

abstract
Autophagy is a major molecular mechanism that eliminates cellular damage in eukaryotic organisms. Basal levels of autophagy are required for maintaining cellular homeostasis and functioning. Defects in the autophagic process are implicated in the development of various age-dependent pathologies including cancer and neurodegenerative diseases, as well as in accelerated aging. Genetic activation of autophagy has been shown to retard the accumulation of damaged cytoplasmic constituents, delay the incidence of age-dependent diseases, and extend life span in genetic models. This implies that autophagy serves as a therapeutic target in treating such pathologies. Although several autophagy-inducing chemical agents have been identified, the majority of them operate upstream of the core autophagic process, thereby exerting undesired side effects. Here, we screened a small-molecule library for specific inhibitors of MTMR14, a myotubularin-related phosphatase antagonizing the formation of autophagic membrane structures, and isolated AUTEN-67 (autophagy enhancer-67) that significantly increases autophagic flux in cell lines and in vivo models. AUTEN-67 promotes longevity and protects neurons from undergoing stress-induced cell death. It also restores nesting behavior in a murine model of Alzheimer disease, without apparent side effects. Thus, AUTEN-67 is a potent drug candidate for treating autophagy-related diseases.
Keywords: age-dependent diseases, aging, AUTEN-67, autophagy induction, drug candidate, EDTP, HeLa cells, LC3B-II, model organism, MTMR14/Jumpy, neuroprotection, SQSTM1/p62
Introduction
The progressive accumulation of cellular damage (such as oxidized, cross-linked, misfolded, or aggregated proteins interfering with cellular functions) over adult life is a feature common to all aging cells, and accompanied by the development of diverse age-dependent degenerative pathologies, including cancer, neuronal demise (e.g., Alzheimer, Parkinson and Huntington diseases), immune deficiency, tissue atrophy, altered lipid metabolism, and diabetes.1-4 Effective elimination of such adverse factors is essential for maintaining cellular homeostasis, and primarily achieved by autophagy (cellular self-eating), which is a highly regulated, evolutionarily conserved self-degradation (catabolic) process of eukaryotic cells. During autophagy, parts of the cytoplasm are delivered into lysosomes for breakdown.1-4 Based on the mechanism of delivery, 3 major types of autophagy can be distinguished: macroautophagy, microautophagy, and chaperone-mediated autophagy. In macroautophagy (hereafter referred to as autophagy), a growing double-membrane structure composed of 2 lipid bilayers (also known as the phagophore) sequesters cytoplasmic constituents destined to be degraded.5,6 The completion of membrane growth results in a vesicle-like structure called the autophagosome.7 Subsequent fusion of an autophagosome with a lysosome generates an autolysosome, in which the enclosed material is broken down by acidic hydrolases.8 The core mechanism of autophagy involves more than 40 Atg/ATG (autophagy-related) proteins (Table S1) that are evolutionarily conserved from yeast to mammals.6 ATG proteins can be classified into functionally distinct complexes such as the autophagy-inducing complex containing ULK1 kinase (the homolog of yeast Atg1) and the class III phosphatidylinositol 3-kinase (PtdIns3K), whose catalytic subunit is PIK3C3 (phosphatidylinositol 3-kinase, catalytic subunit type 3; the mammalian ortholog of yeast Vps34) for vesicle nucleation, as well as a ubiquitin-like conjugation system for vesicle expansion. The ubiquitin-like protein conjugation complex mediates covalent binding of the key autophagy protein MAP1LC3/LC3 (microtubule-associated protein 1 light chain 3; an ortholog of the mammalian family of orthologs and paralogs of yeast Atg8) to the membrane component phosphatidylethanolamine.
When autophagy is downregulated or inhibited, cellular functions are often severely compromised, leading to elevated levels of cell death with apoptotic or nonapoptotic features, depending on the model used.8-14 Hyperactivation of autophagy can also induce massive cell loss.15,16 A natural, age-dependent decline in the autophagic activity can be observed in several organisms and human tissues.17,18 These changes are probably caused by random somatic mutations19,20 in genes involved in the mechanism or regulation of autophagy. Promoting basal levels of autophagy at advanced ages prevents the accumulation of damaged proteins, retards the incidence of degenerative processes, and extends life span.17,21,22 Such observations have prompted basic research and pharma industry to identify autophagy-enhancing drug candidates against a wide range of age-dependent disorders. Although several compounds have been identified, such as the immunosuppressant and antiproliferative drug rapamycin, such attempts have either led to severe undesired side effects or were not followed up with clinical applications, probably due to toxicity.23-28 Rapamycin, for example, is a potent candidate to trigger autophagic activity in patients diagnosed for different types of cancer.29,30 However, this compound enhances autophagy by blocking the MTOR (mechanistic target of rapamycin, [serine/threonine kinase]) kinase system, which in turn affects translation. Perturbation of general protein synthesis is certainly not a desired purpose in medical applications. Indeed, inhibition of MTOR arrests development in animal models.31,32 Hence, there is an urgent demand to identify novel autophagy-enhancing compounds, preferably with no side effect, for treating various age-dependent pathologies.
The growth of the phagophore membrane is driven by a protein complex whose central component is PIK3C3/Vps34 (Fig. 1).33 This enzyme converts the membrane component phosphatidylinositol into phosphatidylinositol 3-phosphate (phosphoinositides have numerous distinct roles in membrane trafficking). The biochemical reaction catalyzed by PIK3C3 is critical for autophagic structure formation, and inhibited by several members of the myotubularin-related phospholipid phosphatase (MTMR) protein family (note that not all of the MTMRs have a catalytically active phosphatase domain).34 The first identified mammalian MTMR capable of blocking autophagy in mammalian cell lines is MTMR14/Jumpy (myotubularin-related protein 14 (MTMR14), the ortholog of Drosophila EDTP (egg-derived tyrosine phosphatase (Table S1).35 The PIK3C3 antagonist MTMR14 prevents the autophagic process from fatal hyperactivation under conditions of cellular stress. In line with this proposal, defects in MTMR function are implicated in myotubular myopathy and Charcot-Marie-Tooth peripheral neuropathy,34 2 groups of diseases that are related to defective autophagy.36-41 In this study, we identified a small molecule, AUTEN-67, which impedes human MTMR14, and potently enhances the autophagic process in HeLa cells, isolated neurons and in vivo models, including Drospohila, zebrafish and mice. Administering AUTEN-67 led to life-span extension in Drosophila, and effectively protected neurons in primary cell cultures from undergoing oxidative-stress induced cell death. Thus, AUTEN-67 serves as a potent drug candidate with antiaging and neuroprotective effects.
Figure 1.
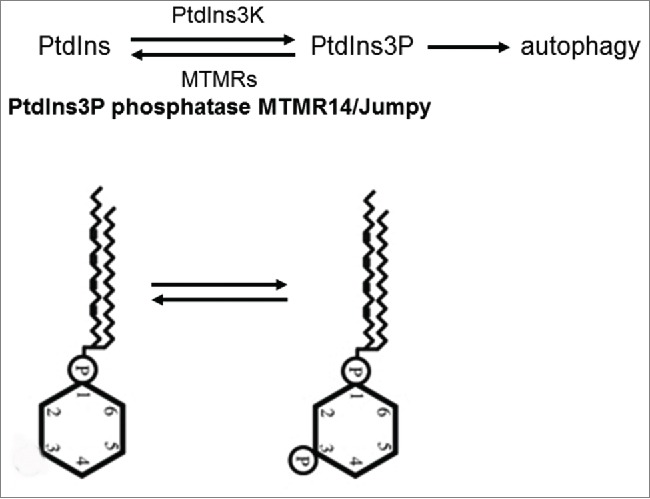
The role of MTMR14 in autophagy control. Formation of the autophagosome membrane depends on the infiltration of phosphatidylinositol-3 phosphate (PtdIns3P) into the growing membrane structure. PtdIns3P is converted from the substrate PtdIns by the class III PtdIns3K. Certain myotubularin phosphatases antagonize PtdIns3K, thereby impeding the autophagic process. The mammalian MTMR protein MTMR14/Jumpy is the ortholog of Drosophila EDTP.
Results
AUTEN-67 inhibits MTMR14 and enhances autophagy in HeLa cells
Chronic or robust upregulation of autophagy under cellular stress conditions frequently leads to the demise of the affected cell. This process is termed as an alternative—i.e., nonapoptotic—type of cell death.12,14 To avoid the harmful hyperactivation of the autophagic process in mammalian cells, MTMR14 cuts down on autophagy initiation via antagonizing PIK3C3, thereby ensuring their long-term survival.35 Therefore, the enzyme may serve as a promising drug target whose specific pharmacological inhibition potentially strengthens autophagy in pathological conditions that are characterized by insufficient levels of cellular self-cleaning.
In this study, we screened a small molecule library for specific inhibitors of human MTMR14 (see Materials and Methods). Several candidates were identified and their structural relatives were also assayed. One of these molecules is N-[3-(benzimidazol-1-yl)-1, 4-dioxonaphthalen-2-yl]-4-nitrobenzenesulfonamide (its identification number in the library is T0501-7132), which we labeled AUTEN-67 (autophagy enhancer-67) (Fig. 2A). This compound reduced the phosphatase activity of MTMR14 in a concentration-dependent manner (Fig. 2B). Applying at concentrations of 2, 10, and 100 µM, AUTEN-67 inhibited MTMR14 by nearly 3%, 25%, and 70%, respectively. So far, this interaction appears to be specific as AUTEN-67 did not influence the activity of either an unrelated protein, the cell cycle regulator CDC25B (cell division cycle 25B), or another phosphatase, PTPN1 (protein tyrosine phosphatase, nonreceptor type 1); there was 0% inhibition in both cases. We next tested whether AUTEN-67 can enhance autophagic activity in HeLa cells expressing an autophagy reporter, RFP-GFP-LC3B (LC3 fused to both the red and green fluorescent proteins); Figs. 2C to F.42 LC3B is a ubiquitin-like protein that is covalently conjugated to the forming autophagosome membrane, thereby labeling autophagic structures unambiguously.43 Moreover, the RFP-GFP-LC3B reporter can be used to assay autophagic flux (i.e., the dynamics of the autophagic process); on fluorescence images autophagosomes and autolysosomes are indicated by yellow and red foci, respectively (GFP protein is unstable in the acidic lumen of autolysosomes).42 By adding AUTEN-67 at 10 and 100 µM concentrations into the cell culture media, the number of autophagic structures (yellow and red structures) increased substantially—by almost 4 times—as compared to untreated control cells (Figs. 2C, F, G). These results indicate that AUTEN-67 acts as a potent enhancer of autophagic flux in HeLa cells. In accordance with these data, AUTEN-67 significantly raised levels of the membrane-bound form of LC3B (LC3B-II) in HeLa cells treated with bafilomycin A1 (Baf) inhibiting autophagosome-lysosome fusion (Fig. S1). Used at 100 µM, the effect of AUTEN-67 on LC3B-II accumulation was similar to, or even stronger than, that generated by 100 nM of rapamycin (Fig. S1B). In addition, levels of the autophagy substrate SQSTM1/p62 (sequestosome 1) were decreased by an extent comparable to that triggered by rapamycin treatment (Fig. S1A).
Figure 2.

AUTEN-67 inhibits MTMR14 and induces autophagic flux in HeLa cells. (A) Chemical structure of AUTEN-67 (AUTophagy ENhancer-67). Its catalog name is T0501-7132 (N-[3-(benzimidazol-1-yl)-1, 4-dioxonaphthalen-2-yl]-4-nitrobenzenesulfon-amide). (B) AUTEN-67 inhibits MTMR14 in a concentration-dependent manner. Treatments were carried out for 3 h (see Materials and Methods), and AUTEN-67 was applied at 2, 10, and 100 µM concentrations. P <0.001, paired Student t test. Untreated cells represent 0% inhibition value (not shown). For treated cells, relative inhibition values are displayed. (C to F) HeLa cells transgenic for a dual-labeled autophagy reporter (RFP-GFP-LC3B)42 were treated with DMSO (control) (C), Baf (which inhibits autophagy at the autophagosme-lysosome fusion stage) (D), rapamycin (an autophagy inducer) (E), and AUTEN-67 (F). Yellow dots correspond to autophagosomal structures, red foci indicate autolysosomes. (G) Quantification of autolysosomes in HeLa cells treated with DMSO only (control), Baf, rapamcin and/or AUTEN-67. The number of red foci was determined in 30 cells, assays were performed in triplicates. Bars represent s.e. Relative to control (DMSO), each treated sample shows a significant difference (P<0.001; paired Student t test). (H) AUTEN-67 enhances viability of HeLa cells (survival rate was determined by the MTT method, see Materials and Methods). The dashed red line represents the mean survival rate of control, untreated cells (100%). Bars represent s.e.m. At each AUTEN-67 concentration tested, the statistical difference: P<0.001 (paired Student t test).
Since physiological levels of autophagy are critical for maintaining cellular homeostasis, we examined the survival of HeLa cells exposed to AUTEN-67 treatment. In this setting, AUTEN-67 effectively elevated cell survival in a concentration-dependent manner (Fig. 2H). When applied at 100 µM, this small molecule inhibited cell loss by almost 40%, as compared with untreated controls. We also assessed whether AUTEN-67 interferes with endocytosis because many ATG genes have been reported to influence this cellular membrane formation-dependent process as well. Following treatment with AUTEN-67, we found no change in endocytic activity in a human cell line transgenic for an EGFR (epidermal growth factor receptor)-GFP reporter (Fig. S2A to D) and in Drosophila fat body cells expressing the early endosome marker Rab5-CFP (cyan fluorescent protein) (Fig. S2E to H). Taken together, AUTEN-67 significantly increases autophagic flux in, and promotes the survival of, HeLa cells.
AUTEN-67 increases the amount of autophagic structures in the Drosophila fat body
MTMR14 negatively regulates autophagy in mammalian cells and zebrafish.35,44 A protein alignment analysis uncovered that human MTMR14 contains some evolutionarily conserved domains between the amino acids 305 and 460 (Fig. S3). This finding prompted us to monitor the inhibitory effect of AUTEN-67 on autophagy in animal genetic models. First, we examined the fat body of feeding L3F stage Drosophila melanogaster larvae transgenic for a mCherry-Atg8a reporter.43 The fat body serves as a tractable tissue model for studying developmentally programmed and stress-induced autophagy. We found that Drosophila EDTP/MTMR14 effectively downregulated the autophagic process in fat body cells (Fig. S4). Clonal inactivation of EDTP caused a significant increase in the amount of autophagic structures in the affected cells, as compared with the corresponding controls (Fig. S4). Both basal and starvation-induced autophagy was negatively regulated by EDTP. Consistent with these results, clonal hyperactivation of EDTP strongly inhibited the formation of autophagic structures in fat body cells exposed to nutrient deprivation (Fig. S5). Thus, EDTP effectively represses both unstressed (basal) and stress-induced autophagy in this organism.
Next, we treated L3F stage Drosophila larvae with AUTEN-67, and examined the amount of autophagic structures in their fat body cells. In untreated control animals, the fat body cells displayed only diffuse red signals, showing no or minimal levels of developmental and housekeeping autophagy (Fig. 3A). In contrast, AUTEN-67 supplemented into the agar media at 100 µM vigorously induced the formation of mCherry-Atg8a-positive red foci corresponding to autophagic structures in the fat body cells of L3F stage larvae (Fig. 3B). The effect of AUTEN-67 treatment in larvae exposed to starvation was also tested. Food deprivation per se significantly increased the number of autophagic structures in the fat body (Fig. 3C). Under such conditions, AUTEN-67 applied only at 10 µM concentration caused a robust upregulation of autophagy (Fig. 3D).
Figure 3.

AUTEN-67 induces autophagy in Drosophila via inhibiting EDTP. (A) Fat body cells from a feeding L3 stage larva (90 h) transgenic for a mCherry-Atg8a reporter show basal levels of autophagic activity. (B) AUTEN-67 (100 µM) treatment results in a massive accumulation of Atg8a-positive structures in the fat body of a L3F larva. (C) Fat body cells of a starved L3 stage larva (90 h) accumulate autophagic structures. (D) AUTEN-67 (10 µM) increases the amount of Atg8a-positive structures in fat body cells of a starved L3 larva, as compared to untreated control. (E to G”) Clonal overexpression of EDTP in fat body cells largely, but not completely, inhibits the formation of autophagic structures (red foci) induced by AUTEN-67 at 10 µM (E and E'), 100 µM (F and F') and 50 µM (G and G') concentrations. EDTP-overexpressing cells are green in the left panels, and outlined by dotted lines in the corresponding right panels. (G”) Quantification of autophagic structures in Drosophila fat body cells treated with AUTEN-67 (50 µM). EDTP-overexpressing (green/outlined) cells contain much fewer autophagic structures than nongreen controls (*: P <0.05, **: P<0.01, ***: P<0.001; paired Student t test). Bars represent s.e. (H) Expression of Atg18a/WIPI1 in fat body cells from a untreated L3 stage larva (control). Red foci label phagophores. (I) Atg18a/WIPI1 abundantly accumulates in fat body cells of an L3 stage larva treated with 100 uM AUTEN-67. In figures A to G, H and I, bars indicate 10 µm, blue coloring (Hoechst staining) labels nuclei.
We also aimed to determine whether the autophagy-enhancing effect of AUTEN-67 in this organism was specific, i.e. whether it occurred through inhibiting EDTP. To this end, we examined L3F larvae whose fat body cells clonally overexpressed EDTP (EDTPEY22967). Upon treatment with AUTEN-67 at different concentrations, the GFP-labeled (green) fat body cells overexpressing EDTP contained many fewer autophagic structures (i.e., mCherry-Atg8a-positive red dots) than those lacking the green fluorescent signal (nonoverexpressing cells) but having otherwise an identical genetic background (Fig. 3E to F'). Quantification of autophagic structures revealed a roughly 30- to 60-fold increase in nongreen cells in response to AUTEN-67 treatment (at 50 µM), and this enhancement was largely inhibited in green (i.e., EDTP-overexpressing) cells (Fig. 3G to G”). It has previously been shown that depletion of MTMR14 causes WIPI1/Atg18 (WD repeat domain, phosphoinositide interacting 1) accumulation on the phagophore. We found that this is also the case with AUTEN-67 treatment (Figs. 3H, I). Together, these results strongly suggest that AUTEN-67 promotes autophagy in the Drosophila fat body via inhibiting EDTP.
We also measured SQSTM1 levels in wild-type (control) versus EDTP defective (caused by the loss-of-function mutant EDTP allele MI08496) fat body samples treated with AUTEN-67 (Fig. 4). Adding the agent to control cells robustly impeded the accumulation of SQSTM1, as compared with untreated controls. However, this inhibitory effect of AUTEN-67 on SQSTM1 levels was completely eliminated in EDTPMI08496 mutant background, implying that EDTP is the sole fly MTMR protein through which AUTEN-67 enhances autophagy. Similar results were obtained by quantifying Atg8a-positive autophagic structures in control vs. EDTPMI08496 mutant genetic backgrounds (Fig. S6).
Figure 4.
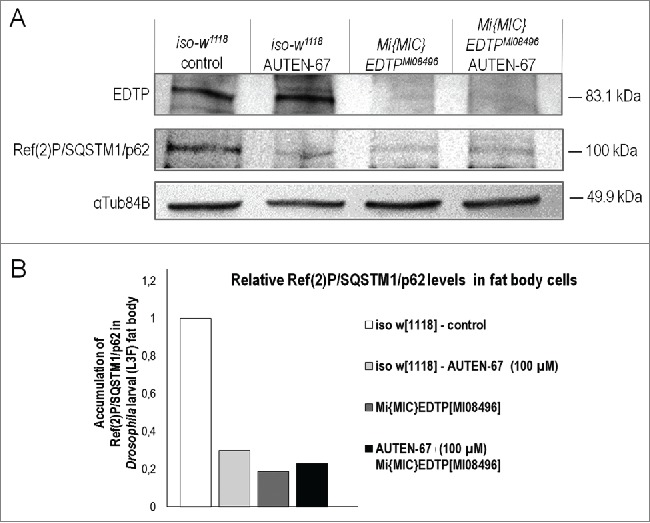
Activation of autophagy by AUTEN-67 depends on EDTP function. (A) Western blot analysis demonstrating Ref(2)P/SQSTM1/p62 protein levels in wild-type (control) and EDTP-deficient (MI08496 mutant) Drosophila fat body samples. EDTP protein is not detectable in EDTPMI08496 mutant samples (upper row). This indicates that EDTPMI08496 is a strong loss-of-function allele. AUTEN-67 cannot further decrease p62/Ref(2)P accumulation in EDTPMI08496 genetic background, as compared with the untreated mutant sample. Thus, EDTP may represent the sole MTMR paralog through which AUTEN-67 induces autophagy. α-Tubulin84B serves as an inner control. (B) Quantification of Ref(2)P/SQSTM1/p62 levels in fat body samples. Band intensities from the western blot (panel A) were measured.
AUTEN-67 increases autophagy in zebrafish and mice
To further confirm the evolutionary conservation of the molecular mechanisms by which AUTEN-67 influences autophagy, we assayed zebrafish transgenic for a Gfp-Lc3 reporter (Fig. 5A to D).45,46 The addition of AUTEN-67 at 50 µM to the samples led to a moderate intensification in Gfp fluorescence, relative to untreated control fish samples (for quantifying Gfp intensity, see the corresponding figure legend). The number of Gfp-positive spots was higher in treated samples (Figs. 5B-D).
Figure 5.
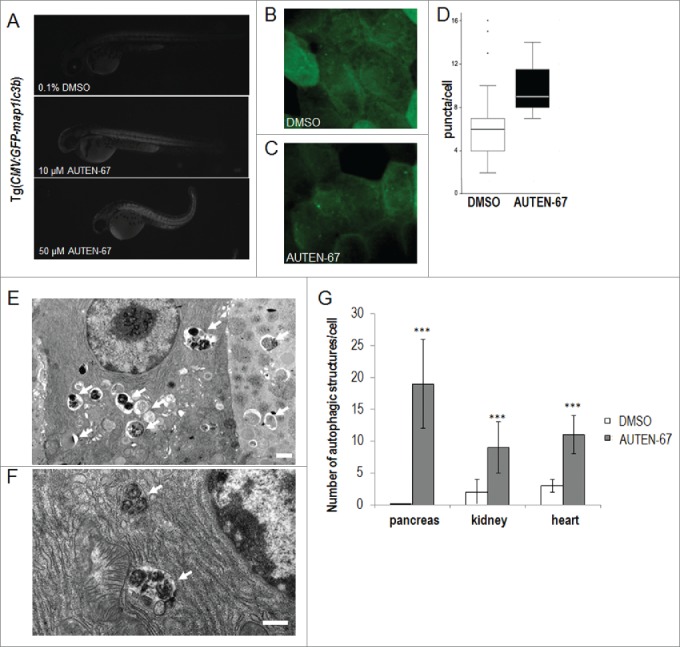
AUTEN-67 enhances autophagy in zebrafish and mice. (A) AUTEN-67 increases the glowing intensity of an autophagy reporter (Gfp-Lc3b) in zebrafish embryos. GFP intensity increases by 1.36- and 1.38-fold upon administration of 10 and 50 µM of AUTEN-67, respectively (P < 0.001, paired Student t test). Fluorescent images. N=4, at both concentrations. (B) Fluorescence image of an untreated (DMSO) Gfp-Lc3 fish sample (large magnification). (C) Fluorescence image of a sample treated with AUTEN-67. Green foci indicate autophagic structures. (D) Quantification of green punctae in untreated vs. treated cells. P < 0.01; paired Student t test. (E) Transmission electron microscopy (TEM) image showing the ultrastructure of exocrine pancreatic tissue from a mouse treated with AUTEN-67. Arrows indicate autophagic structures (autophagosomes and autolysosomes). Note that control cells display almost no sign of autophagic structures observed at the ultrastructural level.47 Scale bar: 1 µm. (F) TEM picture displays the ultrastructure of a pancreatic cell from a mouse administered orally with AUTEN-67. Arrows indicate late autolysosomes, implying that AUTEN-67 induces autophagic degradation. Scale bar: 1 µm. (G) Quantification of autophagic structures in exocrine pancreatic (“pancreas”), kidney epithelial (“kidney”) and heart muscle (“heart”) tissues from mice treated with AUTEN-67 solved in DMSO (red columns) and with DMSO only (blue columns). Bars represent s.e. In each tissue examined, differences between the corresponding treated and untreated samples are statistically significant (***: P < 0.001; paired Student t test).
Finally, we injected AUTEN-67 intraperitoneally into mice, and, after sacrificing the animals 1 h following treatment, examined autophagic structures in different tissues by transmission electron microscopy (EM). Exocrine pancreatic, kidney epithelial, and heart muscle cells from treated animals displayed a massive increase in autophagic activity, as compared to untreated controls (Figs. 5E to G). For example, in untreated pancreatic cells, profiles of autophagic vacuoles can very rarely be seen in EM sections (almost zero level in Fig. 5G).47 In contrast, AUTEN-67 treatment induced the formation of a large number of autophagic structures in this tissue (black arrows in Fig. 5E, and also see Fig. 5G). Many of these structures correspond to late autolysosomes (Fig. 5F), demonstrating that AUTEN-67 acts as a potent stimulator of autophagic flux in vivo. We gained similar data when AUTEN-67 was administered orally. Taken together, these results show that in mice, AUTEN-67 is effectively absorbed, allowing its penetration into different tissues where it enhances autophagic activity.
AUTEN-67 extends life span in Drosophila
The aging process is driven by the lifelong, progressive accumulation of unrepaired cellular damage, leading to a natural decline in the fitness of an organism over time.48 Accumulating evidence indicates that autophagy—a major pathway for eliminating toxic and superfluous cytoplasmic materials—functions as a central regulatory mechanism of aging.8,49-51 In mice, for example, the overexpression of Atg5 significantly promotes longevity.52 Moreover, prolonged expression of Atg8a gene in the nervous system of old adults is sufficient to extend life span in Drosophila.17 Hence, we asked whether AUTEN-67 also has the potential to promote longevity in flies. By applying it from the onset of the adulthood, this compound applied at 30 µM significantly increased the maximum life span of the treated animals (indicated by small arrows on Fig. 6). The maximum life span of treated animals exceeded that of controls by nearly 30%. The life-span-extending effect of AUTEN-67 in this organism was evident mainly at advanced ages when the life-span curves of untreated and treated animals significantly separated from each other. Furthermore, AUTEN-67 increased life span in both sexes (note that on Fig. 6, only cumulative data are shown). These results suggest that at least in Drosophila, AUTEN-67 is not a toxic material and causes no deleterious side effect.
Figure 6.
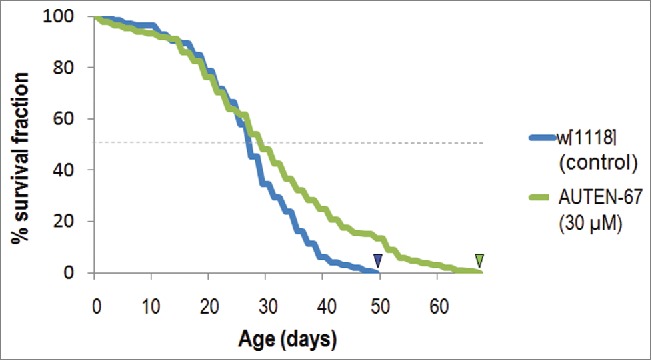
AUTEN-67 extends life span in Drosophila. Newly hatched male and female adults were treated with AUTEN-67 (at 30 µM of final concentration), and dead animals were assayed every 2 d. Kaplan-Meier curves were generated by the SPSS program. Treated animals live significantly longer than untreated controls (P <0.001). Arrowheads indicate maximum life-span data.
AUTEN-67 protects neurons from oxidative stress
By effectively eliminating cellular damage, autophagy protects neurons from undergoing cell death and maintains their normal functions.53 In Drosophila, the capacity of autophagic activity declines progressively as the organism ages.17 Thus, promoting basal levels of autophagy by pharmacological means in the nervous system of patients at advanced ages seems to be a reasonable intervention in the clinics. To test whether it can also exert a neuroprotective effect, we treated mouse cortical neurons in primary cell cultures with AUTEN-67. We found that the molecule indeed causes decreased levels of LC3B-II (the lipid-conjugated form of LC3B) in this in vitro model (Fig. 7A). Since LC3B-I (the soluble form of LC3B) levels were not significantly changed in response to administering AUTEN-67, it is likely that a large portion of LC3B-II proteins became enzymatically degraded in autolysosomes. This is supported by the finding that rapamycin treatment also led to a lowered accumulation of LC3B-II (Fig. 7A). Furthermore, the amount of SQSTM1 markedly decreased when AUTEN-67 was added to the cells. Based on these data we conclude that AUTEN-67 is capable of inducing autophagic flux in neurons. In good agreement with autophagy induction, AUTEN-67 treatment significantly enhanced the viability of isolated neurons in a concentration-dependent manner (Fig. 7B). We also tested the accumulation of SQSTM1 and LC3B-II proteins under conditions where AUTEN-67 was cotreated with the autophagy inhibitor Baf or chloroquine (these inhibitors compromise the fusion of autophagosome with lysosome) (Figs. 7C and C'). Both proteins accumulated considerably when autophagy was blocked even in the presence of AUTEN-67. Thus, AUTEN-67 enhances not only the induction (generation of autophagosomes) but also the degradation (generation of autolysosomes) phases of autophagy.
Figure 7.
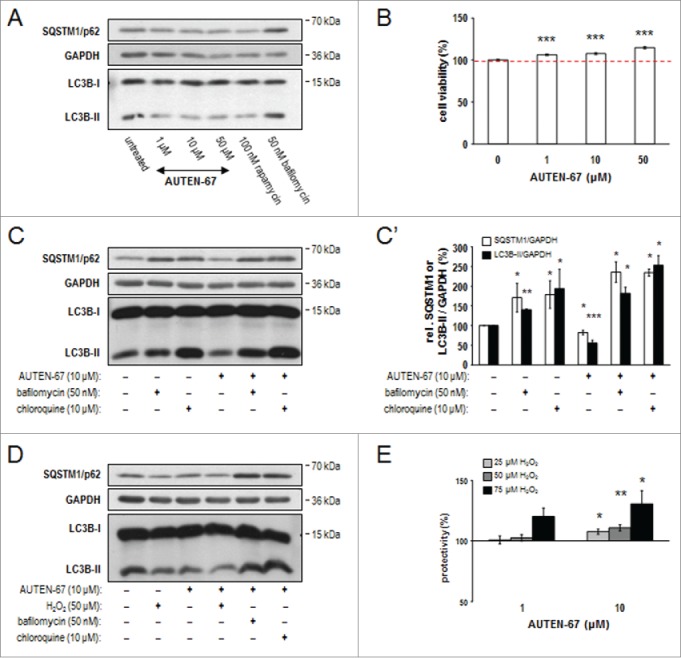
AUTEN-67 induces autophagic flux and increases viability in neurons. (A) Western blotting demonstrates relative SQSTM1/p62 and LC3B-II protein levels in murine primary neuronal cell cultures. Similar to rapamycin treatment, AUTEN-67 decreases the level of both proteins. LC3B-I levels remain nearly constant. (B) AUTEN-67 increases viability of isolated neurons in a concentration dependent manner. The dashed red line indicates mean relative viability of untreated control cells (100%). Survival of cortical neurons from a 7-d-old cell culture prepared from a 15-d-old mouse embryo is significantly increased in response to AUTEN-67 treatment. Survival rate was determined by the MTT method (see Materials and Methods). Data represent the results of 5 independent treatments. Bars represent s.e.m. For each treated sample, P < 0.001 (Student t test). (C) Relative levels of SQSTM1/p62 and LC3B-II proteins (western blot). AUTEN-67 decreases the level of both proteins only in the absence of autophagy inhibitors (Baf and chloroquine). (C') Quantification of band intensities from relevant western blots. Bars represent s.e. *: P < 0.05; **: P < 0.01: ***: P < 0.001, compared to control values (unpaired t test). (D) Western blot analysis showing relative SQSTM1/p62 and LC3B-II levels. AUTEN-67 increases autophagic flux in isolated neurons exposed to oxidative stress. (E) AUTEN-67 strongly enhances the viability of isolated neurons exposed to H2O2 treatment. Survival rates were determined by the MTT method. Data represent relative changes to cells treated with H2O2 only (control). Columns display the results of 4 independent treatments. Bars represent s.e. (Student t test). On panels A, C and D, GAPDH serves as control.
Next, we tested whether AUTEN-67 protects neurons from oxidative stress-induced cell death. Isolated neurons were exposed to 25 to 75 µM hydrogen peroxide (H2O2), an agent that massively causes oxidative stress, and treated with AUTEN-67 at various concentrations (1-10 µM). In this assay, AUTEN-67 markedly enhanced both autophagic flux (Fig. 7D) and the survival of neurons (Fig. 7E). Applying at 10 µM of the compound, the loss of neurons was suppressed by around 30%, relative to cells exposed to 75 µM H2O2 only. Thus, AUTEN-67 exhibits a spectacular neuroprotective power.
AUTEN-67 restores nesting behavior in mice expressing the human APP/amyloid precursor protein
Finally, we tested AUTEN-67 in an in vivo (murine) neurodegenerative model. Mice expressing the human APP (amyloid β [A4] precursor protein) serve as a tractable genetic model of Alzheimer disease.54,55 We assayed nesting behavior in control versus AUTEN-67-treated animals (for quantifying nesting behavior and performing the assay, see the Materials and Methods section; Fig. S7). Upon administering AUTEN-67 for a 3-mo period, impaired nesting behavior was restored by around 30% (Fig. 8A). We also measured the amount of 2 amyloid proteins, β40 and β42, in treated vs. untreated animals, and found a significant (P<0.05; Student t test) decrease in protein levels in the treated samples (Figs. 8B and C). These results suggest that AUTEN-67 improves neuronal functions in mice, probably by reducing levels of toxic proteins. Importantly, AUTEN-67 administered orally was quickly (after 30 min following the treatment) detectable from blood and, to a lesser extent, brain samples (Fig. S8). We conclude that absorption of the molecule from the intestine occurs quite effectively, and that it can cross the blood-brain barrier. Furthermore, general behavior, movement, feeding, body temperature, and weight in the treated animals were comparable with those observed in control (untreated) animals (Fig. S9, and data not shown), implying that application of AUIND-67 for 3 mo does not lead to undesired side effects.
Figure 8.
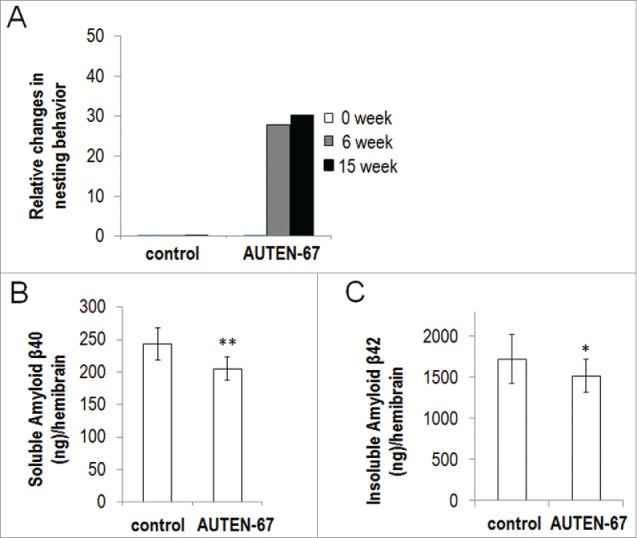
AUTEN-67 restores nesting behavior and decreases APP levels in a mouse model of Alzheimer disease. (A) Relative changes in the ability of nesting behavior, as compared to controls. Animals were classified into 5 distinct nesting groups (see Materials and Methods and the Fig. S7) at 0 (white columns), 6 (gray columns) and 15 (black columns) weeks of treatments. Administration of AUTEN-67 restores nesting ability of mice transgenic for human amyloid precursor protein (APP). For difference between control and treated animals: P < 0.001, unpaired t test. (B, C) AUTEN-67 decreases Amyloid β levels in the hemibrain of mice transgenic for human APP. (B) AUTEN-67 treatment decreases soluble human amyloid β40 protein levels in the hemibrain of mice transgenic for human APP (**: P < 0.01, paired Student t test). (C) AUTEN-67 treatment decreases insoluble human amyloid β42 protein levels in the hemibrain of mice transgenic for human APP (*: P < 0.05, paired Student t test). Bars represent s.e.
Discussion
In this study, we identified a potent autophagy-enhancing small molecule, AUTEN-67 (Fig. 2A). We found that this agent inhibits the phosphatase activity of human MTMR14 (Fig. 2B), a negative regulator of autophagic membrane formation. AUTEN-67 did stimulate autophagic flux (i.e., it increased the number of both autophagosomes and autolysosomes) in human (HeLa) cell lines (Figs. 2C to G), as well as enhanced the amount of autophagic structures in Drosophila, zebrafish, and mice (Figs. 3 and 5). Based on these data we suggest that AUTEN-67 interferes with a functional domain of MTMR14/EDTP that is highly conserved among these species (Fig. S3). Daily treatment of mice with AUTEN-67 for a 3-mo period did not significantly affect the general behavior, activity, and morphology of the animals tested (Fig. S9), implying that it has no apparent side effects. Accordingly, AUTEN-67 was able to extend life span in Drosophila (Fig. 6). Furthermore, AUTEN-67 effectively protected mouse cortical neurons in primary cell cultures against H2O2-induced oxidative stress (Fig. 7E), and fixed nesting behavior in mice expressing the human APP gene (Fig. 8A). What is the significance of these findings? Both the aging process and the incidence of age-dependent diseases are known to result from the lifelong, progressive accumulation of cellular damage.8,48 Autophagy functions as a major catabolic process by which toxic and superfluous cellular constituents are effectively degraded, thereby maintaining the homeostasis of the cytoplasmic milieu.1,2 The capacity of autophagic degradation, however, displays a characteristic age-dependent decline.17 In good accord with these findings, a genetic intervention prolonging Atg8 expression only in neurons protects cells from accumulating ubiquitinated (damaged) proteins, and extends the life span of the organism.17 A similar feature in the age-dependent capacity of autophagy has been observed in humans too: brain samples from elderly patients show elevated levels of SQSTM1 protein serving as a substrate for selective autophagic degradation, as compared to younger ones.56 Based on these data, one can speculate that in humans prolongation of basal autophagy by pharmacological interventions delays substantially the development of a wide range of degenerative pathologies.
Aging is thought to be a collection of seemingly independent degenerative processes leading to disease, and, ultimately, death. Thus, if the cure of such pathology were to be found, it would only increase the risk of an individual to acquire another type of age-dependent disease. Maintaining normal levels of autophagic activity appears to be a potent method of delaying the incidence of various degenerative pathologies and preserving the healthy state of the organism.
The pharmaceutical industry is currently exerting enormous efforts to identify novel autophagy enhancers. However, the majority of the candidates identified so far act upstream of the core autophagic process, thereby generating undesirable side effects. Certain myotubularin phosphatases are known to function as negative regulators of autophagy via antagonizing PtdIns3K, a core component of the process (Fig. 1).35-37 As excessive autophagy often leads to cell death, these enzymes function to inhibit the harmful hyperactivation of basal and stress-induced autophagy. Identified by the present study (Fig. 2A), AUTEN-67 effectively compromised the phosphatase activity of MTMR-14 (Fig. 2B), and stimulated autophagy in both in vitro and in vivo (Figs. 2C to H, 3 and 5). Chronic absence of MTMR14 function, in particular during early development, can be associated with autophagy hyperactivation and the incidence of certain myo- and neuropathies.34 However, moderate activation of cellular self-cleaning by acute or periodic inhibition of MTMR14 in advanced ages may help to maintain cellular homeostasis, enhance the survival of neurons, and protect against the occurrence of diverse human pathologies. Therefore, AUTEN-67 can be used as a potent drug candidate to decelerate the aging process and counter various age-dependent degenerative diseases. Taken together, further development of AUTEN-67, including structural determination of the MTMR14–AUTEN-67 complex, molecular optimization, and a detailed pharmacological analysis, seems to be a reasonable effort.
Materials and methods
Selection of putative MTMR14 inhibitors
A 2-pronged strategy was applied. One method included the collection of known phosphatase inhibitors and running their chemical structures through a similarity test in a diverse compound library. The other method included a chemical microarray screening of 9000 immobilized small molecules (obtained from Enamine Ltd, Ukraine) tested in a binding assay with fluorescently labeled MTMR14 protein in order to identify new active scaffolds. Compounds showing appreciable binding were tested in the described MTMR14 assay. Small Molecule Microarray Screening: AvichemixTM small molecule microarrays (Avicor Ltd. Szeged, Hungary) were used for protein-small molecule interaction studies. Small molecule microarrays were constructed and prepared for screening as previously described.57,58 Briefly, for printing with MicroGrid II mechanical microarray microspotter (BioRobotics, Cambridge, UK) chemically activated glass slides (Avicor Ltd., Szeged, Hungary) were used, which contain a treelike, branched dendrimer structure with reactive functional groups at the terminal position.59 Fluorescent-labeled MTMR14 was prepared with Alexa Fluor 647 carboxylic acid succinimidyl ester (Molecular Probes, A-20106) as previously described.60 The affinity experiments were carried out in a hybridization station (Ventana Discovery, Tucson, AZ, USA). 10 µl of each labeled protein extract was applied in 200 µl phosphate buffered saline [PBS (Sigma, P4417) containing 2% BSA (Sigma, A2153)] (final dilution 1:1000). The slides were incubated at 37°C for 2 h. Afterwards the slides were removed from the hybridization station washed with 2x SSC (Sigma, S0902) with 0.5% Tween 20 (Sigma, P7949) for 1 min. Finally the slides were rinsed with deionized water and dried. The slides were scanned with a ScanArray Lite (GSI Lumonics, Billerica, USA) microarray scanner at 633 nm. Compounds that interacted with MTMR14 were identified as fluorescent intensive spots (4-fold over background average intensity signal). After spotting the microarrays were scanned using an Agilent G2505B Microarray Scanner (Agilent Technologies, Santa Clara, CA, USA) (lasers: 532 nm SH-YAG and 633 nm HeNe) with 10 μm resolution and maximum laser power. Microarray images were analyzed by the Feature Extraction Software (Agilent, Waldbronn, Germany) and GenePix Pro 6.0 (Axon Instruments, Foster City, CA, USA).
Phosphatase assays
MTMR14 assay: MTMR14 protein was purchased from OriGene Technologies (022485). The biochemical assay consisted of purified MTMR14 protein (OriGene Technologies, TP300809) (37 ng/reaction), 100 µM phosphatidylinositol 3-phosphate (Echelon Biosciences, P-3008a) in 25 mM Tris-HCl, pH 6 buffer (Molar Chemicals, 09350-101-190) containing 2 mM DTT (Sigma, 17-1318-01) in 10-µl total volume. The reactions were incubated at room temperature and after 3 h free phosphate was measured with the Malachite green reagent (Sigma, M9015). PTP1B assay: PTP1B protein was purchased from Sino Biological (10304-H07E). Phosphatase activity was measured in a colorimetric biochemical assay using the nonspecific phosphatase substrate para-nitrophenyl-phosphate (pNPP, Sigma, S0942). Each 50 µl reaction contained 71 ng PTP1B enzyme in 30 mM Tris-HCl pH=8 buffer with 1 mM EDTA (Sigma, E5134-500G), 75 mM NaCl (Molar Chemicals, 07220-101-190), 1 mM DTT, 0.033% BSA and 20 mM pNPP (Sigma, 73737). Reactions were incubated at room temperature for 2 h and signals were recorded at 405 nm. CDC25B assay: CDC25B protein was purchased from Bioneer (E-3354). Phosphatase activity was measured in a colorimetric biochemical assay using the non-specific phosphatase substrate para-nitrophenyl-phosphate (pNPP). Each 50 µl reaction contained 150 ng CDC25B enzyme in 30 mM Tris-HCl pH=8 buffer with 1 mM EDTA, 75 mM NaCl, 1 mM DTT, 0.033% BSA and 20 mM pNPP. Reactions were incubated at room temperature for 5 h and signals were recorded at 405 nm.
Assaying autophagy flux in HeLa cells
HeLa cells transgenic for a functional GFP-RFP-LC3B autophagy reporter42 were cultured in DMEM (Dulbecco's modified Eagle's medium; Sigma, D7777) containing 4500 mg/l glucose, 10% heat inactivated fetal calf serum (Merck, 1083421000), 40 µg/ml gentamycin (Hungaropharma, 20130529) and 600 µg/ml G418 (Sigma, G8168). 3 × 104 cells were plated onto 13 mm poly-D-lysine (Sigma, P7405) coated coverslips (Sigma, P0899) in 24-well plates (Greiner, M8812-100EA) 24 h before the treatment. Cells were exposed to 1, 10, and 100 µM AUTEN-67 for 6 h. As controls, 1% DMSO (Sigma, 41640), 200 nM rapamycin (an autophagy inducer; Sigma, R8781) and 100 nM bafilomycin A1/Baf (an autophagy inhibitor; Sigma, B1793) were used. For fluorescence microscopy study, cells were fixed in 4% paraformaldehyde (Taab, P001/0) and mounted in Mowiol 4.88 (Polysciences, 324590) supplemented with bis-benzimide (Sigma, B1155) for nuclear staining. 5 epifluorescent pictures were taken in each condition using a BX51 microscope (Olympus, Unicam, Budapest, Hungary) equipped with a FluoViewII camera and the AnalysisPro software (Olympus), using a 60×/1.4 oil Plan Apochromat objective and the appropriate filter sets (DAPI: BP330-385/DM400/BA420; GFP: BP460-500/DM505/BP510-560; RFP: BP480-550/DM570/BA590). RFP intensity shows both the soluble LC3 molecules and activated LC3 along the whole autophagy process (late stages included). Conversely, GFP intensity shows soluble LC3B molecules and LC3B only in the early stages of autophagy, as GFP fluorescence is bleached by the acidic pH of the lysosome in late autophagosomes. To study autophagic activity, the number of RFP- and GFP-specific foci was measured in each cell by ImageJ software. Data were analyzed with the paired Student t test, statistical significance was set at P<0.05.
Cell viability assays
HeLa cells were seeded into (12-well) cell culture plates (Sigma, M8812) at 105 cells/well density in DMEM (Sigma, D7777) supplemented with 10% fetal calf serum (Life Technologies, 10106-169). Cells were treated with AUTEN-67 (at different concentrations), Baf (50 nM) and rapamycin (100 nM) according to the scientific design for 24 h. Cell viability was measured by the MTT method.61 Briefly, cells grown in 96-well plates were treated with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, Sigma, M2128) in a final concentration of 250 µg/ml. After 2 h of incubation, cells and formazan crystals were dissolved in acidic (0.08 M HCl) isopropanol (Merck, 109634). Optical density was determined at a measuring wavelength of 570 nm against 630 nm as reference with a Multiscan Bia-Rad ELISA reader (Thermo, Bio-science, Budapest, Hungary). Assays were carried out on 6 parallel wells. At least 5 independent viability assays were carried out. Data are shown as average ± s.e.m., compared using the Student t test (P < 0.05).
Culture and drug treatment of fly strains
Flies were raised on standard cornmeal medium (http://flystocks.bio.indiana.edu/Fly_Work/media-recipes/harvardfood.htm) at 18–25°C, with some exceptions that are elsewhere indicated. L3 feeding larvae were treated 3 h prior to dissections. Animals were placed into a suspension consisting of instant yeast medium, supplemented by AUTEN-67 (T0501-7132) solved in DMSO (Sigma, D8418) (or the same volume of DMSO only for untreated, control samples).
Dissection and microscopy of Drosophila larval fat body samples, quantification, and statistics
Preparation of fat bodies was carried out in PBS (Sigma, P4417) solution. Covering was achieved in glycerine (Sigma, G2289)-PBS (8:2) solution containing Hoechst 33342 (Molecular Probes, H-1399) at 10 μM final concentration. Microscopy was performed with a Zeiss AxioImager Z1 epifluorescence microscope equipped with an ApoTome semiconfocal setup with objective Plan-NeoFluar 40x 0.75 NA (Eötvös Loránd University, Budapest). Images were analyzed using the ImageJ 1.45s software. Statistics were calculated by the program MATLAB 7.12.0 (http://en.softonic.com/s/matlab-7.12-full-version).
Measuring autophagy activity in the fat body of feeding Drosophila larvae at the L3 stage
Autophagy activity was assayed in a fly strain with genotype of hsFlp;pAct<CD2<Gal4,UAS-nlsGFP,r4-mCherry-Atg8a, at the feeding L3 larval stage. Without treatment, basal autophagy shows no detectable level of mCherry-Atg8a-positive red foci in the fat body at this developmental stage. Two h prior to treatment, 90-to-94-h-old larvae were placed into a suspension consisting of instant yeast medium (Lesaffre, 59703). AUTEN-67 (T0501-7132) dissolved in DMSO was added into final concentrations of 10 and 100 µM. Larvae were treated for 2 h at 25°C, and compared with untreated control ones with the same age and genotype. To examine the effects of AUTEN-67 on autophagic activity in an EDTP-defective genetic background, we used the EDTP mutant allele Mi{MIC}EDTPMI08496 (Bloomington Drosophila Stock Center – BDSC, 44782). The transposon is located in the first intron of EDTP and serves as a genetrap. Both control w1118 and Mi{MIC}EDTPMI08496 flies were crossed with males carrying r4-mCherry-Atg8a, and a large deletion, Df(2R)BSC161, which overlaps the EDTP genomic region (BDSC, 9596). F1 larvae were raised at 29°C, 74-to-78-h-old L3 feeding larvae were treated with AUTEN-67 (100 μM) as described above. For statistics, unpaired t tests were applied.
Monitoring the accumulation of Atg18a-positive phagophores
To investigate the effect of AUTEN-67 on the phagophore accumulation we used of hsFlp; pAct<CD2<Gal4, UAS-nlsGFP, r4-mCherry-Atg18a strain.62 86-to-90 h-old L3 feeding larvae were treated with AUTEN-67 (100 μM). Unpaired t tests were applied.
Testing autophagic activity in Drosophila fat body cells clonally overexpressing EDTP
Tests were performed in a strain derived from crossing between hsFlp;pAct<CD2<Gal4,UAS-nlsGFP,r4-mCherry-Atg8a62 and y1 w67c23; P{EPgy2}EDTPEY22967 (BDSC, 22600). The fat body of F1 offspring at the L3 larval stage (90 h) overexpresses clonally EDTP, which antagonizes autophagic membrane formation (only the EDTP-overexpressing fat body cells are green). Animals were treated with AUTEN-67 dissolved in 50 µM DMSO. Untreated controls were maintained on media containing DMSO only. Statistics were calculated by the program MATLAB 7.12.0; unpaired and paired t tests were applied.
Testing autophagic activity in Drosophila larval fat bodies defective or hyperactive for EDTP
Tests were performed on animals derived from crossing between hsFlp; pAct<CD2<Gal4, UAS-nlsGFP, r4-mCherry-Atg8a and i) EDTPGL01215 (BDSC, 41633,) ii) P{EPgy2}EDTPEY22967 (BDSC, 22600), iii) EDTPGS9978 (Drosophila Genetic Resource Center, 202239) or iv) UAS-EDTP 14-3 (described in this study). L3F stage larvae (88 to 93 h) were dissected.
Generation of polyclonal anti-EDTP antibody
Partial EDTP (amino acids 1 to 318) coding sequence was PCR amplified and cloned into the vector pEV (kindly provided by Peter Rapali at Eötvös University, Budapest, Hungary) as an NdeI-XhoI fragment. N-terminally 6xHis-tagged protein was then expressed in the E. coli Rosetta strain and purified using Ni-NTA agarose beads (Qiagen, 30230). Recombinant protein was used to immunize rats in-house, following a standard protocol that utilizes Freund adjuvants (Sigma, F5881 and F5506).
Western blotting
With mammalian cells
Western blot samples were obtained by scraping cells from 6-well plates in 100 μl of hot Laemmli buffer. 25 μl samples were run on a 12% SDS-PAGE and blotted onto Immobilon P PVDF membrane (Millipore, ISPN07852). After blocking with 0.5% blocking reagent (Roche, 000007300100119216) in PBS containing 0.1% Tween 20, filters were probed with specific antibodies anti-LC3 (rabbit, 1:1000; Cell Signaling Technology, 2775), anti-SQSTM1/p62 (rabbit, 1:1000; Sigma, P0068) and anti-GAPDH (rabbit, 1:6000; Sigma, G9545). Proteins were visualized using the ECL system (Luminata Crescendo, Millipore, WBLUR0100). Quantification was carried out using ImageStudio (LI-COR Biosciences), by referring the intensity of LC3B-II bands to the corresponding GAPDH intensity. Data are shown as a percentage of intensity ratios obtained from DMSO-only treated control cultures and compared using Student t test (P < 0.05).
With Drosophila samples
Fat body samples from well-fed L3 stage Drosophila larvae were dissected. Filters were probed with anti-Ref(P)2/SQSTM1/p62 (rabbit, 1:2500),63 anti-EDTP (rat, 1:1000, described in this study), anti-rabbit IgG alkaline phosphatase (1:1000, Sigma, A3687), anti-mouse IgG alkaline phosphatase (1:1000, Sigma, A8438) and anti-rat IgG alkaline phosphatase (1:1000, Sigma, A5153), and developed by NBT-BCIP solution (Sigma, 72091).
Life-span assays
The Drosophila canonical control strain (w1118) was used. The autophagy-inducing drug candidate AUTEN-67 was used at 30 µM, and 100 µl solution was dried on normal solid media. Assays were started with 100 to 100 newly hatched male and female imago (day 0) placed into 5 glass vials (around 20 animals per vial). Animals were transferred into new vials (containing fresh media) every 2 d, and the number of dead flies was counted. Tests were carried out at 25°C, control animals were treated with the same way expect from adding AUTEN-67 into the medium. For statistics, the Kaplan-Meier method (http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1114388/) was used, calculated with the SPSS program (http://www-03.ibm.com/software/products/hu/spss-stats-base).64
Fish care
Tg(GFP-Lc3) fish7 stocks were maintained according to standard protocols,45 in the Animal Facility of Eötvös Loránd University, Budapest. All protocols used in this study were approved by the Hungarian National Food Chain Safety Office (Permit Number: XIV-I-001/515-4/2012). Fish embryos were incubated in the respective small molecular compound (or DMSO) at the indicated concentrations, between 90% epiboly and 1 d postfertilization. Embryos were anesthetized using tricaine methanesulfonate (Sigma, MS222) and embedded in 4% methylcellulose (Sigma, M-0387). Fluorescence pictures of live embryos were taken with a Zeiss AxioZoom stereomicroscope, using the manufacturer's software. Fluorescence pictures were analyzed using the ImageJ software package (NIH). Fluorescence intensities were measured in 50x50 pixel areas on the body of embryos, over the yolk sac extension.
Primary neuronal cell cultures
Primary cortical neurons were prepared from 15-d-old mouse embryos.65 Briefly, embryonic cortices were incubated in 0.05% trypsin solution (Gibco, Life Technologies, R001100) for 15 min at 37°C. After a brief centrifugation step, cells were triturated in NeuroBasal media (Gibco, Life Technologies, 21103-049) supplemented with B27 (Gibco, Life Technologies, A24775-01), 0.5 mM Glutamax (Gibco, Life Technologies, 10567014), 40 μg/ml gentamycin and 2.5 μg/ml amphotericin B (Sigma, Y0000005), and filtered through a sterile polyester mesh with 42 μm pore size (EmTek Ltd, S72210-SS). Cells were seeded onto poly-D-lysine (PDL; Sigma, P7405) coated 6 or 96 well plates at 106 or 8x104 cells/well densities, respectively. To prevent the division of nonneuronal cells, cultures were treated with 10 μM cytosine-arabinofuranoside (CAR; Sigma, C6645) on the 2nd d after plating. Oxidative stress was induced on the 7th d of cultivation by 50 or 75 μM H2O2 for 24 h, diluted directly from 30% H2O2 stock solution (Reanal, 12502-0-48-65) kept at 4°C.
Electron microscopy
Adult male BALB/C mice between 18 to 20 g body weight were injected intraperitoneally (i.p.). Stock solutions of the substances were dissolved in DMSO (intraperitoneal) and diluted to the requested concentration with physiological saline for the intraperitoneal injection. Treatments lasted for 90 min. Extermination was carried out by cervical dislocation. The administered doses for AUTEN-67 (T0501-7132) were 50 µmol/g body weight. Controls got physiological saline/DMSO solution. For electron microscopy tissue slices from heart, pancreas and kidney were fixed in cacodylate buffered 2% glutaraldehyde solution. After one d fixation the samples were washed, postfixed in 1% osmium tetroxide, embedded in Araldite (Sigma, A3183), and sectioned with a Reichert Ultracut microtome (Eötvös Loránd University, Budapest). The sections were stained with uranyl acetate and lead citrate, and examined in a JEM 1011 electron microscope (Eötvös Loránd University, Budapest). Quantification of autophagic activity was achieved by counting the number of autophagic structures.
Classification of nesting behavior in mice
Nesting can be divided into the following 5 behavioral classes (1 to 5): class 1: animals do not care or only disassemble the paper handkerchief added; class 2: animals scrunch paper handkerchief into large (>5 cm) pieces; class 3: animals scrunch paper handkerchief into medium-sized (>1 to 2 cm) pieces; class 4: animals scrunch paper handkerchief into small (0.5 to 1 cm) pieces but do not collect them into a nest shape; class 5: animals scrunch paper handkerchief into small (0.5 to 1 cm) pieces and collect them into a nest shape in a nook of the cage. Six-mo-old female transgenic 1xAPP/PS1 (APPswe/PS1dE9; Jackson Laboratory, Bar Harbor, ME, USA) mice were treated with 200 µl solution through a stomach blow-pipe on every Monday, Wednesday and Friday for 96 d of total period. Body weight measurements occurred on the same days, and started from 2th wk of treatment. Each animal was examined individually during the experiment. Solvent was PEG (Sigma, 181986)-Solutiol (BASF, 42966)-water=3:1:8.
Nesting was conducted and assessed,66 and modified slightly. Briefly, mice were individually housed for at least 24 h in clean plastic cages with approximately 1.5 cm of corn cob bedding lining the floor. Individual cages were supplied a commercially available unscented paper handkerchief. Sixteen h later cages were inspected for nest construction. Digital pictures were taken prior to evaluation for documentation. Paper handkerchief nest construction was scored along a 5 point scale: 1=no biting or tears on the paper, 2=moderate biting and/or tears on the paper but spread over the cage, 3=moderate biting with no coherent nest (not grouped into a corner of the cage, 4=the vast majority of paper torn into approximately 1 cm pieces and grouped into a corner of the cage, 5 = a nestled torn >90% and a clear nest crater.
Enzyme-linked immunosorbent assay of human amyloid β40 and β42 proteins
Female APP/PS1 (APPswe/PS1dE9) transgenic mice obtained from Jackson Laboratory (Bar Harbor, ME, USA) were kept and treated at the Biological Research Center (BRC), HAS. Procedures for animal experiments were approved by the Animal Experimentation Committee at BRC. APP/PS1 mice begin to develop Aβ plaques by 4 to 6 mo of age and have robust plaques by 12 mo old. AUTEN-67 (19 mg/kg, 3 times a wk) was administered orally from 6 mo of age for 15 wk. At 9 mo of age, mice were subjected to behavioral tests, and then were sacrificed to obtain brain extract (hemisphere) to assay Aβ (Aβ1→42 and Aβ1→40) levels. Soluble and insoluble (guanidine-extracted) Aβ1-40 and Aβ1-42 levels were quantified in cortical homogenates using commercial colorimetric ELISA kits (Signal SelectTM Human Aβ1–40 and 1–42, BioSource International Inc., Camarillo, CA, USA) as previously described.67 Levels of Aβ were expressed as ng/hemisphere.
Supplementary Material
Abbreviations
- APP
amyloid β (A4) precursor protein
- ATG
autophagy related
- AUTEN
autophagy enhancer
- Baf
bafilomycin A1
- CDC25B
cell division cycle 25B
- CFP
cyan fluorescent protein
- DMSO
dimethyl sulfoxide
- EDTP
egg-derived tyrosine phosphatase
- EGFR
epidermal growth factor receptor
- EM
electron microscopy
- GFP
green fluorescent protein
- H2O2
hydrogen peroxide
- L3F
feeding L3 larval stage
- MAP1LC3B/LC3B
microtubule-associated protein 1 light chain 3 β
- MTMR14
mytobularin related protein 14
- MTOR
mechanistic target of rapamycin (serine/threonine kinase)
- PE
phosphatidylethanolamine
- PIK3C3
phosphatidylinositol 3-kinase, catalytic subunit type 3
- PtdIns
phosphatidylinositol
- PtdIns3K
class III phosphatidylinositol 3-kinase
- PTPN1
protein tyrosine phosphatase, non-receptor type 1
- RFP
red fluorescent protein
- SQSTM1
sequestosome 1
- ULK1
unc-51 like autophagy activating kinase 1
- Vps
vacuolar protein sorting
- WIPI1
WD repeat domain, phosphoinositide interacting 1
Disclosure of potential conflicts of interest
Velgene Ltd is an inventor with patents relating to the use of autophagy induction for treating neurodegenerative and other age-dependent diseases, as well of autophagy markers for detecting early stages of neurodegenerative diseases. The company is grateful for funding from the European Union (grant GOP-1.1.1-11-2012-0405). This sponsor has not reviewed the manuscript.
Acknowledgments
HaCatT cell line (EGFR-eGFP) was kindly provided by Prof. Angel Alonso (Deutsches Krebsforschungszentrum, Heidelberg). We thank Gábor Komlós, Levente Zsembéri and Viktor Nyíri for managing the project, as well as Szilvia Meriáth, Anna Faludi, Tünde Pénzes and Sára Simon for technical and administrative assistance.
Funding
This work was supported by European Union grant GOP-1.1.1-11-2012-0405. K.S. is supported by the OTKA (Hungarian Scientific Research Funds) grant K81934 and by the KTIA_NAP_13-2014-0018 grant (Hungarian Academy of Sciences, National Brain Research Program), L.P. is supported by grants from the National Development Agency of Hungary (KMR_12-1-2012-0072; GOP-1.1.1-11-2011-0003). M.V. is a János Bolyai Fellow of the Hungarian Academy of Sciences. T.V. is supported by the OTKA grant K109349.
References
- 1.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature 2008; 451:1069-75; PMID:18305538; http://dx.doi.org/ 10.1038/nature06639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell 2008; 132:27-42; PMID:18191218; http://dx.doi.org/ 10.1016/j.cell.2007.12.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vellai T, Takács-Vellai K, Sass M, Klionsky DJ. The regulation of aging: does autophagy underlie longevity? Trends Cell Biol 2009; 19:487-94; PMID:19726187; http://dx.doi.org/ 10.1016/j.tcb.2009.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vellai T. Autophagy genes and ageing. Cell Death Differ 2009; 16:94-102; PMID:19079287; http://dx.doi.org/ 10.1038/cdd.2008.126 [DOI] [PubMed] [Google Scholar]
- 5.Eskelinen EL, Kovács AL. Double membranes vs. lipid bilayers, and their significance for correct identification of macroautophagyic structures. Autophagy 2011; 7:931-2; PMID:21642767; http://dx.doi.org/ 10.4161/auto.7.9.16679 [DOI] [PubMed] [Google Scholar]
- 6.Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol 2007; 9:1102-9; PMID:17909521; http://dx.doi.org/ 10.1038/ncb1007-1102 [DOI] [PubMed] [Google Scholar]
- 7.Yang Z, Klionsky DJ. An overview of the molecular mechanism of autophagy. Curr Topics Microbiol Immunol 2009; 335:1-32 PMID:19802558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vellai T, Takács-Vellai K, Sass M, Klionsky DJ. The regulation of aging: does autophagy underlie longevity? Trends Cell Biol 2009; 19:487-94; PMID:19726187; http://dx.doi.org/ 10.1016/j.tcb.2009.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boya P, González-Polo RA, Casares N, Perfettini JL, Dessen P, Larochette N, Métivier D, Meley D, Souquere S, Yoshimori T, et al.. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol 2005; 25:1025-40; PMID:15657430; http://dx.doi.org/ 10.1128/MCB.25.3.1025-1040.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takacs-Vellai K, Vellai T, Puoti A, Passannante M, Wicky C, Streit A, Kovacs AL, Müller F. Inactivation of the autophagy gene bec-1 triggers apoptotic cell death in C. elegans. Curr Biol 2005; 15:1513-7; PMID:16111945; http://dx.doi.org/ 10.1016/j.cub.2005.07.035 [DOI] [PubMed] [Google Scholar]
- 11.Berry DL, Baehrecke EH. Growth arrest and autophagy are required for salivary gland cell degradation in Drosophila. Cell 2007; 131:1137-48; PMID:18083103; http://dx.doi.org/ 10.1016/j.cell.2007.10.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yuan J, Kroemer G. Alternative cell death mechanisms in development and beyond. Genes Dev 2010; 24:2592-602; PMID:21123646; http://dx.doi.org/ 10.1101/gad.1984410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Erdélyi P, Borsos E, Takács-Vellai K, Kovács T, Kovács AL, Sigmond T, Hargitai B, Pásztor L, Sengupta T, Dengg M, et al.. Shared developmental roles and transcriptional control of autophagy and apoptosis in Caenorhabditis elegans. J Cell Sci 2011;124:1510-8; PMID:21502138; http://dx.doi.org/ 10.1242/jcs.080192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Galluzzi L, et al.. Essential versus accessory aspects of cell death: recommendations of the NCCD 2015. Cell Death Differ. 2015;. 22:58-73; PMID:25236395; http://dx.doi.org/ 10.1038/cdd.2014.137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Erdélyi P, Borsos E, Takács-Vellai K, Kovács T, Kovács AL, Sigmond T, Hargitai B, Pásztor L, Sengupta T, Dengg M, et al.. Shared developmental roles and transcriptional control of autophagy and apoptosis in Caenorhabditis elegans. J Cell Sci 2011; 124:1510-1518; PMID:21502138; http://dx.doi.org/ 10.1242/jcs.080192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Borsos E, Erdélyi P, Vellai T. Autophagy and apoptosis are redundantly required for C. elegans embryogenesis. Autophagy (2011); 7:557-9; PMID:21285529; http://dx.doi.org/ 10.4161/auto.7.5.14685 [DOI] [PubMed] [Google Scholar]
- 17.Simonsen A, Cumming RC, Brech A, Isakson P, Schubert DR, Finley KD. Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy 2008; 4:176-84; PMID:18059160; http://dx.doi.org/ 10.4161/auto.5269 [DOI] [PubMed] [Google Scholar]
- 18.Salminen A, Kaarniranta K, Haapasalo A, Hiltunen M, Soininen H, Alafuzoff I. Emerging role of p62/sequestosome-1 in the pathogenesis of Alzheimer's disease. Prog Neurobiol 2012; 96:87-95; PMID:22138392; http://dx.doi.org/ 10.1016/j.pneurobio.2011.11.005 [DOI] [PubMed] [Google Scholar]
- 19.Belancio VP, Roy-Engel AM, Pochampally RR, Deininger P. Somatic expression of LINE-1 elements in human tissues. Nucleic Acids Res 2010; 38:3909-22; PMID:20215437; http://dx.doi.org/ 10.1093/nar/gkq132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baillie JK, Barnett MW, Upton KR, Gerhardt DJ, Richmond TA, De Sapio F, Brennan PM, Rizzu P, Smith S, Fell M, et al.. Somatic retrotransposition alters the genetic landscape of the human brain. Nature 2011; 479:534-7; PMID:22037309; http://dx.doi.org/ 10.1038/nature10531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eisenberg T, Knauer H, Schauer A, Büttner S, Ruckenstuhl C, Carmona-Gutierrez D, Ring J, Schroeder S, Magnes C, Antonacci L, et al.. Induction of autophagy by spermidine promotes longevity. Nat Cell Biol 2009; 11:1305-14; PMID:19801973; http://dx.doi.org/ 10.1038/ncb1975 [DOI] [PubMed] [Google Scholar]
- 22.Zhang C, Cuervo AM. Restoration of chaperone-mediated autophagy in aging liver improves cellular maintenance and hepatic function. Nat Med 2008; 14:959-65; PMID:18690243; http://dx.doi.org/ 10.1038/nm.1851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alayev A, Berger SM, Kramer MY, Schwartz NS, Holz MK. The combination of rapamycin and resveratrol blocks autophagy and induces apoptosis in breast cancer cells. J Cell Biochem 2015; 116:450-7; PMID:25336146; http://dx.doi.org/ 10.1002/jcb.24997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hidvegi T, Ewing M, Hale P, Dippold C, Beckett C, Kemp C, Maurice N, Mukherjee A, Goldbach C, Watkins S, et al.. An autophagy-enhancing drug promotes degradation of mutant alpha1-antitrypsin Z and reduces hepatic fibrosis. Science 2010; 329:229-32; PMID:20522742; http://dx.doi.org/ 10.1126/science.1190354 [DOI] [PubMed] [Google Scholar]
- 25.Zhang L, Yu J, Pan H, Hu P, Hao Y, Cai W, Zhu H, Yu AD, Xie X, Ma D, et al.. Small molecule regulators of autophagy identified by an image-based high-throughput screen. Proc Natl Acad Sci USA 2007; 104:19023-8; PMID:18024584; http://dx.doi.org/ 10.1073/pnas.0709695104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hundeshagen P, Hamacher-Brady A, Eils R, Brady NR. Concurrent detection of autolysosome formation and lysosomal degradation by flowcytometry in a high-content screen for inducers of autophagy. BMC Biol 2011; 9:38; PMID:21635740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lamming DW, Ye L, Sabatini DM, Baur JA. Rapalogs and mTOR inhibitor sas anti-aging therapeutics. J Clin Invest 2013; 123:980-9; PMID:23454761; http://dx.doi.org/ 10.1186/1741-7007-9-38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li BH, Liao SQ, Yin YW, Long CY, Guo L, Cao XJ, Liu Y, Zhou Y, Gao CY, Zhang LL, Li JC. Telmisartan-induced PPARγ activity attenuates lipid accumulation in VSMCs via induction of autophagy. Mol Biol Rep 2014; Epub ahead of print; PMID:25249228; http://dx.doi.org/ 10.1007/s11033-014-3757-6 [DOI] [PubMed] [Google Scholar]
- 29.Kangwan N, Park JM, Kim EH, Hahm KB. Chemoquiescence for ideal cancer treatment and prevention: where are we now? J Cancer Prev 2014; 19:89-6; PMID:25337576; http://dx.doi.org/ 10.15430/JCP.2014.19.2.89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim KW, Myers CJ, Jung DK, Lu B. NVP-BEZ-235 enhances radiosensitization via blockade of the PI3K/mTOR pathway in cisplatin-resistant non-small cell lung carcinoma. Genes Cancer 2014; 5:293-302; PMID:25221647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vellai T, Takacs-Vellai K, Zhang Y, Kovacs AL, Orosz L, Müller F. Influence of TOR kinase on lifespan in C. elegans. Nature 2003; 426:620; PMID:14668850; http://dx.doi.org/ 10.1038/426620a [DOI] [PubMed] [Google Scholar]
- 32.Kapahi P, Zid BM, Harper T, Koslover D, Sapin V, Benzer S. Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr Biol 2004; 14:885-90; PMID:15186745; http://dx.doi.org/ 10.1016/j.cub.2004.03.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Feng Y, He D, Yao Z, Klionsky DJ. The machinery of macroautophagy. Cell Res 2014; 24:24-41; PMID:24366339; http://dx.doi.org/ 10.1038/cr.2013.168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Robinson FL, Dixon JE. Myotubularin phosphatases: policing 3-phosphoinositides. Trends Cell Biol 2006; 16:403-12; PMID:16828287; http://dx.doi.org/ 10.1016/j.tcb.2006.06.001 [DOI] [PubMed] [Google Scholar]
- 35.Vergne I, Roberts E, Elmaoued RA, Tosch V, Delgado MA, Proikas-Cezanne T, Laporte J, Deretic V. Control of autophagy initiation by phosphoinositide 3-phosphatase Jumpy. EMBO J 2009; 28:2244-58; PMID:19590496; http://dx.doi.org/ 10.1038/emboj.2009.159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Al-Qusairi L, Prokic I, Amoasii L, Kretz C, Messaddeq N, Mandel JL, Laporte J. Lack of myotubularin (MTM1) leads to muscle hypotrophy through unbalanced regulation of theautophagy and ubiquitin-proteasome pathways. FASEB J 2013; 27:3384-94; PMID:23695157; http://dx.doi.org/ 10.1096/fj.12-220947 [DOI] [PubMed] [Google Scholar]
- 37.Fetalvero KM, Yu Y, Goetschkes M, Liang G, Valdez RA, Gould T, Triantafellow E, Bergling S, Loureiro J, Eash J, et al.. Defective autophagy and mTORC1 signaling in myotubularin null mice. Mol Cell Biol 2013; 33:98-110; PMID:23109424; http://dx.doi.org/ 10.1128/MCB.01075-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Durieux AC, Vignaud A, Prudhon B, Viou MT, Beuvin M, Vassilopoulos S, Fraysse B, Ferry A, Lainé J, Romero NB, et al.. A centronuclear myopathy-dynamin 2 mutation impairs skeletal muscle structure and function in mice. Hum Mol Genet 2010; 19:4820-36; PMID:20858595; http://dx.doi.org/ 10.1093/hmg/ddq413 [DOI] [PubMed] [Google Scholar]
- 39.Nicks J, Lee S, Harris A, Falk DJ, Todd AG, Arredondo K, Dunn WA Jr, Notterpek L. Rapamycin improves peripheral nerve myelination while it fails to benefit neuromuscular performance in neuropathic mice. Neurobiol Dis 2014; 70:224-36; PMID:25014022; http://dx.doi.org/ 10.1016/j.nbd.2014.06.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Katsetos CD, Koutzaki S, Melvin JJ. Mitochondrial dysfunction in neuromuscular disorders. Semin Pediatr Neurol 2013; 20:202-15; PMID:24331362; http://dx.doi.org/ 10.1016/j.spen.2013.10.010 [DOI] [PubMed] [Google Scholar]
- 41.Chen H, Chan DC. Mitochondrial dynamics–fusion, fission, movement, and mitophagy–in neurodegenerativediseases. Hum Mol Genet 2009; 18:R169-76; PMID:19808793; http://dx.doi.org/ 10.1093/hmg/ddp326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, et al.. TFEB links autophagy to lysosomal biogenesis. Science 2011; 332:1429-33; PMID:21617040; http://dx.doi.org/ 10.1126/science.1204592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al.. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445-544; PMID:22966490; http://dx.doi.org/ 10.4161/auto.19496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dowling JJ, Low SE, Busta AS, Feldman EL. Zebrafish MTMR14 is required for excitation-contraction coupling, developmental motor function and the regulation of autophagy. Hum Mol Genet 2010; 19:2668-81; PMID:20400459; http://dx.doi.org/ 10.1093/hmg/ddq153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.He C, Bartholomew CR, Zhou W, Klionsky DJ. Assaying autophagic activity in transgenic GFP-Lc3 and GFP-Gabarapzebrafish embryos. Autophagy 2009; 5:520-6; PMID:19221467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Varga M, Sass M, Papp D, Takács-Vellai K, Kobolak J, Dinnyés A, Klionsky DJ, Vellai T. Autophagy is required for zebrafish caudal fin regeneration. Cell Death Differ 2014; 21:547-56; PMID:24317199; http://dx.doi.org/ 10.4161/auto.5.4.7768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kovács J, László L, Kovács AL. Regression of autophagic vacuoles in pancreatic acinar, seminal vesicle epithelial, and liver parenchymal cells: a comparative morphometric study of the effect of vinblastine and leupeptin followed by cycloheximide treatment. Exp Cell Res 1988; 174:244-51; PMID:3335225; http://dx.doi.org/ 10.1016/0014-4827(88)90158-9 [DOI] [PubMed] [Google Scholar]
- 48.Kirkwood TB. A systematic look at an old problem. Nature 2008; 451:644-7; PMID:18256658; http://dx.doi.org/ 10.1038/451644a [DOI] [PubMed] [Google Scholar]
- 49.Tóth ML, Sigmond T, Borsos E, Barna J, Erdélyi P, Takács-Vellai K, Orosz L, Kovács AL, Csikós G, Sass M, Vellai T. Longevity pathways converge on autophagy genes to regulate life span in Caenorhabditis elegans. Autophagy 2008; 4:330-8; PMID:18219227; http://dx.doi.org/ 10.4161/auto.5618 [DOI] [PubMed] [Google Scholar]
- 50.Vellai T. Autophagy genes and ageing. Cell Death Differ 2009; 16:94-102; PMID:19079287 [DOI] [PubMed] [Google Scholar]
- 51.Rubinsztein DC, Mariño G, Kroemer G. Autophagy and aging. Cell 2011; 146:682-95; PMID:21884931; http://dx.doi.org/ 10.1016/j.cell.2011.07.030 [DOI] [PubMed] [Google Scholar]
- 52.Pyo JO, Yoo SM, Ahn HH, Nah J, Hong SH, Kam TI, Jung S, Jung YK. Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat Commun 2013; 4:2300; PMID:23939249; http://dx.doi.org/ 10.1038/ncomms3300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rubinsztein DC. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 2006; 443:780-6; PMID:17051204; http://dx.doi.org/ 10.1038/nature05291 [DOI] [PubMed] [Google Scholar]
- 54.Czech C, Masters C, Beyreuther K. Alzheimer's disease and transgenic mice. J Neural Transm Suppl 1994; 44:219-30; PMID:7897394 [DOI] [PubMed] [Google Scholar]
- 55.Khalil Z, Poliviou H, Maynard CJ, Beyreuther K, Masters CL, Li QX. Mechanisms of peripheral microvascular dysfunction in transgenic miceoverexpressing the Alzheimer's disease amyloid Abeta protein. J Alzheimers Dis 2002; 4:467-8; PMID:12515898 [DOI] [PubMed] [Google Scholar]
- 56.Kuusisto E, Kauppinen T, Alafuzoff I. Use of p62/SQSTM1 antibodies for neuropathological diagnosis. Neuropathol Appl Neurobiol 2008; 34:169-80; PMID:17961133; http://dx.doi.org/ 10.1111/j.1365-2990.2007.00884.x [DOI] [PubMed] [Google Scholar]
- 57.Molnár E, Kuntam S, Cingaram PK, Peksel B, Suresh B, Fábián G, Fehér LZ, Bokros A, Medgyesi A, Ayaydin F, Puskás LG. Combination of small molecule microarray and confocal microscopy techniques for live cell staining fluorescent dye discovery. Molecules 2003; 18:9999-10013; PMID:23966084; http://dx.doi.org/ 10.3390/molecules18089999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hackler L, Dormán G, Kele Z, Urge L, Darvas F, Puskás LG. Development of chemically modified glass surfaces for nucleic acid, protein and small molecule microarrays. Mol Divers 2003; 7:25-36; PMID:14768901; http://dx.doi.org/ 10.1023/B:MODI.0000006534.36417.06 [DOI] [PubMed] [Google Scholar]
- 59.Darvas F, Dormán G, Krajcsi P, Puskás LG, Kovári Z, Lörincz Z, Urge L. Recent advances in chemical genomics. Curr Med Chem 2004; 11:3119-45; PMID:15579004; http://dx.doi.org/ 10.2174/0929867043363848 [DOI] [PubMed] [Google Scholar]
- 60.Molnár E, Puskás LG, Fehér LZ, Hackler L, Lorincz Z, Lang C, Urge L, Darvas F. Application of small molecule microarrays in comparative chemical proteomics. QSAR Comb Sci 2006; 25:1020-6; PMID:17094670; http://dx.doi.org/ 10.1002/qsar.20064008017094670 [DOI] [Google Scholar]
- 61.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 1983; 65:55-63; PMID:6606682; http://dx.doi.org/ 10.1016/0022-1759(83)90303-4 [DOI] [PubMed] [Google Scholar]
- 62.Nagy P, Varga A, Kovacs AL, Takats S, Juhasz G. How and why to study autophagy in Drosophila: It's more than just a garbage chute. Methods (San Diego, Calif) 2014; 75:151-61; PMID:25481477; http://dx.doi.org/ 10.1016/j.ymeth.2014.11.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pircs K, Nagy P, Varga A, Venkei Z, Erdi B, Hegedus K, Juhasz G. Advantages and limitations of different p62-based assays for estimating autophagic activity in Drosophila. PloS One 2012; 7:e44214; PMID:22952930; http://dx.doi.org/ 10.1371/journal.pone.0044214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bland JM, Altman DG. The logrank test. BMJ (Clinical research ed) 2004; 328:1073; PMID:15117797; http://dx.doi.org/ 10.1136/bmj.328.7447.1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tárnok K, Kiss E, Luiten PG, Nyakas C, Tihanyi K, Schlett K, Eisel UL. Effects of Vinpocetine on mitochondrial function and neuroprotection in primary cortical neurons. Neurochem Int 2008; 53:289-95; PMID:18793690 [DOI] [PubMed] [Google Scholar]
- 66.Wesson DW, Wilson DA. Age and gene overexpression interact to abolish nesting behavior in Tg2576 amyloid precursor protein (APP) mice. Behav Brain Res 2011; 216:408-13; PMID:20804789; http://dx.doi.org/ 10.1016/j.bbr.2010.08.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lilja AM, Röjdner J, Mustafiz T, Thomé CM, Storelli E, Gonzalez D, Unger-Lithner C, Greig NH, Nordberg A, Marutle A. Age-dependent neuroplasticity mechanisms in Alzheimer Tg2576 mice following modulation of brain amyloid-β levels. PLoS One 2013; 8:e58752; PMID:23554921; http://dx.doi.org/ 10.1371/journal.pone.0058752 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.