

ABSTRACT
Cytokine modulation of autophagy is increasingly recognized in disease pathogenesis, and current concepts suggest that type 1 cytokines activate autophagy, whereas type 2 cytokines are inhibitory. However, this paradigm derives primarily from studies of immune cells and is poorly characterized in tissue cells, including sentinel epithelial cells that regulate the immune response. In particular, the type 2 cytokine IL13 (interleukin 13) drives the formation of airway goblet cells that secrete excess mucus as a characteristic feature of airway disease, but whether this process is influenced by autophagy was undefined. Here we use a mouse model of airway disease in which IL33 (interleukin 33) stimulation leads to IL13-dependent formation of airway goblet cells as tracked by levels of mucin MUC5AC (mucin 5AC, oligomeric mucus/gel forming), and we show that these cells manifest a block in mucus secretion in autophagy gene Atg16l1-deficient mice compared to wild-type control mice. Similarly, primary-culture human tracheal epithelial cells treated with IL13 to stimulate mucus formation also exhibit a block in MUC5AC secretion in cells depleted of autophagy gene ATG5 (autophagy-related 5) or ATG14 (autophagy-related 14) compared to nondepleted control cells. Our findings indicate that autophagy is essential for airway mucus secretion in a type 2, IL13-dependent immune disease process and thereby provide a novel therapeutic strategy for attenuating airway obstruction in hypersecretory inflammatory diseases such as asthma, chronic obstructive pulmonary disease, and cystic fibrosis lung disease. Taken together, these observations suggest that the regulation of autophagy by Th2 cytokines is cell-context dependent.
Keywords: airway epithelial cells, autophagy, IL13, MUC5AC, reactive oxygen species, secretion
Introduction
Autophagy may be induced by stress from starvation, pathogens, or cytokine signals. Of these, cytokines are increasingly recognized as potent modulators of autophagy, capable of both activation and inhibition during inflammation.1,2 Cytokine regulation of autophagy may occur as a component of the immune response to pathogens or in noninfectious disorders such as atherosclerosis or malignancy.3-5 In response to pathogen invasion or a “danger signal” TNF (tumor necrosis factor), IFNA1 (interferon, α 1), IFNB1 (interferon, β 1), IFNG (interferon, gamma), IL6 (interleukin 6) and TGFB1 (transforming growth factor, β 1) stimulate autophagy, while other data suggests that Th2 cytokines, including IL13 (interleukin 13) can inhibit autophagy.2,6 In these cases, modulation of autophagy by IL13 is in the context of the macrophage and infection with mycobacterium or parasites.7,8
The actions of cytokines in the lung are widely investigated because of the interface of the airway with the environment, the susceptibility of the lung to infection, and the burden of chronic inflammatory airway diseases, particularly asthma and chronic obstructive pulmonary disease (COPD).9,10 As an experimental model, the lung is ideal for the study of inflammatory responses due to the ease of direct delivery of cytokines to the airway in vivo and the ability to assay cultured primary airway epithelial cells in vitro. Using approaches similar to these, our group has recently demonstrated that airway epithelial cell IL33 (interleukin 33) leads to IL13 production and lung pathology in a mouse model and in cells obtained from patients with COPD.11-14
IL13 is a Th2-type cytokine that is elevated in COPD as well as in asthma and other lung diseases.11,15-17 In the airway, IL13 regulates the differentiation of epithelial cells to goblet cells via intracellular signaling through the IL4R (interleukin 4 receptor) and IL13RA1 (interleukin 13 receptor, α 1) receptor heterodimer and STAT6 (signal transducer and activator of transcription 6).12,13 Functionally, IL13 has been postulated to increase both intracellular oxidant stress and the secretion of MUC5AC (mucin 5AC, oligomeric mucus/gel forming), the dominant gel forming mucin in airway epithelial cells. Airway epithelial cells treated with IL13 have increased oxidant activity,18,19 higher numbers of MUC5AC-expressing cells and more MUC5AC secreted in the apical media.11,20 The mechanism utilized by the goblet cell to adapt to the stress of IL13-mediated acceleration in activity is not known.
Our current understanding of mucin formation and secretion has been gathered from observation of both nonmammalian and mammalian models and particularly in the intestine.21,22 However, less is known about the regulation of airway-specific secretory processes. In airway epithelial cells, MUC5AC secretion is considered an adaptive response to environmental stimuli such as respiratory viruses,23 airway allergens,24 and cigarette smoke.25 We, and others, have demonstrated a requirement of autophagy for cell secretion of VWF (Von Willebrand factor) from endothelial cells,26 lysozyme from intestinal Paneth cells,27 and MUC2 from colonic goblet cells.28,29 In each case, secretion requires the participation of ATG7 (autophagy-related 7), the ATG12–ATG5 conjugate and ATG16L1 (autophagy-related 16-like 1 [S. cerevisiae]) for autophagosome maturation and fusion. In the colon, autophagy proteins are required for both the generation of lipopolysaccharide-induced reactive oxygen species (ROS) and secretion of MUC2 from goblet cells.
Given the parallel structure and function of airway and colonic goblet cells, we hypothesized that autophagy is essential as a response for IL13-mediated oxidative stress and secretion of MUC5AC in airway epithelial cells. To test this hypothesis, we assayed autophagy proteins in primary-culture human airway epithelial cells in the presence of IL13 and then assessed the effect on goblet secretion by silencing autophagy genes using RNAi. Contrary to the role of IL13 on autophagy in immune cells, our findings indicate that in airway epithelial cells, autophagy is activated. This activity is required for the dominant features of chronic lung disease, IL13-stimulated MUC5AC secretion and ROS generation in airway epithelial cells.
Results
IL33-induced airway goblet cell hypertrophy in ATG16L1-deficient mice
Autophagy is required for colonic epithelial goblet cell secretion.28,29 However, unlike the physiological requirement for goblet cells in the intestine, goblet cell differentiation in the mouse airway must be induced.27 Airway goblet cell formation can be efficiently induced by airway delivery of IL33, which leads to macrophage and lymphocyte production of IL13.14,30-33 To test if autophagy plays a role in cytokine-induced inflammation in vivo, we compared the response of wild-type (WT) and Atg16l1 hypomorphic (HM) mice (Atg16l1HM/HM)27 following intranasal administration of IL33. Airways of control mice (naïve vehicle treated) contained few if any detectable goblet cells. As anticipated, IL33 treatment induced goblet cells in WT and Atg16l1HM/HM airways (Fig. 1A). Quantitative analysis showed increased area of mucus staining per goblet cell in IL33-treated Atg16l1HM/HM mice as compared to similarly treated WT controls (Fig. 1B). In addition, the total area of periodic acid–Schiff (PAS) staining as measured as a percent of total airway epithelium in the Atg16l1HM/HM mice was greater than WT littermate controls (Fig. 1C). Despite the fact that there were more and larger goblet cells in the IL33-treated Atg16l1HM/HM mice, the lavage fluid from these mice actually contained less MUC5AC as compared to WT mice (Fig. 1D). These results suggest the hypothesis that ATG16L1 function plays a role in goblet cell secretion in IL33-induced goblet cell metaplasia as indicated by an increase in airway goblet cells that supplant the normal ciliated and nongoblet secretory cell populations.34 As IL33 is well known to induce goblet cell metaplasia, as a pathological response to IL13,34 this further suggests that IL13 is the factor that acts directly on lung airway epithelial cells.
Figure 1.
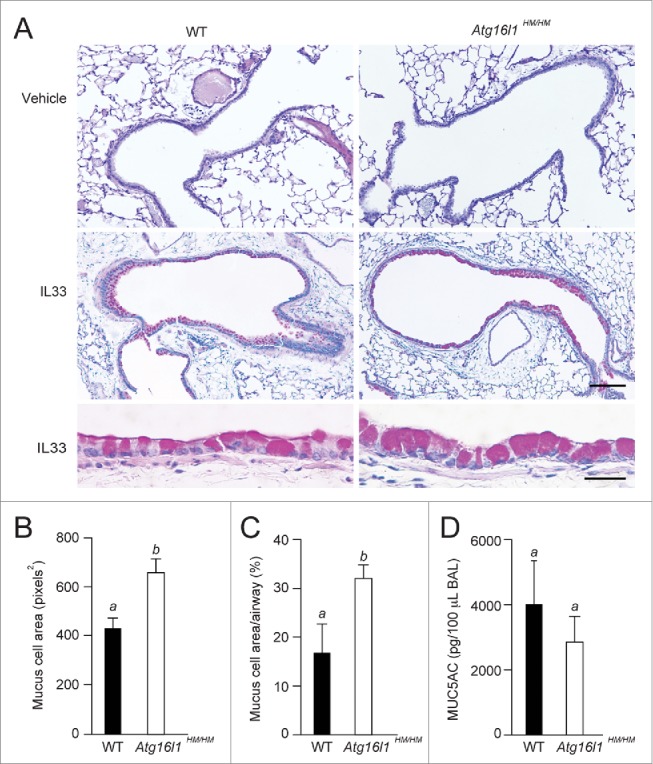
Goblet cell hypertrophy in autophagy-deficient mice. WT and Atg16l1 hypomorphic (Atg16l1HM/HM) mice were administered vehicle or IL33 intranasally. Lungs were evaluated on d 7 post-challenge. (A) Representative photomicrographs of lung sections stained with PAS. Scale bars, top and middle panels: 100 μm; lower panel: 10 μm. (B) Quantification of area per goblet cell (n = 6 mice per group, (C) percentage of PAS staining (on histological sections) of total airway epithelium area (n = 6 mice per group) of IL33-treated WT and Atg16l1HM/HM, and (D) levels of MUC5AC in bronchoalveolar lavage (BAL) fluid from lungs of IL33-treated mice (n = 3 mice per group). (B–D) are graphs of the means ±SEM. Means with different letters are significantly different by the unpaired 2-tailed Student's t-test.
IL13 induces MUC5AC secretion in human tracheobronchial epithelial cells
To probe the mechanism of secretion in epithelial cells in vitro, we utilized a well characterized and widely used model of cultured primary human airway cells from nondiseased lung tissue donated for lung transplantation.35,36 To determine if autophagy was required for airway goblet secretion, we used an established protocol to differentiate goblet cells within cultures of primary hTEC (human tracheobronchial epithelial cells) (Fig. 2A). In this model, airway progenitor cells differentiate into functional ciliated and secretory cells when subjected to an ALI (air-liquid interface) condition for 18–21 d. The best, established model for induction of goblet cells is treatment with IL13.11,37 We devised a protocol whereby treatment of cells with IL13 for 7 or 21 d (Fig. 2A) resulted in an increased number of MUC5AC-positive cells and elevated levels of MUC5AC RNA (Fig. 2B, C) as previously reported.11 We recently showed that an increase in the amount of intracellular reactive oxygen species (ROS) is required for active secretion of colonic goblet epithelial cells.28 IL13 stimulates ROS production in both intestinal and airway epithelial cell lines.18,19,39 We evaluated the influence of IL13 on intracellular ROS levels in the hTEC model (Fig. 2E). Treatment of hTEC preparations for 7 d (ALI d 14 to 21) with IL13 significantly increased intracellular ROS levels as detected by the fluorogenic oxidant probe DCF (CM-H2DCFDA; Fig. 2D). The IL13-induced ROS activity was attenuated by treatment with the NOX (NADPH oxidase) inhibitor DPI (diphenyleneiodonium).40 To test the immediate role of IL13 on MUC5AC secretion, hTEC preparations were treated with IL13 for 21 d, then withdrawn for 2 d.38 We then treated the cells with fresh IL13 for one h and compared MUC5AC levels in the supernatant fractions to those treated with vehicle only. IL13 significantly increased levels of apical MUC5AC secretion in media collected over one h, relative to phosphate-buffered saline (PBS). This effect was only less slightly pronounced relative to the effect of stimulation by ATP-γ–S (100 μM) a well recognized mucin secretagogue38 (Fig. S1). Blocking the action of NOX activity with DPI prior to the addition of IL13 also significantly reduced IL13-mediated MUC5AC secretion (Fig. 2E). Thus IL13 induced both MUC5AC secretion and ROS activity in cultured airway cells.
Figure 2.

IL13 increases MUC5AC expression and secretion. (A) In vitro protocol for IL13 treatment of human tracheal/bronchial epithelial cells (hTEC) differentiated using air-liquid interface conditions (ALI). Cells were assayed at the indicated times. (B) Representative images of hTEC treated with vehicle or IL13 for the indicated periods, then immunostained for MUC5AC and cilia marker acetylated tubulin (acetylated-TUB). Nuclei were stained with DAPI. MC, goblet cell; arrows, secreted MUC5AC on the cell surface. Scale bars: 10 μm. (C) IL13-induced MUC5AC mRNA levels (+) relative to vehicle (−) (n = 6 preparations/condition). (D) ROS production in hTEC cultured with or without IL13 during ALI d 14–21 d. Three h prior to loading the ROS probe DCF (CM-H2DCFDA), cells were treated with combinations of IL13 and the NOX inhibitor DPI (5 μM), or vehicle controls and the resulting mean fluorescent intensity signal was measured from 3 random fields. AU, arbitrary units. (E) MUC5AC secretion in hTEC preparations cultured with or without IL13 for 21 d as in (A) and pre-treated with DPI (5 μM) or DMSO vehicle prior to the MUC5AC secretion assay. Levels of secreted MUC5AC were measured by ELISA after performing a series of washes (to obtain baseline levels) followed by a 1 h incubation with IL13. Values were normalized to the baseline amount of MUC5AC for each condition and reported as fold change (n = 7 for DMSO vehicle pretreatment, n = 4 for DPI pretreatment). Measures in (C–E) were obtained from hTEC preparations obtained from at least 3 unique donors and independent experiments, displayed as the graph of the means ±SEM. In (C), means from each time point were compared to vehicle alone using the paired Student t test. In (D, E), means with different letters are significantly different by ANOVA with Bonferroni's correction.
IL13 activates autophagy in human tracheobronchial epithelial cells in vitro
Based on finding that IL13 activated both secretion and intracellular ROS, we determined whether IL13 was dependent on autophagy as a pathway for MUC5AC secretion. We first monitored the effect of IL13 on MAP1LC3A (microtubule-associated protein 1 light chain 3 α) conversion (LC3-I to LC3-II) by immunoblot during the differentiation of hTEC (Fig. 3A). We observed that prolonged treatment with IL13 for 21 d increased the LC3-II to actin ratio, compared to culture with standard medium, suggesting that IL13 is requirement for autophagy (Fig. 3A, B). ATG5 protein and ATG5 mRNA levels were similar over the course of in vitro differentiation both with and without IL13 treatment (Fig. 3A, Fig. S2B). To confirm that IL13 stimulated autophagy, we performed autophagy flux assays by incubating cell preparations in the presence of IL13 plus chloroquine (Fig. 3C). Bafilomycin A1 was also used as a lysosomal vacuolar-type ATPase inhibitor and had a similar effect (data not shown). Treatment with IL13 for as few as 7 d (which stimulates goblet cell formation; Fig. 2C) significantly increased LC3-II levels in cells (Fig. 3D, E), indicating stimulation of autophagy. This finding was confirmed by demonstrating increased LC3 puncta by immunofluorescent staining after treatment of hTEC with IL13 for 7 d (Fig. 3F, G). Short treatments with IL13 pretreatment, for 3 h, did not significantly stimulate autophagy, suggesting that goblet cell differentiation was required for autophagy activity. In sum, these observations indicate that autophagy was associated with goblet cells that form in response to chronic IL13 treatment.
Figure 3.

IL13 increases autophagy activity. (A) hTEC were cultured in the presence of vehicle or IL13 for 21 d of ALI conditions. Cell lysates were prepared at the indicated time points of ALI culture for immunoblot analysis for ATG5 and LC3. Representative blots are shown. (B) Quantification of immunoblot density of the LC3-II to actin ratio as the mean ±SEM (n = 3 independent experiments each from a preparation of a unique cell donor). (C) Autophagy flux assay to determine the influence of IL13 on autophagy activity in hTEC. (D) Representative LC3-immunoblots of hTEC incubated with vehicle (−) or IL13 (+) for 7 d then treated with chloroquine (Chloro) as shown in (C). Lysates were harvested at ALI 21. (E) Immunoblot image densities of LC3-II to actin ratios shown as the mean ±SEM (n = 5 independent experiments). Mean LC3 to actin band densities with different letters are significantly different by ANOVA with Bonferroni's correction. (F) Representative images of LC3 puncta in hTEC (E) in the presence or absence of IL13 for 7 d, on ALI d 14–21, then treated with chloroquine as shown in (C). Scale bar: 10 μm. (G) Quantification of the number of LC3 puncta as mean ±SEM from triplicate values per high-powered field (hpf) obtained from photomicrographs for each experiment (n = 3 independent experiments). Means with different letters are significantly different by unpaired 2-tailed Student t test.
ATG5 deficiency impairs IL13-induced autophagy in hTEC
To functionally connect autophagy to secretion, we depleted hTECs of ATG5 using gene-specific shRNA sequences transduced by lentiviral vectors. We identified 2 different targeting sequences that achieved significant depletion of ATG5 mRNA with 70% (ATG5 shRNA #1) and 80% (ATG5 shRNA #2) silencing of ATG5 by immunoblot and mRNA expression (Fig. 4A and S2A). For each experiment, cells were transduced with nontargeted (NT) sequences as controls. The reduction of ATG5 mRNA expression correlated with reduced levels of LC3-II protein and increased levels of SQSTM1/p62 (sequestosome 1) protein (Fig. 4A). Moreover, we observed that in response to IL13 stimulation, ATG5-depleted cells had increased the number of SQSTM1 puncta and decreased LC3 puncta formation (Fig. 4B–D).
Figure 4.
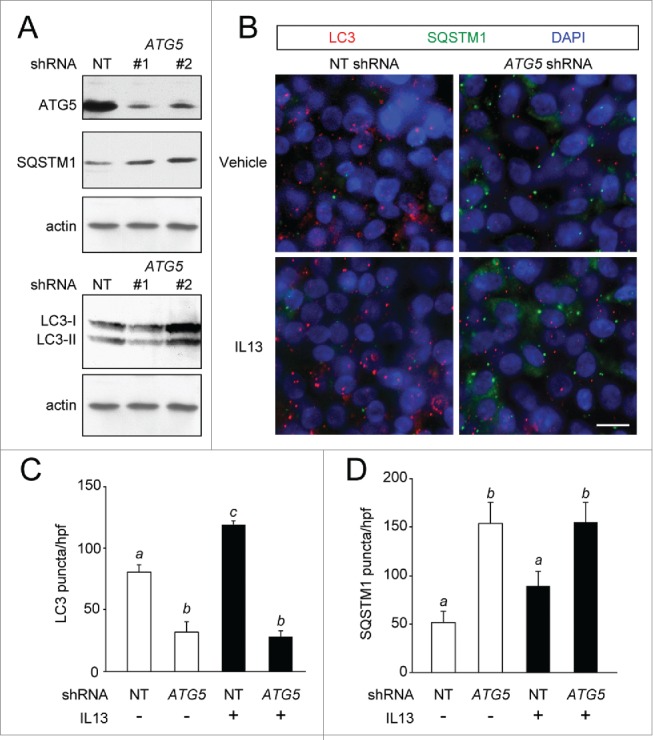
ATG5 depletion reduces autophagy activity in hTEC preparations. hTEC were transduced with nontargeted (NT) or ATG5-specific shRNA sequences. (A) ATG5 levels in representative immunoblots for NT or 2 different ATG5 shRNA sequences (#1 and #2) at ALI d 21 with corresponding protein levels of SQSTM1 and LC3. (B) Representative images of LC3 (red) and SQSTM1 (green) puncta in hTEC treated with vehicle or IL13 in shRNA-transduced cells NT and ATG5 (sequence #2) shRNA. Scale bars: 10 μm. (C and D) Quantification of LC3 and SQSTM1 puncta in transduced cells treated with vehicle (−) or IL13 (+) for 7 d. (C and D) are graphs of number of puncta as the mean ±SEM of triplicate samples from high-powered field images obtained from n = 4 different cell preparations. Means with different letters are significantly different by ANOVA with Bonferroni's multiple comparison tests.
ATG5 deficiency results in diminished IL13-induced goblet cell secretion
We used the ATG5-depleted hTEC to quantify the effect of IL13 on goblet cell formation and intracellular mucin stores (Fig. 5A, B). IL13 had no significant effect on ATG5 mRNA levels (Fig. S2B). As expected, IL13 treatment for 7 d significantly increased the absolute number and percentage of MUC5AC-positive cells in both the NT- and ATG5-transduced cell preparations relative to untreated hTECs. However, ATG5 depletion resulted in a similar percent of MUC5AC-positive cells compared to NT-transduced cells (Fig. 5C). Despite the similar fractions of goblet cells, ATG5-depleted hTEC contained greater amounts of MUC5AC staining per cell compared to control preparations (Fig. 5D, E). This result indicated an intracellular accumulation of mucin in the ATG5-deficient cells.
Figure 5.

Depletion of ATG5 is associated with airway goblet cell hypertrophy. hTEC were transduced with nontargeted (NT) or ATG5-specific shRNA and treated with vehicle or IL13 for 7 d (ALI days 14–21). (A) Representative confocal photomicrographs of hTEC in the x,y plane, stained for MUC5AC and actin using phalloidin. Scale bars: 10 μm. (B) Representative x,z images demonstrating the optical level selected in (A) relative to the apical membrane (dashed line). (C) Quantification of (A) as percent (%) goblet cells of total cell number on the apical surface, (D) MUC5AC-staining area per goblet cell and (E) MUC5AC signal intensity per goblet cell. Graphs in (C–E) are from are the mean ±SEM from triplicate values per high-powered field (hpf) obtained from photomicrographs for each experiment (n = 3–4 independent experiments, each from a unique donor). Means in (C–E) with different letters are significantly different by ANOVA with Bonferroni's multiple comparison test. (F) Representative transmission EM images of goblet cells treated with IL13. Red lines mark cell borders. Scale bars: 2 μm. (G) Quantification of number of mucin granules per cell was performed with 8 goblet cells evaluated from each group. Shown are the means ±SEM with different letters indicating a significant difference by the unpaired 2-tailed Student t test.
We performed ultrastructural analysis of goblet cells under IL13-activated conditions to determine potential cellular mechanisms for the accumulation of intracellular mucins in autophagy protein-depleted hTEC. The mucin granule morphology of goblet cells was similar in ATG5-deficient and control cells. The most obvious difference was that there were a greater number of intracellular mucin granules in the ATG5-deficient cells (Fig. 5 F, G). RNA levels of MUC5AC were similar between these 2 groups (Fig. S3B,C) indicating that there was not a detectable difference in mucin production. Thus, blocking autophagy in hTEC in IL13-treated cells induced goblet cell hypertrophy and suggested a corresponding functional defect in MUC5AC secretion.
One advantage of hTEC preparations is that secretion can be measured using an ELISA for MUC5AC in the apical supernatant. We administered IL13 to control or ATG5-depleted hTEC for 21 d to generate goblet cells. As before, the cytokine was withdrawn for 48 h followed by secretion assays under defined conditions38 (Fig. 6A). The ATG5-deficient hTEC treated with IL13 for one h had significantly less newly secreted MUC5AC than control cells (Fig. 6B). To further confirm the requirement of autophagy for mucin secretion, we prepared hTEC deficient in ATG14, an upstream autophagy regulatory protein not directly responsible for LC3 lipidation (Fig. S3).41 Similar to ATG5 deficiency, ATG14 depletion resulted in reduced MUC5AC secretion following stimulation with IL13 (Fig. 6C). Taken together, these findings indicate a requirement of autophagy proteins for IL13-activated secretion of MUC5AC.
Figure 6.

Depletion of ATG5 and ATG14 reduces IL13-mediated MUC5AC secretion. (A) Scheme of in vitro IL13-mediated MUC5AC secretion protocol in hTEC. Secreted MUC5AC was measured 1 h following the application of fresh IL13 on cells transduced with NT or autophagy gene shRNA. (B) hTEC transduced with ATG5 (sequence #2) shRNA. (C) hTEC transduced with ATG14 shRNA. Graphs in (B and C) are the means ±SEM from cells derived from 4 unique preparations. Means with different letters are significantly different by the unpaired 2-tailed Student t test.
Autophagy is required for IL13-mediated ROS activity
Previously, we showed that MUC2 secretion in colonic epithelial cells requires both ROS and autophagy activity.28 We used parallel sample preparations as used above in secretion assays for the analysis of IL13-activated ROS production. Preparations of ATG5-deficient hTEC treated continuously with IL13 to generate goblet cells had significantly attenuated ROS activity compared to control cells (Fig. 7A, B). Similarly, hTEC preparations rendered deficient in ATG14 also demonstrated reduced IL13-mediated ROS activity (Fig. 7C). Furthermore, pretreatment with the NOX inhibitor DPI reduced IL13 activated MUC5AC secretion in hTECs (Fig. 2E). This finding suggests that both autophagy and ROS are required for mucin secretion. Thus, in airway epithelial cells, the autophagy pathway directs factors required for generation of IL13-mediated ROS generation and is required for ROS production. These findings suggest a new mechanism in which autophagy is stimulated by IL13 to regulate MUC5AC secretion.
Figure 7.
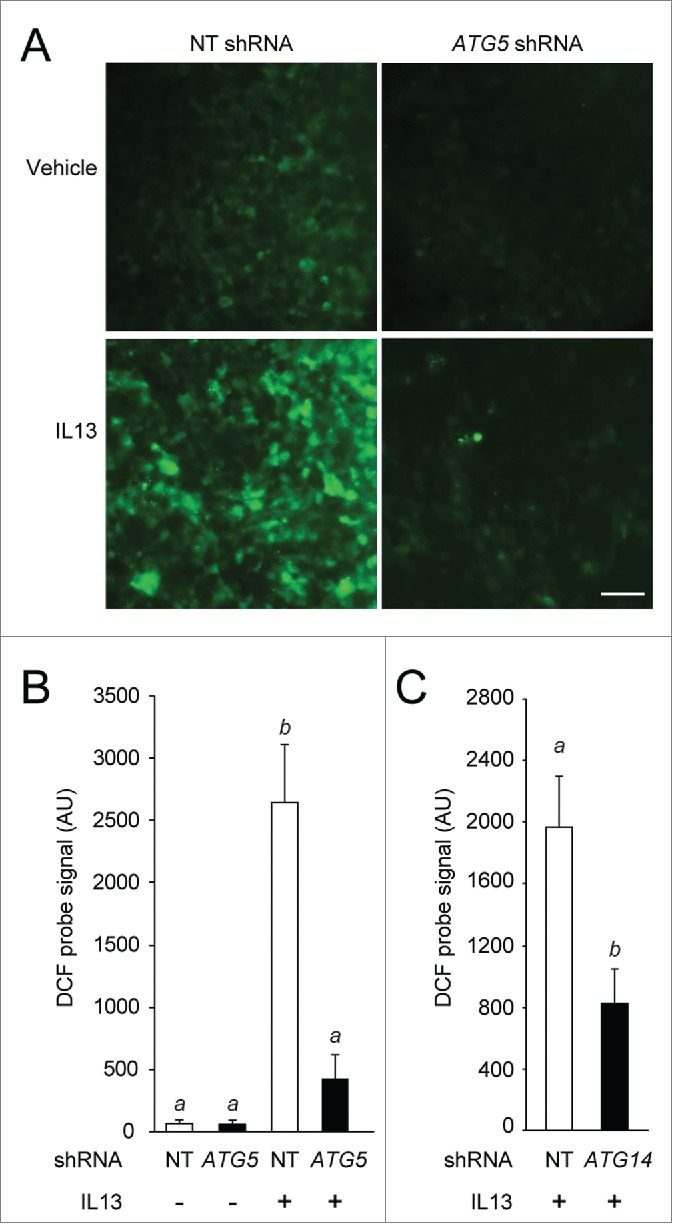
Depletion of ATG5 and ATG14 reduces IL13-mediated ROS activity. hTEC transduced with NT, ATG5 or ATG14 shRNA, treated with vehicle (−) or IL13 (+) for 7 d, ALI d 14–21 as in Figure 5. (A) Representative images of DCF (CM-H2DCFDA) probe fluorescent signal; Scale bar: 100 μm. (B and C) Quantification of ROS signal intensity is shown. Graphs in (B and C) are the means ±SEM of values obtained from images of 3 random fields from 4 unique preparations. Means with different letters are significantly different by ANOVA with Bonferroni's multiple comparison test (B) or the unpaired 2-tailed Student t test (C).
Discussion
Here, we show evidence that IL13 stimulates autophagy and that this signaling has physiological consequences. We capitalized on the ability to study the effects of cytokines on airway epithelial cells that have prominent physiological effects. Using this approach, we demonstrate that IL13 directly induces airway epithelial cell MUC5AC hypersecretion and oxidant stress. In a primary human airway epithelial cell culture system, we found that prolonged treatment of cells with IL13 was required to promote increase of LC3-II relative to actin, indicating increased autophagy activity. Additionally, depleting ATG5 or ATG14 in IL13-treated hTEC resulted in goblet cell hypertrophy and a decrease in MUC5AC secretion. Furthermore, blocking autophagy in this model significantly attenuated IL13-mediated ROS generation. Therefore, these findings provide evidence that autophagy regulates the IL13-mediated inflammatory response in airway epithelial cells characterized by goblet cell secretion and intracellular oxidant generation.
The effect of IL13 to activate autophagy in airway epithelial cells is in contrast to previous reports in macrophages. In these studies, the effect of IL13 has been primarily to modulate starvation- or IFNG-induced autophagy during infection. Harris et al. found that starvation- or IFNG-induced autophagy following infection with mycobacteria was attenuated by IL13 in macrophages.7 Using experiments of similar design, Su et al. showed that IL13 blocked autophagy activity in peritoneal macrophages in a mouse model of enteric bacterial co-infection after intestinal parasitic nematode exposure.8 Data from these studies showed that IL13 inhibits starvation-induced autophagy through the AKT pathway and IFNG-induced autophagy through STAT6-dependent mechanisms. While STAT6 is induced in airway epithelial cells during goblet cell formation, IL13 and STAT6 regulate a remarkably different gene profile in immune compared to epithelial cell types.42-44 The contrasting activities of IL13 and autophagy in infected macrophages and inflamed airway epithelial cells suggest a cell-specific dependence of autophagy.
Previous reports indicate that autophagy might play a more general role in stress response in airway epithelial cells. In COPD the number of autophagosomes and LC3-II levels are higher in samples of lungs from those with the disease, though the role in goblet cells was not investigated.45 The same group has shown that cigarette smoke extract or hyperoxia increase autophagy activity in airway epithelial cells and cilia injury.45-47 However, IL13 was not examined in that context. Thus in epithelial cells, autophagy activity may serve as a functional response to noxious or inflammatory signals such as IL13, which leads to increased ROS generation and MUC5AC secretion.
We have reported the measurement of increased MUC5AC secretion following prolonged stimulation with IL13 relative to untreated cell hTEC preperations.11 In the current study, prolonged treatment with IL13 in a similar model resulted in accumulation of mucus on the cell surface, typically noted after 7–14 d of a 21-d treatment course (e.g., Fig. 2B). We now provide strong evidence that IL13 can act as a secretagogue for MUC5AC in human airway epithelial cells. The magnitude and timing (within an hour) of this effect was similar to the known secretagogue ATP-γ-S. Other cytokines such as IL1B (interleukin 1, β) increase MUC5AC production48 and secretion.49 However, the effect was delayed for hours or days, suggesting that a secondary pathway was utilized for mucus production.
Autophagy is now shown to be required for secretory pathways in mast cells, bone osteoclasts, intestinal Paneth cells, and colonic goblet cells.27,28,50-52 Shared or unique utilization of specific autophagy proteins for secretion is yet to be resolved, however it is possible that proteins that are similarly processed have similar autophagy-dependent mechanisms. For example, the autophagy-dependent secretion of VWF by vascular endothelial cells requires protein dimerization in the ER via disulfide bond linkage and packaging in the Golgi before budding into secretory granules prior to ligand regulated secretion, much the same as MUC5AC.26 In colonic goblet cells, autophagy is required for the formation of amphisomes and the generation of ROS,28 both essential for the secretion of MUC2, similar to MUC5AC.
Our findings indicate that a similar requirement for auto-phagy exists in the mucin secretion of the colonic and airway epithelium. Autophagy is required for both IL13-mediated MUC5AC secretion and ROS activity. In the colonic goblet cells, autophagy and NOX enzymes contribute to constitutive MUC2 secretion via calcium-mediated exocytosis. We speculate that in airway epithelial cells, autophagy offers a similar convergence of cytoplasmic proteins (e.g., endosomes, autophagosomes) allowing for IL13-mediated ROS generation leading to MU5AC secretion. Thus, there is potential therapeutic benefit from short-term targeting of the IL13-activated pathway to modulate ROS activity and MUC5AC secretion using drugs that inhibit autophagy. Known autophagy inhibitors that are FDA approved for alternative indications are now being studied in clinical trials, primarily as cancer therapeutics.53,54 Use of these agents may prove a rapid means to translate our findings to novel treatments of mucin secretion and chronic lung disease.
In summary, we find that IL13 activates autophagy in airway epithelial cells to direct mucin secretion and cell oxidant stress responses, which are major cellular phenotypes in the chronic inflammatory airway diseases asthma and COPD. Identifying enhanced IL13-mediated autophagy in epithelial cells alters the current concept that IL13 broadly inhibits autophagy activity. These findings suggest another view, that IL13 regulation of autophagy is much different in macrophages than in epithelial cells. This finding takes on added importance when analyzing the yet undefined status of autophagy activation in airway epithelial cells from individuals with asthma. The identification of single nucleotide polymorphisms associated with childhood asthma in 2 different populations that lead to functional ATG5 activity in nasal airway epithelial cells suggests that future studies in this area are warranted but must be carefully interpreted in the context of the cytokine environment.55,56
Materials and methods
Mice
Animal studies were approved by the institutional committee. C57BL/6 mice deficient in the autophagy gene Atg16l1 were previously described (Atg16l1HM/HM).27 Mice were administered PBS (Sigma-Aldrich, P3813) vehicle or recombinant mouse IL33 (Peprotech, 210–33), 1.0 µg, intranasal on d 1 and 3. At d 7 post-challenge, mice were euthanized, then underwent lung lavage with 3, one-mL aliquots of sterile PBS, followed by tissue fixation and resection. Recovered lavage fluid was reserved for MUC5AC quantification, while lung tissue was processed for histological examination using the PAS reaction to stain intercellular mucin.
Airway epithelial cell culture
Primary human tracheal-bronchial epithelial cells (hTEC) were isolated from excess surgical tissue of lungs donated for transplantation (exempted from regulated human subjects research) and cultured as previously described.57,58 Briefly, airways were dissected and digested in 1.5 mg/mL pronase (Sigma-Aldrich, 9036-06-0) for 48 to 72 h. Recovered epithelial cells were expanded on dishes coated with type I rat-tail collagen (BD Biosciences, 354236) until 70% to 80% were confluent. Cells were detached by trypsin with EDTA, then seeded on collagen-coated 12-mm Transwell Clear (Corning Life Sciences, 3460) supported membranes (“inserts”) at a density of 1 to 2 ×105/insert. When confluent, the apical medium was withdrawn to create an air-liquid interface. Cells were maintained for 3 to 4 wk and medium in the basal compartment changed every 2 to 3 d. Following the creation of ALI, human recombinant IL13 (Peprotech, 200-13A) (10 ng/mL) was added to the basal compartment with each media change. Cell preparations derived from tracheobronchial tissues from a minimum of 3 different donated lungs were used for each experiment unless indicated.
Immunoblot analysis
Cells were lysed in a modified RIPA buffer (PBS, pH 7.4, 1.0% IPEGAL CA630 [Sigma-Aldrich, I8896]; 0.5% sodium deoxycholate [Sigma-Aldrich, D6750]; 0.1% SDS [Fisher, BP166]) containing freshly added proteinase inhibitors [Roche, 04693116001]). ATG5, ATG14, SQSTM1, and actin were resolved by electrophoresis in a 7.5% Tris-HCl polyacrylamide gel, while a 15% polyacrylamide gel (Bio-Rad Criterion, 345-0019) was used to resolve LC3 and its 2 isoforms LC3-I and LC3-II. Proteins were transferred to polyvinylidene difluoride (PVDF) membranes then blocked in tris-buffered saline (NaCl 137 mM, KCl 2.7 mM, Tris-Cl 25 mM, pH 7.4) containing 0.2% polysorbate (Tween 20; Fisher, BP337) detergent and 5% powdered milk. Membranes were then incubated in this blocking buffer with primary antibodies (and dilutions) including: LC3 (1:750; Sigma-Aldrich, L7543), ATG5 (1:1000; Sigma-Aldrich, A0856), ATG14 (1:500; Cell Signaling Technology, 5504), SQSTM1 (1:1000; Santa Cruz Biotechnology, sc-28359) and pan-actin antibody against all 6 isoforms of actin (1:2000; Millipore, MAB1501R). Primary antibodies were detected by horseradish peroxidase–labeled secondary antibody (Goat anti-rabbit, Pierce 31460; Goat anti-mouse, Pierce, 31430) binding, which was detected using enhanced chemiluminescence.
Immunostaining and microscopy
Cells cultured on inserts were washed in PBS, fixed in a solution of methanol:acetone (50:50, by volume) at −20°C for 15 min, followed by post-fixation in 4% paraformaldehyde for 10 min at room temperature. Fixed cells were blocked with 1% bovine serum albumin (Fisher, BP1605), 0.1% Triton X-100 (Fisher, BP151), and 5% donkey serum (Sigma-Aldrich D9663) in PBS for 30 min prior to immunostaining. Primary antibodies (and dilutions) included: LC3 (1:750; Novus Biologicals, NB600-1384), MUC5AC (1:500; Thermo Scientific, MS145-p1), acetylated tubulin (1:5000; Sigma-Aldrich, T7451), and SQSTM1 (1:500; Santa Cruz Biotechnology, sc-28359). Fluorophore-labeled donkey secondary antibodies were Alexa Fluor 488 or 555 (Life Technologies, A-31570, A21202, A21206, A31572). Actin was stained with phalloidin (1:100; Life Technologies, A12380) and DNA with 4′,6 diamidino-2-phenylindole (DAPI; included in Fluoroshield mounting medium, Sigma-Aldrich, F6057). Images were obtained using a 5000B Leica microscope equipped with a charge-coupled device camera (Retiga 200R) interfaced with QCapture Pro software (Q Imaging). Images were globally adjusted to optimize contrast and brightness, and composed using Photoshop software (Adobe Systems).
Autophagy flux assay
The IL13-induced autophagy flux activity was determined by blocking autophagosome-lysosome fusion using chloroquine (Sigma-Aldrich, 50-63-5) or bafilomycin A1 (Sigma-Aldrich, 88899-55-2) as previously described.28,45,59 Briefly, following IL13 treatment of hTEC cultured at the air-liquid interface, cells were incubated with 50 μM of chloroquine or 100 μM of bafilomycin A1 for 3 h in the absence or presence of IL13. Subsequently, immunoblot analysis was used to quantify the change in LC3-II to actin ratio, and in parallel samples cells were fixed for LC3 puncta immunostaining.
RNA interference
RNA depletion was achieved using short hairpin RNA (shRNA) targeted to specific transcripts and delivered by lentivirus to hTEC as previously described.35,60,61 Gene-specific shRNA sequences subcloned into the pLKO.1 transfer plasmid62 were from the RNAi Consortium collection including a control nontargeted sequence (SHC016), 2 ATG5-specific sequences (#1, TRCN0000330392; #2, TRCN0000330394) and one ATG14-specific sequence (TRCN0000010211), all available from Sigma-Aldrich. Recombinant lentiviruses were produced by co-transfection of the specific pLKO.1 plasmid that includes a cassette for puromycin resistance with plasmids containing packaging (pHR'8.2ΔR) and envelope (pCMV-VSV-G) sequences in HEK293T cells using FuGENE 6 (Promega, E2691) as described.62 Twenty h after transfection, the virus was collected in DMEM/F12 media with 5% fetal bovine serum then passed through a 45-micron filter (Millipore, CE0459). Virus-containing medium was supplemented with protamine sulfate, 10 mg/mL (Sigma-Aldrich, 53597-25-4), INS/insulin (10 μg/mL, Sigma-Aldrich, I6634), TF/transferrin (5 μg/mL, Sigma-Aldrich, T1147), EGF/epidermal growth factor (25 ng/mL, BD Biosciences, 354052), bovine pituitary extract (300 μg/mL total protein, prepared as described57), and cholera toxin (0.1 μg/mL, Sigma-Aldrich, C8052), which were added to hTEC that were seeded on collagen-coated inserts at a density of 2 × 105 cells/insert. Forty-eight to 72 h later, puromycin (Sigma-Aldrich, P8833) 2.5 mg/ml was added for 7 d. When cells were confluent, apical medium was withdrawn, and cells were cultured using ALI conditions.
Quantitative PCR
RNA was isolated from human airway cells using the RNeasy Spin column kit (Qiagen, 74104) then reverse transcribed using the cDNA Reverse Transcription Kit (Applied Biosystems, 4306736). cDNAs were amplified using Fast SYBR Green master mix (Applied Biosystems, 4309155) in a Lightcycler 480 (Roche, Mannheim, Germany). Sequence-specific primers for MUC5AC and ATG5 were used as previously published.11,63 Gene expression was normalized to OAZ1.64
Electron microscopy
Cultured cells on inserts were fixed with a solution containing 4% paraformaldehyde and 0.5% glutaraldehyde and processed for transmission electron microscopy as previously described.28 Preparations were viewed on a JEOL 1200 EX electron microscope (JEOL USA, Peabody, MA) equipped with an AMT 8 megapixel digital camera (Advanced Microscopy Techniques, Woburn, MA).
MUC5AC ELISA
Levels of secreted MUC5AC were quantified by ELISA as previously described.11,38 Briefly, cells were cultured in the presence of IL13 (10 ng/mL) after which IL13 was withdrawn from the medium for 48 h to allow goblet cells to accumulate mucin granules. The culture plate was then transferred to a foam pad to reduce the effect of jarring on goblet secretion.38 To remove residual secreted mucus, the apical surface of the cells was gently washed 4 times, each for a 30-min period, using 200 μl of PBS warmed to 37°C. Following the final wash, fresh IL13 (20 ng/mL) or ATP-γ-S (100 mM) (Sigma-Aldrich, A1388) in warmed PBS was added to the apical surface of the cells. After incubation for 1 h at 37°C, the newly secreted mucus was collected as the “agonist” quantity. For DPI experiments preparations of hTEC were pretreated with DPI (5 μM) or DMSO vehicle. Cells were washed as above with warm PBS and the last wash was saved for normalization. Fresh IL13 (20 ng/mL) was added to the apical surface of the cells and newly secreted mucins were collected from the surface after 1 h.
ROS measurement
ROS activity in hTEC was measured using the oxidant sensitive fluorometric probe DCF (CM-H2DCFDA; Life Technologies, C6827). Three h prior to loading with DCF, cells were exposed to media or IL13 (10 ng/mL). DPI, 10 μM, was used as a NOX inhibitor for each condition. Cells were washed twice with Hank's balanced salt solution then 5 μM of DCF was added to the basal medium compartment and cells incubated at 37°C for 15 min. Following this, cells were washed twice with Hank's balanced salt solution and imaged using fluorescence microscopy to acquire an image intensity using a 20X (0.8 NA) Plan-APOCHROMAT air objective on a Zeiss Axiovert 200 equipped with a Axiocam MRM camera and AxioVision software.
Image analysis and statistical methods
ImageJ software65 was used to quantify immunoblot band intensity. MUC5AC intracellular immunostaining area and intensity were quantified from images of multiple fields at 400X magnification by using a common signal threshold level among all images. The number of SQSTM1 and LC3 puncta were quantified using a monochrome image and the binary watershed command in ImageJ. A common fluorescent detection threshold was applied to all images. DCF detection of ROS activity was quantified from images of multiple fields at 200X magnification using the binary command and a common threshold as used for puncta quantification. For ultrastructural quantification by electron microscopy (EM), a blinded observer, not related to the research project, counted the number of mucin granules per cell on photomicrographs and averaged the result for each group. The unpaired, 2-tailed Student t test was used to compare values for statistical significance between experimental groups. A paired 2-tailed Student t test was used to compare gene expression by qPCR with and without IL13 from the same preparations. Likewise, a paired t-test was used to compare fold-change of MUC5AC secretion following treatment with IL13 or ATP relative to untreated cells. Multiple means were compared using ANOVA with Bonferroni's correction.
Supplementary Material
Abbreviations
- ALI
air-liquid interface
- COPD
chronic obstructive pulmonary disease
- DPI
diphenyleneiodonium
- EM
electron microscopy
- HM
hypomorphic
- hTEC
human tracheal epithelial cells
- IL13
interleukin 13
- NT
nontargeted
- PAS
periodic acid–Schiff
- PBS
phosphate-buffered saline
- ROS
reactive oxygen species
- shRNA
short hairpin RNA
- WT
wild type
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank members of the Stappenbeck Lab for helpful discussions, Wandy Beatty in the Molecular Microbiology Imaging Core for assistance with electron microscopy, Cassie Mikols in the Pulmonary Cell Culture Core for assistance with airway epithelial cell culture, and the Children's Discovery Institute of Washington University School of Medicine and St. Louis Children's Hospital RNAi core.
Funding
Funding for this work was provided by the National Institutes of Health for the Allergic Disease Cooperative Research Center (AADCRC) U19AI070489, Training Grant in Pulmonary Disease T32HL007317, and the Washington University Institute of Clinical and Translational Sciences grant UL1 TR000448; and Children's Discovery Institute of St. Louis Children's Hospital for support of the Pulmonary Epithelial Cell Core.
References
- 1.Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature 2011; 469:323-35; PMID:21248839; http://dx.doi.org/ 10.1038/nature09782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harris J. Autophagy and cytokines. Cytokine 2011; 56:140-4; PMID:21889357; http://dx.doi.org/ 10.1016/j.cyto.2011.08.022 [DOI] [PubMed] [Google Scholar]
- 3.Delgado MA, Elmaoued RA, Davis AS, Kyei G, Deretic V. Toll-like receptors control autophagy. EMBO J 2008; 27:1110-21; PMID:18337753; http://dx.doi.org/ 10.1038/emboj.2008.31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Travassos LH, Carneiro LA, Ramjeet M, Hussey S, Kim YG, Magalhaes JG, Yuan L, Soares F, Chea E, Le Bourhis L, et al.. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol 2010; 11:55-62; PMID:19898471; http://dx.doi.org/ 10.1038/ni.1823 [DOI] [PubMed] [Google Scholar]
- 5.Martinet W, De Meyer GR. Autophagy in atherosclerosis: a cell survival and death phenomenon with therapeutic potential. Circ Res 2009; 104:304-17; PMID:19213965; http://dx.doi.org/ 10.1161/CIRCRESAHA.108.188318 [DOI] [PubMed] [Google Scholar]
- 6.Ma Y, Galluzzi L, Zitvogel L, Kroemer G. Autophagy and cellular immune responses. Immunity 2013; 39:211-27; PMID:23973220; http://dx.doi.org/ 10.1016/j.immuni.2013.07.017 [DOI] [PubMed] [Google Scholar]
- 7.Harris J, De Haro SA, Master SS, Keane J, Roberts EA, Delgado M, Deretic V. T helper 2 cytokines inhibit autophagic control of intracellular Mycobacterium tuberculosis. Immunity 2007; 27:505-17; PMID:17892853; http://dx.doi.org/ 10.1016/j.immuni.2007.07.022 [DOI] [PubMed] [Google Scholar]
- 8.Su CW, Cao Y, Zhang M, Kaplan J, Su L, Fu Y, Walker WA, Xavier R, Cherayil BJ, Shi HN. Helminth infection impairs autophagy-mediated killing of bacterial enteropathogens by macrophages. J Immunol 2012; 189:1459-66; PMID:22732589; http://dx.doi.org/ 10.4049/jimmunol.1200484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barnes PJ. The cytokine network in asthma and chronic obstructive pulmonary disease. J Clin Invest 2008; 118:3546-56; PMID:18982161; http://dx.doi.org/ 10.1172/JCI36130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Holtzman MJ. Asthma as a chronic disease of the innate and adaptive immune systems responding to viruses and allergens. J Clin Invest 2012; 122:2741-8; PMID:22850884; http://dx.doi.org/ 10.1172/JCI60325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alevy YG, Patel AC, Romero AG, Patel DA, Tucker J, Roswit WT, Miller CA, Heier RF, Byers DE, Brett TJ, et al.. IL13-induced airway mucus production is attenuated by MAPK13 inhibition. J Clin Invest 2012; 122:4555-68; PMID:23187130; http://dx.doi.org/ 10.1172/JCI64896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuperman DA, Huang X, Koth LL, Chang GH, Dolganov GM, Zhu Z, Elias JA, Sheppard D, Erle DJ. Direct effects of interleukin-13 on epithelial cells cause airway hyperreactivity and mucus overproduction in asthma. Nat Med 2002; 8:885-9; PMID:12091879 [DOI] [PubMed] [Google Scholar]
- 13.Rolling C, Treton D, Pellegrini S, Galanaud P, Richard Y. IL4 and IL13 receptors share the gamma c chain and activate STAT6, STAT3 and STAT5 proteins in normal human B cells. FEBS Lett 1996; 393:53-6; PMID:8804422; http://dx.doi.org/ 10.1016/0014-5793(96)00835-6 [DOI] [PubMed] [Google Scholar]
- 14.Byers DE, Alexander-Brett J, Patel AC, Agapov E, Dang-Vu G, Jin X, Wu K, You Y, Alevy Y, Girard JP, et al.. Long-term IL33-producing epithelial progenitor cells in chronic obstructive lung disease. J Clin Invest 2013; 123:3967-82; PMID:23945235; http://dx.doi.org/ 10.1172/JCI65570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ingram JL, Kraft M. IL13 in asthma and allergic disease: asthma phenotypes and targeted therapies. J Allergy Clin Immunol 2012; 130:829-42; quiz 43–4; PMID:22951057; http://dx.doi.org/ 10.1016/j.jaci.2012.06.034 [DOI] [PubMed] [Google Scholar]
- 16.Zhen G, Park SW, Nguyenvu LT, Rodriguez MW, Barbeau R, Paquet AC, Erle DJ. IL13 and epidermal growth factor receptor have critical but distinct roles in epithelial cell mucin production. Am J Respir Cell Mol Biol 2007; 36:244-53; PMID:16980555; http://dx.doi.org/ 10.1165/rcmb.2006-0180OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhu Z, Homer RJ, Wang Z, Chen Q, Geba GP, Wang J, Zhang Y, Elias JA. Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J Clin Invest 1999; 103:779-88; PMID:10079098; http://dx.doi.org/ 10.1172/JCI5909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jiang D, Li Q, Kolosov VP, Zhou X. The inhibition of aldose reductase on mucus production induced by interleukin-13 in the human bronchial epithelial cells. Int Immunopharmacol 2012; 12:588-93; PMID:22386909; http://dx.doi.org/ 10.1016/j.intimp.2012.02.007 [DOI] [PubMed] [Google Scholar]
- 19.Harper RW, Xu C, Eiserich JP, Chen Y, Kao CY, Thai P, Setiadi H, Wu R. Differential regulation of dual NADPH oxidases/peroxidases, Duox1 and Duox2, by Th1 and Th2 cytokines in respiratory tract epithelium. FEBS Lett 2005; 579:4911-7; PMID:16111680; http://dx.doi.org/ 10.1016/j.febslet.2005.08.002 [DOI] [PubMed] [Google Scholar]
- 20.Parker JC, Thavagnanam S, Skibinski G, Lyons J, Bell J, Heaney LG, Shields MD. Chronic IL9 and IL13 exposure leads to an altered differentiation of ciliated cells in a well-differentiated paediatric bronchial epithelial cell model. PLoS One 2013; 8:e61023; PMID:23671562; http://dx.doi.org/ 10.1371/journal.pone.0061023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davis CW, Dickey BF. Regulated airway goblet cell mucin secretion. Annu Rev Physiol 2008; 70:487-512; PMID:17988208; http://dx.doi.org/ 10.1146/annurev.physiol.70.113006.100638 [DOI] [PubMed] [Google Scholar]
- 22.Burgess J, Del Bel LM, Ma CI, Barylko B, Polevoy G, Rollins J, Albanesi JP, Kramer H, Brill JA. Type II phosphatidylinositol 4-kinase regulates trafficking of secretory granule proteins in Drosophila. Development 2012; 139:3040-50; PMID:22791894; http://dx.doi.org/ 10.1242/dev.077644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hewson CA, Haas JJ, Bartlett NW, Message SD, Laza-Stanca V, Kebadze T, Caramori G, Zhu J, Edbrooke MR, Stanciu LA, et al.. Rhinovirus induces MUC5AC in a human infection model and in vitro via NF-kappaB and EGFR pathways. Eur Respir J 2010; 36:1425-35; PMID:20525715; http://dx.doi.org/ 10.1183/09031936.00026910 [DOI] [PubMed] [Google Scholar]
- 24.Evans CM, Kim K, Tuvim MJ, Dickey BF. Mucus hypersecretion in asthma: causes and effects. Curr Opin Pulm Med 2009; 15:4-11; PMID:19077699; http://dx.doi.org/ 10.1097/MCP.0b013e32831da8d3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baginski TK, Dabbagh K, Satjawatcharaphong C, Swinney DC. Cigarette smoke synergistically enhances respiratory mucin induction by proinflammatory stimuli. Am J Respir Cell Mol Biol 2006; 35:165-74; PMID:16543607; http://dx.doi.org/ 10.1165/rcmb.2005-0259OC [DOI] [PubMed] [Google Scholar]
- 26.Torisu T, Torisu K, Lee IH, Liu J, Malide D, Combs CA, Wu XS, Rovira II, Fergusson MM, Weigert R, et al.. Autophagy regulates endothelial cell processing, maturation and secretion of von Willebrand factor. Nat Med 2013; 19:1281-7; PMID:24056772; http://dx.doi.org/ 10.1038/nm.3288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cadwell K, Liu JY, Brown SL, Miyoshi H, Loh J, Lennerz JK, Kishi C, Kc W, Carrero JA, Hunt S, et al.. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 2008; 456:259-63; PMID:18849966; http://dx.doi.org/ 10.1038/nature07416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Patel KK, Miyoshi H, Beatty WL, Head RD, Malvin NP, Cadwell K, Guan JL, Saitoh T, Akira S, Seglen PO, et al.. Autophagy proteins control goblet cell function by potentiating reactive oxygen species production. EMBO J 2013; 32:3130-44; PMID:24185898; http://dx.doi.org/ 10.1038/emboj.2013.233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wlodarska M, Thaiss CA, Nowarski R, Henao-Mejia J, Zhang JP, Brown EM, Frankel G, Levy M, Katz MN, Philbrick WM, et al.. NLRP6 inflammasome orchestrates the colonic host-microbial interface by regulating goblet cell mucus secretion. Cell 2014; 156:1045-59; PMID:24581500; http://dx.doi.org/ 10.1016/j.cell.2014.01.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barlow JL, Bellosi A, Hardman CS, Drynan LF, Wong SH, Cruickshank JP, McKenzie AN. Innate IL13-producing nuocytes arise during allergic lung inflammation and contribute to airways hyperreactivity. J Allergy Clin Immunol 2012; 129:191-8.e1-4; PMID:22079492; http://dx.doi.org/ 10.1016/j.jaci.2011.09.041 [DOI] [PubMed] [Google Scholar]
- 31.Kim EY, Battaile JT, Patel AC, You Y, Agapov E, Grayson MH, Benoit LA, Byers DE, Alevy Y, Tucker J, et al.. Persistent activation of an innate immune response translates respiratory viral infection into chronic lung disease. Nat Med 2008; 14:633-40; PMID:18488036; http://dx.doi.org/ 10.1038/nm1770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stolarski B, Kurowska-Stolarska M, Kewin P, Xu D, Liew FY. IL33 exacerbates eosinophil-mediated airway inflammation. J Immunol 2010; 185:3472-80; PMID:20693421; http://dx.doi.org/ 10.4049/jimmunol.1000730 [DOI] [PubMed] [Google Scholar]
- 33.Kondo Y, Yoshimoto T, Yasuda K, Futatsugi-Yumikura S, Morimoto M, Hayashi N, Hoshino T, Fujimoto J, Nakanishi K. Administration of IL33 induces airway hyperresponsiveness and goblet cell hyperplasia in the lungs in the absence of adaptive immune system. Int Immunol 2008; 20:791-800; PMID:18448455; http://dx.doi.org/ 10.1093/intimm/dxn037 [DOI] [PubMed] [Google Scholar]
- 34.Barlow JL, Peel S, Fox J, Panova V, Hardman CS, Camelo A, Bucks C, Wu X, Kane CM, Neill DR, et al.. IL33 is more potent than IL-25 in provoking IL13-producing nuocytes (type 2 innate lymphoid cells) and airway contraction. J Allergy Clin Immunol 2013; 132:933-41; PMID:23810766; http://dx.doi.org/ 10.1016/j.jaci.2013.05.012 [DOI] [PubMed] [Google Scholar]
- 35.Horani A, Nath A, Wasserman MG, Huang T, Brody SL. Rho-associated protein kinase inhibition enhances airway epithelial Basal-cell proliferation and lentivirus transduction. Am J Respir Cell Mol Biol 2013; 49:341-7; PMID:23713995; http://dx.doi.org/ 10.1165/rcmb.2013-0046TE [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fulcher ML, Gabriel S, Burns KA, Yankaskas JR, Randell SH. Well-differentiated human airway epithelial cell cultures. Methods Mol Med 2005; 107:183-206; PMID:15492373 [DOI] [PubMed] [Google Scholar]
- 37.Danahay H, Atherton H, Jones G, Bridges RJ, Poll CT. Interleukin-13 induces a hypersecretory ion transport phenotype in human bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol 2002; 282:L226-36; PMID:11792627; http://dx.doi.org/ 10.1152/ajplung.00311.2001 [DOI] [PubMed] [Google Scholar]
- 38.Abdullah LH, Wolber C, Kesimer M, Sheehan JK, Davis CW. Studying mucin secretion from human bronchial epithelial cell primary cultures. Methods Mol Biol 2012; 842:259-77; PMID:22259142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mandal D, Fu P, Levine AD. REDOX regulation of IL13 signaling in intestinal epithelial cells: usage of alternate pathways mediates distinct gene expression patterns. Cell Signal 2010; 22:1485-94; PMID:20570727; http://dx.doi.org/ 10.1016/j.cellsig.2010.05.017 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 40.Shao MX, Nadel JA. Dual oxidase 1-dependent MUC5AC mucin expression in cultured human airway epithelial cells. Proc Natl Acad Sci U S A 2005; 102:767-72; PMID:15640347; http://dx.doi.org/ 10.1073/pnas.0408932102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fogel AI, Dlouhy BJ, Wang C, Ryu SW, Neutzner A, Hasson SA, Sideris DP, Abeliovich H, Youle RJ. Role of membrane association and Atg14-dependent phosphorylation in beclin-1-mediated autophagy. Mol Cell Biol 2013; 33:3675-88; PMID:23878393; http://dx.doi.org/ 10.1128/MCB.00079-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kuperman DA, Schleimer RP. Interleukin-4, interleukin-13, signal transducer and activator of transcription factor 6, and allergic asthma. Curr Mol Med 2008; 8:384-92; PMID:18691065; http://dx.doi.org/ 10.2174/156652408785161032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thai P, Chen Y, Dolganov G, Wu R. Differential regulation of MUC5AC/Muc5ac and hCLCA-1/mGob-5 expression in airway epithelium. Am J Respir Cell Mol Biol 2005; 33:523-30; PMID:16151054; http://dx.doi.org/ 10.1165/rcmb.2004-0220RC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee JH, Kaminski N, Dolganov G, Grunig G, Koth L, Solomon C, Erle DJ, Sheppard D. Interleukin-13 induces dramatically different transcriptional programs in three human airway cell types. Am J Respir Cell Mol Biol 2001; 25:474-85; PMID:11694453; http://dx.doi.org/ 10.1165/ajrcmb.25.4.4522 [DOI] [PubMed] [Google Scholar]
- 45.Chen ZH, Kim HP, Sciurba FC, Lee SJ, Feghali-Bostwick C, Stolz DB, Dhir R, Landreneau RJ, Schuchert MJ, Yousem SA, et al.. Egr-1 regulates autophagy in cigarette smoke-induced chronic obstructive pulmonary disease. PLoS One 2008; 3:e3316; PMID:18830406; http://dx.doi.org/ 10.1371/journal.pone.0003316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tanaka A, Jin Y, Lee SJ, Zhang M, Kim HP, Stolz DB, Ryter SW, Choi AM. Hyperoxia-induced LC3B interacts with the Fas apoptotic pathway in epithelial cell death. Am J Respir Cell Mol Biol 2012; 46:507-14; PMID:22095627; http://dx.doi.org/ 10.1165/rcmb.2009-0415OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim HP, Wang X, Chen ZH, Lee SJ, Huang MH, Wang Y, Ryter SW, Choi AM. Autophagic proteins regulate cigarette smoke-induced apoptosis: protective role of heme oxygenase-1. Autophagy 2008; 4:887-95; PMID:18769149; http://dx.doi.org/ 10.4161/auto.6767 [DOI] [PubMed] [Google Scholar]
- 48.Fujisawa T, Velichko S, Thai P, Hung LY, Huang F, Wu R. Regulation of airway MUC5AC expression by IL-1beta and IL-17A; the NF-kappaB paradigm. J Immunol 2009; 183:6236-43; PMID:19841186; http://dx.doi.org/ 10.4049/jimmunol.0900614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gray T, Coakley R, Hirsh A, Thornton D, Kirkham S, Koo JS, Burch L, Boucher R, Nettesheim P. Regulation of MUC5AC mucin secretion and airway surface liquid metabolism by IL-1beta in human bronchial epithelia. Am J Physiol Lung Cell Mol Physiol 2004; 286:L320-30; PMID:14527933; http://dx.doi.org/ 10.1152/ajplung.00440.2002 [DOI] [PubMed] [Google Scholar]
- 50.Stappenbeck TS. The role of autophagy in Paneth cell differentiation and secretion. Mucosal Immunol 2010; 3:8-10; PMID:19890269; http://dx.doi.org/ 10.1038/mi.2009.121 [DOI] [PubMed] [Google Scholar]
- 51.DeSelm CJ, Miller BC, Zou W, Beatty WL, van Meel E, Takahata Y, Klumperman J, Tooze SA, Teitelbaum SL, Virgin HW. Autophagy proteins regulate the secretory component of osteoclastic bone resorption. Dev Cell 2011; 21:966-74; PMID:22055344; http://dx.doi.org/ 10.1016/j.devcel.2011.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ushio H, Ueno T, Kojima Y, Komatsu M, Tanaka S, Yamamoto A, Ichimura Y, Ezaki J, Nishida K, Komazawa-Sakon S, et al.. Crucial role for autophagy in degranulation of mast cells. J Allergy Clin Immunol 2011; 127:1267-76.e6; PMID:21333342; http://dx.doi.org/ 10.1016/j.jaci.2010.12.1078 [DOI] [PubMed] [Google Scholar]
- 53.Amaravadi RK, Lippincott-Schwartz J, Yin XM, Weiss WA, Takebe N, Timmer W, DiPaola RS, Lotze MT, White E. Principles and current strategies for targeting autophagy for cancer treatment. Clin Cancer Res 2011; 17:654-66; PMID:21325294; http://dx.doi.org/ 10.1158/1078-0432.CCR-10-2634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gorski SM, Ries J, Lum JJ. Targeting autophagy: the Achilles' heel of cancer. Autophagy 2012; 8:1279-80; PMID:22836583; http://dx.doi.org/ 10.4161/auto.20828 [DOI] [PubMed] [Google Scholar]
- 55.Poon A, Eidelman D, Laprise C, Hamid Q. ATG5, autophagy and lung function in asthma. Autophagy 2012; 8:694-5; PMID:22498476; http://dx.doi.org/ 10.4161/auto.19315 [DOI] [PubMed] [Google Scholar]
- 56.Martin LJ, Gupta J, Jyothula SS, Butsch Kovacic M, Biagini Myers JM, Patterson TL, Ericksen MB, He H, Gibson AM, Baye TM, et al.. Functional variant in the autophagy-related 5 gene promotor is associated with childhood asthma. PLoS One 2012; 7:e33454; PMID:22536318; http://dx.doi.org/ 10.1371/journal.pone.0033454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.You Y, Richer EJ, Huang T, Brody SL. Growth and differentiation of mouse tracheal epithelial cells: selection of a proliferative population. Am J Physiol Lung Cell Mol Physiol 2002; 283:L1315-21; PMID:12388377; http://dx.doi.org/ 10.1152/ajplung.00169.2002 [DOI] [PubMed] [Google Scholar]
- 58.You Y, Brody SL. Culture and differentiation of mouse tracheal epithelial cells. Methods Mol Biol 2013; 945:123-43; PMID:23097105 [DOI] [PubMed] [Google Scholar]
- 59.Marino ML, Pellegrini P, Di Lernia G, Djavaheri-Mergny M, Brnjic S, Zhang X, Hagg M, Linder S, Fais S, Codogno P, et al.. Autophagy is a protective mechanism for human melanoma cells under acidic stress. J Biol Chem 2012; 287:30664-76; PMID:22761435; http://dx.doi.org/ 10.1074/jbc.M112.339127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Horani A, Brody SL, Ferkol TW, Shoseyov D, Wasserman MG, Ta-shma A, Wilson KS, Bayly PV, Amirav I, Cohen-Cymberknoh M, et al.. CCDC65 mutation causes primary ciliary dyskinesia with normal ultrastructure and hyperkinetic cilia. PLoS One 2013; 8:e72299; PMID:23991085; http://dx.doi.org/ 10.1371/journal.pone.0072299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Horani A, Ferkol TW, Shoseyov D, Wasserman MG, Oren YS, Kerem B, Amirav I, Cohen-Cymberknoh M, Dutcher SK, Brody SL, et al.. LRRC6 mutation causes primary ciliary dyskinesia with dynein arm defects. PLoS One 2013; 8:e59436; PMID:23527195; http://dx.doi.org/ 10.1371/journal.pone.0059436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stewart SA, Dykxhoorn DM, Palliser D, Mizuno H, Yu EY, An DS, Sabatini DM, Chen IS, Hahn WC, Sharp PA, et al.. Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 2003; 9:493-501; PMID:12649500; http://dx.doi.org/ 10.1261/rna.2192803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Coffey EE, Beckel JM, Laties AM, Mitchell CH. Lysosomal alkalization and dysfunction in human fibroblasts with the Alzheimer's disease-linked presenilin 1 A246E mutation can be reversed with cAMP. Neuroscience 2014; 263:111-24; PMID:24418614; http://dx.doi.org/ 10.1016/j.neuroscience.2014.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Patel DA, Patel AC, Nolan WC, Zhang Y, Holtzman MJ. High throughput screening for small molecule enhancers of the interferon signaling pathway to drive next-generation antiviral drug discovery. PLoS One 2012; 7:e36594; PMID:22574190; http://dx.doi.org/ 10.1371/journal.pone.0036594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 2012; 9:671-5; PMID:22930834; http://dx.doi.org/ 10.1038/nmeth.2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.