

ABSTRACT
Lipids stored in lipid droplets are hydrolyzed via either cytosolic lipases or a selective form of macroautophagy known as lipophagy. We recently demonstrated that chaperone-mediated autophagy (CMA) is required for the initiation of lipolysis by either of these independent lipolytic pathways. CMA selectively degrades the lipid droplet proteins perilipins (PLIN) 2 and 3 from the lipid droplet surface, thus, facilitating the recruitment of cytosolic lipases and autophagy effector proteins to the lipid droplets. PLIN2 phosphorylation was observed upon induction of lipolysis, but the phosphorylating kinase and the relation of this phosphorylation with CMA of PLIN2 remained unknown. Here, we report that phosphorylation of PLIN2 is dependent on AMPK and occurs after the interaction of PLIN2 with the CMA chaperone HSPA8/Hsc70. Our results highlight a role for posttranslational modifications in priming proteins to be amenable for degradation by CMA.
KEYWORDS: chaperones, lipid droplets, lysosomes, lysosome-associated membrane protein 2A, perilipins, protein degradation
Introduction
Chaperone-mediated autophagy selectively degrades a subset of cytosolic proteins that bear in their amino acid sequence a pentapeptide motif biochemically related to KFERQ.1,2 This motif is recognized by HSPA8/Hsc70 (heat shock 70 kDa protein 8) that targets the protein to lysosomes.3 At the lysosome, the substrate proteins bind to the CMA receptor, LAMP2A (lysosome-associated membrane protein 2A),4 which then organizes into a multimeric complex for translocation of the substrate into the lumen.5 Unfolding of the substrate is required6 before a lumenal form of HSPA8 completes translocation of the substrate into the lysosomal lumen where it is rapidly degraded.7 The subproteome that can undergo degradation by CMA includes proteins involved in diverse cellular functions such as metabolic enzymes, transcription factors, ubiquitin ligases and subunits of proteases, regulatory kinases, and cell cycle modulators, among others.2
We recently reported 2 proteins that associate with the surface of lipid droplets (LD), perilipin (PLIN) 2 and 3, as the first lipid-anchored proteins to be CMA substrates.8 Perilipins act as a shield for the triglyceride core of LD against the hydrophilic surrounding cytosol. We showed that CMA impairment results in LD accumulation both in vitro and in vivo because removal of PLINs via CMA is a prerequisite before initiation of lipolysis via either cytosolic lipases or lipophagy. This finding places CMA upstream of the 2 major pathways that contribute to intracellular lipid mobilization9 and highlights an important role for this selective type of autophagy in the control of lipid metabolism, also supported by the severe hepatosteatosis observed in mice with hepatic blockage of CMA.10
At the mechanistic level, we found that blocking CMA of PLINs impaired the recruitment of the cytosolic lipase PNPLA2/ATGL and the macroautophagy effector proteins (BECN1/Beclin1, ATG12–ATG5, MAP1LC3B/LC3B) to the LD surface.8 Interestingly, association of HSPA8 with the LD, kiss-and-run events between LD and lysosomes, as well as the formation of the limiting membrane of autophagosomes in LD, seem to occur only in discrete areas of the LD surface. These findings suggest that a subset of PLINs on the LD is targeted for degradation via CMA. In our efforts to identify the determinants of this selective removal of only certain PLIN molecules at the LD, we performed a comparative analysis of posttranslational modifications of PLIN2. We found that induction of lipolysis associates with changes in the phosphorylation status of PLIN2 that are not observed in CMA-incompetent cells.8 Here we perform further analysis of the lipolysis-induced phosphorylation of PLIN2 and provide information on the step in CMA when PLIN2 phosphorylation occurs, a kinase required for this phosphorylation, and an unexpected reciprocal interplay between CMA and this kinase.
Results
As we reported recently,8 a phosphorylated form of PLIN2 became more apparent in cultured fibroblasts when lipolysis was induced after a lipogenic stimulus (oleate; OL). Fig. 1A shows conventional electrophoresis and electrophoresis using Phos-tag to facilitate resolution of phosphorylated variants and reveals an increase in total levels of PLIN2 (1.5+0.1 fold) upon OL treatment and a larger increase (5+1.2 fold) of the phosphorylated form. This phosphorylation was almost undetectable when CMA was blocked by knocking down LAMP2A (L2A− cells), even when lipolysis was forced by changing OL-loaded cells to an OL-free medium (regular medium) (OL>RM), as described previously8 (Fig. 1B-D). Phos-tag electrophoresis did not reveal the presence of phosphorylation variants for PLIN3 under the same conditions, but we observed differences in the bidimensional electrophoretic pattern of this protein in lysosomes when compared with lipid droplets (data not shown). These data suggested that posttranslational modifications could act as a trigger for PLINs to be primed for CMA degradation and prompted us to study in detail the contribution of PLIN2 phosphorylation in this process.
Figure 1.

PLIN2 phosphorylation is AMPK dependent. (A) Total cell lysates from cells cultured with or without OL were subjected to Phos-tag gel electrophoresis and immunoblotted for PLIN2. p-PLIN2: black arrow. Top shows immunoblot for PLIN2 without Phos-tag. (B) Total cell lysates from CTR and L2A− cells treated with OL and incubated with regular medium (RM) after OL treatment. ACTB/β-actin was used as loading control. (C) Levels of total, phosphorylated and unphosphorylated PLIN2 in the same cells as in (B). Values are expressed relative to total levels of PLIN2 in control cells that was given an arbitrary value of 1. n=4. (D) Percentage of p-PLIN2 relative to total PLIN2 levels in the same cells as in (B). n=4. (E, F) Total cell lysates from cells cultured with OL, treated with inhibitors (i) or activators (a) of the indicated kinases (pan-Tyrosine, pan tyrosine kinase inhibitor; MAPK8/JNK1; MAPK14/p38α; MAP2K1/MEK1-MAP2K2/MEK2; SRC/Src) were subjected to Phos-tag gel electrophoresis and immunoblotted for PLIN2. p-PLIN2: black arrow. Representative densitometric values of p-PLIN2 relative to untreated cells upon correction by total protein levels (Ponceau staining) are indicated. Differences with the control (CTR) were significant for *, p < 0.05 and **, p < 0.01.
To identify the kinase(s) behind this lipolysis-induced PLIN2 phosphorylation, we screened with a panel of kinase modulators in control cells upon lipolysis induction (subjected to OL challenge) and found that a reduction in PLIN2 phosphorylation was noticeable upon treatment with compound C, an inhibitor of the AMP-activated protein kinase (AMPK) (Fig. 1E; 62.7+13.5% reduction of phosphorylated [p]-PLIN2, n=3). In contrast, an AMPK activator, A-769662, and a MAPK14/p38α inhibitor enhanced PLIN2 phosphorylation (Fig. 1F); since we observed a reciprocal effect of AMPK activation and inhibition on PLIN2 phosphorylation we focused our study on this kinase. The lower effect of the AMPK activator on PLIN2 phosphorylation when compared with the inhibitor could be due to the hyper-phosphorylated state of PLIN2 during lipolysis. AMPK-dependent phosphorylation of PLIN2 may be required for its degradation, as under lipolysis-inducing conditions we found higher levels of PLIN2 and PLIN2-positive LD in cells treated with the AMPK inhibitor than in untreated cells (Fig. 2A, B). In contrast, treatment of L2A− cells with the AMPK inhibitor did not show a further increase in the already elevated levels of PLIN2 in these cells (Fig. 2C). The lack of additive effect of AMPK inhibition in these cells (Fig. 2C) suggests that the stimulatory effect of AMPK on PLIN2 degradation requires functional CMA. Contrary to the well-described inhibitory effect of AMPK on lipolysis in adipose tissue, activation of AMPK in many other tissues leads to increased lipid oxidation.11 We propose that AMPK facilitates this lipid mobilization, at least in part, by triggering PLIN2 degradation by CMA. Interestingly, treatment with the AMPK inhibitor did not prevent colocalization of PLIN2 and HSPA8 in LD; on the contrary, we observed increased PLIN2-HSPA8 colocalization in cells treated with the AMPK inhibitor (Fig. 2C). These results suggest that association of HSPA8 with LD precedes PLIN2 phosphorylation.
Figure 2.

AMPK inhibition blocks PLIN2 CMA. (A) Immunostaining for PLIN2 in cells treated or not with AMPKi. Boxed area is shown at higher magnification on the right. (B) Average puncta/cell. n>50 cells from (A). (C) Immunoblots of total cell lysates from CTR and L2A- cells treated or not with AMPKi. p-PLIN2: black arrow. Representative densitometric values of p-PLIN2 relative to untreated (CTR) cells are indicated. ACTB/β-actin was used as loading control. (D) Coimmunostaining for PLIN2 and HSPA8 in cells treated or not with AMPKi. Colocalized pixels are in white. Boxed area is shown at higher magnification. (E) Percentage colocalization of PLIN2 with HSPA8. n>50 cells from (D). Differences with untreated were significant for *, p < 0.05 and **, p < 0.01.
We found that the AMPK-dependent phosphorylation of PLIN2 is tightly related to the status of CMA. Thus, PLIN2 phosphorylation was almost undetectable even upon induction of lipolysis in cells incompetent for CMA (L2A− cells) (Figs. 1B and 2C) suggesting that AMPK and CMA functions may be interdependent. Although further studies are needed to investigate the reasons of the failure to phosphorylate PLIN2 in the absence of functional CMA, immunoblot for AMPK (catalytic subunits PRKAA1/α1 and PRKAA2/α2) revealed that L2A− cells, even when placed in lipolysis-inducing conditions, displayed markedly reduced levels of phosphorylated AMPK (51.52+8.9% reduction), and to a lesser extent of total AMPK (27.6+6.3% reduction) (Fig. 3A, B). We confirmed that these changes were not due to off-target effects of the L2A knockdown, as we also observed a similar reduction in phosphorylated and total AMPK in embryonic fibroblasts (Fig. 3C; 77.8% and 30.4% reduction in p-AMPK and AMPK, respectively) and liver (Fig. 3D; 41.2% and 26.8% reduction in p-AMPK and AMPK, respectively) of mouse knocked out for L2A when compared to wild-type littermates. The relative phosphorylation of AMPK (upon correcting for total AMPK levels) was reduced by 68.1% and 19.6% in fibroblasts and liver of the L2A knockout (KO) mice, respectively, supporting the conclusion that reduction in AMPK phosphorylation was not merely resulting from the lower levels of the kinase in these cells.
Figure 3.

AMPK in CMA-defective cells. (A) Immunoblots of total cell lysates from CTR and L2A- cells. ACTB/β-actin was used as loading control. (B) Levels of p-AMPK and AMPK relative to CTR cells from (A). Values are expressed relative to control cells that were given an arbitrary value of 1. n=3. (C) Immunoblots of total cell lysates from WT and L2A KO mouse embryonic fibroblast cells. (D) Immunoblots of liver homogenates from 24-h starved WT and L2A KO mice. (E) Levels of the indicated mRNA in livers from WT and L2A KO mice fed or 24-h starved. n=3. (F) Immunoblots of total cell lysates from CTR and L2A- cells treated or not with lactacystin (Lacta). (G) Immunoblots of total cell lysates from CTR and L2A- cells treated or not with lysosomal inhibitors (ammonium chloride and leupeptin, NL). Two independent experiments are shown. Differences with CTR were significant for *, p < 0.05 and **, p < 0.01. Ubq, ubiquitin.
Lower levels of AMPK in L2A− cells are not a consequence of transcriptional downregulation of this kinase, since levels of the 2 mRNA transcripts that code for AMPK (Prkaa1 and Prkaa2) were comparable in livers of L2A KO mice, even when lipolysis was induced by starvation (Fig. 3E; note: we did not find differences in the mRNA levels of the genes that code for the other AMPK catalytic subunit (Prkaca and Prkacb)) or for the 2 AMPK regulatory subunits (isoforms of Prkab and Prkag) (data not shown). Inhibition of either proteasome degradation (Fig. 3F) or lysosomal proteolysis (Fig. 3G) did not reveal accelerated degradation of AMPK in L2A− cells that could explain its lower levels in these cells. In fact, compared with other cytosolic proteins (ubiquitinated proteins and MAP1LC3B-II, shown in Fig. 3F and G, as control of the efficiency of the inhibitors), and in contrast to the rapid degradation of AMPK by the proteasome recently described in tumor cells,12 AMPK was remarkably stable in both groups of cells under our experimental conditions. Future studies are needed to determine the reasons for the reduced levels of p-AMPK and AMPK upon CMA blockage.
Taken together, our studies indicate PLIN2 phosphorylation is dependent on AMPK and occurs after the recognition of PLIN2 by HSPA8 but prior to its degradation via CMA (Fig. 4).
Figure 4.
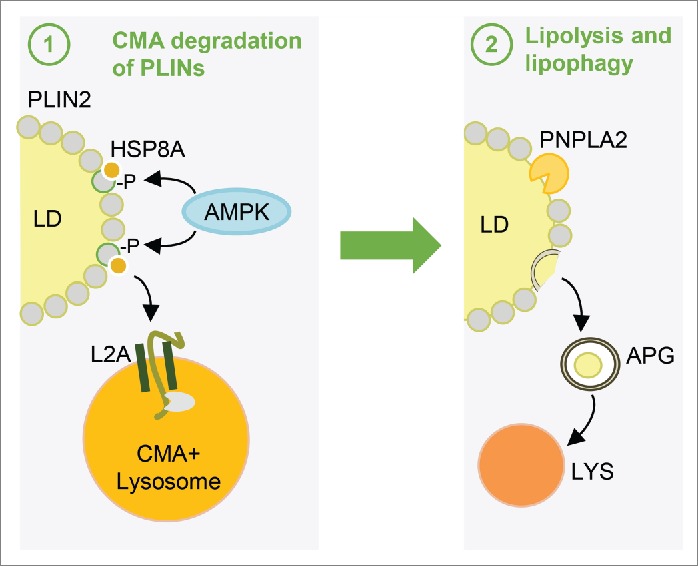
AMPK-dependent PLIN2 phosphorylation precedes lipolysis. Schematic working model depicting the proposed steps in recognition and degradation of PLIN2 by CMA: (1) HSPA8 arrives at the lipid droplet (LD) and we propose that its interaction there with PLIN2 promotes AMPK-mediated phosphorylation of PLIN2. This phosphorylated variant (p-PLIN2) is the one released from the LD and targeted for degradation by CMA. (2) The CMA-dependent removal of PLIN2 now allows the recruitment of a cytosolic lipase (PNPLA2/ATGL) and macroautophagy effectors (ATG proteins). Arrival of these proteins results in the breakdown of triglycerides in the LD (by PNPLA2) or in the delivery via macroautophagy (by ATG proteins) of portions of the LD to lysosomes (Lys) for breakdown. APG, autophagosomes.
Discussion
In recent years, numerous studies have supported tight connections between autophagy and lipid metabolism.9,13 Added to the active contribution of macroautophagy to the direct mobilization of lipid stores, through what is known as lipophagy,14 we have recently demonstrated that CMA can also modulate intracellular lipolysis by selectively removing PLINs from discrete regions from the surface of LDs.8 The follow up studies reported here reveal the requirement of an AMPK-mediated phosphorylation event in this selective degradation of PLINs and subsequent triggering of lipolysis.
The dependence on AMPK activity for the degradation of PLIN2 by CMA and the fact that we found that this degradation is required prior to lipolysis,8 reveal an additional mechanism that contributes to the known role of AMPK in stimulating lipolysis in organs such as liver upon starvation.11 Activation of AMPK under these conditions would facilitate both priming of a subset of PLIN2 molecules for their removal by CMA (Fig. 4) as well as activation of downstream lipolytic mechanisms, such as the recently described AMPK-dependent stimulation of macroautophagy and fatty acid oxidation.11,15,16
The decrease in PLIN2 phosphorylation upon CMA blockage and the recruitment of HSPA8 in a phosphorylation-independent manner were unexpected findings. We originally hypothesized that phosphorylation of PLIN2 may be required to unmask the HSPA8-recognizing motif; however, our data support the idea that HSPA8 is recruited to LDs even when phosphorylation of PLIN2 is inhibited. Our current model is that binding of HSPA8 to PLIN2 may facilitate its AMPK-phosphorylation and subsequent release for lysosomal degradation. If that is the case, HSPA8 could be considered a checkpoint for the release and degradation of PLIN2, and inhibiting HSPA8 binding may be an efficient way to block lipolysis. Due to the multiple cellular functions of HSPA8, studies knocking down or inhibiting this chaperone are difficult to interpret, and future development of inhibitors of only the pool of HSPA8 dedicated to CMA are needed to validate this possible checkpoint role of HSPA8 in lipolysis. However, it is also possible that the initial association of HSPA8 with LD occurs through a different binding partner, and its binding with PLIN2 once at the LD is triggered by PLIN2 phosphorylation.
Although we also predicted that blockage of CMA would lead to accumulation of phosphorylated PLIN2 at the LD, our studies have revealed that this phosphorylation event and CMA activity are interdependent. We propose that if the release of PLINs from the LD into the cytosol for CMA degradation is triggered by the AMPK-phosphorylation, then the decrease in the pool of activated AMPK in absence of competent CMA may be a safety mechanism to prevent accumulation of these lipid-binding PLIN proteins in the cytosol. The basis for AMPK inactivation in CMA-deficient cells requires future investigation. We have discarded changes at the transcriptional level or in the stability of the protein; however, changes in translation, synthesis or posttranslational modifications that could make the protein no longer detectable by the antibodies used in this study, are all plausible explanations. Of high interest, also, is the elucidation of the basis of the interdependence between CMA activity and AMPK-dependent PLIN2 phosphorylation. An attractive possibility in this respect is that signals originating through active CMA degradation are normally sensed by AMPK and/or its modulatory kinases and phosphatases to regulate the level of AMPK activation.
Our study provides further support for a physiological contribution of CMA to the regulation of lipid metabolism and opens the exciting possibility that additional effectors and regulators of lipolysis could also be part of the subproteome degraded via CMA. Blockage of CMA in liver, to reproduce the reduced activity of this pathway observed in aging, causes severe hepatosteatosis10 while pharmacologically activating CMA, at least in vitro, reduces lipid accumulation associated with acute lipogenic challenges.8 Enhancing CMA activity and/or the cue for degradation of PLINs by CMA identified in this study could be attractive approaches to ameliorate steatosis and preserve adequate lipohomeostasis.
Materials and methods
Animals and cell culture
The mouse model with a conditional deletion for LAMP2A (L2A) in the liver was generated as previously described.10 All mice were housed under pathogen-free conditions and handled according to protocols approved by the Institutional Animal Care and Use Committee of Albert Einstein College of Medicine. NIH3T3 mouse fibroblasts (American Type Culture Collection, 1658) were cultured in Dulbecco's modified Eagle's medium (Sigma-Aldrich, D5648) with 10% heat-inactivated newborn calf serum (HyClone, SH30401), 1% penicillin/streptomycin/fungizone (Invitrogen, 15240). Mouse embryonic fibroblasts from L2A KO mice and NIH3T3 clones stably RNA-interfered for L2A (L2A−) were generated as described.17,18
Chemicals and treatments
Cells were treated with 0.06 mM oleate conjugated to albumin (OL; Sigma-Aldrich, O3008). After 24 h, OL was washed off with Hank's balanced salt solution (Invitrogen, 14185052) and replaced by Dulbecco's modified Eagle's medium with serum (regular medium/RM; [OL>RM]) for 16 h. The MTOR inhibitor rapamycin (100 nM) was from Calbiochem (553210) and the AMPK inhibitor compound C (10 μM) was from Selleck Chemicals (S7306). Inhibitors of PRKA/PKA (KT5720, 1 μM), pan-Tyrosine kinase (Apatinib, 2 μM), MAPK8/JNK1 (SP600125, 2 μM), MAPK14/p38α (VX-702, 2 μM), MAP2K1/MEK1-MAP2K2/MEK2 (U0126, 2 μM) and SRC/Src tyrosine kinase (KX2-391, 2 μM) and the activator of AMPK (A769662, 2 μM) were from Selleck Chemicals (L1200) and were added to cultured cells for 16 h. Lactacystin (5 μM; Enzo Life Sciences, BML-PI104-0200) and a mixture of 20 mM ammonium chloride (Sigma-Aldrich, A9434) and 100 μM leupeptin (Fisher Scientific, BP2662100) were added for 12 h. The antibodies used were as follows: anti-PLIN2 (Progen Biotechnik, GP-40), anti-L2A (Invitrogen, 512200), anti- HSPA8/Hsc70 (Novus Biologicals, 13D3), anti-LC3 (Cell Signaling Technology, 2775), anti-PRKAA/AMPKα (Cell Signaling Technology, 2532), anti-p-PRKAA/AMPKα (T172; Cell Signaling Technology, 2531), anti-K48-specific ubiquitin (Millipore, 05-1307), and anti-ACTB/β−actin (Abcam, ab6276).
Immunoblot
Cells were lysed in in RIPA buffer (150 mM NaCl, 1% NP-40 (American Bioanalytical, AB01425), 0.5% NaDoc (Sigma-Aldrich, D6750), 0.1% SDS (American Bioanalytical, AB01920), 50 mM Tris, pH 8) containing protease and phosphatase inhibitors (Sigma-Aldrich, P5726, P0044). After SDS-PAGE, samples were transferred to nitrocellulose membrane and immunoblotting was performed following standard procedures.19 Phos-tag PAGE was performed according to the manufacturer's instructions (Wako) using 100 μM Phos-tag (Fisher Scientific, NC0232095). Proteins recognized by the specific antibodies were visualized using peroxidase-conjugated secondary antibodies and chemiluminescent reagent (PerkinElmer, NEL104001EA) in an LAS-3000 Imaging System (Fujifilm, Tokyo, Japan).
Immunofluorescence microscopy
Cells grown on coverslips were fixed (30 min with 4% paraformaldehyde for PLIN2 staining alone or 2 min with pre-chilled methanol for HSPA8 staining), permeabilized and blocked with 1% BSA (American Bioanalytical, AB00440), 0.01% Triton X-100 (Sigma-Aldrich, X100) in phosphate-buffered saline (137 mM sodium chloride, 3 mM potassium chloride, 7 mM disodium hydrogen phosphate, 11 mM dipotassium hydrogen phosphate, pH 7.4). Coverslips were incubated with primary antibody and secondary antibody conjugated to Alexa Fluor 488 or 647 (Invitrogen, A-11073, A-21238) in 0.1% BSA in phosphate-buffered saline at room temperature for 1 h each and mounted in DAPI-Fluoromount-G (Southern Biotechnology, 0100-20) to stain the nucleus. All images were acquired with an Axiovert 200 fluorescence microscope (Carl Zeiss Microscopy, Thornwood, NY) with ×63 objective and 1.4 numerical aperture, mounted with an ApoTome.2 slider, and prepared using Adobe Photoshop CS3 (Adobe Systems) and ImageJ (NIH). The number of particles/puncta per cell was quantified using the ‘analyze particles' function of ImageJ after thresholding in non-saturated images. The percentage of colocalization using Manders' coefficient was determined by ‘JACoP’ plugin in ImageJ after thresholding of individual frames.20 Where indicated, the colocalized pixels were highlighted using the ‘colocalization’ plugin of ImageJ.
Other methods
For immunoblotting, protein concentration was determined by the Lowry method 21 using bovine serum albumin as a standard. Transcriptional data were obtained from the microarray analysis for livers from fed and 24-h starved mice deposited in the NCBI Gene Expression Omnibus database with accession number GSE49553.
Statistical analyses
All values are reported as mean ± standard error of the mean (SEM) and the statistical significance of the difference between experimental groups was determined using individual values by 2-tailed unpaired Student ttest.
Abbreviations
- AMPK
AMP-activated protein kinase
- CMA
chaperone-mediated autophagy
- HSPA8/HSC70
heat shock 70 kDa protein 8
- KO
knockout
- L2A
LAMP2A
- LAMP
lysosome-associated membrane protein
- LD
lipid droplets
- OL
oleate
- PLIN
perilipin
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by grants from the National Institutes of Health AG021904, AG031782, AG038072, DK098408 and the generous support of Robert and Renée Belfer.
References
- 1.Dice JF. Peptide sequences that target cytosolic proteins for lysosomal proteolysis. Trends Biochem Sci 1990; 15:305-9; PMID:2204156; http://dx.doi.org/ 10.1016/0968-0004(90)90019-8 [DOI] [PubMed] [Google Scholar]
- 2.Kaushik S, Cuervo AM. Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends Cell Biol 2012; 22:407-17; PMID:22748206; http://dx.doi.org/ 10.1016/j.tcb.2012.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chiang H, Terlecky S, Plant C, Dice JF. A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science 1989; 246:382-5; PMID:2799391; http://dx.doi.org/ 10.1126/science.2799391 [DOI] [PubMed] [Google Scholar]
- 4.Cuervo AM, Dice JF. A receptor for the selective uptake and degradation of proteins by lysosomes. Science 1996; 273:501-3; PMID:8662539; http://dx.doi.org/ 10.1126/science.273.5274.501 [DOI] [PubMed] [Google Scholar]
- 5.Bandyopadhyay U, Kaushik S, Varticovski L, Cuervo AM. The chaperone-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol Cell Biol 2008; 28:5747-63; PMID:18644871; http://dx.doi.org/ 10.1128/MCB.02070-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Salvador N, Aguado C, Horst M, Knecht E. Import of a cytosolic protein into lysosomes by chaperone-mediated autophagy depends on its folding state. Journal of Biological Chemistry 2000; 275:27447-56; PMID:10862611 [DOI] [PubMed] [Google Scholar]
- 7.Agarraberes F, Terlecky S, Dice J. An intralysosomal hsp70 is required for a selective pathway of lysosomal protein degradation. J Cell Biol 1997; 137:825-34; PMID:9151685; http://dx.doi.org/ 10.1083/jcb.137.4.825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaushik S, Cuervo AM. Degradation of lipid droplet-associated proteins by chaperone-mediated autophagy facilitates lipolysis. Nat Cell Biol 2015; 17:759-70; PMID:25961502; http://dx.doi.org/ 10.1038/ncb3166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Singh R, Cuervo AM. Autophagy in the cellular energetic balance. Cell Metab 2011; 13:495-504; PMID:21531332; http://dx.doi.org/ 10.1016/j.cmet.2011.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schneider JL, Suh Y, Cuervo AM. Deficient chaperone-mediated autophagy in liver leads to metabolic dysregulation. Cell Metab 2014; 20:417-32; PMID:25043815; http://dx.doi.org/ 10.1016/j.cmet.2014.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Long YC, Zierath JR. AMP-activated protein kinase signaling in metabolic regulation. J Clin Invest 2006; 116:1776-83; PMID:16823475; http://dx.doi.org/ 10.1172/JCI29044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pineda CT, Ramanathan S, Fon Tacer K, Weon JL, Potts MB, Ou YH, White MA, Potts PR. Degradation of AMPK by a cancer-specific ubiquitin ligase. Cell 2015; 160:715-28; PMID:25679763; http://dx.doi.org/ 10.1016/j.cell.2015.01.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaur J, Debnath J. Autophagy at the crossroads of catabolism and anabolism. Nat Rev Mol Cell Biol 2015; 16:461-72; PMID:26177004; http://dx.doi.org/ 10.1038/nrm4024 [DOI] [PubMed] [Google Scholar]
- 14.Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ. Autophagy regulates lipid metabolism. Nature 2009; 458:1131-5; PMID:19339967; http://dx.doi.org/ 10.1038/nature07976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Egan D, Kim J, Shaw RJ, Guan KL. The autophagy initiating kinase ULK1 is regulated via opposing phosphorylation by AMPK and mTOR. Autophagy 2011; 7:643-4; PMID:21460621; http://dx.doi.org/ 10.4161/auto.7.6.15123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Herms A, Bosch M, Reddy BJ, Schieber NL, Fajardo A, Ruperez C, Fernandez-Vidal A, Ferguson C, Rentero C, Tebar F, et al.. AMPK activation promotes lipid droplet dispersion on detyrosinated microtubules to increase mitochondrial fatty acid oxidation. Nat Commun 2015; 6:7176; PMID:26013497; http://dx.doi.org/ 10.1038/ncomms8176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schneider J, Villarroya J, Diaz A, Patel B, Urbanska A, Thi M, Villarroya F, Santambrogio L, Cuervo A. Loss of hepatic chaperone-mediated autophagy accelerates proteostasis failure in aging. Aging Cell 2015; 14(2):249-64; PMID:25620427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Massey AC, Kaushik S, Sovak G, Kiffin R, Cuervo AM. Consequences of the selective blockage of chaperone-mediated autophagy. Proc Nat Acad Sci USA 2006; 103:5905-10; PMID:16585532; http://dx.doi.org/ 10.1073/pnas.0507436103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci 1979; 76:4350-4; PMID:388439; http://dx.doi.org/ 10.1073/pnas.76.9.4350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bolte S, Cordelieres FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc 2006; 224:213-32; PMID:17210054; http://dx.doi.org/ 10.1111/j.1365-2818.2006.01706.x [DOI] [PubMed] [Google Scholar]
- 21.Lowry O, Rosebrough N, Farr A, Randall R. Protein measurement with the Folin phenol reagent. J Biol Chem 1951; 193:265-75; PMID:14907713 [PubMed] [Google Scholar]