

Abstract
In thick ascending limbs (THALs), nitric oxide (NO) decreases NaCl reabsorption via cGMP-mediated inhibition of Na-K-2Cl cotransporter (NKCC2). In angiotensin (ANG II)-induced hypertension, endothelin-1 (ET-1)-induced NO production by THALs is impaired. However, whether this alters NO's natriuretic effects and the mechanisms involved are unknown. In other cell types, ANG II augments phosphodiesterase 5 (PDE5)-mediated cGMP degradation. We hypothesized that NO-mediated inhibition of NKCC2 activity and stimulation of cGMP synthesis are blunted via PDE5 in ANG II-induced hypertension. Sprague-Dawley rats were infused with vehicle or ANG II (200 ng·kg−1·min−1) for 5 days. ET-1 reduced NKCC2 activity by 38 ± 13% (P < 0.05) in THALs from vehicle-treated rats but not from ANG II-hypertensive rats (Δ: −9 ± 13%). A NO donor yielded similar results as ET-1. In contrast, dibutyryl-cGMP significantly decreased NKCC2 activity in both vehicle-treated and ANG II-hypertensive rats (control: Δ−44 ± 15% vs. ANG II: Δ−41 ± 10%). NO increased cGMP by 2.08 ± 0.36 fmol/μg protein in THALs from vehicle-treated rats but only 1.06 ± 0.25 fmol/μg protein in ANG II-hypertensive rats (P < 0.04). Vardenafil (25 nM), a PDE5 inhibitor, restored NO's ability to inhibit NKCC2 activity in THALs from ANG II-hypertensive rats (Δ: −60 ± 9%, P < 0.003). Similarly, NO's stimulation of cGMP was also restored by vardenafil (vehicle-treated: 1.89 ± 0.71 vs. ANG II-hypertensive: 2.02 ± 0.32 fmol/μg protein). PDE5 expression did not differ between vehicle-treated and ANG II-hypertensive rats. We conclude that NO-induced inhibition of NKCC2 and increases in cGMP are blunted in ANG II-hypertensive rats due to PDE5 activation. Defects in the response of THALs to NO may enhance NaCl retention in ANG II-induced hypertension.
Keywords: cGMP, kidney, sodium transport, phosphodiesterase 5, endothelin-1
the thick ascending limb of the loop of Henle (THAL) reabsorbs 20 to 30% of the filtered NaCl load and contributes to the generation of the corticomedullary osmotic gradient and blood pressure regulation (12). Na-K-2Cl cotransporter type 2 (NKCC2) activity accounts for most of the transcellular NaCl reabsorption, transporting 80% of Na and 100% of Cl. The importance of NKCC2 in NaCl reabsorption is underscored by the profuse diuresis induced by loop diuretics like furosemide, which inhibit NKCC2 activity (13). In addition to regulating urine concentration, the THAL plays an important role in the maintenance of blood pressure. Defects in THAL transport that diminish NKCC2 activity cause hypotension (16), whereas increased THAL NaCl reabsorption has been found in hypertension (10, 36).
Angiotensin II (ANG II) is a potent vasoconstrictor and anti-natriuretic hormone. Its importance in blood pressure regulation is underscored by the effectiveness of anti-hypertensive drugs that block ANG II production or actions (5, 43, 44) and by the hypertension induced by chronic ANG II infusion (11, 14). ANG II enhances Na reabsorption directly by increasing Na transport in proximal tubules (6), THALs (40, 45), and collecting ducts (29), and indirectly by reducing the responsiveness to some natriuretic stimuli in collecting ducts (19). However, whether ANG II-induced hypertension also affects THAL response to natriuretic stimuli is not known.
In contrast, nitric oxide (NO) plays an important role in the kidney by inhibiting Na reabsorption in proximal tubules (35), THALs (28, 31), and collecting ducts (42) and by increasing renal blood flow (22). In the THAL many autacoids and hormones, such as endothelin-1 (ET-1), ATP, clonidine, and l-arginine, inhibit NKCC2 via NO (17, 27, 30, 31, 39, 41). Furthermore, infusion of subpressor doses of the nitric oxide synthase (NOS) inhibitor l-NAME induces salt-sensitive hypertension (37), indicating that impaired NO production and/or signaling contributes to high blood pressure.
We recently showed that in ANG II-mediated hypertension ET-1-induced NO production by THALs is impaired and that this correlates with reduced NOS3 expression and altered phosphorylation (33). However, whether ET-1- and NO-induced inhibition of NKCC2 is also impaired or whether changes downstream from NO compensate for this defect remain to be elucidated.
NO inhibits NKCC2 activity via cGMP (27), and cGMP can be degraded by phosphodiesterases (PDEs) (8). In smooth muscle cells, PDE5 reduces NO-induced elevations in cGMP (9, 21), and in the kidney PDE5 regulates Na reabsorption in pregnant rats (38). Interestingly, acute ANG II treatment increases PDE5 levels and activity in smooth muscle cells (18) and ANG II-induced hypertension elevates PDE5 activity in cardiomyocytes (23). However, whether PDE5 plays a role in the THAL in ANG II-induced hypertension is not known. Therefore, we hypothesized that NO-induced inhibition of NKCC2 activity and increases in cGMP are blunted in THALs of ANG II-hypertensive rats and this is mediated in part by PDE5.
MATERIALS AND METHODS
Reagents and solutions.
Most chemicals and reagents were purchased from Sigma (St. Louis, MO). ET-1 was from Bachem (Bubendorf, Switzerland). The NO donor spermine NONOate was from Caymen Chemical (Ann Arbor, MI). Dibutyryl (db)-cGMP was from ENZO Life Sciences (Ann Arbor, MI). Sodium Green was from Invitrogen (Grand Island, NY). Vardenafil was from Bayer AG (Leverkusen, Germany). The cGMP enzyme-immunoassay kit was from Biomedical Technologies (Stoughton, MA). Coomassie Plus protein assay reagent was from Pierce (Rockford, IL). The PDE5A1 antibody was from BD Transduction (San Jose, CA) and the secondary antibody was from GE Healthcare.
Physiological saline contained (in mM) 130 NaCl, 2.5 NaH2PO4, 4 KCl, 1.2 MgSO4, 6 D/l-alanine, 1 trisodium citrate, 5.5 glucose, 2 calcium dilactate, and 10 HEPES (pH 7.4 with NaOH). The NaCl-free solution contained the same ingredients except that NaCl and KCl were omitted, mannitol was added in equi-osmolar amounts, and the solution pH was adjusted with KOH.
ANG II-induced hypertension.
All protocols involving animals were approved by the Institutional Animal Care and Use Committee of Henry Ford Hospital. Male Sprague-Dawley rats weighing 90–120 g were fed a standard chow containing 0.4% Na and infused with either vehicle (0.01 N acetic acid) or ANG II at 200 ng·kg−1·min−1 via osmotic mini-pumps (Durect; model 1007D) for 5 days. Mean arterial blood pressure was measured by a femoral arterial catheter in anesthetized rats (90 mg/kg ketamine, 10 mg/kg xylazine) using a pressure transducer and PowerLab software (ADInstruments, Colorado Springs, CO).
Isolation and perfusion of rat THALs.
THALs were isolated and perfused as we have done before (17). Once the rat was anesthetized, the abdominal cavity was opened, and the kidney was bathed in ice-cold saline and removed. Coronal slices were placed in oxygenated physiological saline. THALs were dissected from the outer medulla under a stereomicroscope at 4°C and transferred to a temperature-regulated chamber, where they were perfused using concentric glass pipettes at 37 ± 1°C.
Measurement of NKCC2 activity.
NKCC2 activity was measured using methods similar to those we reported (28). Briefly, tubules were perfused at 37°C in a chamber on the stage of an inverted microscope with the NaCl-free solution (to inhibit NKCC2 activity) with 100 μM dimethyl amiloride (to inhibit Na/H exchange) and bathed in physiological saline. Tubules were loaded for 20 min with 2 μM Sodium Green in the bath and washed for 20 min with physiological saline. Dye was excited at 490 nm and fluorescence emission was recorded at >500 nm to measure intracellular Na every 5 s for a 12-min control period. Acquired data were processed using Metafluor (Molecular Devices, Sunnyvale, CA). Then, the luminal solution was switched to physiological saline plus dimethyl amiloride to stimulate NKCC2. Twenty minutes after the test compound was added to the bath and the process was repeated. The first five points were used to calculate the initial rate of fluorescence increase. NKCC2 activity was calculated from the initial rate of change normalized by the fluorescence at the time of the solution switch.
In the experiments using ET-1, 100 μM l-arginine was present in the bath. The NO donor spermine NONOate (100 μM) was prepared 2 min before being added to the bath to minimize degradation. When vardenafil (25 nM) was used to inhibit PDE5, it was added to the bath solution during the wash and was present for the remainder of the experiment.
THAL suspensions.
Suspensions were prepared as we reported (32, 34). Briefly, the abdominal cavity was opened, and the kidneys were perfused with 40 ml of ice-cold physiological saline containing 0.1% collagenase (cat. no. C0130, 233 U/mg, Sigma) and 100 U heparin via the aorta. Kidneys were removed, and coronal slices were cut. The inner stripe of the outer medulla was minced and incubated in physiological saline containing 0.1% collagenase for 30 min at 37°C. Every 5 min, the tissue was gently agitated and gassed with 100% oxygen. Tissue was centrifuged at 60 g for 2 min; the pellet was resuspended in cold physiological saline and stirred on ice for 30 min. The suspension was filtered through a 250-μm nylon mesh and centrifuged at 60 g for 2 min. The pellet was washed and centrifuged again to collect THALs.
cGMP measurements.
Briefly, rat THAL suspensions were divided in 4 aliquots. Aliquots were treated with either vehicle or vardenafil (25 nM) for 10 min. Then, THALs were treated with vehicle or 100 μM spermine NONOate for another 10 min. THALs were then lysed with an equal volume of methanol and stored at −80°C for 2–3 h to precipitate proteins. Tubes were centrifuged for 15 min at 15,600 g to precipitate proteins. Supernatants were recovered and dried overnight (Savant SpeedVac Plus SC110A), and pellets were stored at −20°C. Dried samples were resuspended in 120 μl assay buffer and cGMP was assayed following the manufacturer's instructions. Samples of basal cGMP below the minimum detection level of 5 fmol/well were assigned this value. cGMP recovery was 89 ± 11%.
Precipitated proteins were resuspended by mechanical disruption of the pellet in physiological saline by vortexing and sonication. Protein concentration was measured by using a colorimetric assay (Coomassie Plus Protein Assay).
Western blot for PDE5.
Tubules were lysed in lysis buffer containing 20 mM HEPES (pH 7.5), 2 mM EDTA, 0.3 M sucrose, 1.0% Igepal CA-630, 0.1% sodium dodecyl sulfate, 5 μg/ml antipain, 10 μg/ml aprotinin, 5 μg/ml leupeptin, 4 mM benzamidine, 5 μg/ml chymostatin, 5 μg/ml pepstatin A, and 0.116 M pf-block. Debris was removed by centrifugation for 5 min at 5,600 g. Protein concentration was measured using Coomassie Plus Protein Assay. Western blots were performed as done before (32, 34) with the following modification: membranes were blotted in 5% milk-TBST for 1 h at room temperature and then incubated overnight at 4°C with anti-PDE5A1 antibody 1:750 dilution, washed, and incubated with 1:1,000 anti-rabbit antibody.
Statistical analysis.
Results are expressed as means ± SE. Data were analyzed by the Biostatistics and Research Epidemiology Department of Henry Ford Hospital. Paired and unpaired t-tests were used as appropriate as tests of significance.
RESULTS
Rats infused with ANG II had a significantly higher mean arterial blood pressure than vehicle-treated rats (vehicle-treated: 91.5 ± 2.4 mmHg vs. ANG II-hypertensive rats: 106.1 ± 3.4 mmHg, n = 20 and 26, respectively, P < 0.002), demonstrating that infusion of this dose of ANG II for 5 days produces a small but significant increase in blood pressure. Because of our previous findings showing that THAL NO production stimulated by ET-1 is impaired in ANG II-induced hypertension, we first tested whether ET-1-induced NO inhibits NKCC2 activity in THALs from ANG II-hypertensive rats. In THALs from vehicle-treated rats, ET-1 decreased NKCC2 activity by 38 ± 13% (n = 5, P < 0.05; Fig. 1, A and B). However, in ANG II-hypertensive rats, ET-1's inhibitory effect was blunted (Δ: −9 ± 13%, n = 6; N.S.; Fig. 1, C and D). These data indicate that the ability of ET-1 to decrease NKCC2 activity is impaired in THALs from ANG II-induced hypertensive rats.
Fig. 1.

Effect of 1 nM endothelin (ET)-1 on Na-K-2Cl cotransporter (NKCC2) activity. Nai, intracellular Na; AFU/s, arbitrary fluorescence U/s. A: Nai measured as AFU vs. time in isolated and perfused thick ascending limbs (THALs) from vehicle-treated rats (n = 5). B: same data as A but NKCC2 activity is displayed as the rate of increase in Nai in vehicle-treated rats (n = 5). C: Nai measured as AFU vs. time in isolated and perfused THALs from ANG II-induced hypertensive rats (n = 6). D: same data as C but NKCC2 activity is displayed as the rate of increase in Nai in ANG II-induced hypertensive rats (n = 6).
The effects of ET-1 could be diminished due to a reduction in the bioavailability of NO or due to decreased NO signaling. Since reactive oxygen species can reduce NO bioavailability, we tested whether scavenging superoxide with tempol could normalize the response to ET-1. However, pretreatment with 100 μM tempol did not restore the ability of ET-1 to decrease NKCC2 activity in THALs from ANG II-hypertensive rats (Δ in NKCC2 activity: +14 ± 19%, not significantly different, n = 5; Fig. 2). These data indicate that the impaired ability of ET-1 to inhibit NKCC2 activity in ANG II-hypertensive rats is not due to acute elevations of superoxide.
Fig. 2.
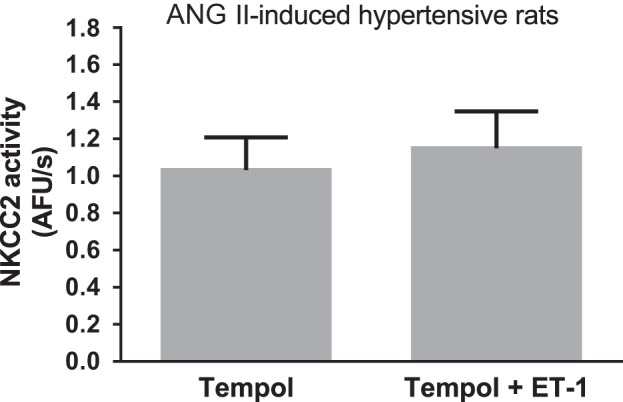
Effect of ET-1 on NKCC2 activity in the presence of tempol in THALs from ANG II-induced hypertensive rats. Effect of 1 nM ET-1 on NKCC2 activity in the presence of 100 μM tempol measured in isolated and perfused THALs (n = 5).
We reported that ET-1 acts via NO in THALs. Thus, we next tested whether inhibition of NKCC2 by exogenously added NO was blunted in ANG II-induced hypertension. In THALs from vehicle-treated rats, treatment with the NO donor spermine NONOate (100 μM) reduced NKCC2 activity by 33 ± 6% (P < 0.003, n = 6; Fig. 3A). In contrast, in THALs from ANG II-hypertensive rats, NKCC2 activity was not significantly decreased by treatment with the NO donor (Δ: +23 ± 14%, n = 6; Fig. 3B). Taken together, these results and the ET-1 data indicate that NO-induced inhibition of NKCC2 is blunted in THALs from ANG II-hypertensive rats.
Fig. 3.
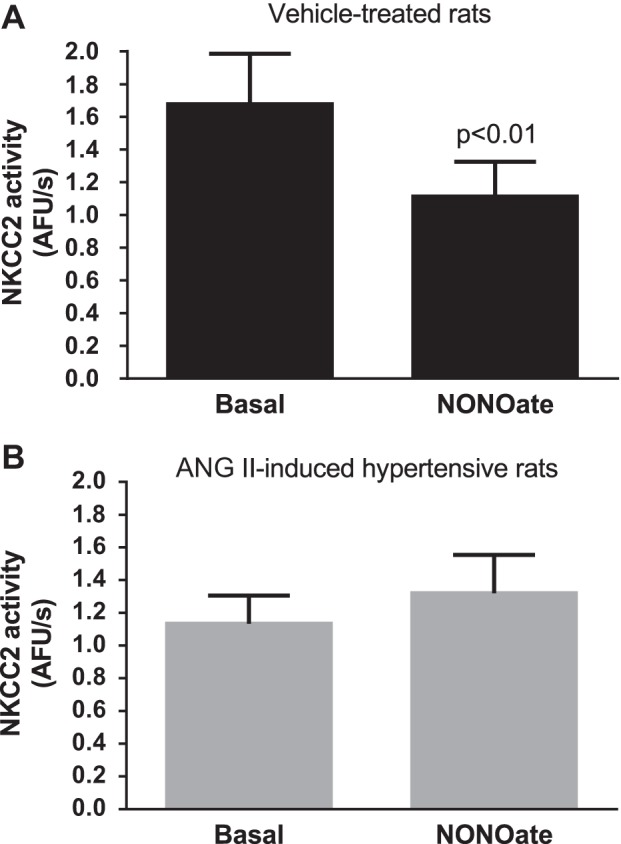
Effect of NONOate (NO) on NKCC2 activity. NKCC2 activity was measured in isolated and perfused THALs before and after treatment with 100 μM spermine NONOate. A: vehicle-treated rats (n = 6). B: ANG II-induced hypertensive rats (n = 6).
NO inhibits NKCC2 activity via cGMP. To test whether the response to cGMP was reduced in THALs from ANG II-induced hypertensive rats, we measured NKCC2 activity before and after incubation with the cGMP analog db-cGMP (500 μM). In vehicle-treated rats, db-cGMP reduced NKCC2 activity by 44 ± 15% (P < 0.05, n = 5; Fig. 4A). Similarly, in ANG II-hypertensive rats db-cGMP reduced NKCC2 activity by 41 ± 10% (P < 0.03, n = 4; Fig. 4B). These data suggest that the signaling cascade downstream from cGMP is intact in THALs from ANG II-induced hypertensive rats.
Fig. 4.
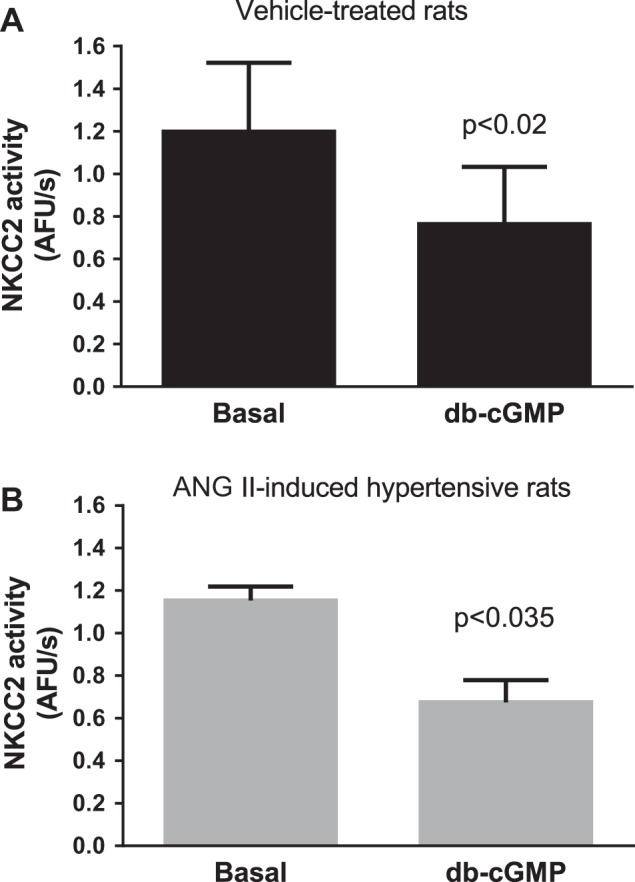
Effect of db-cGMP on NKCC2 activity. NKCC2 activity was measured in isolated and perfused THALs before and after treatment with 500 μM db-cGMP. A: vehicle-treated rats (n = 5). B: ANG II-induced hypertensive rats (n = 4).
We next studied whether NO-induced cGMP levels were diminished in ANG II-hypertensive rats. In THALs from vehicle-treated rats, 100 μM spermine NONOate increased cGMP by 2.08 ± 0.36 fmol/μg protein (n = 6) but only 1.06 ± 0.25 fmol/μg protein in tubules from ANG II-hypertensive rats (n = 8, P < 0.04; Fig. 5). These data indicate that NO-induced increases in cGMP are impaired in THALs from ANG II-induced hypertensive rats.
Fig. 5.
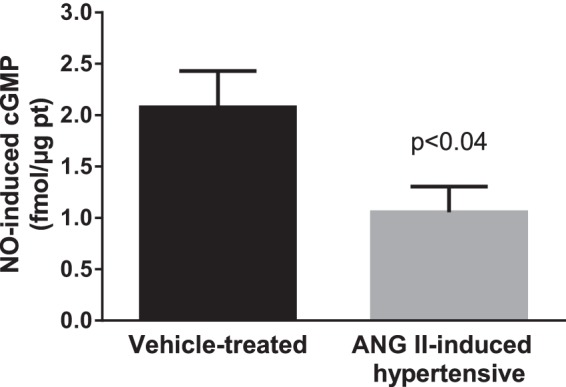
Effect of NONOate (NO) on cGMP levels in THALs from vehicle- and ANG II-induced hypertensive rats. THAL suspensions were incubated in the presence and absence of 100 μM spermine NONOate for 10 min (vehicle n = 6; ANG II n = 8). cGMP was measured by enzyme immunoassay.
PDE5 mediates decreases in cGMP in response to ANG II in other cell types. Thus, we tested whether PDE5 was responsible for the aforementioned results. In the presence of the PDE5 inhibitor vardenafil (25 nM), 100 μM spermine NONOate decreased NKCC2 activity by 45 ± 12% in THALs from vehicle-treated rats (P < 0.03, n = 4; Fig. 6A). Interestingly, vardenafil treatment restored the ability of spermine NONOate to decrease NKCC2 activity in tubules from ANG II-induced hypertensive rats (Δ: −60 ± 9%, P < 0.003, n = 5; Fig. 6B). Time controls with vardenafil showed no significant effect.
Fig. 6.
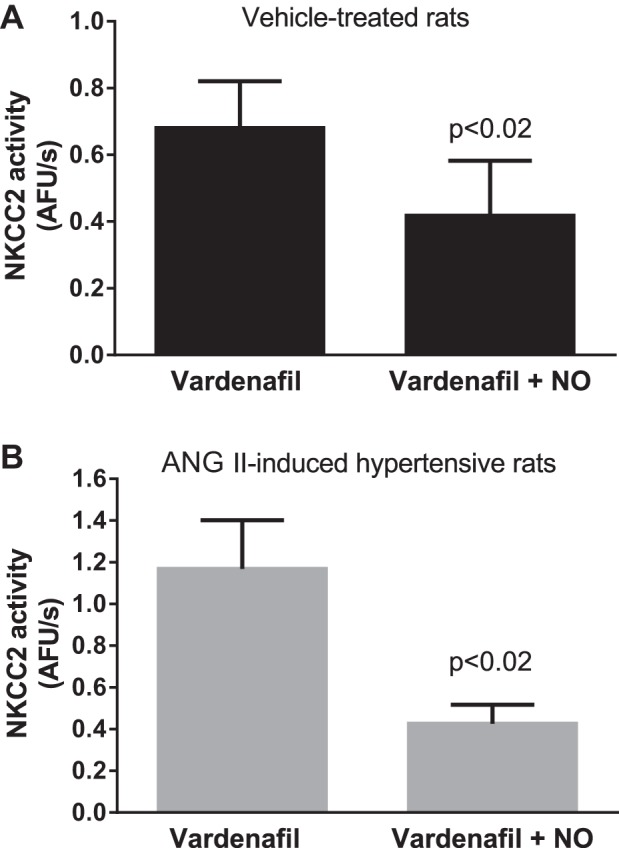
Effect of NO on NKCC2 activity in the presence of the phosphodiesterase 5 (PDE5) inhibitor vardenafil. Effect of 100 μM spermine NONOate on NKCC2 activity in the presence of 25 nM vardenafil in isolated and perfused THALs. A: vehicle-treated rats (n = 4). B: ANG II-induced hypertensive rats (n = 5).
In the presence of vardenafil, 100 μM spermine NONOate increased cGMP to the same extent in THALs from vehicle-treated and ANG II-hypertensive animals [Fig. 7; vehicle-treated: 1.89 ± 0.71 fmol/μg protein (n = 6) vs. ANG II-induced hypertensive: 2.02 ± 0.32 fmol/μg protein (n = 8)]. Basal cGMP levels were no different between vehicle and ANG II-hypertensive rats. These data indicate that in ANG II-induced hypertensive rats PDE5 blunts the ability of NO to inhibit NKCC2 activity by enhancing cGMP degradation.
Fig. 7.
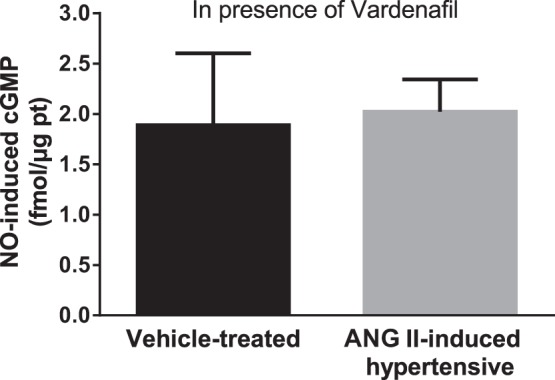
Effect of NO on cGMP levels in the presence of the PDE5 inhibitor vardenafil in THALs from vehicle- and ANG II-induced hypertensive rats. THAL suspensions were preincubated with 25 nM vardenafil for 10 min and then treated with 100 μM spermine NONOate for 10 min (vehicle n = 6; ANG II n = 8). cGMP was measured by enzyme immunoassay.
Finally, to study whether PDE5 expression was different between groups, we performed Western blots. PDE5 expression was not different in THALs from vehicle-treated and ANG II-induced hypertensive rats (PDE5/β-tubulin ratio = 0.55 ± 0.07 in vehicle-treated rats vs. 0.47 ± 0.11 in ANG II-hypertensive rats; n = 5; Fig. 8).
Fig. 8.
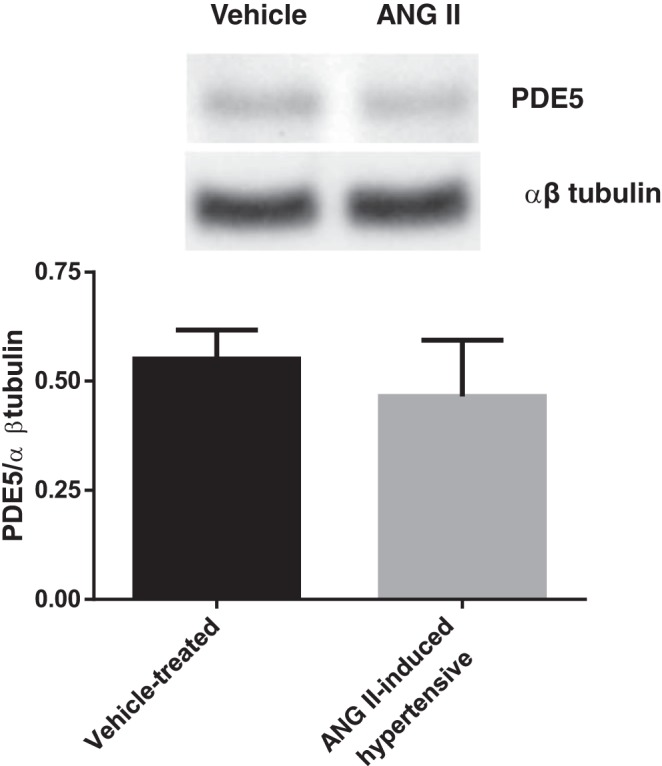
PDE5 protein levels in THALs from vehicle- and ANG II-induced hypertensive rats. Top: representative Western blot for PDE5 and β-tubulin. Bottom: cumulative data showing PDE5/β-tubulin ratio (n = 5).
DISCUSSION
We hypothesized that NO's inhibition of NKCC2 activity and stimulation of cGMP synthesis are blunted via PDE5 in ANG II-induced hypertension. We found that PDE5 reduces NO-induced inhibition of NKCC2 activity and this is likely due to enhanced degradation of cGMP by PDE5 in THALs from ANG II-induced hypertensive rats. This conclusion is based on the following findings: 1) ET-1- and NO-induced inhibition of NKCC2 activity was impaired in THALs from ANG II-induced hypertensive rats; 2) db-cGMP inhibited NKCC2 activity to the same extent in ANG II-induced hypertensive and vehicle-treated rats; 3) NO-induced increases in cGMP were impaired in THALs from ANG II-induced hypertensive rats; and 4) NO-induced decreases in NKCC2 activity and increases in cGMP were restored in THALs from ANG II-induced hypertensive rats by inhibiting PDE5. To our knowledge, this is the first time that impaired NO-induced inhibition of Na reabsorption in THALs has been shown in ANG II-induced hypertension. Moreover, this is the first time that PDE5 has been shown to play a role in NaCl reabsorption in any nephron segment during ANG II-induced hypertension.
We found that ET-1-induced inhibition of THAL NKCC2 activity was reduced in ANG II hypertension. The inability of ET-1 to reduce NKCC2 activity is likely due to two factors: 1) diminished ET-1-induced NO production; and 2) blunted effect of NO. We previously reported that ET-1-induced NO production is blunted in this model (33) and now show that the effects of a NO donor are also reduced. Of note, we did not observe higher absolute values in basal NKCC2 activity in isolated and perfused THALs from rats treated with ANG II when compared with vehicle-treated rats. The reason for this finding is not clear but may be due to different causes. First, the experimental design was aimed to measure NKCC2 activity in response to acute treatments within the same tubule and was not intended to compare NKCC2 activity in different rats which requires special experimental strategies. This specific experimental design is necessary to test for differences between groups because there is high interexperimental variability when measuring NKCC2 activity in isolated and perfused THALs. Additionally, NKCC2 activity was measured under conditions in which NO production was not promoted (except during ET-1 treatment). Thus, differences in NKCC2 activity caused by changes in NO production or effect would not be present. Finally, other signals that are not present in isolated and perfused THALs might be necessary to observe increases in basal NKCC2 activity (3). Regardless of whether basal NKCC2 activity is different in isolated and perfused THALs from vehicle- vs. ANG II-hypertensive rats, our data conclusively demonstrate that NO-mediated inhibition of NKCC2 activity is impaired in this model of hypertension.
The fact that ET-1 has less of an effect on NKCC2 activity in THALs from ANG II-induced hypertensive rats suggests that either bioavailability or NO signaling is reduced. ANG II-induced hypertension increases superoxide production by this nephron segment (40) and superoxide decreases NO bioavailability by reacting with it to form peroxynitrite (24, 26). Thus, we tested whether elevated superoxide was the cause of the diminished ability of ET-1 to inhibit transport. We found that acutely scavenging these reactive oxygen species did not restore the inhibitory effect of ET-1.
To dissect the mechanism by which ANG II-induced hypertension impairs NO's ability to reduce NKCC2 activity, we next studied the ability of db-cGMP to inhibit NKCC2. This cGMP analog is resistant to hydrolysis by PDEs (46). We found that treatment with db-cGMP inhibited NKCC2 activity to the same extent in THALs from normotensive and hypertensive rats. These data indicate that the signaling cascade downstream from cGMP is intact in ANG II-induced hypertensive rats. Since the effect of cGMP was intact but the ability of NO was diminished in THALs from ANG II-induced hypertensive rats, we next measured the ability of the NO donor to elevate cGMP levels. We found that the NO increased cGMP less in THALs from ANG II-induced hypertensive rats compared with tubules from vehicle-treated rats. These data indicate that the cause of NO's inability to reduce NKCC2 activity in THALs from ANG II-induced hypertensive animals is due to a failure to raise cGMP levels.
Reduced cGMP levels in response to NO could be caused either by decreased production or enhanced cGMP degradation. Previously, we reported that degradation is elevated in another model of salt-sensitive hypertension (15) so we focused on cGMP degradation. At the present, there are three known cGMP-specific PDEs (PDE5, PDE6, and PDE9) and five PDEs (PDE1, 2, 3, 10, and 11) with dual specificity (8). PDE5 is present in proximal tubules and collecting ducts (20, 25). Additionally, ANG II increases PDE5 activity in smooth muscle cells (18). Thus, we focused on this isoform.
To show that PDE5 was responsible for blunted NO-induced inhibition of NKCC2, we measured the effects of NO on NKCC2 activity and cGMP in the presence of a PDE5 inhibitor. We found that blockade of PDE5 restored NO's ability to reduce NKCC2 activity and increase cGMP in THALs from ANG II-induced hypertensive rats. These data support the conclusion that PDE5 blunts NO-induced inhibition of cotransport in THALs from ANG II-induced hypertensive rats by reducing cGMP.
Similar to our findings, NO donors only cause natriuresis in pregnant rats in the presence of PDE5 inhibition (38); however, unlike our results in THALs in ANG II hypertension where NO production is also impaired, PDE5 inhibition alone increases natriuresis in pregnant rats. Inhibition of PDE5 in inner medullary collecting ducts from pregnant rats also restores the ability of NO donors to increase cGMP levels to the same extent as in nonpregnant rats (38).
Resistance to NO donors in ANG II hypertension has been reported for other renal tissues. In glomeruli cGMP production in response to sodium nitroprusside was decreased (2). However, the authors attributed the defect to reduced cGMP production rather than enhanced degradation (2).
We used vardenafil to inhibit PDE5 because of its specificity and potency. It has more than 10 times the potency of sildenafil and taldalafil (4). The EC50 for PDE5 is ∼0.4 nM and, except for PDE6A and B, is at least 500 times more selective for PDE5 than for any other PDE discovered so far. At the concentration used in this study (25 nM), only PDE5 and PDE6 would be inhibited; however, PDE6 has only been found in the retinal photoreceptors, pineal gland, and in some melanoma cells (8) but not in the kidney. Although our data do not exclude the possibility that PDEs with dual specificity could be involved, PDE5 inhibition restored the ability of NO to increase cGMP levels and inhibit NKCC2 activity in ANG II-induced hypertensive rats to levels similar to those seen in control rats. Therefore, our data strongly suggest that PDE5 is the PDE responsible for enhanced cGMP degradation in THALs from ANG II-induced hypertensive rats.
ANG II-induced hypertension did not increase PDE5 levels in THALs. These data suggest that posttranslational mechanisms may be responsible for enhanced PDE5-mediated cGMP degradation in this model of hypertension. PDE5 activity is stimulated directly by increases in cGMP which binds PDE5's allosteric N-terminus side and indirectly through protein kinase G- or A-mediated PDE5 phosphorylation (7, 21). In contrast, PDE5 activity can be reduced by protein phosphatase 1 (PP1)-mediated dephosphorylation. PP1 activity in turn can be regulated by interacting with different regulatory subunits, by phosphorylation of those subunits, and by endogenous inhibitors (1).
In conclusion, ANG II-induced hypertension blunts NO-induced inhibition of NKCC2 activity due to enhanced degradation of cGMP by PDE5. The inability of the THAL to respond to this important natriuretic factor may contribute to the enhanced NaCl reabsorption observed in this model of hypertension.
GRANTS
This work was supported by grants from the National Institutes of Health (HL 028982) to J. L. Garvin and from American Heart Association (11PRE7510005) to V. D. Ramseyer.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: V.D.R., P.A.O., O.A.C., and J.L.G. conception and design of research; V.D.R. performed experiments; V.D.R., P.A.O., and J.L.G. analyzed data; V.D.R., P.A.O., O.A.C., and J.L.G. interpreted results of experiments; V.D.R. prepared figures; V.D.R. and J.L.G. drafted manuscript; V.D.R., P.A.O., O.A.C., and J.L.G. edited and revised manuscript; V.D.R., P.A.O., O.A.C., and J.L.G. approved final version of manuscript.
REFERENCES
- 1.Aggen JB, Nairn AC, Chamberlin R. Regulation of protein phosphatase-1. Chem Biol 7: R13–23, 2000. [DOI] [PubMed] [Google Scholar]
- 2.Bae EH, Ma SK, Lee J, Kim SW. Altered regulation of renal nitric oxide and atrial natriuretic peptide systems in angiotensin II-induced hypertension. Regul Pept 170: 31–37, 2011. [DOI] [PubMed] [Google Scholar]
- 3.Baum M. Luminal angiotensin II stimulates rat medullary thick ascending limb chloride transport in the presence of basolateral norepinephrine. Am J Physiol Renal Physiol 310: F294–F299, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blount MA, Beasley A, Zoraghi R, Sekhar KR, Bessay EP, Francis SH, Corbin JD. Binding of tritiated sildenafil, tadalafil, or vardenafil to the phosphodiesterase-5 catalytic site displays potency, specificity, heterogeneity, and cGMP stimulation. Mol Pharmacol 66: 144–152, 2004. [DOI] [PubMed] [Google Scholar]
- 5.Burnier M. Angiotensin II type 1 receptor blockers. Circulation 103: 904–912, 2001. [DOI] [PubMed] [Google Scholar]
- 6.Cogan MG. Angiotensin II: a powerful controller of sodium transport in the early proximal tubule. Hypertension 15: 451–458, 1990. [DOI] [PubMed] [Google Scholar]
- 7.Corbin JD, Turko IV, Beasley A, Francis SH. Phosphorylation of phosphodiesterase-5 by cyclic nucleotide-dependent protein kinase alters its catalytic and allosteric cGMP-binding activities. Eur J Biochem 267: 2760–2767, 2000. [DOI] [PubMed] [Google Scholar]
- 8.Francis SH, Blount MA, Corbin JD. Mammalian cyclic nucleotide phosphodiesterases: molecular mechanisms and physiological functions. Physiol Rev 91: 651–690, 2011. [DOI] [PubMed] [Google Scholar]
- 9.Francis SH, Busch JL, Corbin JD, Sibley D. cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol Rev 62: 525–563, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garcia NH, Plato CF, Stoos BA, Garvin JL. Nitric oxide-induced inhibition of transport by thick ascending limbs from Dahl salt-sensitive rats. Hypertension 34: 508–513, 1999. [DOI] [PubMed] [Google Scholar]
- 11.Gonzalez-Villalobos RA, Janjoulia T, Fletcher NK, Giani JF, Nguyen MT, Riquier-Brison AD, Seth DM, Fuchs S, Eladari D, Picard N, Bachmann S, Delpire E, Peti-Peterdi J, Navar LG, Bernstein KE, McDonough AA. The absence of intrarenal ACE protects against hypertension. J Clin Invest 123: 2011–2023, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Greger R. Ion transport mechanisms in thick ascending limb of Henle's loop of mammalian nephron. Physiol Rev 65: 760–797, 1985. [DOI] [PubMed] [Google Scholar]
- 13.Greger R, Wangemann P. Loop diuretics. Renal Physiol 10: 174–183, 1987. [DOI] [PubMed] [Google Scholar]
- 14.Gurley SB, Riquier-Brison AD, Schnermann J, Sparks MA, Allen AM, Haase VH, Snouwaert JN, Le TH, McDonough AA, Koller BH, Coffman TM. AT1A angiotensin receptors in the renal proximal tubule regulate blood pressure. Cell Metab 13: 469–475, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haque MZ, Ares GR, Ortiz PA. Enhanced phosphodiesterase 5 (PDE5) in thick ascending limbs of Dahl salt sensitive rats blunts NO-induced inhibition of transport. Hypertension 60: A52, 2012. [DOI] [PubMed] [Google Scholar]
- 16.Hebert SC. Bartter syndrome. Curr Opin Nephrol Hypertens 12: 527–532, 2003. [DOI] [PubMed] [Google Scholar]
- 17.Herrera M, Hong NJ, Ortiz PA, Garvin JL. Endothelin-1 inhibits thick ascending limb transport via Akt-stimulated nitric oxide production. J Biol Chem 284: 1454–1460, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim D, Aizawa T, Wei H, Pi X, Rybalkin SD, Berk BC, Yan C. Angiotensin II increases phosphodiesterase 5A expression in vascular smooth muscle cells: a mechanism by which angiotensin II antagonizes cGMP signaling. J Mol Cell Cardiol 38: 175–184, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kittikulsuth W, Pollock JS, Pollock DM. Sex differences in renal medullary endothelin receptor function in angiotensin II hypertensive rats. Hypertension 58: 212–218, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kotera J, Fujishige K, Omori K. Immunohistochemical localization of cGMP-binding cGMP-specific phosphodiesterase (PDE5) in rat tissues. J Histochem Cytochem 48: 685–693, 2000. [DOI] [PubMed] [Google Scholar]
- 21.Lin CS, Lin G, Xin ZC, Lue TF. Expression, distribution and regulation of phosphodiesterase 5. Curr Pharm Des 12: 3439–3457, 2006. [DOI] [PubMed] [Google Scholar]
- 22.Majid DS, Williams A, Kadowitz PJ, Navar LG. Renal responses to intra-arterial administration of nitric oxide donor in dogs. Hypertension 22: 535–541, 1993. [DOI] [PubMed] [Google Scholar]
- 23.Mokni W, Keravis T, Etienne-Selloum N, Walter A, Kane MO, Schini-Kerth VB, Lugnier C. Concerted regulation of cGMP and cAMP phosphodiesterases in early cardiac hypertrophy induced by angiotensin II. PLos One 5: e14227, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mori T, Cowley AW Jr. Angiotensin II-NAD(P)H oxidase-stimulated superoxide modifies tubulovascular nitric oxide cross-talk in renal outer medulla. Hypertension 42: 588–593, 2003. [DOI] [PubMed] [Google Scholar]
- 25.Ni XP, Safai M, Rishi R, Baylis C, Humphreys MH. Increased activity of cGMP-specific phosphodiesterase (PDE5) contributes to resistance to atrial natriuretic peptide natriuresis in the pregnant rat. J Am Soc Nephrol 15: 1254–1260, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ortiz PA, Garvin JL. Interaction of O2− and NO in the thick ascending limb. Hypertension 39: 591–596, 2002. [DOI] [PubMed] [Google Scholar]
- 27.Ortiz PA, Garvin JL. NO inhibits NaCl absorption by rat thick ascending limb through activation of cGMP-stimulated phosphodiesterase. Hypertension 37: 467–471, 2001. [DOI] [PubMed] [Google Scholar]
- 28.Ortiz PA, Hong NJ, Garvin JL. NO decreases thick ascending limb chloride absorption by reducing Na+-K+-2Cl− cotransporter activity. Am J Physiol Renal Physiol 281: F819–F825, 2001. [DOI] [PubMed] [Google Scholar]
- 29.Peti-Peterdi J, Warnock DG, Bell PD. Angiotensin II directly stimulates ENaC activity in the cortical collecting duct via AT(1) receptors. J Am Soc Nephrol 13: 1131–1135, 2002. [DOI] [PubMed] [Google Scholar]
- 30.Plato CF, Garvin JL. Alpha(2)-adrenergic-mediated tubular NO production inhibits thick ascending limb chloride absorption. Am J Physiol Renal Physiol 281: F679–F686, 2001. [DOI] [PubMed] [Google Scholar]
- 31.Plato CF, Stoos BA, Wang D, Garvin JL. Endogenous nitric oxide inhibits chloride transport in the thick ascending limb. Am J Physiol Renal Physiol 276: F159–F163, 1999. [DOI] [PubMed] [Google Scholar]
- 32.Ramseyer VD, Garvin JL. Angiotensin II decreases nitric oxide synthase 3 expression via nitric oxide and superoxide in the thick ascending limb. Hypertension 53: 313–318, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ramseyer VD, Gonzalez-Vicente A, Carretero OA, Garvin JL. Angiotensin II-induced hypertension blunts thick ascending limb NO production by reducing NO synthase 3 expression and enhancing threonine 495 phosphorylation. Am J Physiol Renal Physiol 308: F149–F156, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ramseyer VD, Hong NJ, Garvin JL. Tumor necrosis factor alpha decreases nitric oxide synthase type 3 expression primarily via Rho/Rho kinase in the thick ascending limb. Hypertension 59: 1145–1150, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roczniak A, Burns KD. Nitric oxide stimulates guanylate cyclase and regulates sodium transport in rabbit proximal tubule. Am J Physiol Renal Fluid Electrolyte Physiol 270: F106–F115, 1996. [DOI] [PubMed] [Google Scholar]
- 36.Roman RJ, Kaldunski ML. Enhanced chloride reabsorption in the loop of Henle in Dahl salt-sensitive rats. Hypertension 17: 1018–1024, 1991. [DOI] [PubMed] [Google Scholar]
- 37.Salazar FJ, Alberola A, Pinilla JM, Romero JC, Quesada T. Salt-induced increase in arterial pressure during nitric oxide synthesis inhibition. Hypertension 22: 49–55, 1993. [DOI] [PubMed] [Google Scholar]
- 38.Sasser JM, Ni XP, Humphreys MH, Baylis C. Increased renal phosphodiesterase-5 activity mediates the blunted natriuretic response to a nitric oxide donor in the pregnant rat. Am J Physiol Renal Physiol 299: F810–F814, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Silva G, Beierwaltes WH, Garvin JL. Extracellular ATP stimulates NO production in rat thick ascending limb. Hypertension 47: 563–567, 2006. [DOI] [PubMed] [Google Scholar]
- 40.Silva GB, Garvin JL. Angiotensin II-dependent hypertension increases Na transport-related oxygen consumption by the thick ascending limb. Hypertension 52: 1091–1098, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Silva GB, Garvin JL. Extracellular ATP inhibits transport in medullary thick ascending limbs: role of P2X receptors. Am J Physiol Renal Physiol 297: F1168–F1173, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stoos BA, Garcia NH, Garvin JL. Nitric oxide inhibits sodium reabsorption in the isolated perfused cortical collecting duct. J Am Soc Nephrol 6: 89–94, 1995. [DOI] [PubMed] [Google Scholar]
- 43.Takenaka T, Nobe K, Okayama M, Kojima E, Nodaira Y, Sueyoshi K, Hoshi H, Watanabe Y, Takane H, Suzuki H. Aliskiren reduces morning blood pressure in hypertensive patients with diabetic nephropathy. Clin Exp Hypertens 34: 243–248, 2012. [DOI] [PubMed] [Google Scholar]
- 44.van Vark LC, Bertrand M, Akkerhuis KM, Brugts JJ, Fox K, Mourad JJ, Boersma E. Angiotensin-converting enzyme inhibitors reduce mortality in hypertension: a meta-analysis of randomized clinical trials of renin-angiotensin-aldosterone system inhibitors involving 158,998 patients. Eur Heart J 33: 2088–2097, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu P, Wang M, Luan H, Li L, Wang L, Wang WH, Gu R. Angiotensin II stimulates basolateral 10-pS Cl channels in the thick ascending limb. Hypertension 61: 1211–1217, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zimmerman AL, Yamanaka G, Eckstein F, Baylor DA, Stryer L. Interaction of hydrolysis-resistant analogs of cyclic GMP with the phosphodiesterase and light-sensitive channel of retinal rod outer segments. Proc Natl Acad Sci USA 82: 8813–8817, 1985. [DOI] [PMC free article] [PubMed] [Google Scholar]