also contributes to renal fibrosis in streptozotocin (STZ)-induced type 1 diabetic mice (113). TGF-β1 has been demonstrated to induce EndMT (114). Overexpression of Snail induced EndMT, but suppression of Snail expression abrogated TGF-β2-induced EndMT in mouse endothelial cells, suggesting that Snail mediates TGF-β signaling-induced EndMT (50).
Thus TGF-β1 may be involved in the process of EMT and EndMT in diabetic nephropathy, which decreases the number of functional cells and increases the accumulation of ECM, leading to impairment of renal excretory and reabsorptive function. However, controversy remains over the contribution of each of these processes to renal fibrosis (18).
TGF-β1 Enhances Diabetic Albuminuria Via Both Glomerular and Postglomerular Mechanisms
A number of previous studies have demonstrated that TGF-β1 promotes the development of diabetic albuminuria. Several transgenic animal models overexpressing TGF-β1 exhibited increased urinary excretion of albumin (51, 100). Chronic treatment with the soluble human TGF-β type II receptor reduced albuminuria without changing blood glucose levels in STZ-induced diabetic mice (84). However, several studies did not show a significant effect of the suppression of TGF-β signaling on albuminuria in diabetic animals. For instance, chronic peritoneal infusion of neutralizing anti-TGF-β antibodies did not alter urinary albumin excretion in db/db type 2 diabetic mice (116). Furthermore, genetic deficiency of small mothers against decapentaplegic homolog (Smad) 3, an intracellular signaling molecule which is downstream of TGF-β receptors, did not affect albuminuria in STZ-induced diabetic mice (104).
Microalbuminuria is low-grade albuminuria, which ranges from 30 to 300 mg/day in humans, and is widely established as an early indicator of diabetic nephropathy. However, whether the elevated urinary excretion of albumin plays a causative role in diabetic tubular damage or is merely a consequence of diabetic nephropathy is still controversial (45).
The paradigm that the sieving of albumin at the slit diaphragm is the predominant mechanism in the prevention of albuminuria has been questionable because of the lack of a clearing mechanism of the sieved albumin (91). Indeed, the concentration of albumin at Bowman's capsule estimated with two-photon microscopy has been demonstrated to be within the nephrotic range (1–10 mg/ml) even in normal conditions (86). In contrast, others using a micropuncture technique have reported less concentrations of albumin (20–30 μg/ml) than the nephrotic range in Bowman's capsule (53, 97). Nevertheless, these results indicate that both incomplete glomerular barrier and subsequent tubular reabsorption/degradation are important in preventing albuminuria (10).
Nephrin is abundantly expressed in the slit diaphragm and considered to exert a barrier function against glomerular filtration of albumin (37, 106). The mutations in the nephrin gene lead to urinary overexcretion of high-molecular-weight proteins and albumin and congenital nephrotic syndromes (47, 79). However, tubular handling of protein is important as well. Megalin and cubilin in the brush border of proximal tubular cells contribute to the endocytosis of albumin and low-molecular-weight proteins (9, 22, 58, 67, 111).
The expression of nephrin is decreased in diabetes (12). TGF-β1 decreases renal mRNA levels of nephrin (31) and increases the permeability of albumin in the podocyte (55). Additionally, the presence of diabetes attenuates the expression of megalin (98). TGF-β1 decreases renal megalin mRNA levels in Akita type 1 diabetic mice (31), which might attenuate the albumin endocytosis mediated by megalin (22).
Therefore, it is likely that diabetic albuminuria is the result of a double insult in the kidney's ability to process albumin, increased permeability at the slit diaphragm, and reduced tubular reabsorption. However, in what proportion diabetic albuminuria is caused by tubular dysfunction and/or podocyte dysfunction is controversial (75, 85, 98, 99).
Albuminuria in Akita mice expressing 10% TGF-β1 mRNA was markedly reduced compared with that in Akita mice and was comparable to nondiabetic wild-type mice, despite similar glucose levels and increased systolic blood pressure in the 10% hypomorphic Akita mice (31). In the reverse direction, genetic overexpression of TGF-β1 markedly increased urinary albumin excretion despite no significant changes in blood glucose or blood pressure (31). The amount of urinary albumin excretion in the 10% hypomorphic Akita mice was found to be increased by 20-fold by overexpressing Tgfb1 in proximal tubular cells, but only by 4-fold by overexpressing Tgfb1 in podocytes.
Thus TGF-β1 plays a critical role in developing hyperalbuminuria. These findings demonstrate that attenuated tubular reabsorption of albumin appears to be the predominant factor leading to diabetic albuminuria, rather than enhanced glomerular filtration.
TGF-β1 Inhibits Production and Function of Mineralocorticoids
Plasma levels of aldosterone are increased in diabetic patients (36), and aldosterone has been shown to increase ROS by stimulating NAD(P)H oxidase (83). These findings suggest that the increased action of mineralocorticoids partly contributes to the development of diabetic complications. Indeed, chronic administration of spironolactone, a mineralocorticoid receptor antagonist, decreased albuminuria in patients with diabetes (89).
It has been shown that TGF-β1 potently inhibits the synthesis of aldosterone in vitro (28, 38, 61). TGF-β1 also suppresses the production of androstenedione, corticosterone, and cortisol (38). This broad suppressive effect of TGF-β1 on steroidogenesis may be partly because TGF-β1 decreases the expression of cytochrome P-450 side-chain cleavage (P450scc), adrenodoxin reductase, and adrenodoxin, which controls the initial step of steroidogenesis (69). The mRNA levels for steroidogenic acute regulatory protein (StAR), which supplies cholesterol to P450scc by transporting it from the outer to the inner mitochondrial membrane, is also decreased by TGF-β1 (4). In bovine adrenocortical cells, TGF-β1 also reduced the transcript levels of hydroxyl-delta-5-steroid dehydrogenase, 3β- and steroid delta-isomerase 1 (Hsd3b1), and steroid 17-α-monooxygenase (Cyp17a1) (60).
It has recently been reported that TGF-β1 decreases cortisol and 11-hydroxyandrostenedione production induced by forskolin by 85% and production of aldosterone induced by angiotensin II by 80%. The activity of steroid 11β-hydroxylase (Cyp11b1) and the transcript levels for Cyp11b1 induced by forskolin, as well as the activity of aldosterone synthase (Cyp11b2) and the transcript levels for Cyp11b2 induced by angiotensin II, were strongly inhibited by TGF-β1 in the NCI-H295R cell line (61). TGF-β1 suppressed Cyp11b1 promoter activity, but the Smads-binding sequence was not responsible for the transcriptional inhibition. This result suggests that TGF-β1 indirectly represses Cyp11b1 promoter activity (61). Intriguingly, the mRNA levels of steroidogenic factor 1, the genetic deficiency of which causes the absence of gonadal and adrenal steroidogenic cells (64), were inhibited by TGF-β1 (59). Steroidogenic factor 1 binds to a shared promoter element of steroidogenic enzymes, including Cyp11b1, Cyp11b2, steroid 21-hydroxylase (Cyp21a1), and Cyp11a1 (80), and enhances their expression (52). Thus numerous steroidogenic steps that are upstream of the synthesis of aldosterone are suppressed by TGF-β1.
Indeed, it has been shown that plasma levels of aldosterone and the adrenal expression of several steroidogenic enzymes involved in the production of aldosterone, including Cyp11b1, Cyp11b2, Star, Hsd3b1, and Cyp21a1, are decreased as TGF-β1 expression is increased in mice having graded expression of TGF-β1 (46). The suppressive effect of overexpression of TGF-β1 on plasma levels of aldosterone was also observed in Akita diabetic mice (31).
Although TGF-β1 exerts a suppressive effect on adrenocortical production of aldosterone, the stimulatory effect of aldosterone on the activity of epithelial sodium channel (ENaC) is also inhibited by TGF-β1. Aldosterone expands the volume of extracellular fluid by increasing ENaC activity. ENaC is expressed in the “aldosterone-sensitive distal nephron,” which is composed of collecting ducts, connecting tubules, and late distal convoluted tubules (21, 62). The aldosterone-sensitive distal nephron also expresses two other components required for sodium reabsorption in response to aldosterone: Na+-K+-ATPase and mineralocorticoid receptors.
Although aldosterone is the major regulator of ENaC activity, TGF-β1 also regulates ENaC activity either in combination with aldosterone or independently (6, 62). The aldosterone-induced increase in sodium transport via the ENaC is inhibited by TGF-β1 (40). TGF-β1 enhances β-ENaC internalization, which facilitates destabilization of the cell surface ENaC complex (78). It has also been found that the total activity, functional expression, and open probability of ENaC in mice underexpressing TGF-β1 are all greater than those in wild-type (46).
Protease nexin-1 (PN-1) has recently been found to inhibit prostasin, which augments the activity of ENaC(8, 103). Intriguingly, aldosterone and TGF-β1 reciprocally regulate the expressions of PN-1 and prostasin. Thus, prostasin expression is increased by aldosterone and decreased by TGF-β1, whereas PN-1 expression is decreased by aldosterone and increased by TGF-β1 (103).
Aldosterone and TGF-β1 differentially control the reabsorption of sodium in proximal tubular cells. Na+-K+-ATPase α1-subunits and mineralocorticoid receptors are expressed in the proximal tubules of the mice, rats, and humans (87). The activity of sodium/proton exchange is stimulated, and the expression of sodium/proton exchanger 3 on the cell surface is increased by aldosterone in cultured proximal tubular cells (14). By contrast, Na+-K+-ATPase α- and β-subunit expression and Na+-K+-ATPase activity are dose dependently decreased by TGF-β1 (73, 94).
Thus TGF-β1 has been suggested to inhibit both aldosterone production in the adrenal cortex and aldosterone actions in the kidney. Although TGF-β1 has been shown to directly increase ROS in cultured cells (95), it counteracts production and action of aldosterone, which enhance ROS generation and tissue fibrosis (7, 65). Inhibition of aldosterone by TGF-β1 may be a negative feedback mechanism in diabetic nephropathy.
TGF-β1 Suppresses Reabsorption of Water and Glucose in Proximal Tubules
Notably, the urine volume in TGF-β1 hypomorphic Akita diabetic mice was comparable to that in nondiabetic wild-type mice (31). In addition, the urinary output of glucose in diabetes was substantially decreased by genetic insufficiency of TGF-β1, despite unchanged plasma concentrations of insulin and glucose. The marked reduction in urinary water and glucose excretion by genetic insufficiency of TGF-β1 was also observed in their nondiabetic counterparts (31, 46). In addition, genetic disruption of Smad3, which is the intracellular signaling molecule downstream of TGF-β receptors, also reduced urine volume despite little change in plasma levels of glucose in STZ-induced diabetic mice (19). These findings indicate that TGF-β1 mediates hyperglycemia-induced polyuria and glucosuria, which has been attributed to osmotic diuresis.
The marked decrease in urinary water excretion in TGF-β1 hypomorphic mice cannot be explained merely by enhanced aldosterone function, because chronically high aldosterone levels have been demonstrated to increase urine volume due to impaired urine concentration ability (11). The diminished diuresis of the 10% hypomorphs was found to be fully reversed by overexpressing Tgfb1 in the proximal tubule (31), and partially restored by overexpressing Tgfb1 in the collecting duct cells (our unpublished observations), indicating that TGF-β1 suppresses the reabsorption of water in both proximal tubular cells and collecting ducts.
The finding that glucosuria and polyuria are both absent in the TGF-β1 hypomorphic diabetic mice may be related to the previous observation that the sodium/glucose cotransporters (SGLTs), which the proximal tubule highly expresses, cotransport each sugar molecule with >200 water molecules (63). In concordance with this inference, the urinary output of glucose was partially restored in Akita mice with the expression of Tgfb1 augmented in proximal tubules (31). Furthermore, high glucose, hydrogen peroxide, and TGF-β1 have been shown to decrease the SGLT activity measured with α-methyl-d-glucopyranoside, a metabolically inert analog of d-glucose, and the expression of SGLT 1 and 2 and sodium-hydrogen exchanger (NHE) 1 and 3 in primary cultured rabbit proximal tubule cells, which is associated with genetic expression profile of EMT (57).
Although the chronic efficacy of the inhibitors of SGLT 1 and 2, which are being used for the purpose of controlling blood glucose, on diabetic nephropathy is still controversial (20, 102), several studies suggest that SGLT inhibitors prevent cardiovascular and renal complications via reducing blood glucose levels (49, 70). Thus the facts that both diabetes by itself and TGF-β1 decrease the reabsorption of glucose in proximal tubular cells may be the compensatory mechanism preventing further tissue damage.
Conclusion
A number of features of diabetic nephropathy were absent in the TGF-β1 hypomorphic Akita mice, indicating that decreasing the expression of TGF-β1 may have therapeutic benefits in diabetics. However, the globally low expression of TGF-β1 resulted in primary aldosteronism (31, 46). Consequently, lowering the expression of TGF-β1 throughout the body could be deleterious in patients with diabetes. Nevertheless, these problems of TGF-β1 insufficiency might be circumvented if kidney-specific reduction of the expression of TGF-β1 were achieved.
Experimental results with tissue-specific TGF-β1 overexpression suggest that suppressing TGF-β1 expression in the proximal tubule is more likely to decrease albuminuria and interstitial fibrosis than to prevent the decrease in renal excretory function. In contrast, reducing expression of TGF-β1 in the podocyte may be more effective in avoiding the GFR decrease than in decreasing albuminuria and interstitial fibrosis. Thus manipulations which lead to decreased TGF-β1 expression in the podocyte may be useful for preventing/treating the decline in renal function in diabetic nephropathy.
Further studies are needed to unravel the mechanisms for the TGF-β1-induced accumulation of ECM and for the suppression of the effects of mineralocorticoids by TGF-β1, and this understanding might be useful in developing new therapeutic options for preventing and/or treating diabetic nephropathy.
GRANTS
This work was supported by National Institutes of Health Grants HL49277, HL70523, HL71266, and DK34987 and Career Development Award 2006-102 from the Juvenile Diabetes Research Foundation.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: A.S.C. drafted manuscript; C.K.H. prepared figures; O.S. and M.K. edited and revised manuscript; O.S. and M.K. approved final version of manuscript.
REFERENCES
- 1.Bank N. Mechanisms of diabetic hyperfiltration. Kidney Int 40: 792–807, 1991. [DOI] [PubMed] [Google Scholar]
- 2.Barcellos-Hoff MH, Cucinotta FA. New tricks for an old fox: Impact of TGFbeta on the DNA damage response and genomic stability. Sci Signal 7: re5, 2014. [DOI] [PubMed] [Google Scholar]
- 3.Boak AM, Roy R, Berk J, Taylor L, Polgar P, Goldstein RH, Kagan HM. Regulation of lysyl oxidase expression in lung fibroblasts by transforming growth factor-beta 1 and prostaglandin E2. Am J Respir Cell Mol Biol 11: 751–755, 1994. [DOI] [PubMed] [Google Scholar]
- 4.Brand C, Cherradi N, Defaye G, Chinn A, Chambaz EM, Feige JJ, Bailly S. Transforming growth factor beta1 decreases cholesterol supply to mitochondria via repression of steroidogenic acute regulatory protein expression. J Biol Chem 273: 6410–6416, 1998. [DOI] [PubMed] [Google Scholar]
- 5.Chakrabarty S, Tobon A, Varani J, Brattain MG. Induction of carcinoembryonic antigen secretion and modulation of protein secretion/expression and fibronectin/laminin expression in human colon carcinoma cells by transforming growth factor-beta. Cancer Res 48: 4059–4064, 1988. [PubMed] [Google Scholar]
- 6.Chang CT, Hung CC, Chen YC, Yen TH, Wu MS, Yang CW, Phillips A, Tian YC. Transforming growth factor-beta1 decreases epithelial sodium channel functionality in renal collecting duct cells via a Smad4-dependent pathway. Nephrol Dial Transplant 23: 1126–1134, 2008. [DOI] [PubMed] [Google Scholar]
- 7.Chen D, Chen Z, Park C, Centrella M, McCarthy T, Chen L, Al-Omari A, Moeckel GW. Aldosterone stimulates fibronectin synthesis in renal fibroblasts through mineralocorticoid receptor-dependent and independent mechanisms. Gene 531: 23–30, 2013. [DOI] [PubMed] [Google Scholar]
- 8.Chen LM, Zhang X, Chai KX. Regulation of prostasin expression and function in the prostate. Prostate 59: 1–12, 2004. [DOI] [PubMed] [Google Scholar]
- 9.Christensen EI, Birn H. Megalin and cubilin: synergistic endocytic receptors in renal proximal tubule. Am J Physiol Renal Physiol 280: F562–F573, 2001. [DOI] [PubMed] [Google Scholar]
- 10.Comper WD, Hilliard LM, Nikolic-Paterson DJ, Russo LM. Disease-dependent mechanisms of albuminuria. Am J Physiol Renal Physiol 295: F1589–F1600, 2008. [DOI] [PubMed] [Google Scholar]
- 11.Conte G, Dal Canton A, Balletta M, Meccariello S, Sabbatini M, Andreucci VE. Mechanism of impaired urinary concentrating ability in normokalemic primary aldosteronism. Clin Nephrol 22: 91–96, 1984. [PubMed] [Google Scholar]
- 12.Doublier S, Salvidio G, Lupia E, Ruotsalainen V, Verzola D, Deferrari G, Camussi G. Nephrin expression is reduced in human diabetic nephropathy: evidence for a distinct role for glycated albumin and angiotensin II. Diabetes 52: 1023–1030, 2003. [DOI] [PubMed] [Google Scholar]
- 13.Douthwaite JA, Johnson TS, Haylor JL, Watson P, El Nahas AM. Effects of transforming growth factor-beta1 on renal extracellular matrix components and their regulating proteins. J Am Soc Nephrol 10: 2109–2119, 1999. [DOI] [PubMed] [Google Scholar]
- 14.Drumm K, Kress TR, Gassner B, Krug AW, Gekle M. Aldosterone stimulates activity and surface expression of NHE3 in human primary proximal tubule epithelial cells (RPTEC). Cell Physiol Biochem 17: 21–28, 2006. [DOI] [PubMed] [Google Scholar]
- 15.Fan Y, Li X, Xiao W, Fu J, Harris RC, Lindenmeyer M, Cohen CD, Guillot N, Baron MH, Wang N, Lee K, He JC, Schlondorff D, Chuang PY. BAMBI elimination enhances alternative TGF-beta signaling and glomerular dysfunction in diabetic mice. Diabetes 64: 2220–2233, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fejes-Toth G, Naray-Fejes-Toth A. Early aldosterone-regulated genes in cardiomyocytes: clues to cardiac remodeling? Endocrinology 148: 1502–1510, 2007. [DOI] [PubMed] [Google Scholar]
- 17.Feng YF, Yuan F, Guo H, Wu WZ. TGF-beta1 enhances SDF-1-induced migration and tube formation of choroid-retinal endothelial cells by up-regulating CXCR4 and CXCR7 expression. Mol Cell Biochem 397: 131–138, 2014. [DOI] [PubMed] [Google Scholar]
- 18.Fragiadaki M, Mason RM. Epithelial-mesenchymal transition in renal fibrosis - evidence for and against. Int J Exp Pathol 92: 143–150, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fujimoto M, Maezawa Y, Yokote K, Joh K, Kobayashi K, Kawamura H, Nishimura M, Roberts AB, Saito Y, Mori S. Mice lacking Smad3 are protected against streptozotocin-induced diabetic glomerulopathy. Biochem Biophys Res Commun 305: 1002–1007, 2003. [DOI] [PubMed] [Google Scholar]
- 20.Gangadharan Komala M, Gross S, Mudaliar H, Huang C, Pegg K, Mather A, Shen S, Pollock CA, Panchapakesan U. Inhibition of kidney proximal tubular glucose reabsorption does not prevent against diabetic nephropathy in type 1 diabetic eNOS knockout mice. PLoS One 9: e108994, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garty H, Palmer LG. Epithelial sodium channels: function, structure, and regulation. Physiol Rev 77: 359–396, 1997. [DOI] [PubMed] [Google Scholar]
- 22.Gekle M, Knaus P, Nielsen R, Mildenberger S, Freudinger R, Wohlfarth V, Sauvant C, Christensen EI. Transforming growth factor-beta1 reduces megalin- and cubilin-mediated endocytosis of albumin in proximal-tubule-derived opossum kidney cells. J Physiol 552: 471–481, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gilet A, Zou F, Boumenir M, Frippiat JP, Thornton SN, Lacolley P, Ropars A. Aldosterone up-regulates MMP-9 and MMP-9/NGAL expression in human neutrophils through p38, ERK1/2 and PI3K pathways. Exp Cell Res 331: 152–163, 2015. [DOI] [PubMed] [Google Scholar]
- 24.Gjaltema RA, de Rond S, Rots MG, Bank RA. Procollagen lysyl hydroxylase 2 expression is regulated by an alternative downstream transforming growth factor beta-1 activation mechanism. J Biol Chem 290: 28465–28476, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gonzalez-Ramos M, de Frutos S, Griera M, Luengo A, Olmos G, Rodriguez-Puyol D, Calleros L, Rodriguez-Puyol M. Integrin-linked kinase mediates the hydrogen peroxide-dependent transforming growth factor-beta1 up-regulation. Free Radic Biol Med 61: 416–427, 2013. [DOI] [PubMed] [Google Scholar]
- 26.Gonzalez-Ramos M, Mora I, de Frutos S, Garesse R, Rodriguez-Puyol M, Olmos G, Rodriguez-Puyol D. Intracellular redox equilibrium is essential for the constitutive expression of AP-1 dependent genes in resting cells: studies on TGF-beta1 regulation. Int J Biochem Cell Biol 44: 963–971, 2012. [DOI] [PubMed] [Google Scholar]
- 27.Grafe I, Yang T, Alexander S, Homan EP, Lietman C, Jiang MM, Bertin T, Munivez E, Chen Y, Dawson B, Ishikawa Y, Weis MA, Sampath TK, Ambrose C, Eyre D, Bachinger HP, Lee B. Excessive transforming growth factor-beta signaling is a common mechanism in osteogenesis imperfecta. Nat Med 20: 670–675, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gupta P, Franco-Saenz R, Gentry LE, Mulrow PJ. Transforming growth factor-beta 1 inhibits aldosterone and stimulates adrenal renin in cultured bovine zona glomerulosa cells. Endocrinology 131: 631–636, 1992. [DOI] [PubMed] [Google Scholar]
- 29.Han A, Zhao H, Li J, Pelikan R, Chai Y. ALK5-mediated transforming growth factor beta signaling in neural crest cells controls craniofacial muscle development via tissue-tissue interactions. Mol Cell Biol 34: 3120–3131, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Han WQ, Zhu Q, Hu J, Li PL, Zhang F, Li N. Hypoxia-inducible factor prolyl-hydroxylase-2 mediates transforming growth factor beta 1-induced epithelial-mesenchymal transition in renal tubular cells. Biochim Biophys Acta 1833: 1454–1462, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hathaway CK, Gasim AM, Grant R, Chang AS, Kim HS, Madden VJ, Bagnell CR Jr, Jennette JC, Smithies O, Kakoki M. Low TGFbeta1 expression prevents and high expression exacerbates diabetic nephropathy in mice. Proc Natl Acad Sci USA 112: 5815–5820, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.He BJ, Joiner ML, Singh MV, Luczak ED, Swaminathan PD, Koval OM, Kutschke W, Allamargot C, Yang J, Guan X, Zimmerman K, Grumbach IM, Weiss RM, Spitz DR, Sigmund CD, Blankesteijn WM, Heymans S, Mohler PJ, Anderson ME. Oxidation of CaMKII determines the cardiotoxic effects of aldosterone. Nat Med 17: 1610–1618, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Herman-Edelstein M, Thomas MC, Thallas-Bonke V, Saleem M, Cooper ME, Kantharidis P. Dedifferentiation of immortalized human podocytes in response to transforming growth factor-beta: a model for diabetic podocytopathy. Diabetes 60: 1779–1788, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Higgins DF, Kimura K, Bernhardt WM, Shrimanker N, Akai Y, Hohenstein B, Saito Y, Johnson RS, Kretzler M, Cohen CD, Eckardt KU, Iwano M, Haase VH. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J Clin Invest 117: 3810–3820, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hills CE, Squires PE. The role of TGF-beta and epithelial-to mesenchymal transition in diabetic nephropathy. Cytokine Growth Factor Rev 22: 131–139, 2011. [DOI] [PubMed] [Google Scholar]
- 36.Hollenberg NK, Stevanovic R, Agarwal A, Lansang MC, Price DA, Laffel LM, Williams GH, Fisher ND. Plasma aldosterone concentration in the patient with diabetes mellitus. Kidney Int 65: 1435–1439, 2004. [DOI] [PubMed] [Google Scholar]
- 37.Holzman LB, St. John PL, Kovari IA, Verma R, Holthofer H, Abrahamson DR. Nephrin localizes to the slit pore of the glomerular epithelial cell. Kidney Int 56: 1481–1491, 1999. [DOI] [PubMed] [Google Scholar]
- 38.Hotta M, Baird A. Differential effects of transforming growth factor type beta on the growth and function of adrenocortical cells in vitro. Proc Natl Acad Sci USA 83: 7795–7799, 1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang W, Xu C, Kahng KW, Noble NA, Border WA, Huang Y. Aldosterone and TGF-β1 synergistically increase PAI-1 and decrease matrix degradation in rat renal mesangial and fibroblast cells. Am J Physiol Renal Physiol 294: F1287–F1295, 2008. [DOI] [PubMed] [Google Scholar]
- 40.Husted RF, Sigmund RD, Stokes JB. Mechanisms of inactivation of the action of aldosterone on collecting duct by TGF-β. Am J Physiol Renal Physiol 278: F425–F433, 2000. [DOI] [PubMed] [Google Scholar]
- 41.Ignotz RA, Massague J. Transforming growth factor-beta stimulates the expression of fibronectin and collagen and their incorporation into the extracellular matrix. J Biol Chem 261: 4337–4345, 1986. [PubMed] [Google Scholar]
- 42.Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG Jr. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 292: 464–468, 2001. [DOI] [PubMed] [Google Scholar]
- 43.Ivanova L, Butt MJ, Matsell DG. Mesenchymal transition in kidney collecting duct epithelial cells. Am J Physiol Renal Physiol 294: F1238–F1248, 2008. [DOI] [PubMed] [Google Scholar]
- 44.Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, von Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, Maxwell PH, Pugh CW, Ratcliffe PJ. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 292: 468–472, 2001. [DOI] [PubMed] [Google Scholar]
- 45.Jheng HF, Tsai PJ, Chuang YL, Shen YT, Tai TA, Chen WC, Chou CK, Ho LC, Tang MJ, Lai KT, Sung JM, Tsai YS. Albumin stimulates renal tubular inflammation through an HSP70-TLR4 axis in mice with early diabetic nephropathy. Dis Model Mech 8: 1311–1321, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kakoki M, Pochynyuk OM, Hathaway CM, Tomita H, Hagaman JR, Kim HS, Zaika OL, Mamenko M, Kayashima Y, Matsuki K, Hiller S, Li F, Xu L, Grant R, Bertorello AM, Smithies O. Primary aldosteronism and impaired natriuresis in mice underexpressing TGFbeta1. Proc Natl Acad Sci USA 110: 5600–5605, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kestila M, Lenkkeri U, Mannikko M, Lamerdin J, McCready P, Putaala H, Ruotsalainen V, Morita T, Nissinen M, Herva R, Kashtan CE, Peltonen L, Holmberg C, Olsen A, Tryggvason K. Positionally cloned gene for a novel glomerular protein—nephrin—is mutated in congenital nephrotic syndrome. Mol Cell 1: 575–582, 1998. [DOI] [PubMed] [Google Scholar]
- 48.Kim KH, Park GT, Lim YB, Rue SW, Jung JC, Sonn JK, Bae YS, Park JW, Lee YS. Expression of connective tissue growth factor, a biomarker in senescence of human diploid fibroblasts, is up-regulated by a transforming growth factor-beta-mediated signaling pathway. Biochem Biophys Res Commun 318: 819–825, 2004. [DOI] [PubMed] [Google Scholar]
- 49.Kojima N, Williams JM, Takahashi T, Miyata N, Roman RJ. Effects of a new SGLT2 inhibitor, luseogliflozin, on diabetic nephropathy in T2DN rats. J Pharmacol Exp Ther 345: 464–472, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.KokudoT, Suzuki Y, Yoshimatsu Y, Yamazaki T, Watabe T, Miyazono K. Snail is required for TGFbeta-induced endothelial-mesenchymal transition of embryonic stem cell-derived endothelial cells. J Cell Sci 121: 3317–3324, 2008. [DOI] [PubMed] [Google Scholar]
- 51.Krag S, Osterby R, Chai Q, Nielsen CB, Hermans C, Wogensen L. TGF-beta1-induced glomerular disorder is associated with impaired concentrating ability mimicking primary glomerular disease with renal failure in man. Lab Invest 80: 1855–1868, 2000. [DOI] [PubMed] [Google Scholar]
- 52.Lala DS, Rice DA, Parker KL. Steroidogenic factor I, a key regulator of steroidogenic enzyme expression, is the mouse homolog of fushitarazu-factor I. Mol Endocrinol 6: 1249–1258, 1992. [DOI] [PubMed] [Google Scholar]
- 53.Lazzara MJ, Deen WM. Model of albumin reabsorption in the proximal tubule. Am J Physiol Renal Physiol 292: F430–F439, 2007. [DOI] [PubMed] [Google Scholar]
- 54.Le Bras GF, Taylor C, Koumangoye RB, Revetta F, Loomans HA, Andl CD. TGFbeta loss activates ADAMTS-1-mediated EGF-dependent invasion in a model of esophageal cell invasion. Exp Cell Res 330: 29–42, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee EY, Chung CH, Khoury CC, Yeo TK, Pyagay PE, Wang A, Chen S. The monocyte chemoattractant protein-1/CCR2 loop, inducible by TGF-β, increases podocyte motility and albumin permeability. Am J Physiol Renal Physiol 297: F85–F94, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee HB, Yu MR, Yang Y, Jiang Z, Ha H. Reactive oxygen species-regulated signaling pathways in diabetic nephropathy. J Am Soc Nephrol 14: S241–S245, 2003. [DOI] [PubMed] [Google Scholar]
- 57.Lee YJ, Han HJ. Troglitazone ameliorates high glucose-induced EMT and dysfunction of SGLTs through PI3K/Akt, GSK-3β, Snail1, and β-catenin in renal proximal tubule cells. Am J Physiol Renal Physiol 298: F1263–F1275, 2010. [DOI] [PubMed] [Google Scholar]
- 58.Leheste JR, Rolinski B, Vorum H, Hilpert J, Nykjaer A, Jacobsen C, Aucouturier P, Moskaug JO, Otto A, Christensen EI, Willnow TE. Megalin knockout mice as an animal model of low molecular weight proteinuria. Am J Pathol 155: 1361–1370, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lehmann TP, Biernacka-Lukanty JM, Trzeciak WH, Li JY. Steroidogenic factor 1 gene transcription is inhibited by transforming growth factor beta. Endocr Res 31: 71–79, 2005. [DOI] [PubMed] [Google Scholar]
- 60.Le Roy C, Li JY, Stocco DM, Langlois D, Saez JM. Regulation by adrenocorticotropin (ACTH), angiotensin II, transforming growth factor-beta, and insulin-like growth factor I of bovine adrenal cell steroidogenic capacity and expression of ACTH receptor, steroidogenic acute regulatory protein, cytochrome P450c17, and 3beta-hydroxysteroid dehydrogenase. Endocrinology 141: 1599–1607, 2000. [DOI] [PubMed] [Google Scholar]
- 61.Liakos P, Lenz D, Bernhardt R, Feige JJ, Defaye G. Transforming growth factor beta1 inhibits aldosterone and cortisol production in the human adrenocortical cell line NCI-H295R through inhibition of CYP11B1 and CYP11B2 expression. J Endocrinol 176: 69–82, 2003. [DOI] [PubMed] [Google Scholar]
- 62.Loffing J, Korbmacher C. Regulated sodium transport in the renal connecting tubule (CNT) via the epithelial sodium channel (ENaC). Pflügers Arch 458: 111–135, 2009. [DOI] [PubMed] [Google Scholar]
- 63.Loo DD, Zeuthen T, Chandy G, Wright EM. Cotransport of water by the Na+/glucose cotransporter. Proc Natl Acad Sci USA 93: 13367–13370, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Luo X, Ikeda Y, Parker KL. A cell-specific nuclear receptor is essential for adrenal and gonadal development and sexual differentiation. Cell 77: 481–490, 1994. [DOI] [PubMed] [Google Scholar]
- 65.MatavelliLC, Siragy HM. Reduction of aldosterone production improves renal oxidative stress and fibrosis in diabetic rats. J Cardiovasc Pharmacol 61: 17–22, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McMahon S, Charbonneau M, Grandmont S, Richard DE, Dubois CM. Transforming growth factor beta1 induces hypoxia-inducible factor-1 stabilization through selective inhibition of PHD2 expression. J Biol Chem 281: 24171–24181, 2006. [DOI] [PubMed] [Google Scholar]
- 67.Moestrup SK, Christensen EI, Nielsen S, Jorgensen KE, Bjorn SE, Roigaard H, Gliemann J. Binding and endocytosis of proteins mediated by epithelial gp330. Ann NY Acad Sci 737: 124–137, 1994. [DOI] [PubMed] [Google Scholar]
- 68.Munoz-Espin D, Canamero M, Maraver A, Gomez-Lopez G, Contreras J, Murillo-Cuesta S, Rodriguez-Baeza A, Varela-Nieto I, Ruberte J, Collado M, Serrano M. Programmed cell senescence during mammalian embryonic development. Cell 155: 1104–1118, 2013. [DOI] [PubMed] [Google Scholar]
- 69.Naaman-Reperant E, Hales DB, Durand P. Effect of transforming growth factor beta-1 on the cholesterol side-chain cleavage system in the adrenal gland of sheep fetuses and newborns. Endocrinology 137: 886–892, 1996. [DOI] [PubMed] [Google Scholar]
- 70.Nagata T, Fukuzawa T, Takeda M, Fukazawa M, Mori T, Nihei T, Honda K, Suzuki Y, Kawabe Y. Tofogliflozin, a novel sodium-glucose co-transporter 2 inhibitor, improves renal and pancreatic function in db/db mice. Br J Pharmacol 170: 519–531, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ng DP, Warram JH, Krolewski AS. TGF-beta 1 as a genetic susceptibility locus for advanced diabetic nephropathy in type 1 diabetes mellitus: an investigation of multiple known DNA sequence variants. Am J Kidney Dis 41: 22–28, 2003. [DOI] [PubMed] [Google Scholar]
- 72.Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP, Giardino I, Brownlee M. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 404: 787–790, 2000. [DOI] [PubMed] [Google Scholar]
- 73.Nowak G, Schnellmann RG. Autocrine production and TGF-β1-mediated effects on metabolism and viability in renal cells. Am J Physiol Renal Fluid Electrolyte Physiol 271: F689–F697, 1996. [DOI] [PubMed] [Google Scholar]
- 74.Nyhan KC, Faherty N, Murray G, Cooey LB, Godson C, Crean JK, Brazil DP. Jagged/Notch signalling is required for a subset of TGFbeta1 responses in human kidney epithelial cells. Biochim Biophys Acta 1803: 1386–1395, 2010. [DOI] [PubMed] [Google Scholar]
- 75.Pagtalunan ME, Miller PL, Jumping-Eagle S, Nelson RG, Myers BD, Rennke HG, Coplon NS, Sun L, Meyer TW. Podocyte loss and progressive glomerular injury in type II diabetes. J Clin Invest 99: 342–348, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Park JG, Lee DH, Moon YS, Kim KH. Reversine increases the plasticity of lineage-committed preadipocytes to osteogenesis by inhibiting adipogenesis through induction of TGF-beta pathway in vitro. Biochem Biophys Res Commun 446: 30–36, 2014. [DOI] [PubMed] [Google Scholar]
- 77.Patel A, Scott WR, Lympany PA, Rippin JD, Gill GV, Barnett AH, Bain SC, Warren 3/UK GoKind Study Group . The TGF-beta 1 gene codon 10 polymorphism contributes to the genetic predisposition to nephropathy in Type 1 diabetes. Diabet Med 22: 69–73, 2005. [DOI] [PubMed] [Google Scholar]
- 78.Peters DM, Vadasz I, Wujak L, Wygrecka M, Olschewski A, Becker C, Herold S, Papp R, Mayer K, Rummel S, Brandes RP, Gunther A, Waldegger S, Eickelberg O, Seeger W, Morty RE. TGF-beta directs trafficking of the epithelial sodium channel ENaC which has implications for ion and fluid transport in acute lung injury. Proc Natl Acad Sci USA 111: E374–E383, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Putaala H, Soininen R, Kilpelainen P, Wartiovaara J, Tryggvason K. The murine nephrin gene is specifically expressed in kidney, brain and pancreas: inactivation of the gene leads to massive proteinuria and neonatal death. Hum Mol Genet 10: 1–8, 2001. [DOI] [PubMed] [Google Scholar]
- 80.Rice DA, Mouw AR, Bogerd AM, Parker KL. A shared promoter element regulates the expression of three steroidogenic enzymes. Mol Endocrinol 5: 1552–1561, 1991. [DOI] [PubMed] [Google Scholar]
- 81.Roberts AB, Heine UI, Flanders KC, Sporn MB. Transforming growth factor-beta. Major role in regulation of extracellular matrix. Ann NY Acad Sci 580: 225–232, 1990. [DOI] [PubMed] [Google Scholar]
- 82.Rougier JP, Moullier P, Piedagnel R, Ronco PM. Hyperosmolality suppresses but TGF beta 1 increases MMP9 in human peritoneal mesothelial cells. Kidney Int 51: 337–347, 1997. [DOI] [PubMed] [Google Scholar]
- 83.Rude MK, Duhaney TA, Kuster GM, Judge S, Heo J, Colucci WS, Siwik DA, Sam F. Aldosterone stimulates matrix metalloproteinases and reactive oxygen species in adult rat ventricular cardiomyocytes. Hypertension 46: 555–561, 2005. [DOI] [PubMed] [Google Scholar]
- 84.Russo LM, del Re E, Brown D, Lin HY. Evidence for a role of transforming growth factor (TGF)-beta1 in the induction of postglomerular albuminuria in diabetic nephropathy: amelioration by soluble TGF-beta type II receptor. Diabetes 56: 380–388, 2007. [DOI] [PubMed] [Google Scholar]
- 85.Russo LM, Sandoval RM, Campos SB, Molitoris BA, Comper WD, Brown D. Impaired tubular uptake explains albuminuria in early diabetic nephropathy. J Am Soc Nephrol 20: 489–494, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Russo LM, Sandoval RM, McKee M, Osicka TM, Collins AB, Brown D, Molitoris BA, Comper WD. The normal kidney filters nephrotic levels of albumin retrieved by proximal tubule cells: retrieval is disrupted in nephrotic states. Kidney Int 71: 504–513, 2007. [DOI] [PubMed] [Google Scholar]
- 87.Salyer SA, Parks J, Barati MT, Lederer ED, Clark BJ, Klein JD, Khundmiri SJ. Aldosterone regulates Na+, K+ ATPase activity in human renal proximal tubule cells through mineralocorticoid receptor. Biochim Biophys Acta 1833: 2143–2152, 2013. [DOI] [PubMed] [Google Scholar]
- 88.Sam R, Wanna L, Gudehithlu KP, Garber SL, Dunea G, Arruda JA, Singh AK. Glomerular epithelial cells transform to myofibroblasts: early but not late removal of TGF-beta1 reverses transformation. Transl Res 148: 142–148, 2006. [DOI] [PubMed] [Google Scholar]
- 89.Sato A, Hayashi K, Naruse M, Saruta T. Effectiveness of aldosterone blockade in patients with diabetic nephropathy. Hypertension 41: 64–68, 2003. [DOI] [PubMed] [Google Scholar]
- 90.Sharma K, Jin Y, Guo J, Ziyadeh FN. Neutralization of TGF-beta by anti-TGF-beta antibody attenuates kidney hypertrophy and the enhanced extracellular matrix gene expression in STZ-induced diabetic mice. Diabetes 45: 522–530, 1996. [DOI] [PubMed] [Google Scholar]
- 91.Smithies O. Why the kidney glomerulus does not clog: a gel permeation/diffusion hypothesis of renal function. Proc Natl Acad Sci USA 100: 4108–4113, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Stephen TL, Rutkowski MR, Allegrezza MJ, Perales-Puchalt A, Tesone AJ, Svoronos N, Nguyen JM, Sarmin F, Borowsky ME, Tchou J, Conejo-Garcia JR. Transforming growth factor β-mediated suppression of antitumor T cells requires foxp1 transcription factor expression. Immunity 41: 427–439, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Swetha G, Chandra V, Phadnis S, Bhonde R. Glomerular parietal epithelial cells of adult murine kidney undergo EMT to generate cells with traits of renal progenitors. J Cell Mol Med 15: 396–413, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tang MJ, Wang YK, Lin HH. Butyrate and TGF-beta downregulate Na,K-ATPase expression in cultured proximal tubule cells. Biochem Biophys Res Commun 215: 57–66, 1995. [DOI] [PubMed] [Google Scholar]
- 95.Thannickal VJ, Fanburg BL. Activation of an H2O2-generating NADH oxidase in human lung fibroblasts by transforming growth factor beta 1. J Biol Chem 270: 30334–30338, 1995. [DOI] [PubMed] [Google Scholar]
- 96.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell 139: 871–890, 2009. [DOI] [PubMed] [Google Scholar]
- 97.Tojo A, Endou H. Intrarenal handling of proteins in rats using fractional micropuncture technique. Am J Physiol Renal Fluid Electrolyte Physiol 263: F601–F606, 1992. [DOI] [PubMed] [Google Scholar]
- 98.Tojo A, Onozato ML, Ha H, Kurihara H, Sakai T, Goto A, Fujita T, Endou H. Reduced albumin reabsorption in the proximal tubule of early-stage diabetic rats. Histochem Cell Biol 116: 269–276, 2001. [DOI] [PubMed] [Google Scholar]
- 99.Tucker BJ, Rasch R, Blantz RC. Glomerular filtration and tubular reabsorption of albumin in preproteinuric and proteinuric diabetic rats. J Clin Invest 92: 686–694, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ueberham E, Low R, Ueberham U, Schonig K, Bujard H, Gebhardt R. Conditional tetracycline-regulated expression of TGF-beta1 in liver of transgenic mice leads to reversible intermediary fibrosis. Hepatology 37: 1067–1078, 2003. [DOI] [PubMed] [Google Scholar]
- 101.Vallon V, Richter K, Blantz RC, Thomson S, Osswald H. Glomerular hyperfiltration in experimental diabetes mellitus: potential role of tubular reabsorption. J Am Soc Nephrol 10: 2569–2576, 1999. [DOI] [PubMed] [Google Scholar]
- 102.Vallon V, Rose M, Gerasimova M, Satriano J, Platt KA, Koepsell H, Cunard R, Sharma K, Thomson SC, Rieg T. Knockout of Na-glucose transporter SGLT2 attenuates hyperglycemia and glomerular hyperfiltration but not kidney growth or injury in diabetes mellitus. Am J Physiol Renal Physiol 304: F156–F167, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wakida N, Kitamura K, Tuyen DG, Maekawa A, Miyoshi T, Adachi M, Shiraishi N, Ko T, Ha V, Nonoguchi H, Tomita K. Inhibition of prostasin-induced ENaC activities by PN-1 and regulation of PN-1 expression by TGF-beta1 and aldosterone. Kidney Int 70: 1432–1438, 2006. [DOI] [PubMed] [Google Scholar]
- 104.Wang A, Ziyadeh FN, Lee EY, Pyagay PE, Sung SH, Sheardown SA, Laping NJ, Chen S. Interference with TGF-β signaling by Smad3-knockout in mice limits diabetic glomerulosclerosis without affecting albuminuria. Am J Physiol Renal Physiol 293: F1657–F1665, 2007. [DOI] [PubMed] [Google Scholar]
- 105.Wang YW, Liou NH, Cherng JH, Chang SJ, Ma KH, Fu E, Liu JC, Dai NT. siRNA-targeting transforming growth factor-beta type i receptor reduces wound scarring and extracellular matrix deposition of scar tissue. J Invest Dermatol 134: 2016–2025, 2014. [DOI] [PubMed] [Google Scholar]
- 106.Wartiovaara J, Ofverstedt LG, Khoshnoodi J, Zhang J, Makela E, Sandin S, Ruotsalainen V, Cheng RH, Jalanko H, Skoglund U, Tryggvason K. Nephrin strands contribute to a porous slit diaphragm scaffold as revealed by electron tomography. J Clin Invest 114: 1475–1483, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Welch DR, Fabra A, Nakajima M. Transforming growth factor beta stimulates mammary adenocarcinoma cell invasion and metastatic potential. Proc Natl Acad Sci USA 87: 7678–7682, 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wiener Z, Band AM, Kallio P, Hogstrom J, Hyvonen V, Kaijalainen S, Ritvos O, Haglund C, Kruuna O, Robine S, Louvard D, Ben-Neriah Y, Alitalo K. Oncogenic mutations in intestinal adenomas regulate Bim-mediated apoptosis induced by TGF-beta. Proc Natl Acad Sci USA 111: E2229–E2236, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wong TY, Poon P, Chow KM, Szeto CC, Cheung MK, Li PK. Association of transforming growth factor-beta (TGF-beta) T869C (Leu 10Pro) gene polymorphisms with type 2 diabetic nephropathy in Chinese. Kidney Int 63: 1831–1835, 2003. [DOI] [PubMed] [Google Scholar]
- 110.Yamamoto T, Nakamura T, Noble NA, Ruoslahti E, Border WA. Expression of transforming growth factor beta is elevated in human and experimental diabetic nephropathy. Proc Natl Acad Sci USA 90: 1814–1818, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yammani RR, Sharma M, Seetharam S, Moulder JE, Dahms NM, Seetharam B. Loss of albumin and megalin binding to renal cubilin in rats results in albuminuria after total body irradiation. Am J Physiol Regul Integr Comp Physiol 283: R339–R346, 2002. [DOI] [PubMed] [Google Scholar]
- 112.Zechel J, Gohil H, Lust WD, Cohen A. Alterations in matrix metalloproteinase-9 levels and tissue inhibitor of matrix metalloproteinases-1 expression in a transforming growth factor-beta transgenic model of hydrocephalus. J Neurosci Res 69: 662–668, 2002. [DOI] [PubMed] [Google Scholar]
- 113.Zeisberg EM, Potenta SE, Sugimoto H, Zeisberg M, Kalluri R. Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J Am Soc Nephrol 19: 2282–2287, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT, Roberts AB, Neilson EG, Sayegh MH, Izumo S, Kalluri R. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med 13: 952–961, 2007. [DOI] [PubMed] [Google Scholar]
- 115.Zeisberg M, Hanai J, Sugimoto H, Mammoto T, Charytan D, Strutz F, Kalluri R. BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat Med 9: 964–968, 2003. [DOI] [PubMed] [Google Scholar]
- 116.Ziyadeh FN, Hoffman BB, Han DC, Iglesias-De La Cruz MC, Hong SW, Isono M, Chen S, McGowan TA, Sharma K. Long-term prevention of renal insufficiency, excess matrix gene expression, and glomerular mesangial matrix expansion by treatment with monoclonal antitransforming growth factor-beta antibody in db/db diabetic mice. Proc Natl Acad Sci USA 97: 8015–8020, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]