

ABSTRACT
The common underlying feature of most neurodegenerative diseases such as Alzheimer disease (AD), prion diseases, Parkinson disease (PD), and amyotrophic lateral sclerosis (ALS) involves accumulation of misfolded proteins leading to initiation of endoplasmic reticulum (ER) stress and stimulation of the unfolded protein response (UPR). Additionally, ER stress more recently has been implicated in the pathogenesis of HIV-associated neurocognitive disorders (HAND). Autophagy plays an essential role in the clearance of aggregated toxic proteins and degradation of the damaged organelles. There is evidence that autophagy ameliorates ER stress by eliminating accumulated misfolded proteins. Both abnormal UPR and impaired autophagy have been implicated as a causative mechanism in the development of various neurodegenerative diseases. This review highlights recent advances in the field on the role of ER stress and autophagy in AD, prion diseases, PD, ALS and HAND with the involvement of key signaling pathways in these processes and implications for future development of therapeutic strategies.
KEYWORDS: autophagy, alzheimer disease, amyotrophic lateral sclerosis and HIV-associated neurocognitive disorders, ER stress, neurodegenerative disorders, prion diseases, Parkinson disease
Introduction
Neurodegenerative disorders are characterized by progressive loss of neuronal functions that are closely associated with loss/dysfunction of neuronal cells. Emerging evidence suggests that accumulation of exogenous or abnormal misfolded proteins leads to a state of endoplasmic reticulum (ER) stress in neurons, contributing to the salient pathology associated with neurodegeneration. In order to restore homeostasis within the ER there occurs sequential activation of the unfolded protein response (UPR) involving translational attenuation of global protein synthesis, transcriptional induction of genes functioning as ER chaperones, and the ER-associated degradation (ERAD) of aggregated proteins ultimately leading to autophagy.1,2 Autophagy is an essential catabolic mechanism that delivers misfolded proteins and damaged organelles to the lysosome for degradation.3 Maintaining basal levels of autophagic activity is critical for postmitotic neurons that are unable to dispose of aggregated proteins via cell division.4 As a result, increased dependence of neurons on a higher threshold of constitutive, basal levels of autophagy thus renders these cells more vulnerable to impairments in autophagy, a key feature underlying most neurodegenerative diseases. Accumulating evidence suggests that in chronic neurodegenerative disorders, persistent ER stress often results in stimulation of autophagic activities, likely as a compensatory mechanism, to relieve ER stress; however, in the face of impaired UPR or autophagy, there is inefficient clearance of the accumulated proteins, which in turn, leads to the development and progression of neurodegeneration. This review is an attempt to provide a survey of the major ER stress signaling pathways and the autophagic mechanism(s) and their interplay in the development and progression of neurodegenerative diseases such as AD, prion diseases, PD, ALS and HAND.
ER stress and UPR
ER stress is a pathological state that involves the aberrant aggregation of misfolded proteins, which in turn, results in the UPR that is initially expected to rescue cells from the stress and restore homeostasis within the ER.1 The ER stress stimuli in neurodegenerative disorders include accumulation of exogenous viral proteins,5 aggregation of mutant endogenous proteins6 and depletion of ER calcium stores via ITPR (inositol-1,4,5-trisphosphate receptor)7,8 and RYR (ryanodine receptor).5 Chaperones within the ER, such as HSPA5 (heat shock 70 kDa protein 5 [glucose-regulated protein, 78kDa]), and CALR (calreticulin), require high levels of [Ca2+]ER for binding to paired anionic amino acids to carry out the function of protein folding (Fig. 1).9,10 Thus, depletion of ER calcium store renders ER-resident chaperones inactive for proper protein folding processes, resulting in the accumulation of misfolded proteins.
Figure 1.

ER stress and UPR pathways in neuronal cells. Pathological accumulation of misfolded proteins and/or depletion of ER calcium store via activation of ITPR and RYR leads to ER stress. Dissociation of HSPA5 from 3 ER stress sensors, EIF2AK3, ERN1 and ATF6, results in phosphorylation of EIF2AK3 and ERN1 and translocation of ATF6 to the Golgi apparatus. Activated EIF2AK3 is a serine/threonine protein kinase that phosphorylates EIF2S1. p-EIF2S1 (phosphorylated EIF2S1) inhibits global protein synthesis but selectively upregulates ATF4, PPP1R15A, DDIT3 and ATF3. PPP1R15A provides a negative feedback by dephosphorylating p-EIF2S1. ERN1 cleaves XBP1 mRNA; the spliced form of XBP1 encodes the XBP1 protein. XBP1 increases the expression of genes encoding ER chaperones, ERAD proteins and lipid synthesis to restore the capacity of protein folding. ATF6 is translocated to the Golgi apparatus where it is cleaved by MBTPS1 and MBTPS2, and cleaved ATF6 stimulates the expression of ER chaperones and ERAD proteins. Apoptosis will ensue if upregulation of ER chaperones and ERAD proteins fails to rescue ER stress.
In the central nervous system, the UPR is initiated by 3 major cascading transmembrane sensors: a) EIF2AK3 (eukaryotic translation initiation factor 2-α kinase 3), b) ERN1 (endoplasmic reticulum to nucleus signaling 1) and c) ATF6 (activating transcription factor 6). 11 The most abundant ER chaperone, HSPA5 plays an essential role in initiation of the UPR by 3 major sensors.11 The ER-luminal domain of EIF2AK3, ERN1 and ATF6 interacts with HSPA5 to suppress ER stress under physiological conditions (Fig. 1). Following cellular stress and accumulation of unfolded proteins, there is dissociation of HSPA5 from the ER sensor proteins, leading to the activation of ER stress sensors.12
EIF2AK3 is a serine/threonine protein kinase that is activated by dimerization and trans-autophosphorylation following dissociation from HSPA5 (Fig. 1).13 Phosphorylation of EIF2S1 (eukaryotic translation initiation factor 2, subunit 1 α, 35kDa) mediated by active EIF2AK3 causes a shift in the open reading frames from the true translation initiation site to a site upstream of the coding region of ATF4 (activating transcription factor 4) as well as ATF3 (activating transcription factor 3), DDIT3 (pro-apoptotic transcription factor, DNA-damage-inducible transcript 3) and PPP1R15A (protein phosphatase 1, regulatory subunit 15A), a cofactor for PPP1 (protein phosphatase 1) that serves as a negative-feedback regulator of EIF2S1 phosphorylation.14,15 Activation of EIF2AK3 therefore attenuates global translation of proteins while paradoxically stimulating the expression of stress response genes. However, if the UPR fails to rescue neurons from ER stress, ensuing ER stress culminates in activation of apoptosis.16
ERN1 is a ubiquitous serine/threonine kinase (Fig. 1). Similar to EIF2AK3, following dissociation of HSPA5, the kinase domain of ERN1 is activated. However, unlike EIF2AK3, ERN1 contains a C-terminal endoribonuclease domain that is also activated following accumulation of the unfolded proteins.17 The endoribonuclease domain is responsible for the splicing of the mRNA encoding the transcriptional factor XBP1 (X-box binding protein 1).17 The protein XBP1 translated from XBP1s (spliced form of XBP1 mRNA) in turn stimulates the expression of ER chaperones, ERAD proteins and lipid synthesis to restore protein folding capacity within the ER.18
ATF6 is also an ER stress transmembrane sensor belonging to a family of bZIP transcription factors (Fig. 1).19 In response to the UPR, HSPA5 dissociation releases ATF6 from the ER membrane allowing it to translocate to the Golgi, where it is cleaved by proteolytic cleavage to a soluble form resulting in its trafficking into the nucleus.20-22 Following translocation into the nucleus, ATF6 functions as a transcription factor responsible for recovery from acute stress with tolerance to chronic stress by inducing the expression of ER chaperone genes such as those encoding HSPA5 and PDI (protein disulfide isomerase) folding enzymes and the ERAD components.23
Overall, the activation of ER stress leads to attenuation of global protein synthesis, induction of ER chaperones to increase the capacity of the ER for protein folding and induction of ERAD elements to decrease the ER protein load.18
Autophagy
Macroautophagy (hereafter autophagy) is a tightly regulated catabolic pathway for lysosomal degradation of cytoplasmic organelles or cytosolic components and the recycling of the resulting macromolecules.24 Homeostasis of autophagic activities in the cytoplasm is critical for maintenance of neuronal functioning. There is increasing evidence that failure in clearing the aggregated proteins or impaired organelles contributes to programmed cell death or apoptosis.25 Intriguingly, abnormal autophagic activity has been described in AD,26 HD,27 PD28 and Creutzfeldt-Jakob disease (CJD).29
Autophagy is initiated by the sequestration of targeted substrates within double-membrane vesicles termed phagophores that mature into autophagosomes.30 Prior to autophagosome fusion with the lysosome and degradation of the sequestered cargo, autophagy undergoes a series of key steps including activation by signal transduction, phagophore nucleation, membrane elongation, lysosomal fusion and cargo degradation (Fig. 2).31 Stress stimuli, such as starvation, oxidative stress, and ER stress, result in inhibition of MTORC1 (mechanistic target of rapamycin [serine/threonine kinase] complex 1) and, reciprocally, activation of AMPK (AMP-activated protein kinase),32 both of which lead to activation of the ULK1 (unc-51 like autophagy activating kinase 1) complex by changing the phosphorylation state of certain components in the complex.33 The activated ULK1 complex initiates vesicle nucleation by translocating the BECN1 (Beclin 1, autophagy related)-containing core multiprotein complex and the class III phosphatidylinositol 3-kinase from the cytoskeleton to sites of phagophore assembly.33-36 Vesicle nucleation is followed by membrane elongation. In the process of elongation of autophagic membrane, ATG12 (autophagy-related 12) conjugated to ATG5 (autophagy-related 5) positively regulates the conjugation of phosphatidylethanolamine to MAP1LC3B (microtubule-associated protein 1 light chain 3 β),37,38 followed by the conversion of MAP1LC3B from soluble MAP1LC3B-I into autophagic vesicle-associated MAP1LC3B-II.39 MAP1LC3B-II stably binds to the phagophore and autophagosome membranes.40 Finally, the autophagosomes fuse with the lysosome for degradation of the sequestered cargo.41 Moreover, endocytic recycling is a complicated process correlated with endocytic uptake to regulate the components of the plasma membrane. Regulation of endocytic recycling involves various molecules such as RAB1A, RAB11, RAB8, RAB22, ARHGAP26 (Rho GTPase activating protein 26), EHD1 (EH-domain containing 1), and MICALL1 (MICAL-like 1).42-47 Recent evidence also demonstrates that recycling endosomes promote autophagy.48
Figure 2.

The autophagic cascade in neurodegenerative disorders. Stress stimuli associated with neurodegenerative disorders inhibits MTORC1, resulting in activation of the ULK1 complex. The ULK1 complex can also be activated by AMPK. The ULK1 complex initiates vesicle nucleation by translocating BECN1 and PtdIns3K to phagophores. To elongate the membrane from the phagophore assembly site (PAS), The ATG12–ATG5 conjugate regulates the conjugation of PE to MAP1LC3B, resulting in conversion of MAP1LC3B from soluble MAP1LC3B-I into phagophore and autophagosome-associated MAP1LC3B-II. Once the autophagosome is formed, it fuses with a lysosome for degradation of its sequestered cargo.
Autophagy is highly conserved in all types of eukaryotic cells and is essential for the regulation of basic cellular processes such as growth and apoptosis. Among all the other cell types, neurons exhibit higher basal levels of autophagy. This could likely be attributed to their structure and function: (1) Neurons are the largest cells in the human body (based on their length) and have highly specialized structures such as axons and dendrites that are the sites for synthesis of macromolecules including proteins, RNA and lipids. Appropriate delivery and degradation of these macromolecules is essential for homeostatic maintenance of normal synaptic growth and activity. (2) Neurons are nondividing cells that are extremely sensitive to accumulation of toxic aggregates compared to the other mitotic cells. (3) Neurons require higher energy for execution of their normal functions. In summary, owing to the increased susceptibility of neurons to genetic/environmental insults, they are endowed with tightly regulated and effective intrinsic mechanisms such as autophagy, to execute their normal functions during a pathological insult. Protein quality control by autophagy is thus an essential attribute for neuronal survival and functioning. Dysfunction of the autophagic activities impedes synaptic development,49 hampers axonal function and could underlie the onset and progression of various neurodegenerative disorders.4 Paradoxically, however, once the UPR and autophagy responses are unable to eliminate the damaged proteins or organelles effectively, neurons become highly susceptible to the diseases of protein aggregation underlying the neurodegenerative diseases.
ER stress and autophagy
Emerging studies also demonstrated a mechanistic link between the ER stress and autophagy, in which the activation of EIF2AK3 is essentially required due to the misfolded protein accumulation shown in various diseases to induce autophagy.50,51 Furthermore, the UPR downstream mediators of EIF2AK3 such as ATF4 and DDIT3 were also reported to guide the induction of autophagy gene transcription such as with BECN1, MAP1LC3B, ATG5, ATG7 and ATG12 in response to amino acid starvation or ER stress.52 Hypoxic microenvironments also activate EIF2AK3- and ATF4-DDIT3 signaling, which is ultimately involved in the regulation of MAP1LC3B and ATG5 proteins to induce autophagy.53,54 Another arm of ER stress, involving ERN1, is also involved in autophagy induction via activating AMPK in response to nitric oxide.55 Alternatively, ERN1 induces autophagy through dissociation of BECN1 from its binding with the anti-apoptotic protein BCL2 (B-cell CLL/lymphoma 2)56,57 via MAPK8 (mitogen-activated protein kinase 8)-mediated phosphorylation of BCL2.58,59 Factors downstream of ERN1, such as XBP1, also play an essential role through an interaction with FOXO1 (forkhead box O1) in the negative feedback loop of ER stress-mediated autophagy.60-62 In addition, the soluble ATF6 formed after proteolytic cleavage under ER stress can upregulate the expression level of DAPK1 (death-associated protein kinase 1).63,64 The increased DAPK1 phosphorylates BECN1 leading to the effective dissociation from its negative regulator BCL2, thereby triggering autophagy (Fig. 3).65 Overall, the misfolded/aggregated proteins induce ER stress and shackle the autophagy functions in various diseases including neurodegenerative diseases.
Figure 3.

The interplay between the UPR and autophagy. EIF2AK3-EIF2S1 signaling induces the expression of various autophagy-related proteins via ATF4 and DDIT3. ERN1 triggers autophagy through activation of AMPK and dissociation of BECN1 from BCL2 via MAPK8-dependent phosphorylation of BCL2. Conversely, XBP1 that acts as the downstream mediator of ERN1 provides negative feedback for autophagy by promoting degradation of FOXO1. Upregulation of DAPK1 induced by cleaved ATF6 phosphorylates BECN1 and leads to dissociation of BECN1 from BCL2, resulting in enhanced autophagic activities.
Intracellular protein aggregation is one of the salient features underlying various neurological diseases such as AD and PD.66,67 The balance between protein generation and degradation is crucial for protein homeostasis. Intriguingly, while most of the aggregated proteins result in the common endpoint of neuronal impairment, the mechanism of action of each of the aggregated proteins is distinct. For instance, aggregated Aβ (β-amyloid) that is cleaved from its membrane-bound precursor APP (amyloid precursor protein), is a primary source of ER stress in AD,68,69 whereas in PD dysfunction appears to result from the accumulation of Lewy bodies and intracellular inclusion bodies containing diffuse deposits of misfolded SNCA/α-synuclein (synuclein, α [non A4 component of amyloid precursor]).67 ER stress is likely a secondary response due to oxidative stress and/or inflammation mediated by the aggregates.70-73 Taken together, based on the nature of the disease, the sequence of events mediated by the aggregated proteins can be very different. For example, in the case of AD, Aβ is the primary source of ER stress whereas in PD, the accumulated SNCA first induces cellular responses, which, in turn mediate the secondary ER stress leading to disease pathogenesis.
Similar to the diversity of signaling events observed in AD and PD, differences in the tissue responses in the accumulation of aggregated proteins in the autosomal dominant mutations in PD and AD also show striking differences. Genetic mutations (accounting for autosomal-dominant AD) in APP, PSEN1 (presenilin 1) or PSEN2 (presenilin 2) divert the normal processing of APP toward the amyloidogenic phenotype, leading in turn to enhanced production and deposition of total Aβ and Aβ peptides, that have a greater tendency to aggregate and mediate disease pathogenesis.74 Unlike AD wherein augmented production of aggregated proteins plays a major role in disease pathogenesis, in the autosomal dominant mutations of PD such as SNCA and LRRK2 (leucine-rich repeat kinase 2), protein degradation pathways are impeded, leading to accumulation of faulty aggregated proteins. It has been well documented that mutant SNCA impedes autophagy by inhibiting the GTPase RAB1A, whereas mutations in LRRK2 hinder the autolysosomal degradation pathways in the late stage of autophagic activity.75,76 Intriguingly, therefore, while accumulation of aggregated protein response underlies both the autosomal dominant mutant form of AD and PD, the mechanism(s) by which the protein aggregation occurs (impaired protein degradation in PD vs. increased protein production in AD) leading to disease pathogenesis, is unique for each disease. A better understanding of the tissue response in mediating the aberrant protein aggregation is thus critical in dissecting the mechanism(s) underlying disease pathogenesis and for future development of therapeutic targets.
Alzheimer disease and other tauopathies
AD, one of the most common neurodegenerative diseases in the elderly, is characterized clinically as an ongoing cognitive impairment with typical pathogenic deposits of neurofibrillary tangles containing hyperphosphorylated MAPT/tau (microtubule-associated protein tau) and extracellular accumulations of Aβ plaques.77 Aβ is the cleaved product released from its precursor protein, APP, following exposure to BACE1/β-secretase and γ-secretase.78 The complex of γ-secretase, composed of at least PSEN1 and PSEN2 in the ER, cleaves APP to Aβ of varying lengths—Aβ40 and Aβ42. Genetic mutations in APP, PSEN1 and PSEN2 account for autosomal-dominant AD.79 Another salient feature of AD is pathological aggregation of the MAPT protein.80 MAPT is an axonal cytosolic protein for the stabilization of the microtubule network, but it becomes hyperphosphorylated and aggregated within neurons in the form of neurofibrillary tangles.80
UPR and autophagy play an important role in protecting neurons against accumulation of Aβ and PSEN1 mutations in the familial form of AD. PSEN1 mutations and intracellular Aβ, but not extracellular Aβ, have been reported to trigger ER stress in neurons by releasing ER calcium into the cytosol via ITPR and RYR.68,81,82 Similar to neurons, Aβ also triggers calcium-dependent ER stress in glial cells, which is indicated to play an important role in Aβ-mediated astrogliosis in vivo.69 Interestingly, although Aβ mutation upregulates the expression level of HSPA5 in neurons, HSPA5 plays a neuroprotective role against ER stress-associated cell death by suppressing oxidative stress and maintaining calcium homeostasis.83 Intriguingly, unlike Aβ, mutations of PSEN1 inhibit major ER stress sensors including HSPA5, EIF2AK3, ERN1 and ATF6, rendering neurons vulnerable to ER stress stimuli.84,85 Thus, the UPR plays a neuroprotective role in AD but can be disrupted by either accumulation of Aβ or PSEN1 mutations, possibly sharing a common pathway—release of calcium from the ER into the cytosol via ITPR and RYR. UPR activation is an early event in the AD brain, which enhances autophagy but not the ubiquitin proteasome system in the phosphorylated EIF2AK3-positive neurons via increasing MAP1LC3B, suggesting that autophagy is the predominant pathway for degradation of Aβ or APP.86 Insufficient autophagic function is also suggested to play a significant role in the progression of AD.87 The study by Pickford et al. demonstrated substantial loss of BECN1 in the midfrontal cortex of AD brains in the disease progression.87 Intriguingly, studies by this group also demonstrated that genetic reduction of BECN1 expression results in increased deposition of Aβ deposition and synaptic loss and, reciprocally, increasing BECN1 expression through administration of lentivirus alleviates amyloid pathology in the transgenic AD mouse model.87 In addition to neurons, loss of BECN1 in microglia, leading to impaired phagocytic clearance, is also a contributing factor for exacerbated Aβ accumulation.88 The presence of Aβ also triggers inflammatory responses in microglia.89 Phagocytosis of Aβ crystals by microglia disrupts the integrity of lysosome and the release lysosomal proteolytic enzyme CTSB (cathepsin B) activates NLRP3 (NLR family, pyrin domain containing 3) inflammasomes. This ultimately culminates in activation of the IL1B (interleukin 1, β) pathway with subsequent generation of proinflammatory and neurotoxic factors.89 Reciprocally, enhancing autophagic activity by genetic deletion of CSTB (cystatin B [stefin B]; an endogenous inhibitor of lysosomal cysteine proteases) or temsirolimus (a drug used for the treatment of renal cell carcinoma) reduces Aβ deposition and exerts protective effects in both cellular and mouse models of AD (Fig. 4).90,91 In summary, dysregulated UPR and impaired autophagy fail to eliminate pathogenic accumulation of abnormal proteins, resulting thereby in increased symptomatology of AD. Targeting these pathways could thus have therapeutic relevance for the treatment of AD.
Figure 4.
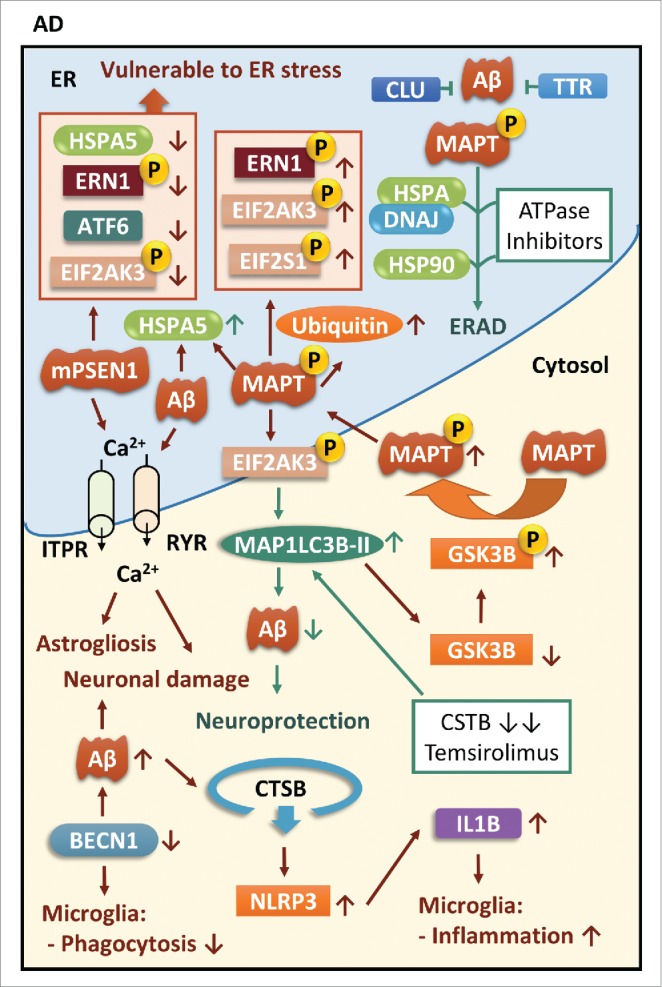
ER stress and autophagy in AD. Both mPSEN1 (mutant PSEN1) and Aβ induce calcium-dependent ER stress, resulting in astrogliosis and neuronal injury. Loss of BECN1 not only contributes to neuronal damage but also debilitates the phagocytic function and induces inflammatory responses in microglia. Interestingly, mPSEN1 renders neurons vulnerable to ER stress by suppressing major UPR mediators while MAPT protein enhances its phosphorylation through activation of UPR mediators, especially EIF2AK3. The EIF2AK3-dependent autophagy pathway is crucial for degradation of Aβ, and autophagic activities can be also enhanced by genetic deletion of CSTB/cystatin B or treatment with temsirolimus. However, autophagy mediated by EIF2AK3 signaling also activates GSK3B by removing its inactive form resulting in subsequent phosphorylation and aggregation of MAPT protein. Moreover, ATPase inhibitors for HSPA/DNAJ complexes and HSP90 multi-component complexes facilitate the triage of MAPT toward the ERAD pathway. CLU and TTR are able to bind to aggregated proteins to prevent their aggregation.
In addition to Aβ accumulation, there is extensive evidence on the aggregation of yet another axonal cytosolic protein, the MAPT that is found hyperphosphorylated as in the brains of patients with AD, frontotemporal dementia (FTD), progressive supranuclear palsy (PSP), corticobasal degeneration, primary age-related tauopathy (PART) and traumatic brain injury (TBI).92-95 Mutations in the gene encoding MAPT are most commonly associated with tauopathies such as FTD, PSP and corticobasal degeneration, whereas other genetic risk factors such as mutations in C9orf72 (chromosome 9 open reading frame 72), GRN (granulin), the gene encoding VCP (valosin containing protein) and CHMP2B (charged multivesicular body protein 2B) affecting UPR and autophagy, play an essential role in frontotemporal lobar degeneration (FTLD) and AD.96 Whereas the neurodegenerative pathology of tauopathies has been well recognized, the relevant molecular mechanism(s) underlying the disease processes remain poorly understood. Increasing evidence suggests that aggregated MAPT can lead to ER stress and activation of the UPR as an early event in tauopathies such as AD, FTD and PSP (Fig. 4).80,97 In AD postmortem brain tissues, activation of UPR mediators such as EIF2AK3, ERN1 and HSPA5 is observed in the pretangled neurons containing diffusely distributed phosphorylated MAPT, but not in the neurons with densely aggregated neurofibrillary tangles.98,99 Direct evidence of MAPT-dependent UPR is found in sporadic tauopathies lacking Aβ.80 For example, a report by Nijholt et al. demonstrated that UPR activation is only observed in the MAPT subtype of FTLD, but not in other aggregate subtypes of FTLD.80 Elegant work by Abisambra et al. using MAPT transgenic mouse brains has also shown that MAPT increases the levels of ubiquitinated proteins in the brain and initiates the activation of the UPR as evidenced by increased levels of phosphorylated EIF2AK3. Findings from this group led to the suggestion that the presence of MAPT interferes with ER protein quality control.93 GSK3B (glycogen synthase kinase 3 β) is an essential kinase for the phosphorylation of MAPT that plays an important role in MAPT aggregation.100 Interestingly, the study by Nijholt et al. has shown that the EIF2AK3 pathway activates GSK3B through selective removal of inactive GSK3B via the autophagy-lysosomal pathway.100 Moreover, findings by van der Harg et al. have also reported that failure to restore the metabolic homeostasis resulting in prolonged UPR activation and MAPT phosphorylation could likely be a contributing factor to AD pathogenesis. Furthermore, these authors also demonstrated the role of UPR-mediated MAPT phosphorylation as part of an adaptive response to metabolic stress, culminating ultimately into the pathogenesis of AD.101
Hyperphosphorylated MAPT is a target client that is recognized by the chaperone system and is processed initially by the HSPA/DNAJ (heat shock 70 kDa proteins/DnaJ (Hsp40) homolog) complex followed by an intermediate complex with HSP90 (heat shock 90 kDa protein) and STIP1 (stress-induced phosphoprotein 1).102 Intriguingly, HSP90 regulates the folding or degradation of its client proteins by forming multicomponent complexes with other chaperones. HSP90 has the ability to cycle between the different multichaperone complexes, from a complex favoring the refolding of abnormal proteins in the presence of ATP to a complex that targets proteins for degradation (in the presence of ADP).103 Inhibitors for heat shock proteins convert the HSP90 complexes from a catalyst for protein folding into one that promotes protein degradation.102 These inhibitors can thus be harnessed to convert the protein folding form to a protein degradation pathway, thereby allowing for the removal of abnormal phosphorylated MAPT. In keeping with this differential function of the chaperone system on toxicity, the extracellular chaperone CLU (clusterin) exhibits paradoxical roles in the pathology of AD depending on the concentration of misfolded proteins.104 On the one hand, under normal conditions CLU can stabilize extracellular misfolded proteins via its binding to them and subsequently guiding them to specific cell surface receptors for uptake and degradation.105 Such extracellular chaperones and their receptors form a critical component of the quality control system to protect the host against abnormal, aggregated proteins such as Aβ.105 On the other hand, there is also evidence that in the presence of high levels of misfolded proteins, CLU can actually exacerbate the pathology.104 Similar to CLU, TTR (transthyretin) can also regulate aggregated protein quality control by sequestering soluble Aβ and thereby preventing the formation of amyloid fibers.106
ER stress in AD is commonly observed in both Aβ and PSEN1 mutation and MAPT pathology. Interestingly, Aβ-induced calcium-dependent ER stress is suggested to be a conserved pathway that both neurons and astrocytes share in common.68,69 Loss of BECN1 impairs autophagy in AD brains and leads to synaptic injury in neurons, and phagocytic deficits in microglia, suggesting that autophagy is a conserved pathway and can affect the functioning of both neurons and glial cells.87,88 Aβ-mediated microglial inflammation further suggests a crucial role of glial cells in modulating AD inflammatory trajectory.89 However, the UPR and autophagy play a protective role in neurotoxicity mediated by Aβ and PSEN1 mutations83,85 but not in MAPT pathology.100 The phosphorylated EIF2AK3-dependent autophagy pathway is considered as the major degradation pathway for Aβ,86 but is also capable of activating GSK3B by degrading its inactive forms, leading to MAPT phosphorylation and aggregation.100 Thus, intervention in the UPR and autophagy in AD can be considered as potential therapeutic targets; however, the genetic profiling and pathology of AD patients should be carefully investigated when designing potential therapeutic strategies based on the perspective of the UPR and autophagy.
Prion diseases
Prion diseases, also known as transmissible fatal neurodegenerative diseases, are characterized by neuronal loss, synaptodendritic degeneration, spongiosis, gliosis and deposition of insoluble PRNP (prion protein) aggregates in the brain.107-109 PRNP exists as a native form of prion protein PRNPC in the noninfected brain, and as a protease-resistant form of prion protein PRNPSC in the prion diseased brain.110 In prion diseases, PRNPC is converted into PRNPSC that is rich in β-sheet and prone to intracellular accumulation, resulting in ER stress.110 The conversion also depletes PRNPC in neurons and stimulates synthesis of PRNPC for further cycles of conversion, thereby exacerbating ER stress.109 A similar template for conformational change is also observed in other prion-like disease-specific proteins including MAPT, Aβ and SNCA.111-113
Accumulating evidence suggests active ER stress responses in prion diseases both in vitro and in vivo (Fig. 5).114-117 Transcriptional analysis of brainstem tissues from 100 confirmed cases of prion diseases in cattle found increased expression of ER chaperones, ERAD components, regulators of ER calcium storage and mediators of ER stress-induced apoptosis.118 Upregulation of ER chaperones HSPA5, PDIA3 (protein disulfide isomerase family A, member 3), and HSP90B1 (heat shock protein 90 kDa β [Grp94], member 1) has been reported in N2a neuroblastoma cells infected with scrapie in vitro and scrapie rodents models in vivo.114-117 Prion proteins also increase the release of ER calcium into cytosol, induce inhibition of global protein synthesis, and cause dysregulation of autophagy. PRNP can induce ER stress-dependent apoptosis through disruption of calcium homeostasis via ITPR and RYR in primary neurons.119 Although overexpression of UPR mediators decreases PRNP aggregation in cell lines in vitro,109,120 the same phenomenon cannot be fully extrapolated in neurons in XBP1 conditional knockout mice.121,122 Studies by Mallucci and Moreno demonstrated that in response to accumulation of PRNP, global protein synthesis in prion-diseased mice is inhibited by phosphorylation of EIF2S1 leading to synaptic failure and neuronal loss.123 This group further demonstrated that targeting the upstream ER stress molecule EIF2S1 is able to reverse cognitive deficits and prevent clinical signs of prion disease at the preclinical stage.124 Intriguingly, the ER-resident protein RTN3 (reticulon 3) that can be activated under ER stress is reported to play an important role in the cytosolic PRNPSC aggregation in N2a cells infected with the prion protein fragment PRNP (90–231). RTN3 restrains autophagic activity by enhancing interaction between BCL2 and BECN1, and knockdown of RTN3 not only promotes the autophagic clearance of PRNPSC, but alleviates the ER stress and improves cellular survival.125
Figure 5.
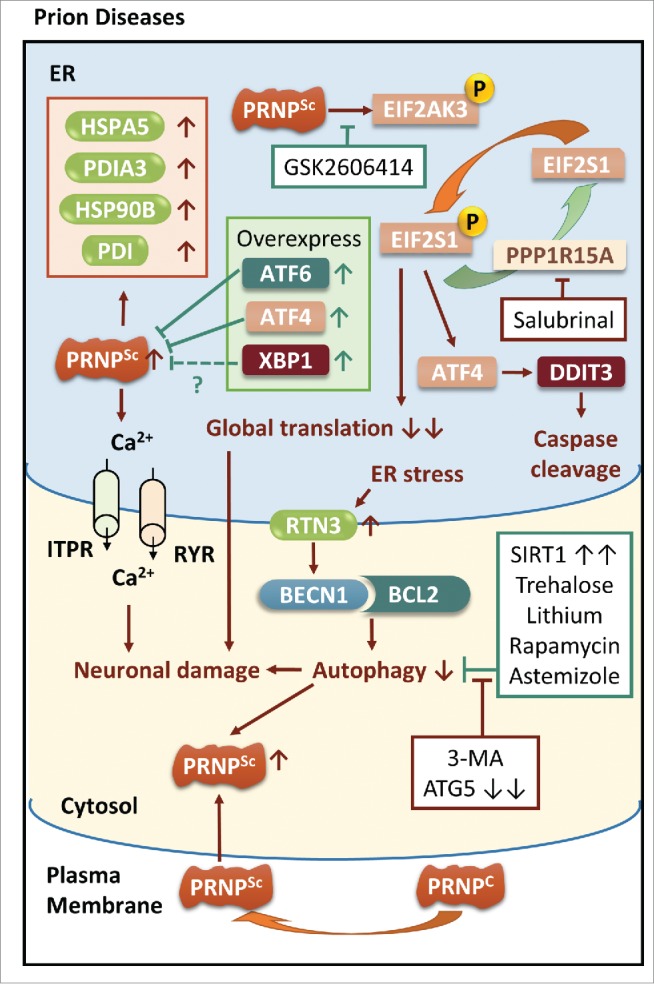
ER stress and autophagy in prion diseases. ER stress induced by disruption of intracellular calcium homeostasis is also observed in prion diseases. Aggregation of PRNP upregulates ER chaperones and activates EIF2AK3-EIF2S1 signaling, resulting in synaptic failure and activation of a caspase cascade, which can be rescued by the EIF2AK3 inhibitor GSK2606414 but not the PPP1R15A inhibitor salubrinal. Overexpression of UPR mediators such as ATF6, ATF4 and XBP1 reduces PRNP aggregation in vitro, but it is not fully extrapolated in XBP1 conditional knockout mice. RTN3 activated by ER stress restrains autophagy by prohibiting dissociation of BCL2 and BECN1. Autophagic activities can be enhanced by overexpression of SIRT1 and autophagic inducers, and can be inhibited by either genetic silencing of ATG5 or in the presence of the autophagic inhibitor 3-MA.
It has been suggested that insufficient cellular clearance in prion diseases can be partially related to dysregulation of autophagy (Fig. 5).126 Intracellular autophagic vacuoles/autophagosomes of different sizes are found located in neuronal perikarya, neurites and synapses in various prion-like diseases.29 It has been reported that adenoviral-mediated SIRT1 (sirtuin 1) overexpression enhances autophagic activity to protect human neuroblastoma SH-SY5Y cells from neurotoxicity, mitochondrial injury and an intrinsic apoptosis pathway that are meditated by the prion peptide PRNP(106–126). Knocking down ATG5 abrogates this effect.127 The autophagic inducers trehalose and lithium diminish accumulation of PRNPSC in ScN2a cells (N2a cells persistently infected with prion strain RML) and this protective effect can be blocked either with 3-methyladenine (3-MA; an inhibitor of phosphatidylinositol 3-kinase that inhibits cellular autophagy) or by genetically silencing ATG5.128,129 Intraperitoneal injection of the classic autophagic inducer rapamycin in a transgenic (PRNP-A116V) Gerstmann-Sträussler-Scheinker mouse model was also demonstrated to decrease PRNPSC accumulation, induce a dose-dependent delay in disease onset, alleviate the disease symptoms and extend the life span.130 Using a PRNP-FRET-enabled high-throughput assay to screen for compounds that decrease PRNP expression, a recent study on CJD demonstrated that another autophagic inducer, astemizole (originally used to treat chronic seasonal allergic rhinitis), prolongs the survival time of prion-infected mice, suggesting thereby the possibility of therapeutic use of astemizole in CJD.131
Taken together, prion protein induces the release of ER calcium into cytosol, induces inhibition of global protein synthesis and causes dysregulation of autophagy. Therefore, maintaining the intracellular calcium homeostasis, counteracting the global translation repression and enhancing autophagic activities can be considered as promising therapeutic strategies for combating prion diseases.
Parkinson disease
PD is the second most common neurodegenerative disorder afflicting the elderly and is usually characterized by hypokinesia, rigidity, and tremor. The extensive degeneration of dopaminergic neurons in the substantia nigra pars compacta accounts for the majority of the symptomatology.132,133 The salient pathological features of PD includes accumulation of Lewy bodies, intracellular inclusion bodies containing diffuse deposits of misfolded SNCA protein.67 Widely accepted PD models mimic apoptosis of dopaminergic neurons through administration of PD-related insults such as 6-hydroxydopamine (6-OHDA), 1-methyl-4-phenyl-pyridinium (MPP+), 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and rotenone.134,135
In PD, phosphorylated forms of the UPR activation marker EIF2AK3 colocalize with the SNCA protein in the human substantia nigra, indicating thereby induction of ER stress and its association with the aggregated SNCA.136 Similar findings have been demonstrated in chemically induced experimental models of PD. Following exposure to 6-OHDA, there is increased phosphorylation of EIF2AK3 and ERN1 in neurons, furthermore, sympathetic neurons from EIF2AK3 null mice exhibit extended sensitivity to 6-OHDA.134 In addition to phosphorylated EIF2AK3, exogenous XBP1 protein also protects dopaminergic neuroblastoma SK-N-SH cells from MPP+ and proteasome inhibitors, and additionally, adenoviral XBP1s suppresses MPTP-mediated degeneration of dopaminergic neurons in mouse models.135 The role of EIF2S1 in the phenotype of α-synucleinopathy, however, remains unclear. Although phosphorylated EIF2S1 is present in the substantia nigra of PD individuals,136 it is not found to be activated in the α-synucleinopathy mouse model (A53TαS Tg).137 Interestingly, DDIT3, a pro-apoptotic mediator in the final stage of ER stress, is a mediator of neuronal cell death in the rat substantia nigra induced by 6-OHDA but not MPTP. Upregulation of DDIT3 is observed in dopaminergic neuronal cell death mediated by both 6-OHDA and MPTP; however, a null mutation of DDIT3 in vivo protects mice from only 6-OHDA-induced apoptosis (Fig. 6).138 Accumulating evidence thus suggests that UPR regulators such as EIF2AK3 and XBP1s play important roles in neuroprotection in PD, however, more evidence is needed to clarify the roles of EIF2S1 and ERN1 in this process. Although DDIT3 is usually associated with ER stress-associated apoptosis, it appears that the nature of the toxic stimulus determines whether DDIT3 will induce neuronal apoptosis in PD.
Figure 6.
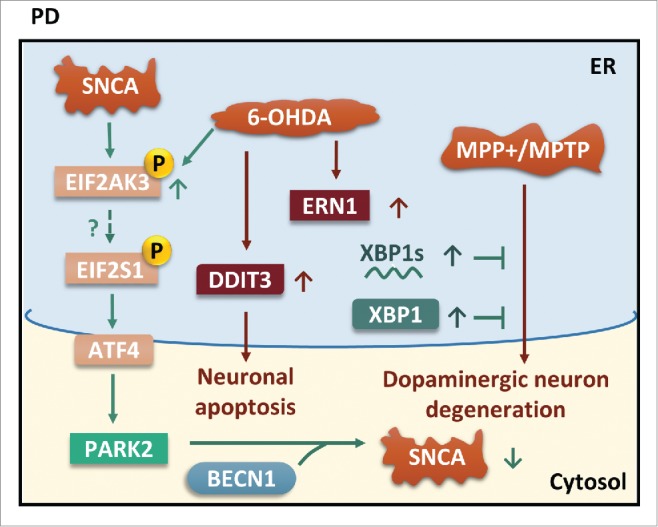
ER stress and autophagy in PD. Phosphorylation of EIF2AK3 can be induced by both SNCA and 6-OHDA. Although phosphorylated EIF2S1 is seen in PD brain but not in the α-synucleinopathy mouse model, EIF2S1-ATF4 signaling upregulates PARK2. Interaction of PARK2 and BECN1 enhances autophagic clearance of SNCA. Moreover, exogenous XBP1s and XBP1 rescue MPP+/MPTP-mediated dopaminergic neuron degeneration.
Chaperone-mediated autophagy is crucial for the clearance of soluble SNCA protein in cultured dopaminergic neurons because the SNCA protein contains a pentapeptide sequence (95VKKDQ99), and its degradation can be blocked by the lysosomal proteolysis inhibitor ammonium chloride.139 PARK2 (parkin RBR E3 ubiquitin protein ligase) is a ubiquitin ligase reported to localize in mitochondria,140 mediate the engulfment of mitochondria by phagophores, selectively eliminate dysfunctional mitochondria141 and rescue the inhibition of mitochondrial fusion induced by SNCA.142 Dopaminergic neuroblastoma-derived SH-SY5Y cells transfected with PARK2 are more resistant to cell death induced by tunicamycin (N-glycosylation inhibitor)-mediated ER stress.143 Silencing EIF2S1 and its downstream target ATF4 reduces PARK2 expression in mouse primary cortical neurons and disrupts mitophagic function, leading to increasing neuronal cell death.144 Interestingly, although PARK2 colocalizes with BECN1 in human mid-brain, it has decreased interaction with BECN1 in sporadic PD cases. Furthermore, there was increased secretion of SNCA from the brain into the blood in PARK2-deficient mice (Fig. 6).145
Different PD insults all trigger ER stress and activate the UPR that is suggested to play a neuroprotective role.134,135 PARK2 is a critical neuroprotective molecule bridging the UPR and autophagy in PD and thus can be considered as a promising target for therapeutic purposes.144 Although upregulation of DDIT3 is a common feature in different PD models, DDIT3 mediates neuronal apoptosis in the presence of 6-OHDA but not in the presence of MPTP,138 an effect that is dependent on the nature of the PD stimulants. Due to the fact that phosphorylated EIF2S1 is present in human PD brains but not in the A53TαS transgenic mouse model, more evidence is needed to clarify the roles of EIF2S1 in this process.136,137
Amyotrophic lateral sclerosis
ALS, often referred to as “Lou Gehrig's disease,” is a progressive and fatal adult-onset neurodegenerative disorder with a selective degeneration and loss of motor neurons, characterized by muscle weakness, atrophy, spasticity, paralysis and premature death.146 Whereas most of the ALS cases are sporadic ALS (sALS), nearly 10% of ALS cases are familial ALS (fALS).146,147 It has been reported that abnormal aggregation of mutant (m)SOD1 (superoxide dismutase 1, soluble) accounts for about 20% of fALS cases6,148 and 1–7% of sALS cases. Additionally, mutations in genes, such as those encoding TARDBP (TAR DNA binding protein), FUS/TLS (FUS RNA binding protein), and OPTN (optineurin) also are reported to be a causative factor of ALS.149-151
Various reports suggest that aggregation of mSOD1 results in prolonged ER stress leading to the loss of motor neurons through the activation of pro-apoptotic factors such as CASP3, ATF3, DDIT3 and BH3-only proteins, in the late stages of SOD1G93A ALS mouse models.152-154 An increase in HSPA5,193 PDI,6 EIF2S1,155 ATF4 and XBP1s156 was also reported in the post-mortem brains of ALS patients. The chronic ER stress in fALS patients is likely to be associated with the suppression of compensatory autophagic clearance of mSOD1. One of the ER-stress regulators, XBP1, suppresses autophagic clearance of mSOD1 possibly via inhibition of MTORC1 (Fig. 7).156 Targeting inhibition of XBP1 and enhancing autophagic clearance can be considered a promising therapeutic strategy for clearing mSOD1 in ALS.
Figure 7.
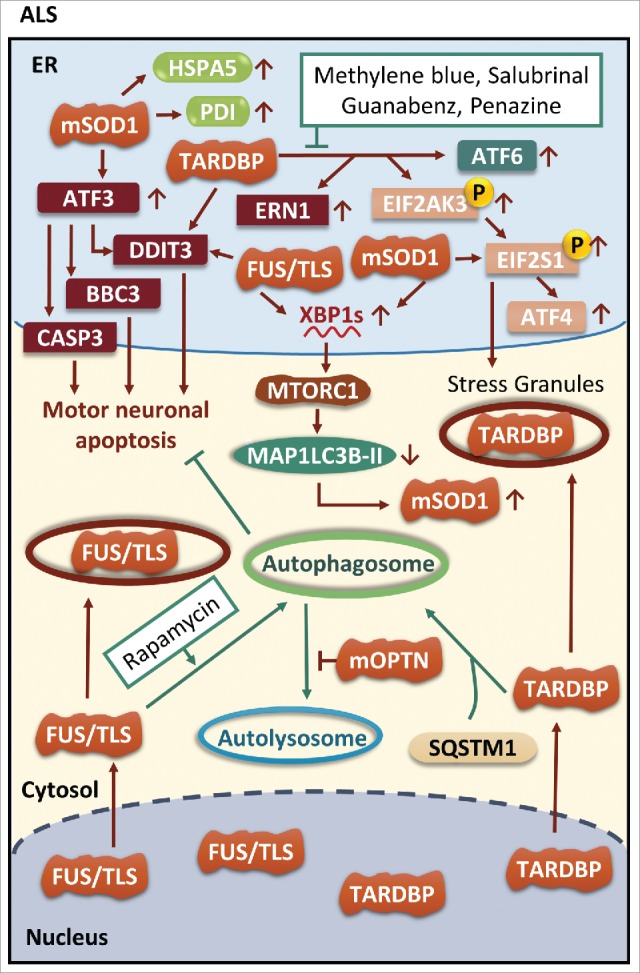
ER stress and autophagy in ALS. Different mutations such as those in SOD1, TARDBP and FUS are all able to induce neuronal apoptosis through activation of DDIT3. Both TARDBP and FUS can be included in stress granules in the cytoplasm. Phosphorylation of EIF2S1 mediated by TARDBP and mSOD1 (mutant SOD1) facilitates stress granules formation. Interestingly, either FUS or mSOD1 increases expression of XBP1s but activated XBP1s inhibits autophagic clearance of mSOD1. Moreover, mOPTN (mutant OPTN) also hampers autophagy by inhibiting fusion of autophagosomes with lysosomes. Enhanced autophagic activity induced by autophagic inducers leads to clearance of FUS and TARDBP. Interestingly, SQSTM1 plays a crucial role in packaging TARDBP into autophagosomes.
TARDBP, a nuclear RNA-binding protein that plays a critical role in RNA processing as well as microRNA biogenesis modulation, continually shuttles between the nucleus and the cytoplasm.157,158 Under normal conditions, TARDBP is processed and degraded by both autophagy and the ubiquitin-proteasome systems. When TARDBP is excluded from the nucleus it forms cytoplasmic aggregates in the brain and spinal cord of ALS patients.159 Over 30 mutations have been reported in the TARDBP gene to cause fALS; such mutations endorse the cytoplasmic mislocalization and accumulation of TARDBP leading to cytoplasmic TARDBP toxicity.159 The aggregated TARDBP also triggers ER stress both in vitro and in vivo.160,161 The aggregated TARDBP can also be phosphorylated, ubiquitinated and degraded by the ubiquitin-proteasome system and autophagy via degradation target proteins such as UBQLN1 (ubiquilin 1) and UBQLN2 (ubiquilin 2), and SQSTM1 (sequestosome 1), respectively (Fig. 7).162,163 Indeed, various studies demonstrated a failure of TARDBP clearance in human ALS. Additionally, in vitro studies demonstrated that the primary mutation of TARDBP confers resistance to its degradation.164
In addition to TARDBP, another RNA metabolism-associated protein, FUS, has been identified as a key constituent within the intracellular protein inclusions in the autopsy tissues of ALS.164 FUS is predominantly restricted in the nucleus with little distribution in the cytoplasm of motor neurons; however, point mutations in the C terminus of FUS cause neuronal cytoplasmic FUS-positive inclusions in ALS patients.165 Hence, FUS reshuffles from the nucleus to the cytoplasm and builds inclusions in affected motor neurons, where it initiates ER stress in motor neuron-like cells expressing mutant FUS. The ER chaperone PDI was observed to colocalize with the mutant FUS in motor-neuron-like NSC-34 cells as well as in the FUS inclusions in human ALS lumbar spinal cords, in both sALS and mutant FUS-linked fALS tissues.161 It was also reported that the point mutation in FUS diminishes the release of FUS from accumulating stress granules in cultured neurons leading to defective autophagy clearance, thereby indicating that activation of autophagic signaling could be a therapeutic approach for FUS mutation-associated ALS pathologies (Fig. 7).166
OPTN is an autophagy receptor protein, and mutations in OPTN have recently been reported as a causative factor for ALS and glaucoma.167 OPTN is primarily involved in autophagosome maturation by fusion with endosomes and its MYO6/myosin VI binding partner.168 OPTN is also considered as a neuroprotective protein, whose accumulation is normally cleared by the proteasome degradation pathway. The OPTN E478G mutation, a causative mutation for ALS, is reported to effectively impair autophagosome recruitment, which further confirms the involvement of OPTN mutation and defective autophagy in the pathology of ALS (Fig. 7).169 Thus, reducing ER stress and maintaining active autophagy can be considered as promising therapeutic targets for ALS.
HIV associated-neurocognitive disorders
In the era of combined anti-retrovirus therapy, the prevalence of HAND is actually on a rise due to a significant increase in the longevity of the infected patients.170,171 Mechanisms underlying HAND pathogenesis, however, remain unclear. Recent emerging studies provide ample evidence indicating that early HIV proteins that are not affected by combined anti-retroviral therapy and that can deregulate the ER stress pathways and autophagy activity play a crucial role in the development of HAND.
Tat (transactivator of transcription), one of the key HIV proteins necessary for virus replication, is capable of activating ER stress pathways in various types of cells in the brain.168-169 Studies from our lab have demonstrated that Tat-induced apoptosis of human brain microvascular endothelial cells involves ER stress activation and mitochondrial dysfunction.172 Tat also upregulates ER stress markers including HSPA5, ATF6 and phosphorylated EIF2S1 in both cortical neurons and astrocytes.173 The fluctuation of cytosolic Ca2+ concentration is the upstream signal for ER activation as evidenced by the depletion of Ca2+ from the ER to the cytoplasm via ITPR and RYR.5,7 Besides Tat, the virus envelope protein gp120 also modulate ER stress in neurons, which in turn, has been suggested to correlate with neuronal injury.174 Activation of ER stress contributes to increased permeability of the blood-brain barrier, and the activation of microglia and astrocytes, thereby resulting in enhanced expression of pro-inflammatory factors, and impairment of brain glutamate homeostasis, leading ultimately to exacerbated brain inflammation, a hallmark feature of HAND pathogenesis.172,173,175 In addition to regulating ER stress activity, HIV proteins also modulate autophagy levels as demonstrated by autophagy dysfunction in HIV brains.170,176-178 HIV gp120 and Nef have the ability to affect autophagy levels by targeting various steps of the autophagy pathway.170,179 For example, BECN1 levels are altered in HIV gp120 transgenic mice,166 whereas Nef blocks the autophagosome maturation step via interaction with BECN1.179 Through the interaction of Nef and BECN1, HIV can decrease autophagic influx in infected cells thereby allowing for its replication.179 HIV Tat also plays a key role in autophagy during the process of HIV infection. Tat suppresses autophagy in macrophages180 and bystander monocytes.181 Noticeably, Tat can also alter neuronal autophagy levels by blocking autophagosome fusion with the lysosome by disrupting lysosome membrane integrity, which is critical for the maintenance of normal lysosomal functioning (Fig. 8).182,183 Autophagy dysfunction in HIV patients has also been linked with aging. Intriguingly, young (<50 y) vs. aging (> 50 y) HIV patients reveal opposite trends in autophagic activity, with increased autophagy observed in younger compared to older HIV-infected individuals. This phenomenon suggests that different autophagy-targeting therapeutic strategies will be needed for young versus the old HIV-infected patients to achieve a beneficial clinical outcome.
Figure 8.
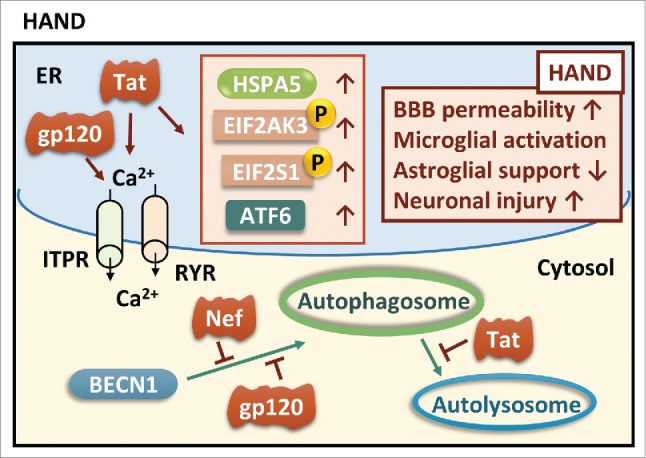
ER stress and autophagy in HAND. Although no direct evidence is found between UPR and autophagy in HAND, it has been suggested that HIV viral proteins induce ER stress and inhibit autophagic clearance. Both gp120 and Tat induce calcium-dependent ER stress, and Tat increases expression of major UPR mediators such as HSPA5, ATF6, phosphorylated EIF2AK3 and EIF2S1. Viral proteins such as Nef and gp120 hamper autophagosome formation initiated by BECN1 while Tat hinders the fusion of lysosomes with autophagosomes. BBB, blood-brain barrier.
Taken together, HIV proteins can affect both ER stress and autophagy activity to contribute to the development of HAND. In most cases, ER stress is the upstream signal for autophagy induction, and autophagy in turn can modulate the tone of ER stress via a feedback mechanism. In HAND, it is still not clear whether HIV proteins can sequentially first upregulate the levels of ER stress followed by induction of autophagy or if they cause the induction of both ER stress and autophagy simultaneously, or whether autophagy upregulation precedes ER stress. Additionally, unlike other neurodegeneration diseases, there is no clear evidence as to whether targeting the autophagy pathway can be considered an effective alternative therapeutic approach to ameliorate the symptoms of HAND. Further investigation is warranted in this area.
Conclusions and future directions
ER stress in neurodegenerative disorders can be triggered by either pathogenic accumulation of misfolded proteins such as Aβ in AD,77,78 PRNPSC in prion diseases,110 SNCA in PD92 and mSOD1 in fALS,152 or depletion of ER calcium stores via the ITPR and RYR channels in HAND5,7 and AD.68 Intriguingly, Aβ-induced calcium-dependent ER stress is also suggested to be a conserved pathway that both neurons and astrocytes share in common that leads to neuronal damage and disruption of trophic support from astrocytes.68,69 Pathogenic accumulation of misfolded proteins leading to activation of the UPR, results in upregulation of mediators such as HSPA5, and the phosphorylated forms of EIF2AK3 and EIF2S1, which have been widely used as indicators of ER stress in various neurodegenerative disorders.5,136,155,184 Generic cells such as HEK293 are powerful tools for studying ER stress. Using a novel methodology that activates the transcription factors XBP1 and/or ATF6 orthogonally by small molecules in the same cell, Shoulders et al. were able to demonstrate remodeling of the ER proteostasis network via the UPR transcriptional programs at the molecular level independent of stress.185 Although most of the experiments in non-neuronal cells can be extrapolatable to neurons in vivo, exceptions do exist and therefore rodent models and human brain tissues are indispensable for investigating neurodegenerative disorders. For instance, overexpression of UPR mediators such as ATF6, ATF4 and XBP1 decrease PRNP aggregation in cell lines in vitro,109,120 but the same phenomenon cannot be fully extrapolated to neurons in XBP1 conditional knockout mice.121,122 Under most circumstances, upregulated expression of UPR mediators is a pathway for protecting neurons against ER stress; however, it must be cautioned that not all upregulated UPR mediators confer neuroprotection. For example, in fALS upregulation of XBP1 mRNA decreases MAP1LC3B-II, thereby inhibiting autophagic clearance of mSOD1.156 In prion diseases, the EIF2S1-dependent prolonged global suppression of protein translation leads to synaptic failure and neuronal loss.123,186-191 In tauopathies, activation of GSK3B by the EIF2AK3 pathway results in phosphorylation of MAPT in the lysosome.86,100 These UPR mediators can thus be envisioned as potential therapeutic targets. Depending on the nature of the toxic stimuli and the signaling pathway, both upregulation and downregulation of UPR mediators can direct the course of disease pathogenesis. For instance, in AD, Aβ induces expression of the ER chaperone HSPA5 to rescue abnormal accumulation of Aβ, whereas a neuronal cell line stably expressing mutant PSEN1 is more vulnerable to ER stress stimuli since mutant PSEN1 inhibits the activity of ER stress sensors HSPA5, ERN1 and ATF6.192,193 Furthermore, maintaining a constant level of ER calcium is essential for the normal functioning of ER-resident molecular chaperones. There is also emerging evidence that pharmacological inhibitors for ITPR and RYR channels can rescue ER-stress associated neuronal apoptosis in HAND7 and AD,68 indicating thereby an important role for loss of the ER calcium store in the neurodegenerative processes.
Emerging evidence suggests that autophagy serves as an intrinsic compensatory mechanism for the UPR to regulate protein homeostasis and clear cell debris and misfolded proteins in neurodegenerative disorders. In the brains of AD patients, increasing MAP1LC3B levels have been observed in the phosphorylated-EIF2AK3-positive neurons.86 The ubiquitin ligase PARK2 plays a vital role in the interplay between ER stress and autophagy. Decreased interaction between PARK2 and BECN1 is involved in increased secretion of SNCA from the brain into the blood in the mouse model of PD.145 Furthermore, dopaminergic neuroblastoma-derived SH-SY5Y cells transfected with PARK2 are more resistant to apoptosis mediated by ER stress.143 Some pathogenic proteins such as HIV Tat can escape autophagic clearance via interference with the endolysosomal pH, inactivation of endolysosomal enzymes and inhibition of autophagy.194 Interestingly, UPR mediators such as XBP1s can also hamper autophagic activity by decreasing the expression levels of MAP1LC3B-II in fALS.156 It is possible that neurons and non-neuronal cells such as microglia share a common pathway in autophagy. For instance, loss of the autophagy initiator BECN1 increases intracellular deposition of Aβ in neurons87 and decreases phagocytic clearing, which exacerbates Aβ in microglial cells.88 Moreover, the dysregulation of autophagy also contributes to neuroinflammation mediated by glial cells. With the loss of BECN1, the capacity of handling Aβ is diminished in microglia88 and aggregated Aβ damages lysosome, releases the lysosomal protease CTSB and activates the IL1B pathway, resulting in a microglia-dependent inflammatory response.89 Hence, glial cells play an indispensable role in mediating neuroinflammation in neurodegenerative disorders. With the discovery of lymphatic vessels in the CNS,195 there will be more emerging critical evidence of neuroinflammation mediated by non-neuronal cells in modulating the trajectory of neurodegeneration.
Although both in vitro and in vivo approaches have advanced our understanding of the molecular pathways underlying ER stress and autophagy, detailed intrinsic molecular mechanisms regulating these processes still remain to be elucidated. Taken together, available evidence suggests a key role for dysregulation of these pathways in various neurodegenerative disorders. Our knowledge is far from complete, particularly in neurons with regard to cell death/dysfunction. Given the diversity of neuronal populations and neurodegenerative disorders, this represents a challenge. The advent and widespread applications of new imaging, gene sequencing and mouse genome engineering tools will contribute significantly to our efforts to dissect the signaling pathways underlying the UPR and autophagy both in development and disease states.
Abbreviations
- 3-MA
3-methyladenine
- 6-OHDA
6-hydroxydopamine
- AD
Alzheimer disease
- ALS
amyotrophic lateral sclerosis
- AMPK
AMP-activated protein kinase
- APP
amyloid β (A4) precursor protein
- ARHGAP26
Rho GTPase activating protein 26
- ATF3
activating transcription factor 3
- ATF4
activating transcription factor 4
- ATF6
activating transcription factor 6
- ATG5
autophagy-related 5
- ATG12
autophagy-related 12
- Aβ
β-amyloid
- BBC3
BCL2 binding component 3
- BCL2
B-cell CLL/lymphoma 2
- BECN1
Beclin 1, autophagy related
- CALR
calreticulin
- CASP3
caspase 3, apoptosis-related cysteine peptidase
- CJD
Creutzfeldt-Jakob disease
- CLU
clusterin
- CSTB
cystatin B (stefin B)
- CTSB
cathepsin B
- DAPK1
death-associated protein kinase 1
- DDIT3
DNA-damage-inducible transcript 3
- DNAJ/HSP40
DnaJ (Hsp40) homolog
- EHD1
EH-domain containing 1
- EIF2AK3
eukaryotic translation initiation factor 2-α kinase 3
- EIF2S1
eukaryotic translation initiation factor 2, subunit 1 α 35kDa
- ER
endoplasmic reticulum
- ERAD
ER-associated degradation
- ERN1
endoplasmic reticulum to nucleus signaling 1
- fALS
familial amyotrophic lateral sclerosis
- FOXO1
forkhead box O1
- FTD
frontotemporal dementia
- FTLD
frontotemporal lobar degeneration
- FUS/TLS
FUS RNA binding protein
- GSK3B
glycogen synthase kinase 3 β
- HAND
HIV-associated neurocognitive disorders
- HD
Huntington disease
- HSF1
heat shock transcription factor 1
- HSP90
heat shock 90 kDa protein
- HSP90B1
heat shock protein 90 kDa β (Grp94), member 1
- HSPA
heat shock 70 kDa protein
- HSPA5
heat shock 70 kDa protein 5 (glucose-regulated protein, 78kDa)
- HTT
huntingtin
- IL1B
interleukin 1 β
- ITPR
inositol-1,4,5-trisphosphate receptor
- MAP1LC3B
microtubule-associated protein 1 light chain 3 β
- MAPK8
mitogen-activated protein kinase 8
- MAPT/tau
microtubule-associated protein tau
- MBTPS1
membrane-bound transcription factor peptidase, site 1
- MBTPS2
membrane-bound transcription factor peptidase, site 2
- MICALL1
MICAL-like 1
- mOPTN
mutant OPTN
- MPP+
1-methyl-4-phenyl-pyridinium
- mPSEN1
mutant PSEN1
- MPTP
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- mSOD1
mutant SOD1
- MTORC1
mechanistic target of rapamycin (serine/threonine kinase) complex 1
- NLRP3
NLR family, pyrin domain containing 3
- OPTN
optineurin
- PARK2
parkin RBR E3 ubiquitin protein ligase
- PARK7
parkinson protein 7
- PART
primary age-related tauopathy
- PD
Parkinson disease
- PDI
protein disulfide isomerase
- PDIA3
protein disulfide isomerase family A, member 3
- PE
phosphatidylethanolamine
- PINK1
PTEN induced putative kinase 1
- PPP1
protein phosphatase 1
- PPP1R15A
protein phosphatase 1, regulatory subunit 15A
- PRNP
prion protein
- PRNPC
native PRNP
- PRNPSC
protease-resistant PRNP
- PSEN1
presenilin 1
- PSEN2
presenilin 2
- PSP
progressive supranuclear palsy
- RTN3
reticulon 3
- RYR
ryanodine receptor
- sALS
sporadic amyotrophic lateral sclerosis
- SIRT1
sirtuin 1
- SNCA/α-synuclein
synuclein, α (non A4 component of amyloid precursor)
- SOD1
superoxide dismutase 1, soluble
- SQSTM1
sequestosome 1
- STIP1
stress-induced phosphoprotein 1
- TARDBP
TAR DNA binding protein
- Tat
HIV-1 transactivator of transcription
- TBI
traumatic brain injury
- TTR
transthyretin
- UBQLN1
ubiquilin 1
- UBQLN2
ubiquilin 2
- ULK1
unc-51 like autophagy activating kinase 1
- UPR
unfolded protein response
- XBP1
X-box binding protein 1
- XBP1s
spliced form of XBP1
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
We would like to acknowledge support from National Institutes of Health (NIH) grants number DA033150, DA035203 and DA036157 (SB).
References
- 1.Buchberger A, Bukau B, Sommer T. Protein quality control in the cytosol and the endoplasmic reticulum: brothers in arms. Mol Cell 2010; 40:238-52; PMID:20965419; http://dx.doi.org/ 10.1016/j.molcel.2010.10.001 [DOI] [PubMed] [Google Scholar]
- 2.Matus S, Lisbona F, Torres M, Leon C, Thielen P, Hetz C. The stress rheostat: an interplay between the unfolded protein response (UPR) and autophagy in neurodegeneration. Curr Mol Med 2008; 8:157-72; PMID:18473817; http://dx.doi.org/ 10.2174/156652408784221324 [DOI] [PubMed] [Google Scholar]
- 3.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell 2011; 147:728-41; PMID:22078875; http://dx.doi.org/ 10.1016/j.cell.2011.10.026 [DOI] [PubMed] [Google Scholar]
- 4.Son JH, Shim JH, Kim KH, Ha JY, Han JY. Neuronal autophagy and neurodegenerative diseases. Exp Mol Med 2012; 44:89-98; PMID:22257884; http://dx.doi.org/ 10.3858/emm.2012.44.2.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Norman JP, Perry SW, Reynolds HM, Kiebala M, De Mesy Bentley KL, Trejo M, Volsky DJ, Maggirwar SB, Dewhurst S, Masliah E, et al.. HIV-1 Tat activates neuronal ryanodine receptors with rapid induction of the unfolded protein response and mitochondrial hyperpolarization. PloS One 2008; 3:e3731; PMID:19009018; http://dx.doi.org/ 10.1371/journal.pone.0003731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Atkin JD, Farg MA, Walker AK, McLean C, Tomas D, Horne MK. Endoplasmic reticulum stress and induction of the unfolded protein response in human sporadic amyotrophic lateral sclerosis. Neurobiol Dis 2008; 30:400-7; PMID:18440237; http://dx.doi.org/ 10.1016/j.nbd.2008.02.009 [DOI] [PubMed] [Google Scholar]
- 7.Haughey NJ, Holden CP, Nath A, Geiger JD. Involvement of inositol 1,4,5-trisphosphate-regulated stores of intracellular calcium in calcium dysregulation and neuron cell death caused by HIV-1 protein tat. J Neurochem 1999; 73:1363-74; PMID:10501179; http://dx.doi.org/ 10.1046/j.1471-4159.1999.0731363.x [DOI] [PubMed] [Google Scholar]
- 8.Leissring MA, LaFerla FM, Callamaras N, Parker I. Subcellular mechanisms of presenilin-mediated enhancement of calcium signaling. Neurobiol Dis 2001; 8:469-78; PMID:11442355; http://dx.doi.org/ 10.1006/nbdi.2001.0382 [DOI] [PubMed] [Google Scholar]
- 9.Ma Y, Hendershot LM. ER chaperone functions during normal and stress conditions. J Chem Neuroanatomy 2004; 28:51-65; http://dx.doi.org/ 10.1016/j.jchemneu.2003.08.007 [DOI] [PubMed] [Google Scholar]
- 10.Lucero HA, Kaminer B. The role of calcium on the activity of ERcalcistorin/protein-disulfide isomerase and the significance of the C-terminal and its calcium binding. A comparison with mammalian protein-disulfide isomerase. J Biol Chem 1999; 274:3243-51; PMID:9915866; http://dx.doi.org/ 10.1074/jbc.274.5.3243 [DOI] [PubMed] [Google Scholar]
- 11.Kaufman RJ. Orchestrating the unfolded protein response in health and disease. J Clin Invest 2002; 110:1389-98; PMID:12438434; http://dx.doi.org/ 10.1172/JCI0216886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schroder M, Kaufman RJ. The mammalian unfolded protein response. Ann Rev Biochem 2005; 74:739-89; PMID:15952902; http://dx.doi.org/ 10.1146/annurev.biochem.73.011303.074134 [DOI] [PubMed] [Google Scholar]
- 13.Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999; 397:271-4; PMID:9930704; http://dx.doi.org/ 10.1038/16729 [DOI] [PubMed] [Google Scholar]
- 14.Lu PD, Harding HP, Ron D. Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J Cell Biol 2004; 167:27-33; PMID:15479734; http://dx.doi.org/ 10.1083/jcb.200408003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hotamisligil GS. Endoplasmic reticulum stress and atherosclerosis. Nat Med 2010; 16:396-9; PMID:20376052; http://dx.doi.org/ 10.1038/nm0410-396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Paschen W, Doutheil J. Disturbances of the functioning of endoplasmic reticulum: a key mechanism underlying neuronal cell injury? J Cerebral Blood Flow Metab 1999; 19:1-18; http://dx.doi.org/ 10.1097/00004647-199901000-00001 [DOI] [PubMed] [Google Scholar]
- 17.Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, et al.. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 2002; 415:92-6; PMID:11780124; http://dx.doi.org/ 10.1038/415092a [DOI] [PubMed] [Google Scholar]
- 18.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 2007; 8:519-29; PMID:17565364; http://dx.doi.org/ 10.1038/nrm2199 [DOI] [PubMed] [Google Scholar]
- 19.Hetz C, Glimcher LH. Fine-tuning of the unfolded protein response: Assembling the IRE1alpha interactome. Mol Cell 2009; 35:551-61; PMID:19748352; http://dx.doi.org/ 10.1016/j.molcel.2009.08.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell 1999; 10:3787-99; PMID:10564271; http://dx.doi.org/ 10.1091/mbc.10.11.3787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen X, Shen J, Prywes R. The luminal domain of ATF6 senses endoplasmic reticulum (ER) stress and causes translocation of ATF6 from the ER to the Golgi. J Biol Chem 2002; 277:13045-52; PMID:11821395; http://dx.doi.org/ 10.1074/jbc.M110636200 [DOI] [PubMed] [Google Scholar]
- 22.Sato Y, Nadanaka S, Okada T, Okawa K, Mori K. Luminal domain of ATF6 alone is sufficient for sensing endoplasmic reticulum stress and subsequent transport to the Golgi apparatus. Cell Structure Function 2011; 36:35-47; PMID:21150130; http://dx.doi.org/ 10.1247/csf.10010 [DOI] [PubMed] [Google Scholar]
- 23.Wu J, Rutkowski DT, Dubois M, Swathirajan J, Saunders T, Wang J, Song B, Yau GD, Kaufman RJ. ATF6alpha optimizes long-term endoplasmic reticulum function to protect cells from chronic stress. Dev Cell 2007; 13:351-64; PMID:17765679; http://dx.doi.org/ 10.1016/j.devcel.2007.07.005 [DOI] [PubMed] [Google Scholar]
- 24.Wong E, Cuervo AM. Autophagy gone awry in neurodegenerative diseases. Nat Neurosci 2010; 13:805-11; PMID:20581817; http://dx.doi.org/ 10.1038/nn.2575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Green DR, Levine B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell 2014; 157:65-75; PMID:24679527; http://dx.doi.org/ 10.1016/j.cell.2014.02.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, Cuervo AM. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol 2005; 64:113-22; PMID:15751225 [DOI] [PubMed] [Google Scholar]
- 27.Kegel KB, Kim M, Sapp E, McIntyre C, Castano JG, Aronin N, DiFiglia M. Huntingtin expression stimulates endosomal-lysosomal activity, endosome tubulation, and autophagy. J Neurosci 2000; 20:7268-78; PMID:11007884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anglade P, Vyas S, Javoy-Agid F, Herrero MT, Michel PP, Marquez J, Mouatt-Prigent A, Ruberg M, Hirsch EC, Agid Y. Apoptosis and autophagy in nigral neurons of patients with Parkinson disease. Histol Histopathol 1997; 12:25-31; PMID:9046040 [PubMed] [Google Scholar]
- 29.Sikorska B, Liberski PP, Giraud P, Kopp N, Brown P. Autophagy is a part of ultrastructural synaptic pathology in Creutzfeldt-Jakob disease: a brain biopsy study. Int J Biochem Cell Biol 2004; 36:2563-73; PMID:15325593; http://dx.doi.org/ 10.1016/j.biocel.2004.04.014 [DOI] [PubMed] [Google Scholar]
- 30.Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nature cell biology 2007; 9:1102-9; PMID:17909521; http://dx.doi.org/ 10.1038/ncb1007-1102 [DOI] [PubMed] [Google Scholar]
- 31.Liang C. Negative regulation of autophagy. Cell Death Differ 2010; 17:1807-15; PMID:20865012; http://dx.doi.org/ 10.1038/cdd.2010.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell 2010; 40:280-93; PMID:20965422; http://dx.doi.org/ 10.1016/j.molcel.2010.09.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mizushima N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol 2010; 22:132-9; PMID:20056399; http://dx.doi.org/ 10.1016/j.ceb.2009.12.004 [DOI] [PubMed] [Google Scholar]
- 34.Di Bartolomeo S, Corazzari M, Nazio F, Oliverio S, Lisi G, Antonioli M, Pagliarini V, Matteoni S, Fuoco C, Giunta L, et al.. The dynamic interaction of AMBRA1 with the dynein motor complex regulates mammalian autophagy. J Cell Biol 2010; 191:155-68; PMID:20921139; http://dx.doi.org/ 10.1083/jcb.201002100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Suzuki K, Kubota Y, Sekito T, Ohsumi Y. Hierarchy of Atg proteins in pre-autophagosomal structure organization. Genes Cells 2007; 12:209-18; PMID:17295840; http://dx.doi.org/ 10.1111/j.1365-2443.2007.01050.x [DOI] [PubMed] [Google Scholar]
- 36.Sinha S, Levine B. The autophagy effector Beclin 1: a novel BH3-only protein. Oncogene 2008; 27 Suppl 1:S137-48; PMID:19641499; http://dx.doi.org/ 10.1038/onc.2009.51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ichimura Y, Kirisako T, Takao T, Satomi Y, Shimonishi Y, Ishihara N, Mizushima N, Tanida I, Kominami E, Ohsumi M, et al.. A ubiquitin-like system mediates protein lipidation. Nature 2000; 408:488-92; PMID:11100732; http://dx.doi.org/ 10.1038/35044114 [DOI] [PubMed] [Google Scholar]
- 38.Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med 2013; 19:983-97; PMID:23921753; http://dx.doi.org/ 10.1038/nm.3232 [DOI] [PubMed] [Google Scholar]
- 39.Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol 2010; 22:124-31; PMID:20034776; http://dx.doi.org/ 10.1016/j.ceb.2009.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tanida I, Ueno T, Kominami E. LC3 and Autophagy. Meth Mol Biol 2008; 445:77-88; http://dx.doi.org/ 10.1007/978-1-59745-157-4_4 [DOI] [PubMed] [Google Scholar]
- 41.He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 2009; 43:67-93; PMID:19653858; http://dx.doi.org/ 10.1146/annurev-genet-102808-114910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Grant BD, Donaldson JG. Pathways and mechanisms of endocytic recycling. Nat Rev Mol Cell Biol 2009; 10:597-608; PMID:19696797; http://dx.doi.org/ 10.1038/nrm2755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hattula K, Furuhjelm J, Tikkanen J, Tanhuanpaa K, Laakkonen P, Peranen J. Characterization of the Rab8-specific membrane traffic route linked to protrusion formation. J Cell Sci 2006; 119:4866-77; PMID:17105768; http://dx.doi.org/ 10.1242/jcs.03275 [DOI] [PubMed] [Google Scholar]
- 44.Maldonado-Baez L, Donaldson JG. Hook1, microtubules, and Rab22: mediators of selective sorting of clathrin-independent endocytic cargo proteins on endosomes. Bioarchitecture 2013; 3:141-6; PMID:24284901; http://dx.doi.org/ 10.4161/bioa.26638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cai B, Xie S, Caplan S, Naslavsky N. GRAF1 forms a complex with MICAL-L1 and EHD1 to cooperate in tubular recycling endosome vesiculation. Front Cell Dev Biol 2014; 2:22; PMID:25364729; http://dx.doi.org/ 10.3389/fcell.2014.00022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cai B, Xie S, Liu F, Simone LC, Caplan S, Qin X, Naslavsky N. Rapid degradation of the complement regulator, CD59, by a novel inhibitor. J Biol Chem 2014; 289:12109-25; PMID:24616098; http://dx.doi.org/ 10.1074/jbc.M113.547083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xie S, Naslavsky N, Caplan S. Diacylglycerol Kinase α Regulates Tubular Recycling Endosome Biogenesis and Major Histocompatibility Complex Class I Recycling. J Biol Chem 2014; 289:31914-26; PMID:25248744; http://dx.doi.org/ 10.1074/jbc.M114.594291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Longatti A, Lamb CA, Razi M, Yoshimura S, Barr FA, Tooze SA. TBC1D14 regulates autophagosome formation via Rab11- and ULK1-positive recycling endosomes. J Cell Biol 2012; 197:659-75; PMID:22613832; http://dx.doi.org/ 10.1083/jcb.201111079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tang G, Gudsnuk K, Kuo SH, Cotrina ML, Rosoklija G, Sosunov A, Sonders MS, Kanter E, Castagna C, Yamamoto A, et al.. Loss of mTOR-Dependent Macroautophagy Causes Autistic-like Synaptic Pruning Deficits. Neuron 2014; 83(5):1131-43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Adolph TE, Tomczak MF, Niederreiter L, Ko HJ, Bock J, Martinez-Naves E, Glickman JN, Tschurtschenthaler M, Hartwig J, Hosomi S, et al.. Paneth cells as a site of origin for intestinal inflammation. Nature 2013; 503:272-6; PMID:24089213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kouroku Y, Fujita E, Tanida I, Ueno T, Isoai A, Kumagai H, Ogawa S, Kaufman RJ, Kominami E, Momoi T. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ 2007; 14:230-9; PMID:16794605; http://dx.doi.org/ 10.1038/sj.cdd.4401984 [DOI] [PubMed] [Google Scholar]
- 52.B'Chir W, Maurin AC, Carraro V, Averous J, Jousse C, Muranishi Y, Parry L, Stepien G, Fafournoux P, Bruhat A. The eIF2alpha/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res 2013; 41:7683-99; PMID:23804767; http://dx.doi.org/ 10.1093/nar/gkt563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rouschop KM, van den Beucken T, Dubois L, Niessen H, Bussink J, Savelkouls K, Keulers T, Mujcic H, Landuyt W, Voncken JW, et al.. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J Clin Invest 2010; 120:127-41; PMID:20038797 http://dx.doi.org/ 10.1172/JCI40027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rzymski T, Milani M, Pike L, Buffa F, Mellor HR, Winchester L, Pires I, Hammond E, Ragoussis I, Harris AL. Regulation of autophagy by ATF4 in response to severe hypoxia. Oncogene 2010; 29:4424-35; PMID:20514020; http://dx.doi.org/ 10.1038/onc.2010.191 [DOI] [PubMed] [Google Scholar]
- 55.Meares GP, Hughes KJ, Naatz A, Papa FR, Urano F, Hansen PA, Benveniste EN, Corbett JA. IRE1-dependent activation of AMPK in response to nitric oxide. Mol Cell Biol 2011; 31:4286-97; PMID:21896783; http://dx.doi.org/ 10.1128/MCB.05668-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Deegan S, Saveljeva S, Gorman AM, Samali A. Stress-induced self-cannibalism: on the regulation of autophagy by endoplasmic reticulum stress. Cell Mol Life Sci 2013; 70:2425-41; PMID:23052213; http://dx.doi.org/ 10.1007/s00018-012-1173-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell 2008; 30:678-88; PMID:18570871; http://dx.doi.org/ 10.1016/j.molcel.2008.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, Ron D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000; 287:664-6; PMID:10650002; http://dx.doi.org/ 10.1126/science.287.5453.664 [DOI] [PubMed] [Google Scholar]
- 59.Haberzettl P, Hill BG. Oxidized lipids activate autophagy in a JNK-dependent manner by stimulating the endoplasmic reticulum stress response. Redox Biol 2013; 1:56-64; PMID:24024137; http://dx.doi.org/ 10.1016/j.redox.2012.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vidal RL, Figueroa A, Court FA, Thielen P, Molina C, Wirth C, Caballero B, Kiffin R, Segura-Aguilar J, Cuervo AM, et al.. Targeting the UPR transcription factor XBP1 protects against Huntington's disease through the regulation of FoxO1 and autophagy. Hum Mol Genet 2012; 21:2245-62; PMID:22337954; http://dx.doi.org/ 10.1093/hmg/dds040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhao Y, Li X, Cai MY, Ma K, Yang J, Zhou J, Fu W, Wei FZ, Wang L, Xie D, et al.. XBP-1u suppresses autophagy by promoting the degradation of FoxO1 in cancer cells. Cell Res 2013; 23:491-507; PMID:23277279; http://dx.doi.org/ 10.1038/cr.2013.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhou Y, Lee J, Reno CM, Sun C, Park SW, Chung J, Lee J, Fisher SJ, White MF, Biddinger SB, et al.. Regulation of glucose homeostasis through a XBP-1-FoxO1 interaction. Nat Med 2011; 17:356-65; PMID:21317886; http://dx.doi.org/ 10.1038/nm.2293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gade P, Manjegowda SB, Nallar SC, Maachani UB, Cross AS, Kalvakolanu DV. Regulation of the death-associated protein kinase 1 expression and autophagy via ATF6 requires apoptosis signal-regulating kinase 1. Mol Cell Biol 2014; 34:4033-48; PMID:25135476; http://dx.doi.org/ 10.1128/MCB.00397-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kalvakolanu DV, Gade P. IFNG and autophagy: a critical role for the ER-stress mediator ATF6 in controlling bacterial infections. Autophagy 2012; 8:1673-4; PMID:22874566; http://dx.doi.org/ 10.4161/auto.21403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zalckvar E, Berissi H, Mizrachy L, Idelchuk Y, Koren I, Eisenstein M, Sabanay H, Pinkas-Kramarski R, Kimchi A. DAP-kinase-mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl-XL and induction of autophagy. EMBO Rep 2009; 10:285-92; PMID:19180116; http://dx.doi.org/ 10.1038/embor.2008.246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Price DL, Tanzi RE, Borchelt DR, Sisodia SS. Alzheimer disease: genetic studies and transgenic models. Ann Rev Genet 1998; 32:461-93; PMID:9928488; http://dx.doi.org/ 10.1146/annurev.genet.32.1.461 [DOI] [PubMed] [Google Scholar]
- 67.Pollanen MS, Dickson DW, Bergeron C. Pathology and biology of the Lewy body. J Neuropathol Exp Neurol 1993; 52:183-91; PMID: 7684074; http://dx.doi.org/ 10.1097/00005072-199305000-00001 [DOI] [PubMed] [Google Scholar]
- 68.Suen KC, Lin KF, Elyaman W, So KF, Chang RC, Hugon J. Reduction of calcium release from the endoplasmic reticulum could only provide partial neuroprotection against β-amyloid peptide toxicity. J Neurochem 2003; 87:1413-26; PMID:14713297; http://dx.doi.org/ 10.1111/j.1471-4159.2003.02259.x [DOI] [PubMed] [Google Scholar]
- 69.Alberdi E, Wyssenbach A, Alberdi M, Sanchez-Gomez MV, Cavaliere F, Rodriguez JJ, Verkhratsky A, Matute C. Ca(2+) -dependent endoplasmic reticulum stress correlates with astrogliosis in oligomeric amyloid β-treated astrocytes and in a model of Alzheimer disease. Aging Cell 2013; 12:292-302; PMID:23409977; http://dx.doi.org/ 10.1111/acel.12054 [DOI] [PubMed] [Google Scholar]
- 70.Imaizumi Y, Okada Y, Akamatsu W, Koike M, Kuzumaki N, Hayakawa H, Nihira T, Kobayashi T, Ohyama M, Sato S, et al.. Mitochondrial dysfunction associated with increased oxidative stress and α-synuclein accumulation in PARK2 iPSC-derived neurons and postmortem brain tissue. Mol Brain 2012; 5:35; PMID:23039195; http://dx.doi.org/ 10.1186/1756-6606-5-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hashimoto M, Hsu LJ, Xia Y, Takeda A, Sisk A, Sundsmo M, Masliah E. Oxidative stress induces amyloid-like aggregate formation of NACP/α-synuclein in vitro. Neuroreport 1999; 10:717-21; PMID:10208537; http://dx.doi.org/ 10.1097/00001756-199903170-00011 [DOI] [PubMed] [Google Scholar]
- 72.Frank-Cannon TC, Tran T, Ruhn KA, Martinez TN, Hong J, Marvin M, Hartley M, Treviño I, O'Brien DE, Casey B, et al.. Parkin deficiency increases vulnerability to inflammation-related nigral degeneration. J Neurosci 2008; 28:10825-34; PMID:18945890; http://dx.doi.org/ 10.1523/JNEUROSCI.3001-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gao HM, Zhang F, Zhou H, Kam W, Wilson B, Hong JS. Neuroinflammation and α-synuclein dysfunction potentiate each other, driving chronic progression of neurodegeneration in a mouse model of Parkinson disease. Environ Health Perspect 2011; 119:807-14; PMID:21245015; http://dx.doi.org/ 10.1289/ehp.1003013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Suarez-Calvet M, Belbin O, Pera M, Badiola N, Magrane J, Guardia-Laguarta C, Muñoz L, Colom-Cadena M, Clarimón J, Lleó A. Autosomal-dominant Alzheimer disease mutations at the same codon of amyloid precursor protein differentially alter Abeta production. J Neurochem 2014; 128:330-9; PMID:24117942; http://dx.doi.org/ 10.1111/jnc.12466 [DOI] [PubMed] [Google Scholar]
- 75.Lynch-Day MA, Mao K, Wang K, Zhao M, Klionsky DJ. The role of autophagy in Parkinson disease. Cold Spring Harb Perspect Med 2012; 2:a009357; PMID:22474616; http://dx.doi.org/ 10.1101/cshperspect.a009357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gomez-Suaga P, Churchill GC, Patel S, Hilfiker S. A link between LRRK2, autophagy and NAADP-mediated endolysosomal calcium signalling. Biochem Soc Trans 2012; 40:1140-6; PMID:22988879; http://dx.doi.org/ 10.1042/BST20120138 [DOI] [PubMed] [Google Scholar]
- 77.Selkoe DJ. Alzheimer disease: genes, proteins, and therapy. Physiol Rev 2001; 81:741-66; PMID:11274343 [DOI] [PubMed] [Google Scholar]
- 78.Chen W, Gamache E, Rosenman DJ, Xie J, Lopez MM, Li YM, Wang C. Familial Alzheimer mutations within APPTM increase Abeta42 production by enhancing accessibility of epsilon-cleavage site. Nat Commun 2014; 5:3037; PMID:24390130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Agyemang AF, Harrison SR, Siegel RM, McDermott MF. Protein misfolding and dysregulated protein homeostasis in autoinflammatory diseases and beyond. Semin Immunopathol 2015; 37(4):335-47; PMID:25994946; http://dx.doi.org/ 10.1007/s00281-015-0496-2 [DOI] [PubMed] [Google Scholar]
- 80.Nijholt DA, van Haastert ES, Rozemuller AJ, Scheper W, Hoozemans JJ. The unfolded protein response is associated with early tau pathology in the hippocampus of tauopathies. J Pathol 2012; 226:693-702; PMID:22102449; http://dx.doi.org/ 10.1002/path.3969 [DOI] [PubMed] [Google Scholar]
- 81.Briggs CA, Schneider C, Richardson JC, Stutzmann GE. β amyloid peptide plaques fail to alter evoked neuronal calcium signals in APP/PS1 Alzheimer disease mice. Neurobiol Aging 2013; 34:1632-43; PMID:23337342; http://dx.doi.org/ 10.1016/j.neurobiolaging.2012.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.D'Adamio L, Castillo PE. Presenilin-ryanodine receptor connection. Proc Natl Acad Sci U S A 2013; 110:14825-6; PMID:23995445; http://dx.doi.org/ 10.1073/pnas.1313996110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yu Z, Luo H, Fu W, Mattson MP. The endoplasmic reticulum stress-responsive protein GRP78 protects neurons against excitotoxicity and apoptosis: suppression of oxidative stress and stabilization of calcium homeostasis. Exp Neurol 1999; 155:302-14; PMID:10072306; http://dx.doi.org/ 10.1006/exnr.1998.7002 [DOI] [PubMed] [Google Scholar]
- 84.Katayama T, Imaizumi K, Manabe T, Hitomi J, Kudo T, Tohyama M. Induction of neuronal death by ER stress in Alzheimer disease. J Chem Neuroanatomy 2004; 28:67-78; http://dx.doi.org/ 10.1016/j.jchemneu.2003.12.004 [DOI] [PubMed] [Google Scholar]
- 85.Katayama T, Imaizumi K, Sato N, Miyoshi K, Kudo T, Hitomi J, Morihara T, Yoneda T, Gomi F, Mori Y, et al.. Presenilin-1 mutations downregulate the signalling pathway of the unfolded-protein response. Nat Cell Biol 1999; 1:479-85; PMID:10587643; http://dx.doi.org/ 10.1038/70265 [DOI] [PubMed] [Google Scholar]
- 86.Nijholt DA, de Graaf TR, van Haastert ES, Oliveira AO, Berkers CR, Zwart R, Ovaa H, Baas F, Hoozemans JJ, Scheper W. Endoplasmic reticulum stress activates autophagy but not the proteasome in neuronal cells: implications for Alzheimer disease. Cell Death Differ 2011; 18:1071-81; PMID:21252911; http://dx.doi.org/ 10.1038/cdd.2010.176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pickford F, Masliah E, Britschgi M, Lucin K, Narasimhan R, Jaeger PA, Small S, Spencer B, Rockenstein E, Levine B, et al.. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid β accumulation in mice. J Clin Invest 2008; 118:2190-9; PMID:18497889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lucin KM, O'Brien CE, Bieri G, Czirr E, Mosher KI, Abbey RJ, Mastroeni DF, Rogers J, Spencer B, Masliah E, et al.. Microglial beclin 1 regulates retromer trafficking and phagocytosis and is impaired in Alzheimer disease. Neuron 2013; 79:873-86; PMID:24012002; http://dx.doi.org/ 10.1016/j.neuron.2013.06.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, Fitzgerald KA, Latz E, Moore KJ, Golenbock DT. The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nat Immunol 2008; 9:857-65; PMID:18604209; http://dx.doi.org/ 10.1038/ni.1636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yang DS, Stavrides P, Mohan PS, Kaushik S, Kumar A, Ohno M, Schmidt SD, Wesson D, Bandyopadhyay U, Jiang Y, et al.. Reversal of autophagy dysfunction in the TgCRND8 mouse model of Alzheimer disease ameliorates amyloid pathologies and memory deficits. Brain 2011; 134:258-77; PMID:21186265; http://dx.doi.org/ 10.1093/brain/awq341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jiang T, Yu JT, Zhu XC, Tan MS, Wang HF, Cao L, Zhang QQ, Shi JQ, Gao L, Qin H, et al.. Temsirolimus promotes autophagic clearance of amyloid-β and provides protective effects in cellular and animal models of Alzheimer disease. Pharmacol Res 2014; 81:54-63; PMID:24602800; http://dx.doi.org/ 10.1016/j.phrs.2014.02.008 [DOI] [PubMed] [Google Scholar]
- 92.Crary JF, Trojanowski JQ, Schneider JA, Abisambra JF, Abner EL, Alafuzoff I, Arnold SE, Attems J, Beach TG, Bigio EH, et al.. Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathologica 2014; 128:755-66; PMID:25348064http://dx.doi.org/ 10.1007/s00401-014-1349-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Abisambra JF, Jinwal UK, Blair LJ, O'Leary JC 3rd, Li Q, Brady S, Wang L, Guidi CE, Zhang B, Nordhues BA, et al.. Tau accumulation activates the unfolded protein response by impairing endoplasmic reticulum-associated degradation. J Neurosci 2013; 33:9498-507; PMID:23719816; http://dx.doi.org/ 10.1523/JNEUROSCI.5397-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Taylor JP, Hardy J, Fischbeck KH. Toxic proteins in neurodegenerative disease. Science 2002; 296:1991-5; PMID:12065827; http://dx.doi.org/ 10.1126/science.1067122 [DOI] [PubMed] [Google Scholar]
- 95.Abisambra JF, Scheff S. Brain injury in the context of tauopathies. J Alzheimer Dis 2014; 40:495-518 [DOI] [PubMed] [Google Scholar]
- 96.Sieben A, Van Langenhove T, Engelborghs S, Martin JJ, Boon P, Cras P, De Deyn PP, Santens P, Van Broeckhoven C, Cruts M. The genetics and neuropathology of frontotemporal lobar degeneration. Acta Neuropathologica 2012; 124:353-72; PMID:22890575; http://dx.doi.org/ 10.1007/s00401-012-1029-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hoozemans JJ, van Haastert ES, Nijholt DA, Rozemuller AJ, Scheper W. Activation of the unfolded protein response is an early event in Alzheimer and Parkinson disease. Neurodegener Dis 2012; 10:212-5; PMID:22302012; http://dx.doi.org/ 10.1159/000334536 [DOI] [PubMed] [Google Scholar]
- 98.Hoozemans JJ, van Haastert ES, Nijholt DA, Rozemuller AJ, Eikelenboom P, Scheper W. The unfolded protein response is activated in pretangle neurons in Alzheimer disease hippocampus. Am J Pathol 2009; 174:1241-51; PMID:19264902; http://dx.doi.org/ 10.2353/ajpath.2009.080814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hoozemans JJ, Veerhuis R, Van Haastert ES, Rozemuller JM, Baas F, Eikelenboom P, Scheper W. The unfolded protein response is activated in Alzheimer disease. Acta Neuropathologica 2005; 110:165-72; PMID:15973543; http://dx.doi.org/ 10.1007/s00401-005-1038-0 [DOI] [PubMed] [Google Scholar]
- 100.Nijholt DA, Nolle A, van Haastert ES, Edelijn H, Toonen RF, Hoozemans JJ, Scheper W. Unfolded protein response activates glycogen synthase kinase-3 via selective lysosomal degradation. Neurobiol Aging 2013; 34:1759-71; PMID:23415837; http://dx.doi.org/ 10.1016/j.neurobiolaging.2013.01.008 [DOI] [PubMed] [Google Scholar]
- 101.van der Harg JM, Nolle A, Zwart R, Boerema AS, van Haastert ES, Strijkstra AM, Hoozemans JJ, Scheper W. The unfolded protein response mediates reversible tau phosphorylation induced by metabolic stress. Cell Death Dis 2014; 5:e1393; PMID:25165879; http://dx.doi.org/ 10.1038/cddis.2014.354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Jinwal UK, Koren J, O'Leary JC, Jones JR, Abisambra JF, Dickey CA. Hsp70 ATPase Modulators as Therapeutics for Alzheimer and other Neurodegenerative Diseases. Mol Cell Pharmacol 2010; 2:43-6; PMID:20523917 [PMC free article] [PubMed] [Google Scholar]
- 103.Hartl FU, Bracher A, Hayer-Hartl M. Molecular chaperones in protein folding and proteostasis. Nature 2011; 475:324-32; PMID:21776078; http://dx.doi.org/ 10.1038/nature10317 [DOI] [PubMed] [Google Scholar]
- 104.Wilson MR, Yerbury JJ, Poon S. Potential roles of abundant extracellular chaperones in the control of amyloid formation and toxicity. Mol Biosyst 2008; 4:42-52; PMID:18075673; http://dx.doi.org/ 10.1039/B712728F [DOI] [PubMed] [Google Scholar]
- 105.Wu ZC, Yu JT, Li Y, Tan L. Clusterin in Alzheimer disease. Adv Clin Chem 2012; 56:155-73; PMID:22397031; http://dx.doi.org/ 10.1016/B978-0-12-394317-0.00011-X [DOI] [PubMed] [Google Scholar]
- 106.Sousa JC, Cardoso I, Marques F, Saraiva MJ, Palha JA. Transthyretin and Alzheimer disease: where in the brain? Neurobiol Aging 2007; 28:713-8; PMID:16698124; http://dx.doi.org/ 10.1016/j.neurobiolaging.2006.03.015 [DOI] [PubMed] [Google Scholar]
- 107.Mead S. Prion disease genetics. Eur J Hum Genet 2006; 14:273-81; PMID:16391566; http://dx.doi.org/ 10.1038/sj.ejhg.5201544 [DOI] [PubMed] [Google Scholar]
- 108.Dearmond SJ, Bajsarowicz K. PrPSc accumulation in neuronal plasma membranes links Notch-1 activation to dendritic degeneration in prion diseases. Mol Neurodegeneration 2010; 5:6; http://dx.doi.org/ 10.1186/1750-1326-5-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hetz C, Castilla J, Soto C. Perturbation of endoplasmic reticulum homeostasis facilitates prion replication. J Biol Chem 2007; 282:12725-33; PMID:17329244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hetz CA, Soto C. Stressing out the ER: a role of the unfolded protein response in prion-related disorders. Curr Mol Med 2006; 6:37-43; PMID:16472111; http://dx.doi.org/ 10.2174/156652406775574578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Petkova AT, Leapman RD, Guo Z, Yau WM, Mattson MP, Tycko R. Self-propagating, molecular-level polymorphism in Alzheimer β-amyloid fibrils. Science 2005; 307:262-5; PMID:15653506; http://dx.doi.org/ 10.1126/science.1105850 [DOI] [PubMed] [Google Scholar]
- 112.Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, Spencer B, Masliah E, Lee SJ. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of α-synuclein. Proc Natl Acad Sci U S A 2009; 106:13010-5; PMID:19651612; http://dx.doi.org/ 10.1073/pnas.0903691106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Frost B, Jacks RL, Diamond MI. Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem 2009; 284:12845-52; PMID:19282288; http://dx.doi.org/ 10.1074/jbc.M808759200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hetz C, Russelakis-Carneiro M, Maundrell K, Castilla J, Soto C. Caspase-12 and endoplasmic reticulum stress mediate neurotoxicity of pathological prion protein. EMBO J 2003; 22:5435-45; PMID:14532116; http://dx.doi.org/ 10.1093/emboj/cdg537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ferreiro E, Costa R, Marques S, Cardoso SM, Oliveira CR, Pereira CM. Involvement of mitochondria in endoplasmic reticulum stress-induced apoptotic cell death pathway triggered by the prion peptide PrP(106-126). J Neurochem 2008; 104:766-76; PMID:17995926 [DOI] [PubMed] [Google Scholar]
- 116.Wang SB, Shi Q, Xu Y, Xie WL, Zhang J, Tian C, Guo Y, Wang K, Zhang BY, Chen C, et al.. Protein disulfide isomerase regulates endoplasmic reticulum stress and the apoptotic process during prion infection and PrP mutant-induced cytotoxicity. PloS One 2012; 7:e38221; PMID:22685557; http://dx.doi.org/ 10.1371/journal.pone.0038221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hetz C, Russelakis-Carneiro M, Walchli S, Carboni S, Vial-Knecht E, Maundrell K, Castilla J, Soto C. The disulfide isomerase Grp58 is a protective factor against prion neurotoxicity. J Neurosci 2005; 25:2793-802; PMID:15772339; http://dx.doi.org/ 10.1523/JNEUROSCI.4090-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Tang Y, Xiang W, Terry L, Kretzschmar HA, Windl O. Transcriptional analysis implicates endoplasmic reticulum stress in bovine spongiform encephalopathy. PloS one 2010; 5:e14207; PMID:21151970; http://dx.doi.org/ 10.1371/journal.pone.0014207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ferreiro E, Resende R, Costa R, Oliveira CR, Pereira CM. An endoplasmic-reticulum-specific apoptotic pathway is involved in prion and amyloid-β peptides neurotoxicity. Neurobiol Dis 2006; 23:669-78; PMID:16844381; http://dx.doi.org/ 10.1016/j.nbd.2006.05.011 [DOI] [PubMed] [Google Scholar]
- 120.Orsi A, Fioriti L, Chiesa R, Sitia R. Conditions of endoplasmic reticulum stress favor the accumulation of cytosolic prion protein. J Biol Chem 2006; 281:30431-8; PMID:16908519; http://dx.doi.org/ 10.1074/jbc.M605320200 [DOI] [PubMed] [Google Scholar]
- 121.Torres M, Castillo K, Armisen R, Stutzin A, Soto C, Hetz C. Prion protein misfolding affects calcium homeostasis and sensitizes cells to endoplasmic reticulum stress. PloS One 2010; 5:e15658; PMID:21209925; http://dx.doi.org/ 10.1371/journal.pone.0015658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hetz C, Lee AH, Gonzalez-Romero D, Thielen P, Castilla J, Soto C, Glimcher LH. Unfolded protein response transcription factor XBP-1 does not influence prion replication or pathogenesis. Proc Natl Acad Sci U S A 2008; 105:757-62; PMID:18178615; http://dx.doi.org/ 10.1073/pnas.0711094105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Moreno JA, Radford H, Peretti D, Steinert JR, Verity N, Martin MG, Halliday M, Morgan J, Dinsdale D, Ortori CA, et al.. Sustained translational repression by eIF2alpha-P mediates prion neurodegeneration. Nature 2012; 485:507-11; PMID:22622579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Moreno JA, Halliday M, Molloy C, Radford H, Verity N, Axten JM, Ortori CA, Willis AE, Fischer PM, Barrett DA, et al.. Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci Transl Med 2013; 5:206ra138; PMID:24107777; http://dx.doi.org/ 10.1126/scitranslmed.3006767 [DOI] [PubMed] [Google Scholar]
- 125.Chen R, Jin R, Wu L, Ye X, Yang Y, Luo K, Wang W, Wu D, Ye X, Huang L, et al.. Reticulon 3 attenuates the clearance of cytosolic prion aggregates via inhibiting autophagy. Autophagy 2011; 7:205-16; PMID:21119307; http://dx.doi.org/ 10.4161/auto.7.2.14197 [DOI] [PubMed] [Google Scholar]
- 126.Rubinsztein DC, DiFiglia M, Heintz N, Nixon RA, Qin ZH, Ravikumar B, Stefanis L, Tolkovsky A. Autophagy and its possible roles in nervous system diseases, damage and repair. Autophagy 2005; 1:11-22; PMID:16874045; http://dx.doi.org/ 10.4161/auto.1.1.1513 [DOI] [PubMed] [Google Scholar]
- 127.Jeong JK, Moon MH, Lee YJ, Seol JW, Park SY. Autophagy induced by the class III histone deacetylase Sirt1 prevents prion peptide neurotoxicity. Neurobiol Aging 2013; 34:146-56; PMID:22575359; http://dx.doi.org/ 10.1016/j.neurobiolaging.2012.04.002 [DOI] [PubMed] [Google Scholar]
- 128.Heiseke A, Aguib Y, Riemer C, Baier M, Schatzl HM. Lithium induces clearance of protease resistant prion protein in prion-infected cells by induction of autophagy. J Neurochem 2009; 109:25-34; PMID:19183256; http://dx.doi.org/ 10.1111/j.1471-4159.2009.05906.x [DOI] [PubMed] [Google Scholar]
- 129.Aguib Y, Heiseke A, Gilch S, Riemer C, Baier M, Schatzl HM, Ertmer A. Autophagy induction by trehalose counteracts cellular prion infection. Autophagy 2009; 5:361-9; PMID:19182537; http://dx.doi.org/ 10.4161/auto.5.3.7662 [DOI] [PubMed] [Google Scholar]
- 130.Cortes CJ, Qin K, Cook J, Solanki A, Mastrianni JA. Rapamycin delays disease onset and prevents PrP plaque deposition in a mouse model of Gerstmann-Straussler-Scheinker disease. J Neurosci 2012; 32:12396-405; PMID:22956830; http://dx.doi.org/ 10.1523/JNEUROSCI.6189-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Karapetyan YE, Sferrazza GF, Zhou M, Ottenberg G, Spicer T, Chase P, Fallahi M, Hodder P, Weissmann C, Lasmézas CI. Unique drug screening approach for prion diseases identifies tacrolimus and astemizole as antiprion agents. Proc Natl Acad Sci U S A 2013; 110:7044-9; PMID:23576755; http://dx.doi.org/ 10.1073/pnas.1303510110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Lang AE, Lozano AM. Parkinson disease. First of two parts. N Eng J Med 1998; 339:1044-53; http://dx.doi.org/ 10.1056/NEJM199810083391506 [DOI] [PubMed] [Google Scholar]
- 133.Lang AE, Lozano AM. Parkinson disease. Second of two parts. N Eng J med 1998; 339:1130-43; http://dx.doi.org/ 10.1056/NEJM199810153391607 [DOI] [PubMed] [Google Scholar]
- 134.Ryu EJ, Harding HP, Angelastro JM, Vitolo OV, Ron D, Greene LA. Endoplasmic reticulum stress and the unfolded protein response in cellular models of Parkinson disease. J Neurosci 2002; 22:10690-8; PMID:12486162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Sado M, Yamasaki Y, Iwanaga T, Onaka Y, Ibuki T, Nishihara S, Mizuguchi H, Momota H, Kishibuchi R, Hashimoto T, et al.. Protective effect against Parkinson disease-related insults through the activation of XBP1. Brain Res 2009; 1257:16-24; PMID:19135031; http://dx.doi.org/ 10.1016/j.brainres.2008.11.104 [DOI] [PubMed] [Google Scholar]
- 136.Hoozemans JJ, van Haastert ES, Eikelenboom P, de Vos RA, Rozemuller JM, Scheper W. Activation of the unfolded protein response in Parkinson disease. Biochem Biophy Res Commun 2007; 354:707-11; http://dx.doi.org/ 10.1016/j.bbrc.2007.01.043 [DOI] [PubMed] [Google Scholar]
- 137.Colla E, Coune P, Liu Y, Pletnikova O, Troncoso JC, Iwatsubo T, Schneider BL, Lee MK. Endoplasmic reticulum stress is important for the manifestations of α-synucleinopathy in vivo. J Neurosc 2012; 32:3306-20; http://dx.doi.org/ 10.1523/JNEUROSCI.5367-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Silva RM, Ries V, Oo TF, Yarygina O, Jackson-Lewis V, Ryu EJ, Lu PD, Marciniak SJ, Ron D, Przedborski S, et al.. CHOP/GADD153 is a mediator of apoptotic death in substantia nigra dopamine neurons in an in vivo neurotoxin model of parkinsonism. J Neurochem 2005; 95:974-86; PMID:16135078; http://dx.doi.org/ 10.1111/j.1471-4159.2005.03428.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant α-synuclein by chaperone-mediated autophagy. Science 2004; 305:1292-5; PMID:15333840; http://dx.doi.org/ 10.1126/science.1101738 [DOI] [PubMed] [Google Scholar]
- 140.Kuroda Y, Mitsui T, Kunishige M, Shono M, Akaike M, Azuma H, Matsumoto T. Parkin enhances mitochondrial biogenesis in proliferating cells. Hum Mol Genet 2006; 15:883-95; PMID:16449237; http://dx.doi.org/ 10.1093/hmg/ddl006 [DOI] [PubMed] [Google Scholar]
- 141.Ashrafi G, Schwarz TL. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ 2013; 20:31-42; PMID:22743996; http://dx.doi.org/ 10.1038/cdd.2012.81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kamp F, Exner N, Lutz AK, Wender N, Hegermann J, Brunner B, Nuscher B, Bartels T, Giese A, Beyer K, et al.. Inhibition of mitochondrial fusion by α-synuclein is rescued by PINK1, Parkin and DJ-1. EMBO J 2010; 29:3571-89; PMID:20842103; http://dx.doi.org/ 10.1038/emboj.2010.223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Imai Y, Soda M, Takahashi R. Parkin suppresses unfolded protein stress-induced cell death through its E3 ubiquitin-protein ligase activity. J Biol Chem 2000; 275:35661-4; PMID:10973942; http://dx.doi.org/ 10.1074/jbc.C000447200 [DOI] [PubMed] [Google Scholar]
- 144.Zhang X, Yuan Y, Jiang L, Zhang J, Gao J, Shen Z, Zheng Y, Deng T, Yan H, Li W, et al.. Endoplasmic reticulum stress induced by tunicamycin and thapsigargin protects against transient ischemic brain injury: Involvement of PARK2-dependent mitophagy. Autophagy 2014; 10:1801-13; PMID:25126734; http://dx.doi.org/ 10.4161/auto.32136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Lonskaya I, Desforges NM, Hebron ML, Moussa CE. Ubiquitination increases parkin activity to promote autophagic α-synuclein clearance. PloS One 2013; 8:e83914; PMID:24386307; http://dx.doi.org/ 10.1371/journal.pone.0083914 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 146.Pasinelli P, Brown RH. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci 2006; 7:710-23; PMID:16924260; http://dx.doi.org/ 10.1038/nrn1971 [DOI] [PubMed] [Google Scholar]
- 147.Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 1994; 264:1772-5; PMID:8209258; http://dx.doi.org/ 10.1126/science.8209258 [DOI] [PubMed] [Google Scholar]
- 148.Strong MJ, Kesavapany S, Pant HC. The pathobiology of amyotrophic lateral sclerosis: a proteinopathy? J Neuropathol Exp Neurol 2005; 64:649-64; PMID:16106213; http://dx.doi.org/ 10.1097/01.jnen.0000173889.71434.ea [DOI] [PubMed] [Google Scholar]
- 149.Del Bo R, Ghezzi S, Corti S, Pandolfo M, Ranieri M, Santoro D, Ghione I, Prelle A, Orsetti V, Mancuso M, et al.. TARDBP (TDP-43) sequence analysis in patients with familial and sporadic ALS: identification of two novel mutations. Eur J Neurol 2009; 16:727-32; PMID:19236453; http://dx.doi.org/ 10.1111/j.1468-1331.2008.02424.x [DOI] [PubMed] [Google Scholar]
- 150.Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Vande Velde C, et al.. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet 2008; 40:572-4; PMID:18372902; http://dx.doi.org/ 10.1038/ng.132 [DOI] [PubMed] [Google Scholar]
- 151.Christensen LA, Fallingborg J, Abildgaard K, Jacobsen BA, Sanchez G, Hansen SH, Bondesen S, Hvidberg EF, Rasmussen SN. Topical and systemic availability of 5-aminosalicylate: comparisons of three controlled release preparations in man. Aliment Pharmacol Ther 1990; 4:523-33; PMID:2129640; http://dx.doi.org/ 10.1111/j.1365-2036.1990.tb00499.x [DOI] [PubMed] [Google Scholar]
- 152.Atkin JD, Farg MA, Turner BJ, Tomas D, Lysaght JA, Nunan J, Rembach A, Nagley P, Beart PM, Cheema SS, et al.. Induction of the unfolded protein response in familial amyotrophic lateral sclerosis and association of protein-disulfide isomerase with superoxide dismutase 1. J Biol Chem 2006; 281:30152-65; PMID:16847061; http://dx.doi.org/ 10.1074/jbc.M603393200 [DOI] [PubMed] [Google Scholar]
- 153.Vlug AS, Teuling E, Haasdijk ED, French P, Hoogenraad CC, Jaarsma D. ATF3 expression precedes death of spinal motoneurons in amyotrophic lateral sclerosis-SOD1 transgenic mice and correlates with c-Jun phosphorylation, CHOP expression, somato-dendritic ubiquitination and Golgi fragmentation. Eur J Neurosci 2005; 22:1881-94; PMID:16262628; http://dx.doi.org/ 10.1111/j.1460-9568.2005.04389.x [DOI] [PubMed] [Google Scholar]
- 154.Kieran D, Woods I, Villunger A, Strasser A, Prehn JH. Deletion of the BH3-only protein puma protects motoneurons from ER stress-induced apoptosis and delays motoneuron loss in ALS mice. Proc Natl Acad Sci U S A 2007; 104:20606-11; PMID:18077368; http://dx.doi.org/ 10.1073/pnas.0707906105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Ilieva EV, Ayala V, Jove M, Dalfo E, Cacabelos D, Povedano M, Bellmunt MJ, Ferrer I, Pamplona R, Portero-Otín M. Oxidative and endoplasmic reticulum stress interplay in sporadic amyotrophic lateral sclerosis. Brain 2007; 130:3111-23; PMID:17716997; http://dx.doi.org/ 10.1093/brain/awm190 [DOI] [PubMed] [Google Scholar]
- 156.Hetz C, Thielen P, Matus S, Nassif M, Court F, Kiffin R, Martinez G, Cuervo AM, Brown RH, Glimcher LH. XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev 2009; 23:2294-306; PMID:19762508; http://dx.doi.org/ 10.1101/gad.1830709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Ayala YM, Zago P, D'Ambrogio A, Xu YF, Petrucelli L, Buratti E, Baralle FE. Structural determinants of the cellular localization and shuttling of TDP-43. J Cell Sci 2008; 121:3778-85; PMID:18957508; http://dx.doi.org/ 10.1242/jcs.038950 [DOI] [PubMed] [Google Scholar]
- 158.Kawahara Y, Mieda-Sato A. TDP-43 promotes microRNA biogenesis as a component of the Drosha and Dicer complexes. Proc Natl Acad Sci U S A 2012; 109:3347-52; PMID:22323604; http://dx.doi.org/ 10.1073/pnas.1112427109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Barmada SJ, Skibinski G, Korb E, Rao EJ, Wu JY, Finkbeiner S. Cytoplasmic mislocalization of TDP-43 is toxic to neurons and enhanced by a mutation associated with familial amyotrophic lateral sclerosis. J Neurosci 2010; 30:639-49; PMID:20071528; http://dx.doi.org/ 10.1523/JNEUROSCI.4988-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Walker AK, Soo KY, Sundaramoorthy V, Parakh S, Ma Y, Farg MA, Wallace RH, Crouch PJ, Turner BJ, Horne MK, et al.. ALS-associated TDP-43 induces endoplasmic reticulum stress, which drives cytoplasmic TDP-43 accumulation and stress granule formation. PloS One 2013; 8:e81170; PMID:24312274; http://dx.doi.org/ 10.1371/journal.pone.0081170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Farg MA, Soo KY, Walker AK, Pham H, Orian J, Horne MK, Warraich ST, Williams KL, Blair IP, Atkin JD. Mutant FUS induces endoplasmic reticulum stress in amyotrophic lateral sclerosis and interacts with protein disulfide-isomerase. Neurobiol Aging 2012; 33:2855-68; PMID:22459602; http://dx.doi.org/ 10.1016/j.neurobiolaging.2012.02.009 [DOI] [PubMed] [Google Scholar]
- 162.Majcher V, Goode A, James V, Layfield R. Autophagy receptor defects and ALS-FTLD. Mol Cell Neurosci 2015; 66:43-52; PMID:25683489; http://dx.doi.org/ 10.1016/j.mcn.2015.01.002 [DOI] [PubMed] [Google Scholar]
- 163.Bose JK, Huang CC, Shen CK. Regulation of autophagy by neuropathological protein TDP-43. J Biol Chem 2011; 286:44441-8; PMID:22052911; http://dx.doi.org/ 10.1074/jbc.M111.237115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Ling SC, Albuquerque CP, Han JS, Lagier-Tourenne C, Tokunaga S, Zhou H, Cleveland DW. ALS-associated mutations in TDP-43 increase its stability and promote TDP-43 complexes with FUS/TLS. Proc Natl Acad Sci U S A 2010; 107:13318-23; PMID:20624952; http://dx.doi.org/ 10.1073/pnas.1008227107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Dormann D, Rodde R, Edbauer D, Bentmann E, Fischer I, Hruscha A, Than ME, Mackenzie IR, Capell A, Schmid B, et al.. ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J 2010; 29:2841-57; PMID:20606625; http://dx.doi.org/ 10.1038/emboj.2010.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Ryu HH, Jun MH, Min KJ, Jang DJ, Lee YS, Kim HK, Lee JA. Autophagy regulates amyotrophic lateral sclerosis-linked fused in sarcoma-positive stress granules in neurons. Neurobiol Aging 2014; 35:2822-31; PMID:25216585; http://dx.doi.org/ 10.1016/j.neurobiolaging.2014.07.026 [DOI] [PubMed] [Google Scholar]
- 167.Maruyama H, Kawakami H. Optineurin and amyotrophic lateral sclerosis. Geriatr Gerontol Int 2013; 13:528-32; PMID:23279185; http://dx.doi.org/ 10.1111/ggi.12022 [DOI] [PubMed] [Google Scholar]
- 168.Tumbarello DA, Waxse BJ, Arden SD, Bright NA, Kendrick-Jones J, Buss F. Autophagy receptors link myosin VI to autophagosomes to mediate Tom1-dependent autophagosome maturation and fusion with the lysosome. Nat Cell Biol 2012; 14:1024-35; PMID:23023224; http://dx.doi.org/ 10.1038/ncb2589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Wong YC, Holzbaur EL. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc Natl Acad Sci U S A 2014; 111:E4439-48; PMID:25294927; http://dx.doi.org/ 10.1073/pnas.1405752111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Fields J, Dumaop W, Rockenstein E, Mante M, Spencer B, Grant I, Ellis R, Letendre S, Patrick C, Adame A, et al.. Age-dependent molecular alterations in the autophagy pathway in HIVE patients and in a gp120 tg mouse model: reversal with beclin-1 gene transfer. J Neurovirol 2013; 19: 89-101; PMID:23341224; http://dx.doi.org/ 10.1007/s13365-012-0145-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Yang L, Yao H, Chen X, Cai Y, Callen S, Buch S. Role of Sigma Receptor in Cocaine-Mediated Induction of Glial Fibrillary Acidic Protein: Implications for HAND. Mol Neurobiol 2015; [Epub ahead of print]; PMID:25631712; http://dx.doi.org/21883374 10.1007/s12035-015-9094-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Ma R, Yang L, Niu F, Buch S. HIV Tat-Mediated Induction of Human Brain Microvascular Endothelial Cell Apoptosis Involves Endoplasmic Reticulum Stress and Mitochondrial Dysfunction. Mol Neurobiol 2014; 2016; 53:132-42; http://dx.doi.org/ 10.1007/s12035-014-8991-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Akay C, Lindl KA, Shyam N, Nabet B, Goenaga-Vazquez Y, Ruzbarsky J, Wang Y, Kolson DL, Jordan-Sciutto KL. Activation status of integrated stress response pathways in neurones and astrocytes of HIV-associated neurocognitive disorders (HAND) cortex. Neuropathol Appl Neurobiol 2012; 38:175-200; PMID:21883374; http://dx.doi.org/ 10.1111/j.1365-2990.2011.01215.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Caporello E, Nath A, Slevin J, Galey D, Hamilton G, Williams L, Steiner JP, Haughey NJ. The immunophilin ligand GPI1046 protects neurons from the lethal effects of the HIV-1 proteins gp120 and Tat by modulating endoplasmic reticulum calcium load. J Neurochem 2006; 98:146-55; PMID:16805804; http://dx.doi.org/ 10.1111/j.1471-4159.2006.03863.x [DOI] [PubMed] [Google Scholar]
- 175.Guo ML, Liao K, Periyasamy P, Yang L, Cai Y, Callen SE, Buch S. Cocaine mediated microglial activation involves the ER stress-Autophagy axis. Autophagy 2015; 11(7):995-1009; PMID:26043790; http://dx.doi.org/18772620 10.1080/15548627.2015.1052205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Alirezaei M, Kiosses WB, Fox HS. Decreased neuronal autophagy in HIV dementia: a mechanism of indirect neurotoxicity. Autophagy 2008; 4:963-6; PMID:18772620; http://dx.doi.org/ 10.4161/auto.6805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Alirezaei M, Kiosses WB, Flynn CT, Brady NR, Fox HS. Disruption of neuronal autophagy by infected microglia results in neurodegeneration. Plos One 2008; 3:e2906; http://dx.doi.org/ 10.1371/journal.pone.0002906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Zhou D, Masliah E, Spector SA. Autophagy is increased in postmortem brains of persons with HIV-1-associated encephalitis. J Infect Dis 2011; 203:1647-57; PMID:21592995; http://dx.doi.org/ 10.1093/infdis/jir163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Kyei GB, Dinkins C, Davis AS, Roberts E, Singh SB, Dong C, Wu L, Kominami E, Ueno T, Yamamoto A, et al.. Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J Cell Biol 2009; 186:255-68; PMID:19635843; http://dx.doi.org/ 10.1083/jcb.200903070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Li JC, Au KY, Fang JW, Yim HC, Chow KH, Ho PL, Lau AS. HIV-1 trans-activator protein dysregulates IFN-gamma signaling and contributes to the suppression of autophagy induction. Aids 2011; 25:15-25; PMID:21099673; http://dx.doi.org/ 10.1097/QAD.0b013e328340fd61 [DOI] [PubMed] [Google Scholar]
- 181.Van Grol J, Subauste C, Andrade RM, Fujinaga K, Nelson J, Subauste CS. HIV-1 Inhibits Autophagy in Bystander Macrophage/Monocytic Cells through Src-Akt and STAT3. PloS One 2010; 5:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Hui L, Chen XS, Haughey NJ, Geiger JD. Role of endolysosomes in HIV-1 Tat-induced neurotoxicity. Asn Neuro 2012; 4:243-52; PMID:22591512; http://dx.doi.org/ 10.1042/AN20120017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Fields J, Dumaop W, Elueteri S, Campos S, Serger E, Trejo M, Kosberg K, Adame A, Spencer B, Rockenstein E, et al.. HIV-1 Tat Alters Neuronal Autophagy by Modulating Autophagosome Fusion to the Lysosome: Implications for HIV-Associated Neurocognitive Disorders. J Neurosci 2015; 35:1921-38; PMID:25653352; http://dx.doi.org/ 10.1523/JNEUROSCI.3207-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Sasaki S. Endoplasmic reticulum stress in motor neurons of the spinal cord in sporadic amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 2010; 69:346-55; PMID:20448480; http://dx.doi.org/ 10.1097/NEN.0b013e3181d44992 [DOI] [PubMed] [Google Scholar]
- 185.Shoulders MD, Ryno LM, Genereux JC, Moresco JJ, Tu PG, Wu C, Yates JR 3rd, Su AI, Kelly JW, Wiseman RL. Stress-independent activation of XBP1s and/or ATF6 reveals three functionally diverse ER proteostasis environments. Cell Reports 2013; 3:1279-92; PMID:23583182; http://dx.doi.org/ 10.1016/j.celrep.2013.03.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Simon-Sanchez J, Dopper EG, Cohn-Hokke PE, Hukema RK, Nicolaou N, Seelaar H, de Graaf JR, de Koning I, van Schoor NM, Deeg DJ, et al.. The clinical and pathological phenotype of C9ORF72 hexanucleotide repeat expansions. Brain 2012; 135:723-35; PMID:22300876; http://dx.doi.org/ 10.1093/brain/awr353 [DOI] [PubMed] [Google Scholar]
- 187.Van Deerlin VM, Leverenz JB, Bekris LM, Bird TD, Yuan W, Elman LB, Clay D, Wood EM, Chen-Plotkin AS, Martinez-Lage M, et al.. TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet Neurol 2008; 7:409-16; PMID:18396105; http://dx.doi.org/ 10.1016/S1474-4422(08)70071-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Gitcho MA, Strider J, Carter D, Taylor-Reinwald L, Forman MS, Goate AM, Cairns NJ. VCP mutations causing frontotemporal lobar degeneration disrupt localization of TDP-43 and induce cell death. J Biol Chem 2009; 284:12384-98; PMID:19237541; http://dx.doi.org/ 10.1074/jbc.M900992200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Deng HX, Chen W, Hong ST, Boycott KM, Gorrie GH, Siddique N, Yang Y, Fecto F, Shi Y, Zhai H, et al.. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 2011; 477:211-5; PMID:21857683; http://dx.doi.org/ 10.1038/nature10353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Hortobagyi T, Troakes C, Nishimura AL, Vance C, van Swieten JC, Seelaar H, King A, Al-Sarraj S, Rogelj B, Shaw CE. Optineurin inclusions occur in a minority of TDP-43 positive ALS and FTLD-TDP cases and are rarely observed in other neurodegenerative disorders. Acta Neuropathologica 2011; 121:519-27; PMID:21360076; http://dx.doi.org/ 10.1007/s00401-011-0813-3 [DOI] [PubMed] [Google Scholar]
- 191.Dickson DW, Kouri N, Murray ME, Josephs KA. Neuropathology of frontotemporal lobar degeneration-tau (FTLD-tau). J Mol Neurosci 2011; 45:384-9; PMID:21720721; http://dx.doi.org/ 10.1007/s12031-011-9589-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Katayama T, Imaizumi K, Honda A, Yoneda T, Kudo T, Takeda M, Mori K, Rozmahel R, Fraser P, George-Hyslop PS, et al.. Disturbed activation of endoplasmic reticulum stress transducers by familial Alzheimer disease-linked presenilin-1 mutations. J Biol Chem 2001; 276:43446-54; PMID:11551913; http://dx.doi.org/ 10.1074/jbc.M104096200 [DOI] [PubMed] [Google Scholar]
- 193.Kudo T, Katayama T, Imaizumi K, Yasuda Y, Yatera M, Okochi M, Tohyama M, Takeda M. The unfolded protein response is involved in the pathology of Alzheimer disease. Ann N Y Acad Sci 2002; 977:349-55; PMID:12480772; http://dx.doi.org/ 10.1111/j.1749-6632.2002.tb04837.x [DOI] [PubMed] [Google Scholar]
- 194.Hui L, Chen X, Haughey NJ, Geiger JD. Role of endolysosomes in HIV-1 Tat-induced neurotoxicity. ASN neuro 2012; 4:243-52; PMID:22591512; http://dx.doi.org/ 10.1042/AN20120017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, Derecki NC, Castle D, Mandell JW, Lee KS, et al.. Structural and functional features of central nervous system lymphatic vessels. Nature 2015; 523(7560):337-41; PMID:26030524; http://dx.doi.org/ 10.1038/nature14432 [DOI] [PMC free article] [PubMed] [Google Scholar]