

ABSTRACT
During proteasomal stress, cells can alleviate the accumulation of polyubiquitinated proteins by targeting them to perinuclear aggresomes for autophagic degradation, but the mechanism underlying the activation of this compensatory pathway remains unclear. Here we report that PINK1-s, a short form of Parkinson disease (PD)-related protein kinase PINK1 (PTEN induced putative kinase 1), is a major regulator of aggresome formation. PINK1-s is extremely unstable due to its recognition by the N-end rule pathway, and tends to accumulate in the cytosol during proteasomal stress. Overexpression of PINK1-s induces aggresome formation in cells with normal proteasomal activities, while loss of PINK1-s function leads to a significant decrease in the efficiency of aggresome formation induced by proteasomal inhibition. PINK1-s exerts its effect through phosphorylation of the ubiquitin-binding protein SQSTM1 (sequestosome 1) and increasing its ability to sequester polyubiquitinated proteins into aggresomes. These findings pinpoint PINK1-s as a sensor of proteasomal activities that transduces the proteasomal impairment signal to the aggresome formation machinery.
KEYWORDS: Aggresome, autophagy, PINK1, proteasomal inhibition, SQSTM1
Introduction
Efficient elimination of misfolded and unwanted proteins in cells is critical for cell survival. In normal physiological conditions, this elimination is mainly achieved through polyubiquitination and subsequent degradation of the proteins by proteasomes. However, in cells with reduced proteasomal functions, ubiquitinated proteins accumulate and concentrate into perinuclear structures called aggresomes before they are degraded through autophagy.1 It has long been recognized that formation of aggresomes and their subsequent autophagic clearance represent a compensatory protein degradation mechanism for proteasomal failure,2-4 but the signaling events controlling the activation of these processes remain largely unclear.
Two UB (ubiquitin)-binding proteins, SQSTM1/p62 (sequestosome 1) and HDAC6 (histone deacetylase 6), have been identified as the key mediators of aggresome formation.4,5 SQSTM1 is a multifunctional scaffold protein that participates in the formation and activation of MTOR (mechanistic target of rapamycin [serine/threonine kinase]), TNFRSF1A/TNFR (tumor necrosis factor receptor superfamily member 1A), NOD2 (nucleotide binding oligomerization domain containing 2) and other signaling complexes,6-8 but in response to proteasomal inhibition, it appears to detach from its usual interactors.9 Instead, it assembles the polyubiquitinated proteins into microaggregates through binding to their UB chains with its C-terminal UBA domain (UB-associated domain) and oligomerizing through its N-terminal PB1 domain (Phox and Bem1p domain).10,11 These microaggregates are then recognized by HDAC6, loaded onto dynein motors and transferred through a microtubule network to the microtubule organization center (MTOC), forming a large protein aggregate, the aggresome.5 As the critical molecules mediating aggresome formation, both SQSTM1 and HDAC6 display distinct changes in their subcellular distributions and biochemical activities during proteasomal inhibition.5,12 However, none of these proteins appears to be the direct target of proteasomal degradation, suggesting that activation of their aggresome-related functions during proteasomal stress requires additional regulatory factors.
PINK1 (PTEN-induced putative kinase 1) is a Ser/Thr protein kinase with a mitochondria-targeting sequence (MTS) and a transmembrane domain (TMD) in its N terminus.13 Mutations in the PINK1 gene are associated with a hereditary form of Parkinson disease (PD).14,15 Functional studies have revealed that PINK1 protein can induce the elimination of damaged mitochondria through selective macroautophagy (mitophagy). After its synthesis in the cytosol, the full-length PINK1, PINK1-l (also referred to as the 66 kDa PINK1), is transported to the TOM and TIM translocase complexes in the mitochondrial membranes, a process mediated by the MTS. In healthy mitochondria, the N-terminal 103 amino acid peptide of PINK1-l is efficiently imported until its TMD is recognized and cleaved by the inner membrane protease PARL (presenilin associated rhomboid-like).16-18 The resultant PINK1 fragment, PINK1-s (also referred to as 52 kDa, 54 kDa, 55 kDa or ΔN PINK1), is then shuttled back to the cytosol and degraded by the proteasome through the N-end rule pathway.19 In damaged mitochondria, however, the reduction in the Δψ of the inner membrane halts PINK1-l import. Without PARL-mediated cleavage, PINK1-l is stuck in the pore of the TOM complex, with its kinase domain exposed at the surface of mitochondria. Through recruitment of another PD-related protein, PARK2/PRKN/PARKIN (parkin RBR E3 ubiquitin protein ligase), from the cytosol, PINK1-l orchestrates the ubiquitination of proteins at the surface of mitochondria, which activates the mitophagy process.20-22 Consistent with this model, loss of PINK1 causes the accumulation of damaged mitochondria in both Drosophila and mammalian cells,23-26 suggesting that it is required for the maintenance of healthy mitochondria.
In addition to their reduced mitochondrial function, mutant PINK1 cells also display other less obvious defects. Klinkenberg et al. have found that fibroblasts from a PD patient who is homozygous for the mutant PINK1 gene show a marked increase in cell death induced by the proteasomal inhibitor MG132, as compared with those from heterozygous and wild-type siblings.27 However these cells display essentially the same sensitivity toward other chemicals known to activate mitochondria-dependent apoptosis, such as staurosporine and etopside,27 suggesting that their hypersensitivity toward MG132 is unlikely due to their mitochondrial defects. Interestingly, proteasomal inhibition also induces the translocation of PINK1 protein into perinuclear aggresomes and smaller SQSTM1-containing protein aggregates.19,28 This raised the possibility that it might reduce the damage caused by proteasomal stress through the activation of the aggresome-autophagy pathway. However, transient overexpression of the wild-type and PD-related mutant PINK1-l in cultured cells fails to reveal any obvious effect on the rate of aggresome formation.28
Here we report that PINK1 suppressed proteasomal stress-induced necrosis through the PINK1-s isoform localized in the cytosol. Extremely unstable under physiological conditions, PINK1-s rapidly accumulated in cells with reduced proteasomal activities. Through phosphorylation of SQSTM1, PINK1-s increased its UB chain-binding activity and promoted the packaging of polyubiquitinated proteins into aggregates and aggresomes, which were degraded by autophagy. This study establishes PINK1-s as a major mediator of proteasomal stress-induced activation of the aggresome-autophagy pathway.
Results
Overexpression of PINK1-s stimulates aggresome formation
By cell fractionation and immunofluorescence staining, we confirmed that moderate proteasomal inhibition, such as a 50% reduction in proteasomal activities, was sufficient to induce the accumulation of PINK1-s in aggresomes, as suggested by other studies (Fig. S1A to E).19,28 To test whether PINK1-s regulated the formation of this organelle, we first overexpressed EGFP-tagged PINK1-s (Fig. 1A and B) in AD293 cells by transient transfection and examined whether it could induce aggresome formation in the absence of proteasomal inhibitors. As expected, PINK1-s with its N terminus fused to EGFP was stable because it could no longer be recognized by the N-end rule pathway. Unlike overexpressed EGFP, which was diffusely distributed in the cytosol and nucleus, EGFP-PINK1-s was concentrated in cytosolic aggregates in 30.4% of the transfected cells and in perinuclear aggresomes in 32.4% of the transfected cells. These aggregates and aggresomes did not show strong colocalization with ER marker HSPA5/Grp78 (heat shock protein family A [Hsp70] member 5) or mitochondrial marker TOMM20 (translocase of outer mitochondrial membrane 20 homolog [yeast]), suggesting EGFP-PINK1-s did not accumulate in these organelles (Fig. S2A and B). Immunofluorescence staining with an antibody specific for K48-linked UB chain, UB(K48) chain, revealed that these aggregates and aggresomes were enriched with polyubiquitinated proteins (Fig. 1B), similar to those formed during proteasomal inhibition.
Figure 1.
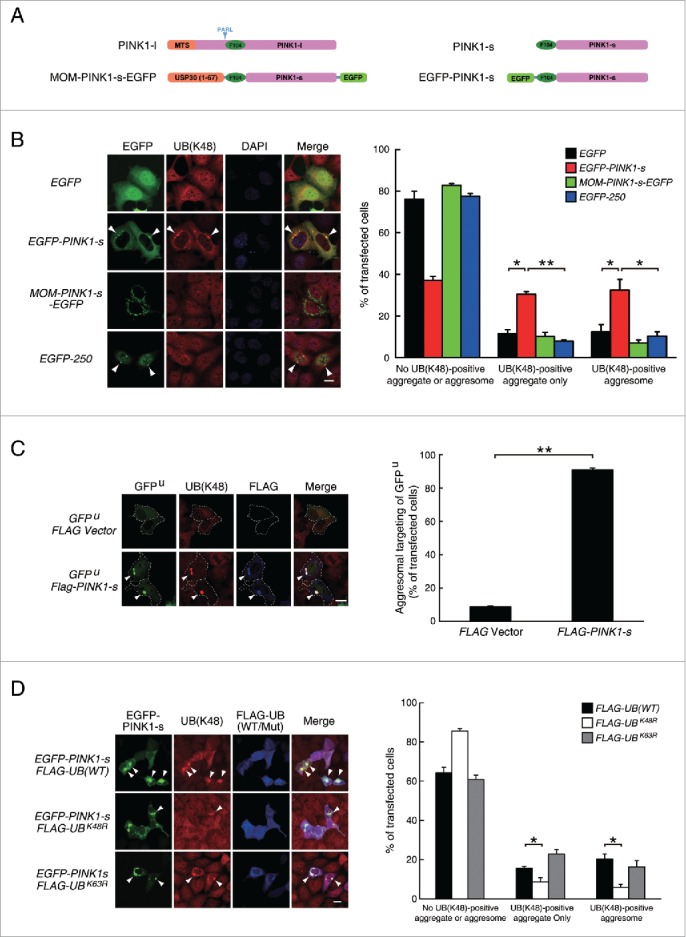
Overexpression of PINK1-s induces the formation of aggregates and aggresomes enriched with K48-linked UB chains. (A) Schematic illustration of the EGFP-tagged PINK1 proteins mimicking the endogenous cytosolic PINK1-s and mitochondrial PINK1-l. A 67-amino acid peptide from the mitochondrial outer membrane (MOM) protein USP30 was fused to the N terminus of PINK1-s-EGFP to target it to the MOM, mimicking the PINK1-l accumulation on the outer membrane of damaged mitochondria. The PARL cleavage site is indicated by arrowheads. (B) Aggregate and aggresome formation in AD293 cells transiently overexpressing EGFP, EGFP-PINK1-s, MOM-PINK1-s-EGFP or EGFP-250 for 24 h. UB-containing aggregates and aggresomes were visualized by immunofluorescence staining with a UB antibody specific for K48-linked chain (UB(K48) antibody). Aggresomes are indicated by arrowheads. Scale bar: 20 μm. Quantification results are shown as mean ± SEM of 3 independent experiments. *, P < 0.05; **, P < 0.01. (C) Aggresome formation in AD293 cells transiently expressing GFPU (Control) or GFPU and FLAG-PINK1-s for 24 h. GFPU, ubiquitinated proteins and FLAG-PINK1-s were detected by immunofluorescence staining with EGFP, UB(K48) and FLAG antibodies, respectively. Aggresomes are indicated by arrowheads. Scale bar: 20 μm. Qualification results are shown as mean ± SEM of 3 independent experiments. **, P < 0.01. (D) Aggresome formation in AD293 cells transiently expressing EGFP-PINK1-s together with FLAG-UB(WT), FLAG-UBK48R or FLAG-UBK63R for 24 h. EGFP-PINK1-s, K48-linked ubiquitin chain, FLAG-UB were detected by immunofluorescence staining with EGFP, UB(K48) and FLAG antibodies, respectively. Aggresomes are indicated by arrowheads. Scale bar: 20 μm. Qualification results are shown as mean ± SEM of 3 independent experiments. *, P < 0.05.
To our knowledge, this property of PINK1 has not been described before, so we decided to examine whether the full-length PINK1 isoform, PINK1-l, had the same function. PINK1-l is also an unstable protein. It only accumulates on the outer membrane of damaged mitochondria. In normal cells, PINK1-l levels are very low. To closely mimic the localization and function of PINK1-l, we targeted PINK1-s to the mitochondrial outer membrane (MOM) using the signal sequence of the MOM protein USP30 (ubiquitin specific peptidase 30, Fig. 1A). Immunofluorescence staining of cells expressing MOM-PINK1-s-FLAG confirmed that there was strong colocalization with the mitochondrial protein TOMM20, suggesting that it was transported to the MOM after its synthesis (Fig. S3A). In PINK1−/− cells, expression of MOM-PINK1-s-FLAG could effectively induce the mitochondrial translocation of EGFP-PARK2, suggesting that it was functionally equivalent to the endogenous PINK1-l that accumulated on the MOM after mitochondrial damage (Fig. S3B). Unlike EGFP-PINK1-s, transient overexpression of MOM-PINK1-s-EGFP did not induce the formation of UB(K48) chain-containing aggregates or aggresomes, suggesting that this was an isoform-specific property of PINK1-s (Fig. 1B). Overexpression of PINK1-s also induced aggregate and aggresome formation in cell types other than AD293 cells, including SH-SY5Y, PC12, L02 and 293T cells. Interestingly, PARK2 was not expressed or was expressed at very low levels in AD293, 293T and L02 cells, so in these cases, PINK1-s induced aggregate and aggresome formation through a signaling pathway that did not require PARK2 (Fig. S3C).
One possible explanation for PINK1-s-induced aggregate and aggresome formation was that it might inhibit proteasomes. However, cells overexpressing FLAG-PINK1-s did not show any significant change in proteasomal activities (Fig. S4A and B). Another possibility is that PINK1-s is prone to misfolding and therefore underwent ubiquitination before being targeted to aggresomes. However, misfolded proteins tend to be tagged with K63-linked UB chains and their overexpression usually does not cause concomitant enrichment of the UB(K48) chain in aggresomes.29,30 For example, overexpression of EGFP-250, a misfolded chimera of EGFP and the first 252 amino acids of human USO1/P115 (USO1 vesicle transport factor),31 led to the formation of perinuclear aggresomes that were not labeled by UB(K48) antibody (Fig. 1B). These results led us to hypothesize that the UB(K48) chains in EGFP-PINK1-s-induced aggregates and aggresomes came from polyubiquitinated proteins normally targeted to proteasomes.
To test this hypothesis, we used GFPU, a proteasomal degron-fused EGFP that usually undergoes rapid ubiquitination and proteasomal degradation after its synthesis.32 Expression of GFPU alone resulted in weak GFP fluorescence in most transfected cells. However, coexpression of FLAG-PINK1-s led to the strong accumulation of GFPU in FLAG-PINK1-s-positive aggregates and aggresomes in 91% of the transfected cells, suggesting the PINK1-s overexpression was sufficient to reroute proteasomal substrates like GFPU to aggregates and aggresomes (Fig. 1C).
To further verify that the UB chain in PINK1-s-induced aggregates and aggresomes was K48-linked, we coexpressed a UBK48R mutant with EGFP-PINK1-s to block K48 chain formation and found that it reduced the percentage of cells with UB(K48)-positive aggregates and aggresomes from 35.8% to 14.4%. In contrast, expression of UBK63R, the UB mutant that can block K63 chain formation, did not have obvious effects (Fig. 1D). These results confirmed the specificity of the UB(K48) antibody we used and suggested that PINK1-s could divert K48-ubiquitinated proteins from proteasomes to aggresomes without the UB(K63) chain.
PINK1-s-induced aggregates and aggresomes are degraded by autophagy
Large protein aggregates and aggresomes formed by misfolded proteins or during proteasomal inhibition are mainly degraded by lysosomes through autophagy. To determine whether the aggregates and aggresomes induced by PINK1-s overexpression were also targeted to the autophagy pathway, we examined the sensitivity of their degradation to lysosomal inhibitor bafilomycin A1 (BafA1). Ubiquitinated proteins in aggregates and aggresomes could not be solubilized in cell lysis buffer containing 1% of a mild detergent, such as NP40, so they could be quantified by western blot analysis of ubiquitinated proteins in NP40-insoluble fractions. Overexpression of FLAG-PINK1-s alone did not significantly change the levels of ubiquitinated proteins in the NP40-soluble fraction, but in the insoluble fraction, it increased the levels of ubiquitinated proteins by 126.0%. Inhibition of autophagic flow with BafA1 for 12 h increased the levels of insoluble ubiquitinated proteins in the control and FLAG-PINK1-s-expressing cells by 141.0% and 68.9%, respectively, suggesting that K48-ubiquitinated proteins in the aggregates and aggresomes were degraded through autophagy (Fig. 2A). Consistent with the results in Fig. 1B, results from western blot analysis also indicated that overexpression of MOM-PINK1-s-EGFP did not significantly change the levels of insoluble ubiquitinated proteins (Fig. 2A).
Figure 2.
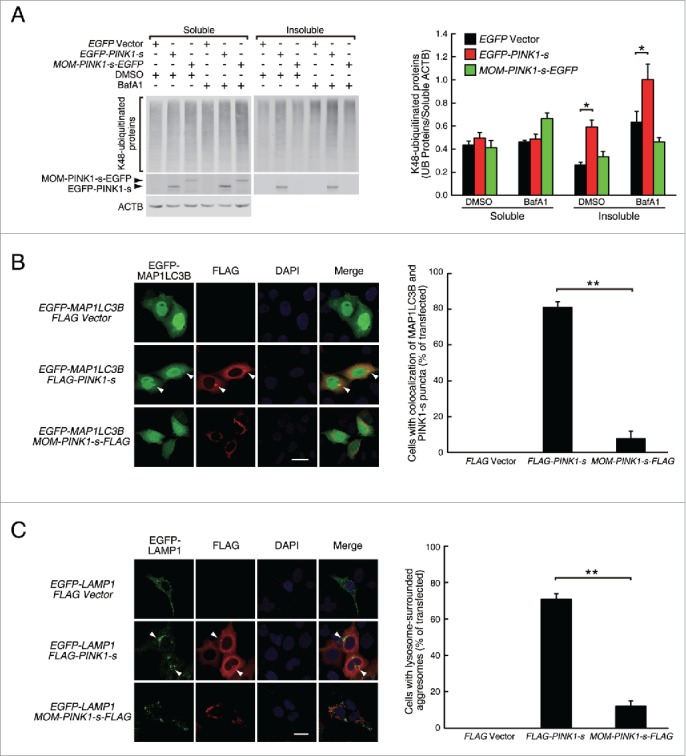
PINK1-s-induced aggresomes are targeted for autophagic degradation. (A) Western blot analysis of the soluble and insoluble K48-ubiquitinated proteins in AD293 cells transiently overexpressing EGFP, EGFP-PINK1-s or MOM-PINK1-s-EGFP. Twenty-four h after transfection, cells were treated with DMSO (control) or BafA1 (100 nM) for 12 h to inhibit the autophagic degradation of the aggregates and aggresomes. Sixteen percent of the soluble fraction and 50% of the insoluble fraction of each sample were loaded for SDS-PAGE and the levels of the ubiquitinated proteins and overexpressed PINK1 were detected by western blot analysis with UB(K48) and EGFP antibodies, respectively. Soluble ACTB (actin β) was detected and used as the loading control. Quantification results are shown as mean ± SEM of 3 independent experiments. *, P < 0.05. (B) Colocalization of EGFP-MAP1LC3 with PINK1-s-induced aggregate and aggresome. AD293 cells were transfected with the indicated plasmids for 24 h. FLAG-PINK1-s and MOM-PINK1-s-FLAG were detected by immunofluorescence staining with FLAG antibody. The nuclei were detected by DAPI staining. Scale bar: 20 μm. Quantification results are shown as mean ± SEM of 3 independent experiments. *, P < 0.05; NS, nonsignificant. (C) PINK1-s-induced aggresomes were surrounded by lysosomes. AD293 cells were transiently transfected with plasmids as indicated for 24 h. FLAG-PINK1-s and MOM-PINK1-s-FLAG were detected by immunofluorescence staining with FLAG antibody. The nuclei were detected by DAPI staining. Scale bar: 20 μm. Quantification results are shown as mean ± SEM of 3 independent experiments. **, P < 0.01.
To test whether PINK1-s-induced aggregates and aggresomes underwent autophagy, we coexpressed EGFP-tagged MAP1LC3B/LC3B (microtubule associated protein 1 light chain 3 β) with FLAG-PINK1-s in cells to label autophagosomes.33 Compared with the control or cells expressing MOM-PINK1-s-FLAG, cells expressing FLAG-PINK1-s had a significant increase in the number of EGFP-MAP1LC3 puncta. In 81.1% of the cotransfected cells, the FLAG-PINK1-s-positive aggregates and aggresomes showed strong colocalization with the EGFP-MAP1LC3 puncta, suggesting that autophagosomes formed around these aggregates and aggresomes (Fig. 2B).
We also coexpressed lysosome marker EGFP-tagged LAMP1 (lysosomal-associated membrane protein 1) with FLAG-PINK1-s to examine the relative distribution of lysosomes and PINK1-s-induced aggregates and aggresomes. The PINK1-s-positive aggregates did not show strong colocalization with the lysosomes, but the perinuclear aggresomes were surrounded by lysosomes in 70.9% of the cells examined, suggesting that after autophagosome formation, lysosome fusion also took place around these aggresomes (Fig. 2C).
Surprisingly, overexpression of PINK1-s did not significantly change the protein levels of MAP1LC3-I, MAP1LC3-II, or their ratio, even in the presence of BafA1, suggesting the PINK1-s did not stimulate the overall rates of autophagosome formation or maturation (Fig. S5). So the autophagosome formation around the PINK1-s-positive aggregates and aggresomes might result from a redistribution of the autophagy machinery in the cells.
Cytosolic PINK1-s protects cells from proteasomal stress-induced cell death
The ability of PINK1-s to target ubiquitinated proteins to the aggresome-autophagy pathway strongly suggests that it is part of the proteasomal stress response in cells. To test whether PINK1-s can reduce the cell death caused by proteasomal inhibition, we established an AD293 cell line stably expressing FLAG-PINK1-s. Compared with the control cells, AD293(FLAG-PINK1-s) cells showed stronger resistance to the treatment with proteasomal inhibitors, such as MG132. After a 20 h treatment with 1 μM MG132, FLAG-PINK1-s expression reduced the apoptosis rate by 60.0% (8.33% vs. 3.33%, P = 0.0045) and the necrosis rate by 38.8% (1.86% vs. 1.14%, P = 0.0155, Fig. 3A). Inhibition of lysosomal functions with BafA1 abolished this protective effect, suggesting that PINK1-s-mediated reduction in cell death requires the degradation of aggresomes by autophagy. The protective effect appeared to be specific to proteasomal inhibition. For other types of stress, such as hypoxia, oxidative stress and ER (endoplasmic reticulum) stress, FLAG-PINK1-s expression did not provide any benefit (Fig. S6A to C).
Figure 3.
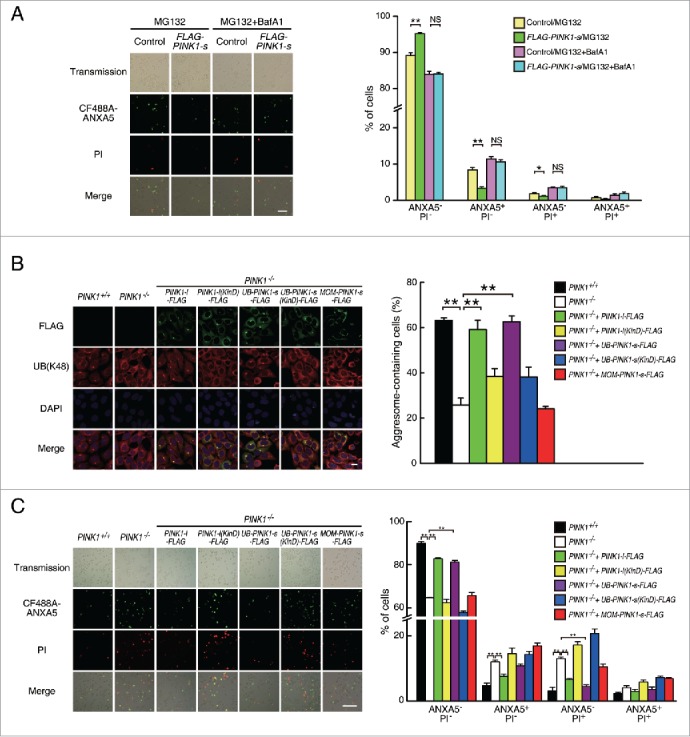
Cytosolic PINK1-s protects cells from proteasomal stress-induced cell death. (A) Cell death analysis of AD293 (control) and AD293 (FLAG-PINK1-s) cells after a 20-h treatment with MG132 (1 μM) or MG132 (1 μM)/BafA1 (100 nM). Cells were stained with CF488A-ANXA5 (green) and propidium iodide (PI, red) to detect dead cells. Live Cells, ANXA5− PI−; early apoptotic cells, ANXA5+PI−; necrotic cells, ANXA5−PI+; late apoptotic cells, ANXA5+PI+. Scale bar: 100 μm. Quantification results are shown as mean±SEM of 3 independent experiments. *, P < 0.05; **, P < 0.01. (B) Aggresome formation in PINK1+/+, PINK1−/− and its rescue cell lines after a 14-h treatment with MG132 (1.5 μM). UB-containing aggresomes and exogenous PINK1 were detected by immunofluorescence staining with UB(K48) and FLAG antibodies, respectively. The nuclei were detected by DAPI staining. Scale bar: 20 μm. Quantification results are shown as mean±SEM of 3 independent experiments. **, P < 0.01. (C) Cell death analysis of PINK1+/+, PINK1−/− and the rescue cell lines after a 20-h treatment with MG132 (1 μM). Cells were stained with CF488A-ANXA5 (green) and propidium iodide (PI, red) to detect dead cells. Live Cells, ANXA−PI−; early apoptotic cells, ANXA5+PI−; necrotic cells, ANXA5−PI+; late apoptotic cells, ANXA5+ PI+. Scale bar: 100 μm. Quantification results are shown as mean±SEM of 3 independent experiments. **, P < 0.01.
Since PINK1-s is a cleavage product of PINK1-l, the ideal approach to test the functions of endogenous PINK1-s is to generate a knock-in cell line expressing a PARL-resistant PINK1-l mutant. Previous studies have identified PINK1-lR98F, a point mutant of PINK1 with reduced cleavage efficiency when overexpressed at high levels by transient transfection.17 However, we found that in cells stably expressing PINK1-lR98F-FLAG at low levels, the protein still underwent efficient cleavage and aggresome-targeting during proteasomal inhibition as revealed by western blot analysis and immunofluorescence staining, making it unsuitable for the dissection of PINK1-s-specific functions (Fig. S7A and B).
As an alternative approach, we generated PINK1-knockout cell lines from AD293 with CRISPR/Cas9 system34-36 and reintroduced different PINK1 isoforms back to the mutant cells by lentiviral transduction (Fig. S7C and D). Loss of PINK1 significantly reduced aggresome formation efficiency. A 14-h treatment with 1.5 μM MG132 induced aggresome formation in 63% of PINK1+/+ cells, but in PINK1−/− cells, the efficiency was reduced to 25% (Fig. 3B). Expression of FLAG-tagged wild-type PINK1-l in these mutant cells completely rescued their efficiency in aggresome formation, while expression of PINK1-l(KinD), a “kinase dead” triple point mutant with 74% reduction in kinase activity,37 only rescued aggresome formation efficiency to 38%, suggesting that PINK1 promotes aggresome formation through phosphorylation of its downstream targets during proteasomal stress (Fig. 3B).
To verify that cytosolic PINK1-s is the isoform mediating aggresome formation, we re-expressed the cytosolic and MOM-targeted PINK1-s-FLAG in the PINK1−/− cells and examined their ability to rescue the decreased aggresome formation efficiency. To closely mimic the unstable nature of the endogenous PINK1-s, we used the UB-fusion technique to express cytosolic PINK1-s.38 Briefly, FLAG-tagged PINK1-s was fused to the C terminus of UB and expressed in PINK1−/− cells by lentiviral transduction (Fig. S7C). Cotranslational cleavage of UB from the fusion protein by deubiquitylating enzymes (DUBs) led to the generation of PINK1-s-FLAG with a Phe104 residue at its N terminus, mimicking the endogenous PINK1-s produced through PARL-mediated cleavage. As a result, it would be targeted for proteasomal degradation by the N-end rule pathway under normal physiological conditions and only accumulate under proteasomal stress (Fig. S7D). We found that UB-PINK1-s-FLAG could fully rescue the aggresome formation defect of PINK1−/− in a kinase activity-dependent manner, while MOM-PINK1-s-FLAG had no rescue activity, suggesting the cytosolic PINK1-s that accumulated during proteasomal stress was the isoform responsible for the full activation of the aggresome formation machinery (Fig. 3B). A similar result was obtained with another aggresome marker, SQSTM1 (Fig. S8A).
We also examined proteasomal stress-induced aggresome formation in other cell lines, including COS7, L02, PC12 and SH-SY5Y cells. MG132 treatment could efficiently induce aggresome formation in COS7 and L02 cells, but not in PC12 and SH-SY5Y cells, possibly due to the protocol we used. Since proteasomal stress-induced aggresome formation had been reported in the liver,39 we decided to use the hepatic cell line L02 for the further analysis of PINK1-s function.40 A 20-h treatment with 1.5 μM MG132 induced aggresome formation in 46.6% of L02 cells and loss of PINK1 reduced this rate to 15.8%. As in AD293 cells, this defect could be rescued by UB-PINK1-s-FLAG, but not by MOM-PINK1-s-FLAG (Fig. S8B). These results suggest that although PINK1-s-induced aggresome formation may not be a universal phenomenon, it is an important proteasomal stress response pathway in many tissues.
Loss of PINK1 in AD293 cells also significantly increased their vulnerability toward proteasomal inhibition-triggered cell death. A 20-h treatment with 1 μM MG132 induced apoptosis (ANXA5/Annexin V+PI−) in 4.7% and necrosis (ANXA5− PI+) in 3.1% of the PINK1+/+ cells, but in PINK1−/− cells, the apoptosis and necrosis rates increased to 12.0% and 13.0% respectively. Interestingly, overexpression of UB-PINK1-s-FLAG in PINK1−/− cells reduced the necrosis rate to 4.5%, but only reduced the apoptosis rate marginally, suggesting the PINK1-s-mediated full activation of aggresome formation was mainly required to protect cells from proteasomal stress-induced necrosis, but not apoptosis (Fig. 3C).
PINK1-s phosphorylates SQSTM1 and enhances its UB-binding affinity
Because PINK1-s is a Ser/Thr protein kinase, it likely promotes the targeting of ubiquitinated proteins to aggregates and aggresomes by phosphorylating key factors in this process. A good candidate for phosphorylation is SQSTM1, which is not only a marker for aggregates and aggresomes, but also a major UB receptor responsible for the sequestration and targeting of polyubiquitinated proteins. Immunoprecipitation experiment showed that HA-SQSTM1 and MYC-PINK1-s overexpressed in the same cells bound each other with high affinity (Fig. 4A). This raised a possibility that SQSTM1 was a substrate of PINK1-s. Previous studies suggested that the efficiency of SQSTM1-mediated microaggregate formation could be regulated through regulation of its UB chain-binding affinity.41,42 To test whether PINK1-s regulated SQSTM1 through a similar mechanism, we coexpressed MYC-PINK1-s with HA-SQSTM1 and FLAG-UB(K48,[6KR]), a mutant UB that could only form a K48-linked chain, and assessed the association of HA-SQSTM1 with ubiquitinated proteins by immunoprecipitation. Compared with the control cells, the levels of HA-SQSTM1-bound ubiquitinated proteins in MYC-PINK1-s-expressing cells were increased by 42.8% (Fig. 4B), supporting the hypothesis that PINK1-s promoted aggregate and aggresome formation by increasing the UB-binding affinity of SQSTM1. Coexpression of PINK1(KinD) did not have a significant effect, suggesting that PINK1-s promoted the UB-binding of SQSTM1 through direct phosphorylation.
Figure 4.
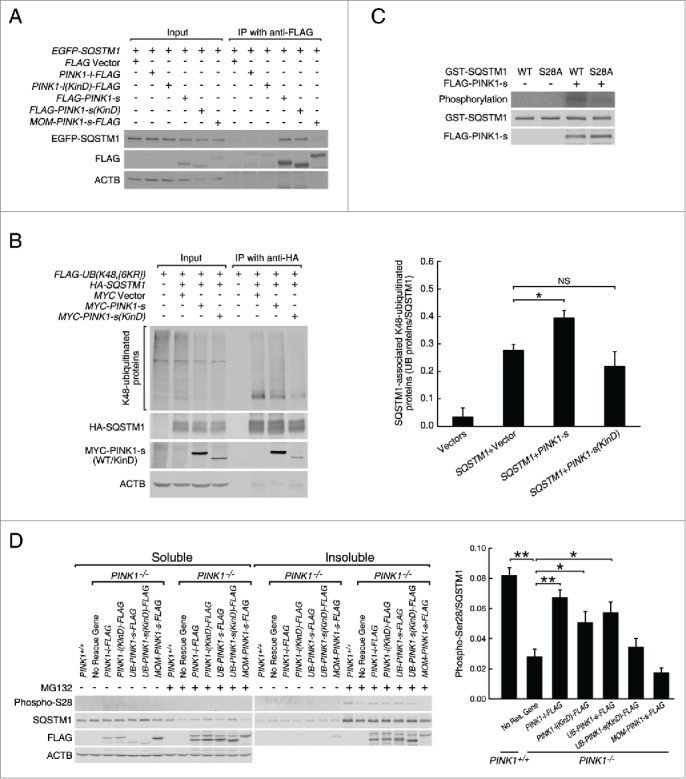
PINK1-s phosphorylates SQSTM1 at Ser28 during proteasomal stress. (A) Immunoprecipitation and western blot analysis of PINK1-SQSTM1 interaction. AD293 cells were transfected with the indicated plasmids. The FLAG-tagged PINK1 proteins were immunoprecipitated with FLAG antibody and the associated EGFP-SQSTM1 was detected with EGFP antibody. (B) Western blot analysis of SQSTM1-bound, K48-ubiquitinated proteins in control cells and cells expressing MYC-PINK1-s or MYC-PINK1-s(KinD). Cells were transfected with the indicated plasmids and immunoprecipitated with HA antibody. HA-SQSTM1-bound ubiquitinated proteins were detected with FLAG antibody. Quantification results are shown as mean±SEM of 3 independent experiments. *, P < 0.05; NS, non-significant. (C) In vitro phosphorylation assay of the wild-type (WT) and S28A mutant GST-SQSTM1 by PINK1-s. GST-SQSTM1(WT) and GST-SQSTM1S28A were expressed in bacteria and affinity purified with glutathione beads. FLAG-PINK1-s was expressed in AD293 cells and immunopurified with FLAG antibody. GST-SQSTM1(WT) or GST-SQSTM1S28A was incubated with FLAG-PINK1-s in the presence of γ-32P-ATP to label the phosphorylated proteins. GST-SQSTM1 and FLAG-PINK1-s in the reactions were detected by western blot analysis with GST and FLAG antibodies, respectively. (D) Western blot analysis of SQSTM1 Ser28 phosphorylation in the soluble and insoluble fractions of PINK1+/+, PINK1−/− and rescue cells after a 12-h treatment with DMSO or MG132 (1 μM). Eight percent of the soluble and 20% of the insoluble fractions of each sample were loaded for SDS-PAGE and western blot analysis. Ser28 phosphorylation was detected with a custom-made antibody. Soluble ACTB levels were detected as the loading control. Quantification results are shown as mean±SEM of 3 independent experiments. *, P < 0.05; **, P < 0.01.
Deletion analysis indicated that PINK1-s could interact with 2 different parts of SQSTM1, an N-terminal fragment F1 composed of the PB1 domain and a central fragment F2 composed of ZZ, LB and TRAF6 domains (Fig. S9A and B).10 To further determine which interaction(s) was relevant to the regulation of SQSTM1 function by PINK1-s, we coexpressed F1 or F2 with PINK1-S to see which fragment could function as a dominant-negative mutant of SQSTM1 and attenuate the aggresome-promoting effect of PINK1-s. As expected, overexpression of PINK1-s together with EGFP (control) led to a 242% increase in the levels of insoluble ubiquitinated proteins (Fig. S9C). Whereas PINK1-s had a similar effect when coexpressed with EGFP-F2, its ability to promote the accumulation of ubiquitinated proteins in the insoluble fractions was significantly reduced when coexpressed with EGFP-F1 (a 64% increase vs. 242% increase in control cells, P=0.0061; Fig. S9C). PINK1-s also induced the accumulation of endogenous SQSTM1 in the insoluble fractions. This was also significantly repressed by EGFP-F1 (a 64% increase vs. 210% increase in control cells, P=0.0183; Fig. S9C) but not by EGFP-F2. These results suggest that F1 acts as a dominant-negative mutant of SQSTM1 and thus, PINK1-s most likely promotes aggresome formation through its interaction with the PB1 domain.
To identify the PINK1-s phosphorylation site in SQSTM1, we coexpressed FLAG-SQSTM1 with either MYC tag or MYC-PINK1-s in AD293 cells and immunopurified FLAG-SQSTM1 for mass spectrometry analysis. In addition to 6 common phosphorylation sites identified in both samples, FLAG-SQSTM1 coexpressed with MYC-PINK1-s also contained a unique phosphorylation site at amino acid 28, a serine residue in PB1 domain (Fig. S9D and E). Sequence alignment of SQSTM1 from different species indicated that Ser28 and its surrounding sequence are well conserved in mammals (Fig. S9E). An in vitro phosphorylation assay with immunopurified FLAG-PINK1-s showed that it could effectively phosphorylate GST-tagged wild-type SQSTM1 protein, but not the mutant SQSTM1 protein with Ser28 replaced by the nonphosphorylatable Ala (S28A mutant, Fig. 4C). This result confirmed Ser28 of SQSTM1 is the major phosphorylation site of PINK1-s. To facilitate the detection of Ser28 phosphorylation in cells, we generated a phospho-Ser28-specific antibody (Fig. S9F). Western blot analysis revealed that Ser28 phosphorylation was not detectable in cells under normal physiological conditions, but significantly increased in the insoluble fraction of the cells treated with MG132, and this increase was reduced by 65.8% in PINK1−/− cells, suggesting that PINK1 was the major kinase that mediated Ser28 phosphorylation during proteasomal stress. Expression of UB-PINK1-s-FLAG in PINK1−/− cells increased Ser28 phosphorylation by 104.5%, whereas MOM-PINK1-s-FLAG did not have the same effect, suggesting that PINK1-s was the isoform responsible for SQSTM1 phosphorylation (Fig. 4D).
Phosphorylation of SQSTM1 at Ser28 is required for efficient aggresome formation during proteasomal stress
To explore the significance of Ser28 phosphorylation, we mutated Ser28 to a phosphomimetic Glu residual (S28E mutation) or nonphosphorylatable Ala residual (S28A mutation), and examined the effects on the UB chain-binding capability of SQSTM1. Because overexpressed SQSTM1 tended to oligomerize and form insoluble protein aggregates, another point mutation that could suppress its oligomerization, D69A, was also introduced to ensure the mutant proteins remained in the soluble fractions.43 Immunoprecipitation with HA antibody from cells coexpressing FLAG-UB(K48,[6KR]) and one of the SQSTM1 single or double mutants, HA-SQSTM1D69A, HA-SQSTM1D69A,S28E or HA-SQSTM1D69A,S28A, showed that the S28E mutation increased the levels of SQSTM1D69A-bound ubiquitinated proteins by 36%, whereas the S28A mutation did not have a significant effect, strongly suggesting that PINK1-s enhances SQSTM1 activity by phosphorylating Ser28 (Fig. 5A).
Figure 5.
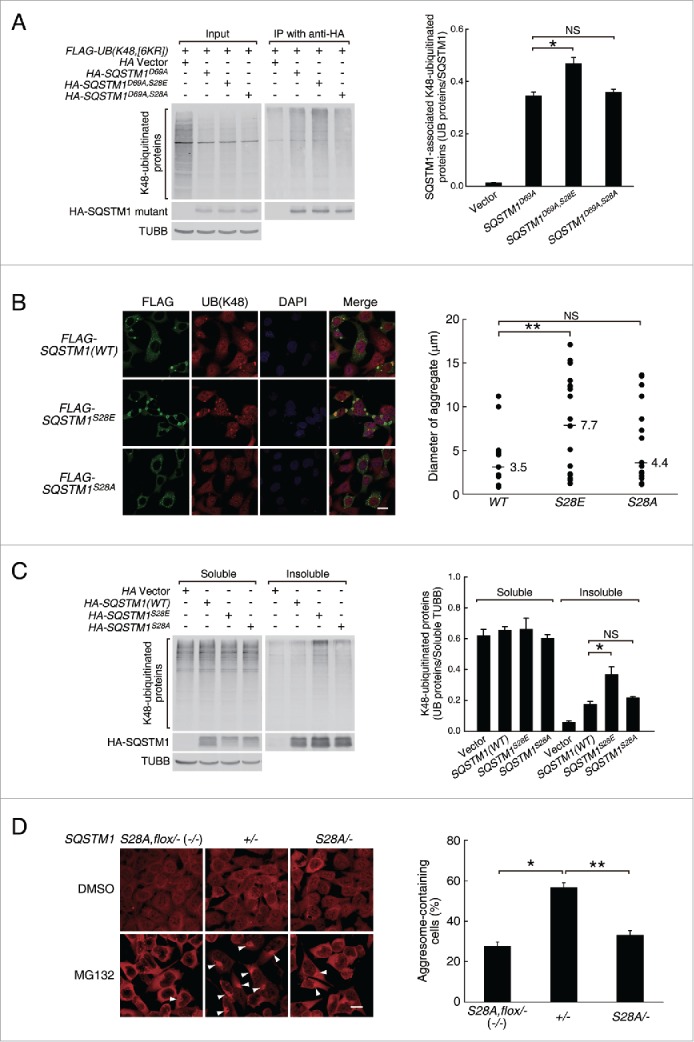
Ser28 phosphorylation in SQSTM1 was required for efficient aggresome-targeting of ubiquitinated proteins during proteasomal inhibition. (A) Coimmunoprecipitation and western blot analysis of the K48-ubiquitinated proteins bound by the WT and Ser28 mutant SQSTM1. AD293 cells were transfected with the indicated plasmids, immunoprecipitated with HA antibody and the associated ubiquitinated proteins were detected with FLAG antibody. The D69A mutation was introduced into SQSTM1 to reduce self-oligomerization and thus increase its solubility. Soluble TUBB (tubulin β class I) was detected as the loading control. Quantification results are shown as mean ± SEM of 3 independent experiments. *, P < 0.05; NS, nonsignificant. (B) Aggregate formation in AD293 cells expressing the WT or the S28E or S28A mutant SQSTM1. The overexpressed SQSTM1 and ubiquitinated proteins were detected by immunofluorescence staining with FLAG and UB(K48) antibodies, respectively. Scale bar: 20 μm. Quantification results are shown as mean ± SEM of 3 independent experiments. **, P < 0.01; NS, non-significant. (C) Western blot analysis of the soluble and insoluble K48-ubiquitinated proteins in AD293 cells expressing the WT or mutant SQSTM1. Cells were transfected with the indicated plasmids. Sixteen percent of the soluble proteins and 50% of the insoluble proteins of each sample were loaded for SDS-PAGE. UB(K48) antibody was used to detect the soluble and insoluble ubiquitinated proteins. HA antibody was used to detect the WT and mutant HA-SQSTM1. TUBB was used as the loading control. Quantification results are shown as mean ± SEM of 3 independent experiments. *, P < 0.05; NS, nonsignificant. (D) Aggresome formation in SQSTM1S28A,flox/− (SQSTM1−/−), SQSTM1+/− and SQSTM1S28A/− cells after a 14-h treatment with DMSO or MG132 (1.5 μM). Ubiquitinated proteins were detected by immunofluorescence staining with UB(K48) antibody. The nuclei were visualized by DAPI staining. Aggresomes are indicated by arrowheads. Scale bar: 20 μm. Quantification results are shown as mean ± SEM of 3 independent experiments. *, P < 0.05; **, P < 0.01.
Overexpressed SQSTM1 usually undergoes self-oligomerization and forms UB-positive aggregates and aggresomes. This feature provides a simple assay for the effects of Ser28 mutations on SQSTM1-mediated aggregate and aggresome formation. Compared with the control cells expressing HA-SQSTM1(WT), HA-SQSTM1S28E-expressing cells tended to form larger aggregates and aggresomes with the average diameter increased by 120% (7.7 vs. 3.5 μm, P=0.001277) (Fig. 5B). Biochemical analysis indicated that the insoluble ubiquitinated proteins in HA-SQSTM1S28E-expressing cells also increased by 109%, suggesting that the increase in the aggregate and aggresome sizes was caused by sequestration of a larger amount of ubiquitinated proteins (Fig. 5C). Consistent with this, neither the S28E nor the S28A mutation changed the oligomerization capability of SQSTM1, as assayed by immunoprecipitation (Fig. S10A).
Since SQSTM1 is a major UB receptor, its activation is a critical event in proteasomal inhibition-induced aggregate and aggresome formation. Recently, 2 other protein kinases, CSNK2/CK2 (casein kinase 2) and ULK1 (unc-51 like autophagy activating kinase 1), have been reported to promote the UB binding of SQSTM1 through phosphorylating 2 serine residues, Ser403 and Ser407 (also referred to as Ser409 by Lim et al. 2015),42 in its UBA domain.41,42 Interestingly, phosphorylation of Ser407 by ULK1 facilitates the subsequent phosphorylation of Ser403 by CSNK2, suggesting that these 2 kinases function in the same pathway to activate SQSTM1.42 Puzzlingly, the PINK1-s phosphorylation site Ser28 is localized in the PB1 domain, so its phosphorylation must enhance the UB affinity of SQSTM1 indirectly. One possibility was that it might also facilitate Ser403 phosphorylation. To test this hypothesis, we compared the levels of Ser403 phosphorylation in cells expressing WT SQSTM1 or the SQSTM1S28E and SQSTM1S28A mutants, but we did not detect a significant difference between them (Fig. S10B). This result suggests that Ser28 phosphorylation enhances the UB affinity of SQSTM1 through a different mechanism.
A critical test for the significance of Ser28 phosphorylation was to determine whether replacing the endogenous SQSTM1 with SQSTM1S28A had any effects on proteasomal stress-induced aggresome formation. Using transcription activator-like effector nucleases (TALEN),44 we generated SQSTM1+/− cells, which retained one copy of the wild-type SQSTM1 gene, SQSTM1S28A/− cells, which contained one copy of SQSTM1S28A mutant gene, and SQSTM1S28A,flox/− cells, which were essentially SQSTM1-null mutants due to the blockage of SQSTM1S28A expression by a floxed Blasticidin S-resistance gene. A 14-h treatment with 1.5 μM MG132 induced aggresome formation in 56.7% of SQSTM1+/− cells and 27.6% of the SQSTM1S28A,flox/− (SQSTM1−/−) cells, respectively, consistent with the current model that SQSTM1 is a critical regulator of aggresome formation during proteasomal stress. The same treatment induced aggresome formation in 33.1% of the SQSTM1S28A/− cells, confirming that Ser28 phosphorylation is required for efficient aggresome formation during proteasomal stress (Fig. 5D).
Discussion
The packaging of ubiquitinated proteins into aggresomes and their subsequent autophagic degradation constitutes a major cytoprotective pathway in response to proteasomal impairment,2,3 but the mechanism that activates this cytoprotective pathway is still poorly understood. In this report, we provide evidence that PINK1-s, a short form of mitochondria-targeted Ser/Thr protein kinase PINK1, promotes SQSTM1-mediated sequestration of ubiquitinated proteins into aggregates and aggresomes (Fig. S11). The levels of cytosolic PINK1-s strongly correlate with the degree of proteasomal inhibition, indicating that PINK1-s functions as a sensor that links the proteasomal deficiency signal to the aggresome formation process.
PINK1-s is generated from the full-length PINK1-l through PARL-mediated proteolytic cleavage. With an “unstable” amino acid Phe104 at its N terminus, it is recognized by the N-end rule pathway and is rapidly degraded by proteasomes.19 In fact, the steady-state levels of PINK1-s are so low under physiological conditions that it is undetectable by western blot analysis in many cell types. This labile nature makes it a good candidate to serve as an upstream modulator in proteasomal stress response. In this study, we tested this hypothesis by examining the functions of PINK1-s that accumulates under moderate proteasomal inhibition and found that it functioned as an activator of the aggresome formation process. First, overexpression of PINK1-s induced the formation of aggregates and aggresomes without affecting proteasomal activities. Unlike the aggregates and aggresomes formed by EGFP-250, a misfolded EGFP fusion protein, PINK1-s-induced aggregates and aggresomes also contained high levels of K48-linked ubiquitinated proteins, suggesting that it directed the assembly of aggregate and aggresome formation machinery and activated the sequestration of ubiquitinated proteins. Second, overexpression of PINK1-s caused the sequestration of GFPU, a model proteasomal substrate, into aggregates and aggresomes, suggesting that high levels of PINK1-s were sufficient to reroute proteasomal substrates to the autophagy pathway. Third, PINK1 knockout cells displayed reduced efficiency in aggresome formation and this phenotype could only be suppressed by the expression of PINK1-s, but not by MOM-PINK1-s, which mimicked PINK1-l on the MOM, suggesting that the reduced aggresome formation efficiency in PINK1 mutant cells is due to their lack of PINK1-s activity in the cytosol. Lastly, PINK1-s promoted the association of SQSTM1 with ubiquitinated proteins through the phosphorylation of its Ser28 residue, an important modification for endogenous SQSTM1 during aggresome formation. Collectively, these results strongly support a major role of PINK1-s in the transduction of proteasomal impairment signal to the downstream aggresome formation machinery.
An unusual feature of this signaling pathway is that it does not rely on another PD-related protein, PARK2. So far, PARK2 has been regarded as a close partner of PINK1 and a crucial UB E3 ligase in multiple autophagy processes. During mitophagy, it cooperates with PINK1-l at the surface of damaged mitochondria to induce the K63 and K27-ubiquitination of MOM proteins, which serves as a signal for mitochondrial translocation and initiation of autophagy. 20-22 Also through its ability to catalyze the formation of K63 UB chain, PARK2 can induce the aggresome-targeting of misfolded proteins, such as PARK7L166P/DJ-1L166P.30 Surprisingly, PARK2 is dispensable for PINK1-s-induced aggresome formation. In AD293 and L02 cells, which we used to study PINK1-s-induced aggresome formation, PARK2 expression is extremely low or undetectable. Consistent with this result, suppression of UB(K48) chain formation in PINK1-s-expressing cells dramatically reduced the levels of ubiquitinated proteins in the aggregates and aggresomes, whereas suppression of UBK63 chain formation had no effect, confirming that UBK63 chain is not the targeting signal for proteins sequestered in PINK1-s-induced aggregates and aggresomes. Our subsequent analysis revealed that PINK1-s induced aggresome formation through a mechanism distinct from PINK1-l-induced mitophagy. Instead of generating the targeting signal through the formation of the UBK63 chain, PINK1-s directly phosphorylates SQSTM1 and enhances its UB(K48) chain-binding affinity, making the formation of new UB(K63) chain unnecessary.
Although PINK1-s is an important mediator of aggresome formation, our study clearly indicates that additional activators are required for this process. Loss of PINK1 did not completely abolish proteasomal inhibition-induced Ser28 phosphorylation in SQSTM1, suggesting that other Ser/Thr protein kinase(s) also participate in the phosphorylation of this site during proteasomal stress. Furthermore, replacement of the endogenous SQSTM1 with the S28A mutant did not completely abolish proteasomal inhibition-induced aggresome formation, suggesting that additional SQSTM1-independent pathway(s) might participate in the activation of this process. Recently, Minoia et al. have found that BAG3 (BCL2 associated athanogene 3), a proteasomal stress-induced chaperone, can sequester ubiquitinated proteins into cytosolic aggregates for autophagic degradation.45 However, it is unclear whether it exerts its aggregate-promoting effects through activation of the PINK1-s-SQSTM1 pathway.
SQSTM1-mediated aggregate and aggresome formation has been initially identified as a compensatory protein degradation pathway, but studies have also revealed that they could play critical roles in the maintenance of protein homeostasis at basal conditions. Recently, Milan et al. have found that in multiple myeloma (MM) cells with active IgG synthesis, which have proteasomes under heavy burden, the SQSTM1-mediated aggresome-autophagy pathway is responsible for the turnover of a significant portion of the ubiquitinated proteins.46 Interestingly, MM cells from different patients exhibit large variance in their aggresome formation and autophagic clearance efficiency. As a result, the activity of the SQSTM1-mediated aggresome-autophagy pathway is a major determinant of sensitivity to bortezomib, the first proteasomal inhibitor drug used for MM treatment. Given that PINK1-s is a major activator of SQSTM1 during aggregate and aggresome formation, it is likely that MM cells could gain their resistance to bortezomib treatment through overexpression and stabilization of PINK1-s. If this is true, PINK1-s could serve as a new pharmacological target for the development of future MM treatments.
Neurons are another cell type in which basal autophagy is critical for protein homeostasis. Mice with their key autophagy genes, such as Atg5 (autophagy-related 5) or Atg7 (autophagy-related 7), knocked out in CNS show neurodegeneration early in their lives, with the accumulation of UB-containing protein aggregates in their neurons.47,48 One mechanism of autophagy inhibition-induced cell damage is the stabilization of SQSTM1. Since autophagy is the main route for SQSTM1 degradation, its prolonged inhibition leads to the excessive accumulation of SQSTM1, which blocks the proteasome-targeting of ubiquitinated proteins and causes indirect proteasomal inhibition.49 Puzzlingly, loss of Sqstm1 in neuron-specific Atg7-knockout mice suppresses aggregate formation and presumably relieves proteasomal inhibition in neurons, but it does not rescue the neurodegeneration phenotypes.50 A plausible explanation is that in the absence of basal autophagy, proteasomes are not sufficient for the degradation of ubiquitinated proteins and maintenance of neuronal health. If this is true, the SQSTM1-mediated aggresome-autophagy pathway must be constantly active in neurons. Consistent with this notion, cytosolic PINK1-s is present in neurons even in the absence of proteasomal dysfunction, presumably due to an uncharacterized stabilization mechanism.51 It would be interesting to determine whether PINK1-s protects neurons by promoting the aggresome-targeting of ubiquitinated proteins through the activation of SQSTM1.
Although PINK1-s does not possess a MTS, it can still be targeted to mitochondria. Cell fractionation analysis suggested that when overexpressed in cells, a small fraction of PINK1-s is loosely associated with the outer membrane of mitochondria, presumably through interaction with other MOM proteins.52 This mitochondrial pool of PINK1-s sometimes functions in a similar fashion as PINK1-l. For example, like PINK1-l, which can inhibit the transport of damaged mitochondria in neuronal axons by inducing the proteasomal degradation of RHOT1/MIRO1 (ras homolog family member T1) and RHOT2/MIRO2 (ras homolog family member T2), overexpressed PINK1-s can also cause mitochondrial arrest.53,54 Recently, Dogda et al. have found that PINK1 promotes the dendritic arborization in developing neurons through enhancing the anterograde transport of mitochondria. Interestingly this effect is mediated exclusively by PINK1-s but not PINK1-l, suggesting that PINK1-s plays an important regulatory role in mitochondrial transport.55 In our study, we have not carefully examined the mitochondria-related function of PINK1-s, but we did observe its mitochondrial localization occasionally in cells treated with proteasomal inhibitors. Since proteasomal inhibition induces the “collapse” of mitochondria to the nuclear periphery, it is highly possible that in addition to promoting aggresome formation, PINK1-s stabilized during proteasomal stress also induces the redistribution of mitochondria in cells.
Proteasomal and mitochondrial dysfunctions have been regarded as the 2 interrelated mechanisms in the pathogenesis of PD. In this context, it is intriguing that PINK1 gene, which is mutated in some PD patients, can produce 2 protein isoforms that activate distinct response pathways to stress caused by proteasomal and mitochondrial damages, respectively. It is tempting to hypothesize that like PINK1-l-mediated activation of mitophagy, PINK1-s-mediated activation of aggresome formation is also critical in the maintenance of neuronal health. Unfortunately, although aggresome formation in neurons and neuronal cell lines has been described before,56 we have not been able to reliably achieve that with moderate proteasomal inhibition, the experimental condition that we have successfully used to study PINK1-s functions in AD293 and L02 cells, so it is unclear whether neuronal PINK1-s plays a similar role during proteasomal stress. Future studies with transgenic and knock-in animals will address whether loss of PINK1-s function will comprise the efficiency in the autophagic clearance of ubiquitinated proteins and contribute to the death of dopaminergic neurons.
Materials and methods
Cell culture
AD293 cell line was used for most of the study. It was obtained from Stratagene/Agilent Technologies Inc.. It was cultured at 37°C in Dulbecco's modified Eagle's medium (DMEM, Corning, 10-013-CVR) supplemented with 10% fetal bovine serum (FBS; Biowest, S01520), 100 units/ml penicillin and 100 μg/ml streptomycin. HEK293FT cell line (Invitrogen/Life Technologies, R700-07) was used for lentivirus production, and was cultured at 37°C in DMEM supplemented with 10% FBS, 2 mM L-glutamine, 1x Non-Essential Amino Acids (NEAA), 100 units/ml penicillin and 100 μg/ml streptomycin. L02 cell line was cultured at 37°C in RPMI-1640 medium supplemented with 10% FBS, 2 mM L-glutamine, 100 units/ml penicillin and 100 μg/ml streptomycin.
Chemical reagents
Carbonyl cyanide 3-chlorophenylhydrazone (CCCP) was obtained from Tocris Bioscience (0452). MG132, rotenone and tunicamycin were obtained from EMD Millipore (474790, 557368, 654380). Bortezomib (Velcade, Bort) was obtained from Selleck Chemicals (S1013). Bafilomycin A1 (BafA1) was obtained from EMD Millipore (196000) and Selleck Chemicals (S1413).
Quantification of insoluble ubiquitinated proteins
Cells were grown on 24-well plates. After predesigned treatments, cells were incubated on ice with 100 μl/well NP40 Alternative-containing cell lysis buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1mM EDTA, 1% NP40 Alternative [EMD Millipore, 492016]) for 15 min. The lysates were transferred to microcentrifuge tubes and centrifuged at 14,000 g for 15 min. The 100 μl supernatant of each sample was mixed with 25 μl 5x SDS sample buffer, and the pellet was solubilized with 40 μl 1x SDS buffer. Both NP40-soluble and insoluble samples were boiled for 5 min, and 20 μl of each was used for SDS-PAGE. The relative levels of ubiquitinated proteins in different samples were determined by western blot analysis with a K48-linked UB chain-specific antibody (EMD Millipore, 05–1307).
Immunoprecipitation
AD293 cells were transfected with the indicated plasmids. After 24 h, the cells were lysed in IP Buffer for 10 min on ice. Cell lysates were centrifuged at 4°C for 15 min to remove insoluble materials. The supernatant fraction from each sample next underwent a 2-h incubation with 2 μl of an IP antibody followed by a 1-h incubation with 10 μl protein G agarose beads (EMD Millipore, 16–266). The beads were then spun down, washed 3 times with wash buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1% NP40 Alternative) and resuspended in 1x SDS protein sample buffer. After a 5-minute boiling, the samples were analyzed by SDS-PAGE and western blot analysis with a detection antibody.
Western blot analysis
Protein samples in SDS Sample Buffer were separated by SDS-PAGE and transferred to Immobilon-FL PVDF membrane (EMD Millipore, IPFL00010). The membrane was next blocked with 5% nonfat dry milk in PBST (PBS, pH 7.4, 0.2% Tween-20) and incubated at 4°C overnight with primary antibodies. The membrane was then washed 3 times with PBST and incubated with Dylight 680 and/or Dylight 800-conjugated secondary antibodies (KPL) in the dark for 2 h. After 3 washes with PBST, the image was acquired by Li-Cor Odyssey Clx Infrared Imaging System (LI-COR Biotechnology, Lincoln, NE, USA).
The following primary antibodies were used for western blot analysis in this study: PINK1 (Abgent, AM6406a), SQSTM1 (Proteintech, 18420-1-AP), Phospho-SQSTM1 (Ser403; EMD Millipore, MABC186), TOMM40 (Proteintech, 18409-1-AP), VDAC1 (Mitosciences, MSA03), UB (Biolegend, MMS-264R-100), K48-linked UB chain-specific (EMD Millipore, 05-1307), ATG5 (Cell Signaling Technology, 12994), MAP1LC3A/B (Cell Signaling Technology, 4108), FLAG Tag (Prospec, ANT-146-b), HA Tag (Proteintech, 51064-2-AP), Myc Tag (Biolegend, MMS-150R), GFP (Immunology Consultants Laboratory, RGFP-45A), GAPDH (Zen Bioscience, 200306), TUBB (Zen Bioscience, 200608), and ACTB (Proteintech, 60008-1-Ig).
Immunocytochemistry
Cells grown on poly-D-Lysine-coated cover slips were washed with PBS and fixed with 4% paraformaldehyde in PBS for 15 min at room temperature. They were then permeabilized with 0.2% Triton X-100 (EMD Millipore, 9410-1L) in PBS for 15 min, blocked with 5% goat serum (Jackson ImmunoResearch Laboratories, 005-000-121) for 1 h and incubated with primary antibodies at room temperature for 2 h or 4°C overnight. After washing with PBS, cells were incubated with Alexa Flour 488 or 568-conjugated secondary antibodies (Invitrogen/Life Technologies, A11034, A11029, A11031, A11036) for 2 h, stained with DAPI solution (EMD Millipore, 508741) for 5 min and mounted for fluorescence microscopy. The following primary antibodies were used: HDAC6 Antibody (Abgent, AP1106a), SQSTM1 Antibody (Proteintech, 18420-1-AP), FLAG Antibody (Propec, ant-146-b), TOMM20 Antibody (Proteintech, 11802-1-AP), TOMM40 Antibody (Proteintech, 18409-1-AP) and K48-linked UB chain-specific Antibody (EMD Millipore, 05–1307).
Generation of AD293 PINK1−/− and its rescue cell lines
PINK1−/− cell lines were generated from AD293 cells with a CRISPR/Cas9 system35 modified by our lab. Briefly, 2 pairs of oligos targeting the CCGCCGCTGCCGCCACCAC sequence in the 5′UTR and GGCCGGGTCCTAAGCCGAG sequence in the 1st intron of the PINK1 gene were annealed and cloned into the gRNA expression vector, respectively. The resultant plasmids, gRNA-PINK1A and gRNA-PINK1B, were transfected into AD293 cells with Cas9 expression vector. After 24 h, the cells were diluted and seeded in 96-well plates at 1 cell/well to isolate monoclonal cell lines. PINK1−/− cell lines were isolated by western blot screen for the failure of PINK1-s accumulation after a 12 h treatment with 1 μM MG132 (EMD Millipore, 474790) and verified by genomic PCR.
PINK1−/− rescue cell lines were generated by transduction of PINK1−/− cells with lentiviruses harboring PINK1-l-FLAG, PINK1-l(KinD)-FLAG, UB-PINK1-s-FLAG, UB-PINK1-s(KinD)-FLAG or MOM-PINK1-s-FLAG rescue genes, and a puromycin resistance gene. After 48 h, the transduced cells were selected by growing cells in the DMEM growth medium containing 0.5 μg/ml puromycin (InvivoGen, ant-pr-5), and the expression of the rescue gene was verified by western blot analysis and immunofluorescence staining.
Generation of AD293 SQSTM1+/− , SQSTM1S28A,flox/− (SQSTM1−/−) and SQSTM1S28A/− cell lines
SQSTM1+/−, SQSTM1S28A,flox/− and SQSTM1S28A/− cells were generated from AD293 cells with TALEN designed by Viewsolid Biotechnology Co. Ltd., China. To generate SQSTM1+/− cells, AD293 cells were first transfected with expression vectors of a pair of TALEN, SQSTM1-T2L and SQSTM1-T2R, which targeted the GCGCTGGCTGCTCCCT and GGCCGAACTGGGGACCC sequences in Intron 1 of SQSTM1 gene, respectively. After 24 h, the cells were diluted and seeded in 96-well plates at 1 cell/well to isolate monoclonal SQSTM1+/− cell lines with about 50% reduction in SQSTM1 expression as determined by western blot analysis.
To generate SQSTM1S28A/− cells, a knockin vector, pBSK- SQSTM1N573-Exon1(S28A)-LoxP-Blar-LoxP-SQSTM1C521, was first generated by Gibson Assembly. It consists of a 573-bp genomic sequence upstream of the ATG start codon of SQSTM1, Exon 1 of SQSTM1 gene with the S28A mutation, a 113 bp intron 1 sequence upstream of the SQSTM1-T2L site, a floxed blasticidin S-resistance gene (Blar) expression cassette and a 521-bp genomic sequence downstream of the SQSTM1-T2R site. The knockin vector, together with SQSTM1-T2L and SQSTM1-T2R expression vectors, was transfected into SQSTM1+/− cells. After 24 h, the cells were transferred into growth medium with 10 μg/ml blasticidin S (InvivoGen, ant-bl-5). Resistant cells were diluted and seeded in 96-well plates at 1 cell/well to isolate monoclonal cell lines with no SQSTM1 expression, as determined by western blot analysis. The resultant SQSTM1S28A,flox/− (SQSTM1−/−)cell lines were infected with a Cre lentivirus to remove the Blar gene. Cell lines with restored SQSTM1S28A expression after Cre-mediated recombination were further characterized by PCR amplification of the first exon in the SQSTM1 gene followed by sequencing to verify the integration of the S28A mutation.
Cell death assay
The cell death-inducing effects of proteasomal inhibition were measured with CF488A-Annexin V (ANXA5) and PI Apoptosis Kit (Biotium, 30061). Cells were grown on 10-cm dishes. After reaching 70% confluency, they were treated with DMSO or 1 μM MG132 for 20 h. They were then harvested by digestion with 0.05% trypsin-EDTA solution, washed twice with ANXA5-Binding Buffer and resuspended in 100 μl of the same buffer. To each sample, 15 μl CF488A-ANXA5 and 5 μl propidium iodide (PI) solutions were added and incubated in the dark for 30 min at room temperature. After the unbound dyes were washed off with the binding buffer, the cells were mounted onto slides and images were acquired with a Nikon fluorescence microscope.
To measure the cell sensitivity to hypoxia, rotenone or tunicamycin, cells were grown in 0.2% O2, 5% CO2 for 48 h, or treated with 50 μM rotenone (EMD Millipore, 557368) or 10 μg/ml tunicamycin (EMD Millipore, 654380) for 24 h before they were harvested for the cell death analysis described above.
Identification of phosphorylation sites by mass spectrometry
AD293 cells were cultured in 2 10-cm dishes and transfected with MYC Vector and FLAG-SQSTM1 as well as MYC-PINK1-s and FLAG-SQSTM1 respectively. After 24 h of expression, cells in each dish were lysed on ice with 2 ml IP buffer (50 mM Tris-HCl pH8.0, 150 mM NaCl, 1 mM EDTA, 1% NP40 Alternative, Halt Protease Inhibitor Cocktail [Thermo Scientific, 78430] and Halt Phosphatase Inhibitor Cocktail [Thermo Scientific, 78428]), and centrifuged to remove insoluble debris. From each supernatant, FLAG-SQSTM1 was immunopurified with 20 μl FLAG antibody (Prospec, ant-146-b) and 200 μl protein G agarose beads (EMD Millipore, 16–266), further separated from other contaminant proteins by SDS-PAGE and identified by staining with 0.25% Coomassie brilliant blue R-250. The FLAG-SQSTM1 protein bands were cut out and digested with MS-grade trypsin. Peptides were analyzed by MALDI-TOF mass spectrometry at the Laboratory of Proteomics, Institute of Biophysics, Chinese Academy of Sciences. The phosphorylation sites were identified by Mascot database search (Matrix Science).
In vitro kinase assay
Two 10-cm dishes of AD293 cells were transfected with FLAG-PINK1-s expression plasmid. Twenty-four h later, the cells were washed with phosphate-buffered saline (PBS, 137 mM NaCl, 2.7 mM KCl, Na2HPO4 10 mM, KH2PO4 1.8 mM, pH 7.4) and lysed with IP Buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1 mM EDTA, 1% NP40 Alternative, Halt Protease Inhibitor Cocktail, Halt Phosphatase Inhibitor Cocktail) for 15 min on ice. Cell lysates were centrifuged at 4°C for 15 min to remove insoluble materials. The supernatant fraction next underwent a 2-h incubation with 20 μg FLAG antibody (followed by a 1-h incubation with 200 μl protein G agarose beads. The beads were then spun down, washed 3 times with wash buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1% NP40 Alternative), once with the phosphorylation buffer (50 mM Tris, pH 7.5, 10 mM MgCl2, 2 mM EGTA, 5 mM DTT), and resuspended in 100 μl of the phosphorylation buffer.
For each kinase reaction, 50 μl of the control or FLAG-PINK1-s-bound protein G bead slurry was mixed with 2 μg of purified GST-SQSTM1 or GST-SQSTM1 (S28A) protein, 5 μCi of γ-32P-ATP and incubated at 30°C for 1 h with gentle agitation. The reaction was stopped by the addition of 8 μl 5x SDS sample buffer followed by a 5-min boiling. After the separation of proteins by SDS-PAGE, the gel was covered with Saran wrap and analyzed with a Cyclone Plus Phosphor Imager (PerkinElmer, Waltham, MA, USA). The proteins on the gel were then transferred to a PVDF membrane, and FLAG-PINK1-s and GST-SQSTM1 were detected by western blot analysis with FLAG tag (ProSpec, ant-146-b) and GST tag (Proteintech, 10000-0-AP) antibodies.
Detection of Ser28 phosphorylation by western blot analysis
Rabbit polyclonal antibody for phosphorylated Ser28 of SQSTM1 was generated and purified by PhosphoSolutions Inc.
To analyze Ser28 phosphorylation during proteasomal inhibition, AD293 PINK1+/+, PINK1−/− and its rescue cells were grown on 6-well plates and treated with DMSO or 1 μM MG132 for 12 h. They were next incubated on ice with 200 μl/well NP40 Alternative-containing cell lysis buffer for 20 min. The lysates were collected, and the soluble and insoluble fractions were separated by centrifugation at 14,000 g, 4°C for 15 min. The 200 μl supernatant fraction of each sample was mixed with 50 μl 5x SDS sample buffer, and the pellet fraction was solubilized with 100 μl 1x SDS buffer. After boiling, 20 μl of each sample was separated by SDS-PAGE and the proteins were transferred to Immobilon-FL PVDF membrane (EMD Millipore, IPFL00010). The membrane was first blocked with 1x Western Blocking Reagent (Roche Life Science, 11921673001) for one h, and then incubated overnight with pSer28 antibody diluted in the same blocking buffer at 1:100. After 3 washes with TBST (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.2% Tween 20 [EMD Millipore, 655206]), the blot was incubated with Dylight 680-conjugated secondary antibodies (KPL, 072-06-15-16) in the dark for 2 h. The blot was then washed again with TBST and the image was acquired with a Li-Cor Odyssey Clx Infrared Imaging System (LI-COR Biotechnology, Lincoln, NE, USA).
Statistics
Each experiment was performed at least 3 times. Fluorescence images from aggresome formation and cell death assay results were quantified manually. The sizes of aggresomes and aggregates were quantified with ImageJ. Results are expressed as mean±SEM. Statistical significance was determined by the Student t test.
Supplementary Material
Abbreviations
- ACTB
actin, β
- BafA1
bafilomycin A1
- BAG3
BCL2 associated athanogene 3
- Bort
Bortezomib
- CRISPR
clustered regularly interspaced short palindromic repeats
- CSNK2/CK2
casein kinase 2
- DAPI
4′,6-diamidino-2-phenylindole
- DUB
deubiquitinating enzyme
- EGFP
enhanced green fluorescent protein
- ER
endoplasmic reticulum
- GFPU
GFP-based UPS reporter
- HDAC6
histone deacetylase 6
- HSPA5/Grp78
heat shock protein family A (Hsp70) member 5
- KinD
kinase dead
- LAMP1
lysosomal-associated membrane protein 1
- MAP1LC3B/LC3B
microtubule associated protein 1 light chain 3 β
- MM
multiple myeloma
- MOM
mitochondrial outer membrane
- MTOC
microtubule organization center
- MTOR
mechanistic target of rapamycin (serine/threonine kinase)
- MTS
mitochondria-targeting sequence
- NOD2
nucleotide binding oligomerization domain containing 2
- PARK2/PRKN/PARKIN
parkin RBR E3 ubiquitin protein ligase
- PARK7/DJ-1
parkinson protein 7
- PARL
presenilin associated, rhomboid-like
- PB1 domain
Phox and Bem1P domain
- PD
Parkinson disease
- PI
propidium iodide
- PINK1
PTEN induced putative kinase 1
- PINK1-l
long isoform of PINK1 protein, full-length PINK1 protein
- PINK1-s
short isoform of PINK1 protein
- RHOT1/MIRO1
ras homolog family member T1
- RHOT2/MIRO2
ras homolog family member T2
- SQSTM1
sequestosome 1
- TALEN
transcription activator-like effector nucleases
- TMD
transmembrane domain
- TNFRSF1A/TNFR
tumor necrosis factor receptor superfamily member 1A
- TOMM20
translocase of outer mitochondrial membrane 20 homolog (yeast)
- TRAF6
TNF receptor associated factor 6
- TUBB
tubulin β class I
- UB
ubiquitin
- UBA domain
ubiquitin-associated domain
- UB(K48,[6KR])
mutant ubiquitin containing only a single lysine at position 48, with all other lysines mutated to arginines
- ULK1
unc-51 like autophagy activating kinase 1
- USO1/p115
USO1 vesicle transport factor
- USP30
ubiquitin-specific peptidase 30
Disclosure of potential conflicts of interest
The authors declare that there is no potential conflict of interest.
Funding
This work was supported by research grants from the Ministry of Science and Technology of China (973 Basic Research Program grant 2009CB941404 to C.J. and 2011CB510002 to Z.Z.), Shenzhen Key Laboratory for Molecular Biology of Neural Development (ZDSY20120617112838879 to H.L.), Shenzhen Peacock Plan (KQCX20130628112914292 to H.L.), the National Natural Science Foundation of China (grant 30871032 to C.J. and grant 81330016 to D.M.), the Ministry of Education of China (Program for New Century Excellent Talents in University grant NCET-08-0372 to C.J., Program for Changjiang Scholars and Innovative Research Teams in University grant IRT0935 to D.M.), the Chinese Academy of Sciences (The Hundred Talent Program to C.J.), the Department of Science and Technology of Sichuan Province (Young Scientific Innovation Team in Neurological Disorders grant 2011JTD0005 to C.J.).
References
- [1].Johnston JA, Ward CL, Kopito RR. Aggresomes: a cellular response to misfolded proteins. J Cell Biol 1998; 143:1883-98; PMID:9864362; http://dx.doi.org/ 10.1083/jcb.143.7.1883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Fortun J, Dunn WA Jr, Joy S, Li J, Notterpek L. Emerging role for autophagy in the removal of aggresomes in Schwann cells. J Neurosci 2003; 23:10672-80; PMID:14627652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Taylor JP, Tanaka F, Robitschek J, Sandoval CM, Taye A, Markovic-Plese S, Fischbeck KH. Aggresomes protect cells by enhancing the degradation of toxic polyglutamine-containing protein. Hum Mol Genet 2003; 12:749-57; PMID:12651870; http://dx.doi.org/ 10.1093/hmg/ddg074 [DOI] [PubMed] [Google Scholar]
- [4].Kirkin V, McEwan DG, Novak I, Dikic I. A role for ubiquitin in selective autophagy. Mol Cell 2009; 34:259-69; PMID:19450525; http://dx.doi.org/ 10.1016/j.molcel.2009.04.026 [DOI] [PubMed] [Google Scholar]
- [5].Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao TP. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 2003; 115:727-38; PMID:14675537; http://dx.doi.org/ 10.1016/S0092-8674(03)00939-5 [DOI] [PubMed] [Google Scholar]
- [6].Duran A, Serrano M, Leitges M, Flores JM, Picard S, Brown JP, Moscat J, Diaz-Meco MT. The atypical PKC-interacting protein p62 is an important mediator of RANK-activated osteoclastogenesis. Dev Cell 2004; 6:303-9; PMID:14960283; http://dx.doi.org/ 10.1016/S1534-5807(03)00403-9 [DOI] [PubMed] [Google Scholar]
- [7].Linares JF, Duran A, Yajima T, Pasparakis M, Moscat J, Diaz-Meco MT. K63 polyubiquitination and activation of mTOR by the p62-TRAF6 complex in nutrient-activated cells. Mol Cell 2013; 51:283-96; PMID:23911927; http://dx.doi.org/ 10.1016/j.molcel.2013.06.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Park S, Ha SD, Coleman M, Meshkibaf S, Kim SO. p62/SQSTM1 enhances NOD2-mediated signaling and cytokine production through stabilizing NOD2 oligomerization. PLoS One 2013; 8:e57138; PMID:23437331; http://dx.doi.org/ 10.1371/journal.pone.0057138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ang E, Pavlos NJ, Rea SL, Qi M, Chai T, Walsh JP, Ratajczak T, Zheng MH, Xu J. Proteasome inhibitors impair RANKL-induced NF-κB activity in osteoclast-like cells via disruption of p62, TRAF6, CYLD, and IκBα signaling cascades. J Cell Physiol 2009; 220:450-9; http://dx.doi.org/ 10.1002/jcp.21787 [DOI] [PubMed] [Google Scholar]
- [10].Lin X, Li S, Zhao Y, Ma X, Zhang K, He X, Wang Z. Interaction domains of p62: a bridge between p62 and selective autophagy. DNA Cell Biol 2013; 32:220-7; PMID:23530606; http://dx.doi.org/ 10.1089/dna.2012.1915 [DOI] [PubMed] [Google Scholar]
- [11].Seibenhener ML, Babu JR, Geetha T, Wong HC, Krishna NR, Wooten MW. Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol Cell Biol 2004; 24:8055-68; PMID:15340068; http://dx.doi.org/ 10.1128/MCB.24.18.8055-8068.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Viiri J, Hyttinen JM, Ryhanen T, Rilla K, Paimela T, Kuusisto E, Siitonen A, Urtti A, Salminen A, Kaarniranta K. p62/sequestosome 1 as a regulator of proteasome inhibitor-induced autophagy in human retinal pigment epithelial cells. Mol Vis 2010; 16:1399-414; PMID:20680098 [PMC free article] [PubMed] [Google Scholar]
- [13].Sim CH, Gabriel K, Mills RD, Culvenor JG, Cheng HC. Analysis of the regulatory and catalytic domains of PTEN-induced kinase-1 (PINK1). Hum Mut 2012; 33:1408-22; PMID:22644621; http://dx.doi.org/ 10.1002/humu.22127 [DOI] [PubMed] [Google Scholar]
- [14].Valente EM, Salvi S, Ialongo T, Marongiu R, Elia AE, Caputo V, Romito L, Albanese A, Dallapiccola B, Bentivoglio AR. PINK1 mutations are associated with sporadic early-onset parkinsonism. Ann Neurol 2004; 56:336-41; PMID:15349860; http://dx.doi.org/ 10.1002/ana.20256 [DOI] [PubMed] [Google Scholar]
- [15].Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, et al.. Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science 2004; 304:1158-60; PMID:15087508; http://dx.doi.org/ 10.1126/science.1096284 [DOI] [PubMed] [Google Scholar]
- [16].Deas E, Plun-Favreau H, Gandhi S, Desmond H, Kjaer S, Loh SH, Renton AE, Harvey RJ, Whitworth AJ, Martins LM, et al.. PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum Mol Genet 2011; 20:867-79; PMID:21138942; http://dx.doi.org/ 10.1093/hmg/ddq526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, Youle RJ. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol 2010; 191:933-42; PMID:21115803; http://dx.doi.org/ 10.1083/jcb.201008084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Meissner C, Lorenz H, Weihofen A, Selkoe DJ, Lemberg MK. The mitochondrial intramembrane protease PARL cleaves human Pink1 to regulate Pink1 trafficking. J Neurochem 2011; 117:856-67; PMID:21426348; http://dx.doi.org/ 10.1111/j.1471-4159.2011.07253.x [DOI] [PubMed] [Google Scholar]
- [19].Yamano K, Youle RJ. PINK1 is degraded through the N-end rule pathway. Autophagy 2013; 9:1758-69; PMID:24121706; http://dx.doi.org/ 10.4161/auto.24633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, Sou YS, Saiki S, Kawajiri S, Sato F, et al.. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol 2010; 189:211-21; PMID:20404107; http://dx.doi.org/ 10.1083/jcb.200910140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 2010; 8:e1000298; PMID:20126261; http://dx.doi.org/ 10.1371/journal.pbio.1000298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Vives-Bauza C, Zhou C, Huang Y, Cui M, de Vries RL, Kim J, May J, Tocilescu MA, Liu W, Ko HS, et al.. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Pro Natl Acad Sci USA 2010; 107:378-83; http://dx.doi.org/ 10.1073/pnas.0911187107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 2006; 441:1162-6; PMID:16672981; http://dx.doi.org/ 10.1038/nature04779 [DOI] [PubMed] [Google Scholar]
- [24].Liu W, Vives-Bauza C, Acin-Perez R, Yamamoto A, Tan Y, Li Y, Magrane J, Stavarache MA, Shaffer S, Chang S, et al.. PINK1 defect causes mitochondrial dysfunction, proteasomal deficit and α-synuclein aggregation in cell culture models of Parkinson's disease. PLoS One 2009; 4:e4597; PMID:19242547; http://dx.doi.org/ 10.1371/journal.pone.0004597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM, et al.. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 2006; 441:1157-61; PMID:16672980; http://dx.doi.org/ 10.1038/nature04788 [DOI] [PubMed] [Google Scholar]
- [26].Sandebring A, Thomas KJ, Beilina A, van der Brug M, Cleland MM, Ahmad R, Miller DW, Zambrano I, Cowburn RF, Behbahani H, et al.. Mitochondrial alterations in PINK1 deficient cells are influenced by calcineurin-dependent dephosphorylation of dynamin-related protein 1. PLoS One 2009; 4:e5701; PMID:19492085; http://dx.doi.org/ 10.1371/journal.pone.0005701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Klinkenberg M, Thurow N, Gispert S, Ricciardi F, Eich F, Prehn JH, Auburger G, Kogel D. Enhanced vulnerability of PARK6 patient skin fibroblasts to apoptosis induced by proteasomal stress. Neuroscience 2010; 166:422-34; PMID:20045449; http://dx.doi.org/ 10.1016/j.neuroscience.2009.12.068 [DOI] [PubMed] [Google Scholar]
- [28].Muqit MM, Abou-Sleiman PM, Saurin AT, Harvey K, Gandhi S, Deas E, Eaton S, Payne Smith MD, Venner K, Matilla A, et al.. Altered cleavage and localization of PINK1 to aggresomes in the presence of proteasomal stress. J Neurochem 2006; 98:156-69; PMID:16805805; http://dx.doi.org/ 10.1111/j.1471-4159.2006.03845.x [DOI] [PubMed] [Google Scholar]
- [29].Lim KL, Dawson VL, Dawson TM. Parkin-mediated lysine 63-linked polyubiquitination: a link to protein inclusions formation in Parkinson's and other conformational diseases? Neurobiol Aging 2006; 27:524-9; PMID:16213628; http://dx.doi.org/ 10.1016/j.neurobiolaging.2005.07.023 [DOI] [PubMed] [Google Scholar]
- [30].Olzmann JA, Li L, Chudaev MV, Chen J, Perez FA, Palmiter RD, Chin LS. Parkin-mediated K63-linked polyubiquitination targets misfolded DJ-1 to aggresomes via binding to HDAC6. J Cell Biol 2007; 178:1025-38; PMID:17846173; http://dx.doi.org/ 10.1083/jcb.200611128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Garcia-Mata R, Bebok Z, Sorscher EJ, Sztul ES. Characterization and dynamics of aggresome formation by a cytosolic GFP-chimera. J Cell Biol 1999; 146:1239-54; PMID:10491388; http://dx.doi.org/ 10.1083/jcb.146.6.1239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Bence NF, Sampat RM, Kopito RR. Impairment of the ubiquitin-proteasome system by protein aggregation. Science 2001; 292:1552-5; PMID:11375494; http://dx.doi.org/ 10.1126/science.292.5521.1552 [DOI] [PubMed] [Google Scholar]
- [33].Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al.. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445-544; PMID:22966490; http://dx.doi.org/ 10.4161/auto.19496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012; 337:816-21; PMID:22745249; http://dx.doi.org/ 10.1126/science.1225829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. RNA-guided human genome engineering via Cas9. Science 2013; 339:823-6; PMID:23287722; http://dx.doi.org/ 10.1126/science.1232033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al.. Multiplex genome engineering using CRISPR/Cas systems. Science 2013; 339:819-23; PMID:23287718; http://dx.doi.org/ 10.1126/science.1231143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Beilina A, Van Der Brug M, Ahmad R, Kesavapany S, Miller DW, Petsko GA, Cookson MR. Mutations in PTEN-induced putative kinase 1 associated with recessive parkinsonism have differential effects on protein stability. Proc Natl Acad Sci USA 2005; 102:5703-8; PMID:15824318; http://dx.doi.org/ 10.1073/pnas.0500617102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Varshavsky A. Ubiquitin fusion technique and related methods. Method Enzymol 2005; 399:777-99; http://dx.doi.org/ 10.1016/S0076-6879(05)99051-4 [DOI] [PubMed] [Google Scholar]
- [39].Kageyama S, Sou YS, Uemura T, Kametaka S, Saito T, Ishimura R, Kouno T, Bedford L, Mayer RJ, Lee MS, et al.. Proteasome dysfunction activates autophagy and the Keap1-Nrf2 pathway. J Biol Chem 2014; 289:24944-55; PMID:25049227; http://dx.doi.org/ 10.1074/jbc.M114.580357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].An J, Yang DY, Xu QZ, Zhang SM, Huo YY, Shang ZF, Wang Y, Wu DC, Zhou PK. DNA-dependent protein kinase catalytic subunit modulates the stability of c-Myc oncoprotein. Mol Cancer 2008; 7:32; PMID:18426604; http://dx.doi.org/ 10.1186/1476-4598-7-32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Matsumoto G, Wada K, Okuno M, Kurosawa M, Nukina N. Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins. Mol Cell 2011; 44:279-89; PMID:22017874; http://dx.doi.org/ 10.1016/j.molcel.2011.07.039 [DOI] [PubMed] [Google Scholar]
- [42].Lim J, Lachenmayer ML, Wu S, Liu W, Kundu M, Wang R, Komatsu M, Oh YJ, Zhao Y, Yue Z. Proteotoxic stress induces phosphorylation of p62/SQSTM1 by ULK1 to regulate selective autophagic clearance of protein aggregates. PLoS Genet 2015; 11:e1004987; PMID:25723488; http://dx.doi.org/ 10.1371/journal.pgen.1004987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 2005; 171:603-14; PMID:16286508; http://dx.doi.org/ 10.1083/jcb.200507002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Bogdanove AJ, Voytas DF. TAL effectors: customizable proteins for DNA targeting. Science 2011; 333:1843-6; PMID:21960622; http://dx.doi.org/ 10.1126/science.1204094 [DOI] [PubMed] [Google Scholar]
- [45].Minoia M, Boncoraglio A, Vinet J, Morelli FF, Brunsting JF, Poletti A, Krom S, Reits E, Kampinga HH, Carra S. BAG3 induces the sequestration of proteasomal clients into cytoplasmic puncta: implications for a proteasome-to-autophagy switch. Autophagy 2014; 10:1603-21; PMID:25046115; http://dx.doi.org/ 10.4161/auto.29409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Milan E, Perini T, Resnati M, Orfanelli U, Oliva L, Raimondi A, Cascio P, Bachi A, Marcatti M, Ciceri F, et al.. A plastic SQSTM1/p62-dependent autophagic reserve maintains proteostasis and determines proteasome inhibitor susceptibility in multiple myeloma cells. Autophagy 2015; 11:1161-78; PMID:26043024; http://dx.doi.org/ 10.1080/15548627.2015.1052928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, et al.. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006; 441:885-9; PMID:16625204; http://dx.doi.org/ 10.1038/nature04724 [DOI] [PubMed] [Google Scholar]
- [48].Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, et al.. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006; 441:880-4; PMID:16625205; http://dx.doi.org/ 10.1038/nature04723 [DOI] [PubMed] [Google Scholar]
- [49].Korolchuk VI, Mansilla A, Menzies FM, Rubinsztein DC. Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol Cell 2009; 33:517-27; PMID:19250912; http://dx.doi.org/ 10.1016/j.molcel.2009.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, et al.. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 2007; 131:1149-63; PMID:18083104; http://dx.doi.org/ 10.1016/j.cell.2007.10.035 [DOI] [PubMed] [Google Scholar]
- [51].Haque ME, Thomas KJ, D'Souza C, Callaghan S, Kitada T, Slack RS, Fraser P, Cookson MR, Tandon A, Park DS. Cytoplasmic Pink1 activity protects neurons from dopaminergic neurotoxin MPTP. Proc Natl Acad Sci USA 2008; 105:1716-21; PMID:18218782; http://dx.doi.org/ 10.1073/pnas.0705363105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Weihofen A, Thomas KJ, Ostaszewski BL, Cookson MR, Selkoe DJ. Pink1 forms a multiprotein complex with Miro and Milton, linking Pink1 function to mitochondrial trafficking. Biochemistry 2009; 48:2045-52; PMID:19152501; http://dx.doi.org/ 10.1021/bi8019178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Wang X, Winter D, Ashrafi G, Schlehe J, Wong YL, Selkoe D, Rice S, Steen J, LaVoie MJ, Schwarz TL. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 2011; 147:893-906; PMID:22078885; http://dx.doi.org/ 10.1016/j.cell.2011.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Matenia D, Hempp C, Timm T, Eikhof A, Mandelkow EM. Microtubule affinity-regulating kinase 2 (MARK2) turns on phosphatase and tensin homolog (PTEN)-induced kinase 1 (PINK1) at Thr-313, a mutation site in Parkinson disease: effects on mitochondrial transport. J Biol Chem 2012; 287:8174-86; PMID:22238344; http://dx.doi.org/ 10.1074/jbc.M111.262287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Dagda RK, Pien I, Wang R, Zhu J, Wang KZ, Callio J, Banerjee TD, Dagda RY, Chu CT. Beyond the mitochondrion: cytosolic PINK1 remodels dendrites through protein kinase A. J Neurochem 2014; 128:864-77; PMID:24151868; http://dx.doi.org/ 10.1111/jnc.12494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Bang Y, Kang BY, Choi HJ. Preconditioning stimulus of proteasome inhibitor enhances aggresome formation and autophagy in differentiated SH-SY5Y cells. Neurosci Let 2014; 566:263-8; http://dx.doi.org/ 10.1016/j.neulet.2014.02.056 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.