

ABSTRACT
Meiosis is a special type of cellular renovation that involves 2 successive cell divisions and a single round of DNA replication. Two major degradation systems, the autophagy-lysosome and the ubiquitin-proteasome, are involved in meiosis, but their roles have yet to be elucidated. Here we show that autophagy mainly affects the initiation of meiosis but not the nuclear division. Autophagy works not only by serving as a dynamic recycling system but also by eliminating some negative meiotic regulators such as Ego4 (Ynr034w-a). In a quantitative proteomics study, the proteasome was found to be significantly upregulated during meiotic divisions. We found that proteasomal activity is essential to the 2 successive meiotic nuclear divisions but not for the initiation of meiosis. Our study defines the roles of autophagy and the proteasome in meiosis: Autophagy mainly affects the initiation of meiosis, whereas the proteasome mainly affects the 2 successive meiotic divisions.
KEYWORDS: autophagy, meiosis, proteasome, quantitative proteomics, yeast
Introduction
Meiosis is a special type of cell division that is necessary for sexual reproduction in eukaryotes. After a single round of DNA replication following 2 successive cell divisions, meiosis results in highly specific gametes or spores. Yeast sporulation involves 2 overlapping processes, meiosis and spore morphogenesis. The process of yeast sporulation is very similar to that of gametogenesis in mammals. The budding yeast Saccharomyces cerevisiae is a very good model for deciphering the molecular mechanism underlying gametogenesis, particularly for the evolutionarily conserved meiotic process. Meiosis is regulated by various genes at the transcriptional, translational, and posttranslational levels.1,2 Ubiquitination and ubiquitin-like modifications are 2 important protein post-translational modifications that play very important roles in meiosis. The well-known anaphase-promoting complex/cyclosome is an E3 ubiquitin ligase that mediates the multiple ubiquitination of substrates and triggers their proteolysis through the 26S proteasome during meiosis.3,4 In addition, many other ubiquitin-related modifications and ubiquitin-like modifications are involved in the regulation of meiosis.5,6 Thus, proteolysis or cellular degradation is a key mechanism that drives the events of meiosis.
There are 2 major cellular degradation systems in eukaryotic cells, the ubiquitin-proteasome system (UPS) and the autophagy–lysosome system. Usually, the UPS is the major cellular pathway for the degradation of short-lived proteins and nonfunctional misfolded proteins, whereas autophagy is regarded as the primary intracellular catabolic mechanism for degrading and recycling long-lived proteins and organelles.7,8 Depending on the pathway by which the cargo is delivered to the lysosome or vacuole, autophagy is divided into roughly 3 main types: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA).9 Among these pathways, macroautophagy (hereafter referred to as autophagy) is thought to be the major type of autophagy, and it has been investigated more extensively than the other 2 types. During autophagy, a small portion of cytoplasmic proteins, organelles, or other materials are sequestered by phagophores, which expand and close to form autophagosomes. After fusion with the lysosome, the cytoplasmic cargos are degraded by the resident hydrolases of autolysosomes. In budding yeast, more than 40 autophagy-related (ATG) genes have been identified,10 and most of them are conserved from yeast to human. The ATG genes were originally identified as genes that are involved in yeast sporulation, which has been confirmed by large-scale screening.11,12 Actually, before the molecular mechanism of autophagy was uncovered, it was found that vacuolar protease activity is required for yeast meiosis, thus hinting that autophagy might also be involved in sporulation.13 However, up to now, the role of Atg proteins in sporulation and how these 2 degradation systems coordinately drive meiotic progression have not be elucidated.
In this study, we found that autophagic activity was higher at early meiosis, and some ATG genes, such as ATG8 and ATG12, are essential to budding yeast sporulation. Knocking out ATG12 prevents premeiotic DNA replication. To determine the role of autophagy in meiosis, we applied quantitative proteomics to a yeast strain that could undergo both meiotic divisions synchronously. A total of 381 differentially expressed proteins were found at various meiotic stages. Among these proteins, the degradation of 4 proteins was found to be dependent on autophagy during the premeiotic DNA replication process, and the overexpression of one (EGO4) of these genes significantly slowed down the meiotic progression, indicating that this gene has a negative effect on meiosis. We further confirmed that Ego4 was selectively degraded by autophagy at the prophase of meiosis I. During the prophase of meiosis, Ego4 mainly interacts with Acs1, suggesting that it might negatively regulate premeiotic DNA replication by either decreasing histone acetylation or changing the central carbon metabolism. In addition, the quantitative proteomics results showed that almost all proteasome components were significantly upregulated after the pachytene stage, which was further confirmed by western blotting. Functional study results suggested that proteasomal activity is essential to the 2 successive meiotic nuclear divisions but not for the initiation of meiosis. Together, our studies define the roles of 2 major degradation systems in meiosis: autophagy mainly takes part in premeiotic DNA replication by eliminating some of the negative regulators of meiosis, while the proteasome mainly affects the 2 successive meiotic divisions. Both of these 2 systems coordinately drive meiotic progression.
Results
Autophagy mainly participates in the initial stage of yeast meiosis
Although there was evidence to show that autophagy is required for yeast meiosis,2,8,13,14 the functional role of autophagy in yeast meiosis is still unclear. To understand the role of autophagy in yeast meiosis, a SK1 background yeast strain which can sporulate with pretty high efficiency15,16 was used to investigate the autophagic activity during yeast meiosis. The meiosis of this strain could be synchronized with an inducible allele of NDT80.16 Briefly, the open reading frame of a transcription factor (NDT80) that is required for progression out of the pachytene stage and into meiosis I was placed under the control of an inducible GAL1-10 promoter, thus, transcription from the GAL1-10 promoter could only be induced by the addition of estrogen to the medium in a strain expressing the Gal4-estrogen receptor fusion protein (Gal4.ER). After transfer into sporulation medium (SPM, 1% KAc) without β-estradiol, GAL4.ER GAL-NDT80 cells failed to undergo any meiotic divisions and arrested in the pachytene stage. When 1 μM β-estradiol was added into the medium, GAL4.ER GAL-NDT80 cells underwent both meiotic divisions synchronously (0 h, meiosis initiation; 6 h, pachytene; 7.5 h, metaphase I; 8 h, anaphase I; 8.5 h, metaphase II; and 9.5 h, anaphase II) (Fig. S1).16
To monitor the autophagic activity during yeast meiosis, p1K-GFP-ATG8 plasmid was transformed into this yeast strain. After being labeled the pre-sporulated cells with FM 4–64 (vacuole membrane dye), cells were diluted into 1% KAc for sporulation, samples were collected from different sporulation time points, and GFP-Atg8 signal was then detected by fluorescence microscopy. The puncta-like GFP-Atg8 was sequestered into the vacuole after transferring into the sporulation medium, and GFP-Atg8 intensity increased very quickly at the prophase of meiosis (before 6 h). While entering the meiotic division process (after 7.5 h), the GFP signal intensity decreased and went back to the initial stage at around 9.5 h (Fig. 1A). GFP-Atg8 processing assay17 showed that the free GFP was quickly accumulated when yeast cells were transferred into SPM, peaked at pachytene stage (6 h), then decreased to some extent during meiotic nuclear division (Fig. 1B). These results suggest that it is most likely autophagy participates in the early stage of yeast meiosis.
Figure 1.
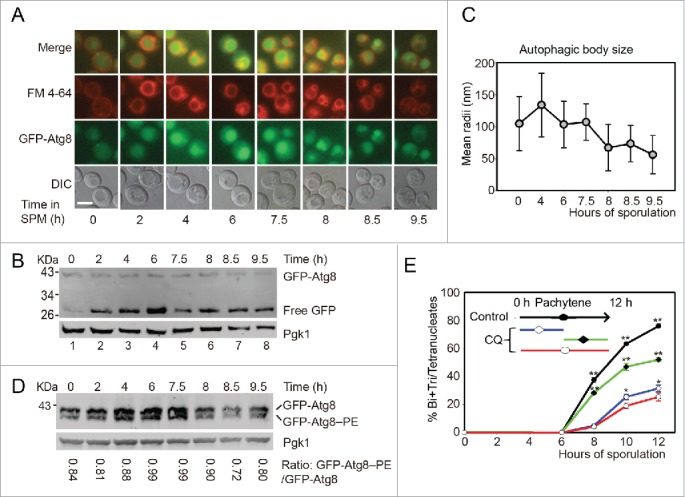
Autophagy mainly participates in the initial stage of yeast meiosis. (A) GFP-Atg8 localization during meiosis. The p1K-GFP-ATG8 plasmid under the control of the ATG8 promoter was introduced into the SK1 background strain. Cells were labeled with FM 4–64 for 5 min then transferred into SPM for sporulation. Samples were collected at different time points and washed 3 times with PBS, then immediately observed by fluorescence microscopy. Scale bar: 5 μm. DIC, differential interference contrast. (B) GFP-Atg8 processing during meiosis. Yeast cells harboring the p1K-GFP-ATG8 plasmid were sporulated and collected from different sporulation time points, and used to generate protein extracts; GFP-Atg8 and free GFP were detected by western blotting with anti-GFP antibody. Lanes 1–8 represent samples at 0, 2, 4, 6, 7.5, 8, 8.5, and 9.5 h in SPM, respectively. (C) Estimation of autophagic body size formed at the indicated sporulation time points. The autophagic body size estimation was followed by a protocol described in the methods. The error bars were the standard deviation (SD) of the radius. (D) GFP-Atg8–PE conjugation during meiosis. The p1K-GFP-ATG8 plasmid was introduced into the SK1 background pep4Δ strain. After sporulation and sample collection, the PE conjugated (GFP-Atg8–PE) and unconjugated (GFP-Atg8) forms were detected by western blotting with anti-GFP antibody. The ratios of conjugated and unconjugated GFP-Atg8 are shown on the bottom of each lane. (E) Effect of CQ treatment on yeast sporulation at different meiotic stages. 200 mM CQ was added into SPM at 0 h then cells were washed with fresh SPM after 5 h (blue line) induction, or CQ was added after 5 h induction (green line). The red line shows the sporulation rate of cells treated with CQ from 0 h to 12 h. Data are presented as the mean ± SD. Asterisk indicates statistically significant difference in comparison with the 0–12 h treated samples. *, P < 0 .05; **, P < 0 .01.
To get a detailed view of the vacuole, transmission electron microscopy (TEM) was used to monitor the dynamics of vacuole and autophagosome of yeast cells during meiosis. Since autophagic bodies (AB) were quickly degraded in wild-type yeast strains, we cannot directly monitor its dynamics during meiosis. Pep4 is a vacuolar aspartyl protease, which is required for the breakdown of autophagic bodies, the deletion of this gene causes AB accumulation in the vacuole lumen,18 and we found that the meiotic division was not affected by the deletion of this gene (data not shown). We then monitored the AB dynamics during meiosis in this pep4Δ strain. Our TEM observation showed that the vacuole was almost empty in rich medium (YPD, 1% yeast extract, 2% peptone, 2% glucose) (Fig. S2A). After transferring into SPM, ABs formed and accumulated quickly at the early stages of meiosis. The mean radii of ABs decreased when yeast cells entered into the meiotic division stages (Fig. 1C, Fig. S2, S3). In addition, we found that the lipid phosphatidylethanolamine (PE) conjugated form of Atg8 (GFP-Atg8–PE) was higher at the beginning of meiosis, while the ratio of GFP-Atg8–PE/GFP-Atg8 was decreased during meiotic division (Fig. 1D). So consistent with previous fluorescent microscopic observation, these results also support how autophagy participates in the early stages of yeast meiosis.
To examine whether autophagy is functionally important to the early meiotic stage, we treated yeast cells with 200 μM chloroquine (CQ, an inhibitor of autophagy-lysosome pathway) at different meiotic stages. CQ treatment for first 5 h then washed the CQ off with fresh medium (Fig. 1E, blue line) resulted in very similar sporulation rate to those CQ treatments for 12 h (Fig. 1E, red line). While 6 h CQ treatment from pachytene stage only resulted in the sporulation efficiency dropping from 76.03% to 52.02%, which was significantly higher than those first 5 h or 12 h treatments (Fig. 1E, green line). These results suggest that the autophagic activity is mainly required during the early stage of yeast meiosis but not the meiotic division stages.
ATG12 deletion mainly affects premeiotic DNA replication
Because most of the screenings for meiosis-related genes were carried out in an S288C background yeast strain,11,12 and the sporulation rate of this strain is not very high, we created ATGs deletion strains in the SK1 background, and then tested the effect of ATGs deletion on yeast meiosis. Atg12 and Atg8 are 2 ubiquitin-like proteins that are essential for autophagy.19 After transferring wild-type strains into liquid SPM at various times, the yeast began to sporulate very quickly, an almost 100% sporulation rate was achieved in approximately 24 h, but either ATG12 or ATG8 deletion significantly repressed meiosis (Fig. 2A, Fig. S4A). The deletion of ATG12 resulted in GFP-Atg8 protein could not be transferred into the vacuole during the meiosis induction process (Fig. 2B) and the processing of GFP-Atg8 was significantly retarded (Fig. S4B), suggesting that the autophagic flux was disrupted in this strain. Although approximately 20% of atg12Δ cells finished one or 2 rounds of meiotic nuclear division after 24 h (Fig. 2A), the sporulated cells could not form mature spores (Fig. 2C). These results are in agreement with the previously reported data, which stated that ATG12 knockout leads to the absence of spore formation.11 Thus, we concluded that ATG12 is essential to yeast sporulation.
Figure 2.
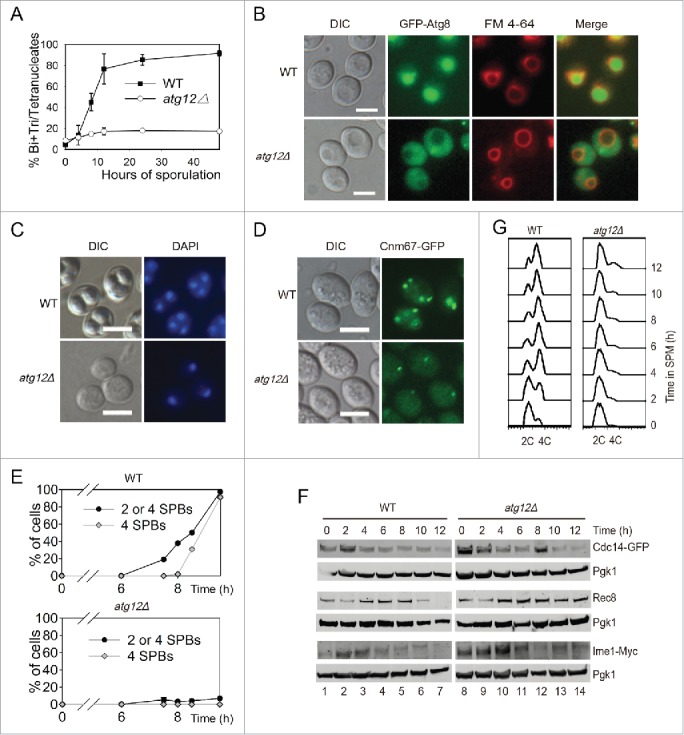
ATG12 deletion yeast cells arrested at the early stage of meiosis. (A-B) Sporulation defects in atg12Δ cells. (A) WT and atg12Δ strains were induced to sporulate at 30°C by transferring into SPM at the indicated times, and cells with 2–4 nuclei were counted as sporulated. (B) GFP-Atg8 localization after transfer into SPM. WT and atg12Δ cells expressing GFP-Atg8 was labeled by FM 4–64 then sporulated in SPM. After transfer into SPM for 4 h, cells were collected and immediately observed by fluorescence microscopy. Scale bars: 5 μm. DIC, differential interference contrast (C) DNA content of WT and atg12Δ strains during meiosis. Samples were collected 48 h after transfer to SPM. Scale bars: 10 µm. (D, E) SPBs failed to segregate in atg12Δ strains during meiosis. (D) SPBs were studied by fluorescence microscopy in WT and atg12Δ strains during meiosis. Scale bars: 5 µm. (E) Quantitative analysis of the SPBs during WT and atg12Δ cell meiosis. The error bar shows the SD of 3 independent experiments. (F) Effect of ATG12 knockout on Cdc14, Rec8 and Ime1 expression. Cdc14 was fused with GFP and detected with a GFP antibody. Ime1 was tagged with MYC for western blotting. Cells were transferred into SPM, and the same amount of samples was collected for protein extraction. Cdc14-GFP, Rec8 and Ime1-MYC were analyzed by western blotting. Lanes 1–14 represent samples at 0, 2, 4, 6, 8, 10, 12 h in SPM for WT and atg12Δ cells, respectively. (G) Premeiotic DNA replication was retarded in the atg12Δ strain during meiosis. The DNA content of WT and atg12Δ cells was analyzed by flow cytometry at different sporulation time points.
Because most of the atg12Δ cells only contained a single nucleus even after 24 h induction (Fig. 2B), the first meiotic division did not occur. We then tested whether ATG12 knockout affected the formation of the spindle pole bodies (SPBs) or not. We fused green fluorescent protein (GFP) with the SPB component CNM67 at the C terminus in either WT or atg12Δ strains and then counted the number of SPBs by fluorescent microscopy (Fig. 2D, E). In the WT strains, after 6 h induction, 2–4 SPBs in a single cell appeared very quickly with more than 90% of the cells exhibiting SPBs at 9.5 h. In contrast, almost no cells had 2–4 SPBs in the atg12Δ strains even after 9.5 h induction. This result suggests that autophagy plays a role in meiosis prior to the formation of SPBs.
Prophase in meiosis is very different from prophase in mitosis. Unique key events occur during this stage, including meiosis initiation, premeiotic DNA replication, homolog pairing and meiotic recombination. Homolog pairing and meiotic recombination are governed by the pachytene checkpoint. Pch2 is the key pachytene checkpoint protein, and it is involved in the initiation of meiotic recombination.20 Knocking out PCH2 can rescue some meiotic recombination-deficient mutants, allowing them to go through the pachytene stage and finish the sporulation process even if there were defects such as abnormal crossover. To test whether ATG12 knockout triggered meiotic recombination defects, we created a PCH2 and ATG12 double knockout strain to determine whether knocking out PCH2 could rescue the meiotic defect of the ATG12 knockout strain. However, no rescue phenotype was observed in the sporulation process (Fig. S5). This result suggests that the major role of autophagy may occur even earlier than the pachytene stage.
Rec8 is essential for sister-chromatid cohesion during meiosis in budding yeast, but it is lost from chromosome arms during meiosis I and lost from centromeres at meiosis II.21,22 In the WT strain, Rec8 was expressed from the beginning of sporulation, peaked at around 4–6 h, then decreased through 8 h and was totally lost by 12 h. In the atg12Δ stain, the expression level of Rec8 was similar to that of the WT during the early stage, but the amount of Rec8 did not decrease, even after 12 h induction (Fig. 2F). This result confirms that there is no sister chromatid segregation during sporulation in atg12Δ cells and indicates that the initiation of meiosis is not disrupted. This was further supported by the expression of Cdc14 and Ime1. The release of the phosphatase Cdc14 from the nucleolus is controlled by the signal transduction cascade known as the mitotic exit network (MEN) in late anaphase in mitosis. It is the final step that allows cells to complete mitosis and enter meiosis.23 In budding yeast, entry into the pre-meiotic S phase is promoted by a principal regulator (Ime1) of meiotic initiation upon nutrient limitation.24 The expression patterns of Cdc14 and Ime1 in atg12Δ cells were very similar to that in WT cells after transfer into SPM (Fig. 2F top and bottom panels). These results indicate that the mitotic exit network and meiosis initiation are not affected by ATG12 knockout.
After cells have committed to the meiotic cell cycle, they undergo pre-meiotic DNA replication to prepare for the next 2 rounds of cell division.25 We therefore determined whether pre-meiotic DNA replication was completed in the atg12Δ strain. We sporulated WT and atg12Δ strains in SPM medium, collected samples at 0–12 h in SPM and fixed the cells in 70% ethanol. The yeast cells were then stained with Sytox Green and analyzed by flow cytometry. In the WT strain, approximately 4–6 h after induction, almost all of the cells had finished pre-meiotic DNA replication. However, in the atg12Δ strain, even 12 h after induction, only a very small fraction of the cells had finished pre-meiotic DNA replication (Fig. 2G). These results suggest that the major function of autophagy during meiosis is to promote pre-meiotic DNA replication, most likely by the degradation of some specific substrates.
Quantitative proteomics study of meiosis
To test the working hypothesis that autophagy participates in meiosis by eliminating negative meiotic regulators, quantitative proteomics was used to identify differentially expressed proteins and potential autophagic substrates during meiosis. Synchronized yeast cells were collected 0 h, 6 h, 7.5 h, 8 h, 8.5 h and 9.5 h after transfer into SPM and were prepared for the quantitative proteomics study. These samples corresponded to cells in meiosis initiation, pachytene, metaphase I, anaphase I, metaphase II and anaphase II stages, respectively (Fig. S1). The proteins from the above samples were digested into peptides, labeled with tandem mass tag (TMT 6 plex), and then separated by strong cation exchange chromatography (SCX). Each of the 17 fractions was analyzed using mass spectrometry (MS) (Fig. 3A). Finally, we identified 2376 proteins from 3 independent replications (Fig. 3B). Then, 381 proteins (Fig. 3C, Table S1) that exhibited statistically significant changes across stages (1.5-fold) were selected for further bioinformatic analysis. The reproducibility of our mass spec data is quite good, as most of the pearson correlation coefficients of the 381 regulated proteins between replicates were more than 0.77 (Fig. S6). These differentially regulated proteins were clustered into 9 groups using Cluster software version 3.0 and visualized by TreeView software (Fig. 3D, and Fig. S7). This analysis showed that functionally related proteins exhibit similar expression patterns.
Figure 3.
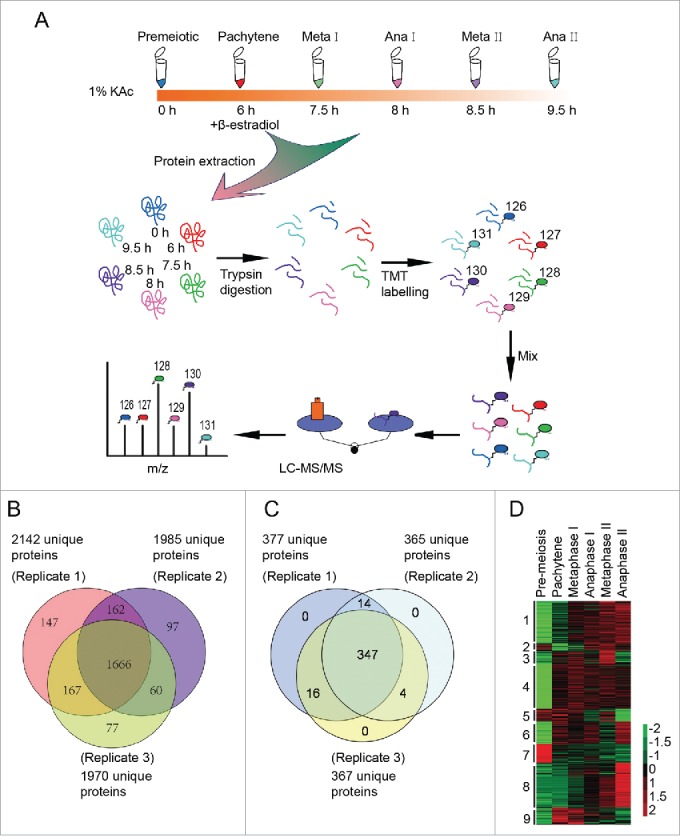
Three hundred eighty one differentially expressed proteins during meiosis were identified by quantitative proteomics. (A) Workflow of the proteomic study of yeast meiosis. Yeast cells (A14201) were induced to sporulate at 30°C by transferring into SPM. After 6 h, 1 μM β-estradiol was added. Samples were collected at 0, 6, 7.5, 8, 8.5, and 9.5 h, and proteins were extracted. After trypsin digestion, peptides were labeled with isobaric TMT and mixed in equal ratios. The labeled mixture was then subjected to an orthogonal first-dimension separation with SCX. Seventeen fractions were subsequently analyzed on an LTQ Orbitrap Velos mass spectrometer coupled with nano-RP HPLC. Biological replicates were performed in triplicate. The spectra were analyzed with MaxQuant software. (B) Protein identification across the 3 biological replicates. (C) Venn plot of 381 differentially regulated proteins. (D) Cluster analysis of 381 differentially expressed proteins. All MS data were normalized and then analyzed for cluster analysis. Cluster results were visualized by TreeView software.
Validation of quantitative proteomics data
To test the reliability of our mass spectrometry data, we first compared our data with that reported in the literature. Twenty-six of the 381 differentially regulated proteins (Table S2) were previously reported to be meiotic essential genes.11 For example, the levels of Pch2 (checkpoint protein required for meiotic double-stranded break formation), Red1 (component of the synaptonemal complex; involved in chromosome segregation during the first meiotic division) and Hop1 (meiosis-specific protein required for chromosome synapsis) increased during the pachytene stage (6 h in SPM) and then decreased as expected.26-28 Sps1, Spr1, Spr3, Spr28, Cda1, Cda2, Sps2 and Smk1 were reported to be necessary for spore wall formation in yeast sporulation29-34 and were found to be highly expressed only after the second meiotic division (Fig. S8). Thus, our results are consistent with those reported by others.
To further validate our data, representative genes from different clusters were selected for knocking-in with 3HA-tag to their C-terminal, their expression levels at different meiotic stages were then detected by western blotting with anti-HA antibody. Fifteen genes were successfully knocked in with 3HA-tag and their expression could be tested by western blotting. Although there were some discrepancies due to technique issue, most of the protein expression patterns were roughly consistent with the MS data (Fig. S9). These results suggest that the data quality of our protein profiling in yeast meiosis is pretty good.
Protein levels in cluster 2 and cluster 7 decreased at pachytene stage significantly (Fig. 4A), and they might be degraded by the autophagy pathway. To simplify the screening, some of them were selected to be tagged with GFP on their C-terminus in the SK1 background (Fig. 4B). The expression levels of these genes were then determined based on the intensity of GFP. Twelve genes were successfully fused with GFP. These strains were sporulated, and the GFP intensity was measured by flow cytometry and western blotting with anti-GFP antibody at 0 h and 6 h after transfer to SPM. Twelve of them could be verified by both flow cytometry and western blotting (Fig. 4C), and the GFP intensities of all strains decreased after 6 h induction (Table S3), which was consistent with our quantitative proteomics data. These results provide solid basis for autophagic substrate screening.
Figure 4.
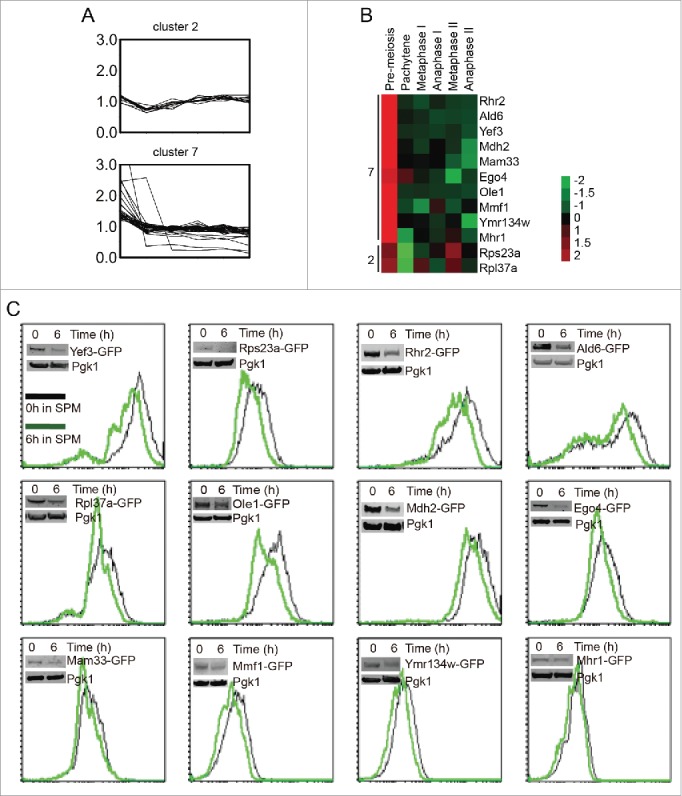
Pachytene stage downregulation of cluster 2 and cluster 7 proteins. (A) The expression pattern of cluster 2 and cluster 7 proteins during meiosis. (B). The expression patterns of 12 selected proteins from cluster 2 and cluster 7. The expression values of each protein were normalized across time points to have an average value of 0 and a standard deviation of 1, to better show the expression trend in color mode. (C) The selected genes were fused with GFP on their C-termini, and the intensity of GFP represented the amount of those proteins. Yeast cells were collected and fixed at 0 h and 6 h after transfer into SPM, then analyzed by flow cytometry and western blotting. Most of the flow cytometry and western blotting results were consistent with the MS data.
Ego4 protein is one of the negative regulators of meiosis
To test whether the degradation of the 12 verified proteins is depended on autophagy, we fused GFP to the C-terminus of the 12 genes in the ATG12 knockout background. After 6 h induction in SPM, we compared their expression levels with that at 0 h by western blotting. The decreasing of most of those GFP-tagged proteins (8 of 12) were not affected by ATG12 deletion, but the decreasing in the expression level of Yef3-GFP, Rpl37a-GFP, Ego4-GFP and Ald6-GFP were retarded in atg12Δ cells compared to WT (Fig. 5A-D), among them, Ald6 has been reported to be selectively degraded by autophagy,35 these results suggest that the other 3 proteins may also be autophagic substrates. Because GFP protein is relatively resistant to hydrolysis in the vacuole, the appearance of free GFP is a canonical characteristic of the GFP fused autophagic substrate.36 Consistent with the above results, along with the degradation of these 4 proteins, there were free GFP accumulations at pachytene stage but not in ATG12 knockout strain (Fig. 5A, B, C and D). So, we conclude that Ego4, Rpl37a, Yef3 and Ald6 are autophagic substrates and all of them need to be degraded by autophagic pathway at the early stage of meiosis.
Figure 5.
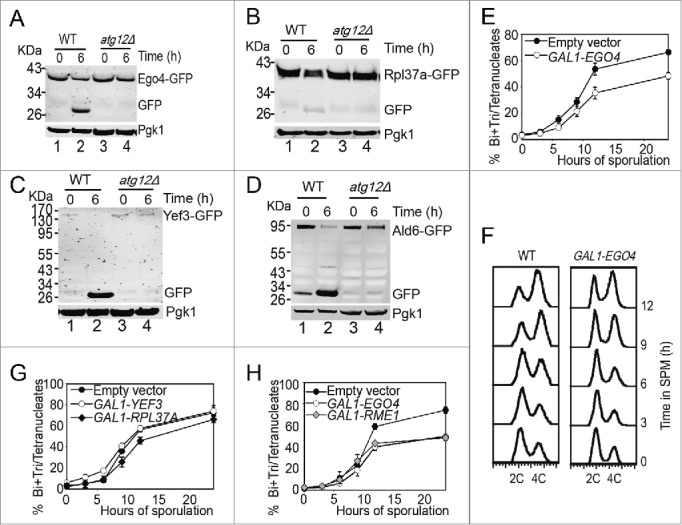
Ego4 protein is identified as a negative meiotic regulator. (A-D) Free GFP accumulation after 6 h sporulation in EGO4-GFP (A), RPL37A-GFP (B), YEF3-GFP (C) and ALD6-GFP (D) WT strains but not atg12Δ strains. WT and atg12Δ strains were sporulated, and samples at 0 h and 6 h sporulation were collected for protein extraction and western blotting analysis. (E, G, H) EGO4, but not RPL37A or YEF3 overexpression inhibited yeast sporulation. EGO4 (E), RPL37A and YEF3 (G) were overexpressed under the control of the GAL1 promoter, and sporulation was induced by 0.5% galactose. Cells with empty vectors (EV) were used as a control. Samples collected at different time points were fixed for checking their sporulation rates under microscopy. The inhibitory effect of Ego4 on meiosis was compared with overexpression of a known negative meiotic regulator, Rme1 (H). (F) Ego4 overexpression delayed premeiotic DNA replication. Premeiotic DNA replications were detected by flow cytometry at different time points when Ego4 was overexpressed.
We then tested whether the degradation of these 3 newly found autophagic substrate (Ego4, Rpl37a and Yef3) proteins was necessary for meiosis by overexpressing these genes under the control of the GAL1 promoter. After induction for 24 h in SPM in the presence of 0.5% galactose, most of the cells in the control group completed sporulation, but the overexpression of Ego4 inhibited yeast sporulation by ˜20% (Fig. 5E) and caused delay in premeiotic DNA replication (Fig. 5F). But overexpression of the other 2 proteins (Rpl37a and Yef3) almost have no effect on sporulation (Fig. 5G). To assess the inhibitory effect of Ego4, we compared the sporulation efficiency of Ego4 overexpression with Rme1 overexpression, which is a known meiotic repressor.37 We found that the inhibitory effect of Ego4 overexpression was comparable with Rme1 overexpression (Fig. 5H). Thus, we concluded that Ego4 is a negative regulator of meiosis, and it must be eliminated at the early stages of meiosis.
Ego4 is selectively degraded by autophagy at the prophase of meiosis I
To exclude that the ubiquitin-proteasome degradation pathway also contributed to the degradation of Ego4, the EGO4-GFP strain was induced to sporulate in the presence of 100 μM MG132 (proteasome inhibitor) or 200μM CQ, and cells were collected 0 h and 6 h after transfer into SPM. After analysis by flow cytometry and western blotting, we found that the decreasing of the GFP signal was not affected by MG132 treatment but significantly inhibited by CQ treatment (Fig. 6A, B). Since EGO4 mRNA level dropped significantly at the pachytene stage (Fig. 6C), we conclude that Ego4 is controlled at both transcriptional and posttranslational level in the early stage of meiosis, and the degradation of this protein is solely dependent on the autophagy pathway but not the ubiquitin-proteasome pathway.
Figure 6.
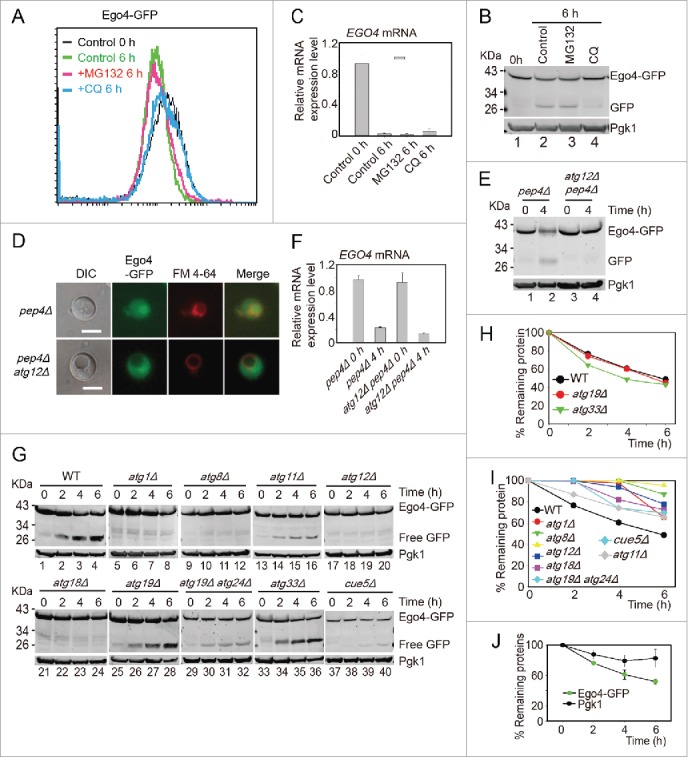
Ego4 is selectively degraded by autophagy. (A-C) The degradation of Ego4 was not dependent on the proteasome but dependent on autophagy. The EGO4-GFP strain was transferred into SPM and treated with or without 100 μM MG132 or 200 μM CQ between 0 to 6 h. The GFP intensity and EGO4 mRNA expression level were analyzed by flow cytometry (A), western blotting (B) and quantitative real time PCR (C). (D-F) Ego4-GFP was transferred into the vacuole at early stage of meiosis, which was dependent on autophagy. pep4Δ or pep4Δ atg12Δ cells were prepared as in Fig. 1A. The 4-h samples from SPM were visualized by fluorescence microscopy (D) and protein changes were detected by western blotting (E). Scale bar: 5 μm. DIC, differential interference contrast. The mRNA expression levels of those samples were analyzed by real-time PCR (F). (G) Ego4-GFP was selectively degraded by autophagy. GFP-tagged Ego4 proteins in WT, atg1Δ, atg8Δ, atg12Δ, atg18Δ, atg19Δ, atg33Δ, atg11Δ, atg19Δ atg24Δ and cue5Δ strains were analyzed by western blotting (lanes 1-40). Samples were collected at 0, 2, 4 and 6 h after transferring into SPM. (H-I) Quantitative analysis of Ego4-GFP degradation in ATG knockout strains in (G). The degradation rates of Ego4-GFP in WT, atg19Δ, atg33Δ (H) atg1Δ, atg8Δ, atg11Δ, atg12Δ, atg18Δ, atg19Δ atg24Δ and cue5Δ (I) strains. (J) Degradation of Ego4-GFP was faster than Pgk1. The EGO4-GFP strain was sporulated and samples were collected at 0, 2, 4, and 6 h for western blotting. The percentages of remaining protein were the value of the protein level divided by their protein levels at 0 h.
To study how Ego4-GFP was degraded by autophagy, we checked the localization of Ego4-GFP in either pep4Δ or pep4Δ atg12Δ double-mutant strains during meiosis. After transfer into SPM for 4 h, the Ego4-GFP was transferred into the vacuole in pep4Δ strain, while in pep4Δ atg12Δ double mutant strain, Ego4-GFP still distributed in the cytosol, it could not be transferred into the vacuole (Fig. 6D). Western blotting result showed that Ego4-GFP was stabilized in pep4Δ atg12Δ double-mutant strain but not in pep4Δ strain (Fig. 6E), and the stabilization was not caused by the increasing of its mRNA level (Fig. 6F). Thus we conclude that the degradation of Ego4 protein occurs in vacuole, and the transfer into the vacuole of Ego4 protein is dependent on autophagy.
To further investigate which autophagy pathway is involved in Ego4-GFP degradation, we deleted ATG1, ATG8, ATG12, ATG18 (all of them are involved in starvation induced autophagy),10 ATG11 (scaffold protein for pexophagy and the cytoplasm-to-vacuole targeting (CVT) pathway,38 ATG19, ATG24 (CVT pathway),39,40 ATG33 (mitophagy specific gene),41 and CUE5 (ubiquitin-Atg8 receptor in ubiquitin-dependent autophagy),42 then tested their effects on Ego4-GFP protein degradation and free GFP accumulation. Samples were collected from 0 to 6 h after transfer into SPM. The deletion of ATG1, ATG8, ATG11, ATG12, ATG18, ATG19-ATG24 and CUE5 stabilized Ego4-GFP protein and decreased free GFP accumulation. While in the wild-type, ATG19 and ATG33 deletion strains, there were clearly free GFP accumulation and the degradation of Ego4-GFP protein were not delayed after transferring into the SPM (Fig. 6G, H, I). All these results suggest that Ego4-GFP protein is selectively degraded by autophagy in a ubiquitin-dependent manner during the prophase of meiosis I. To our surprise, Pgk1-GFP was degraded by both selective during prophase of meiosis I (Fig. S10) and nonselective bulk autophagy,43 but the degradation rate of Pgk1 was much lower than Ego4-GFP (Fig. 6J), suggesting it still could be used as a relative good loading control during this dramatic cellular renovation process.
Ego4 interacts with Acs1 during the prophase of meiosis I
To explore the functional role of Ego4 protein in meiosis, we generated a EGO4-TAP strain. Yeast cells were collected at 0, 3 and 6 h after transfer into the SPM, after purification, trypsin digestion and TMT labeling, samples were applied for mass spectrometry analysis (Fig. 7A). Eventually, we identified 7 candidate proteins that might interact with Ego4 protein (Table S4 and Fig. 7B). Among them, the abundantly expressed proteins such as ribosome-related proteins and chaperone protein might be nonspecific binding proteins, this possibility was then tested by TAP affinity isolation experiment, after pulling down TAP-tagged Ego4 protein, we found almost equal amounts of Ssa1 were copurified from either EGO4-TAP or the control strain (Fig. 7C). We then analyzed the remaining proteins, and only ACS1 was found to be involved in yeast meiosis.44 So we focused on the interaction between Ego4 protein and Acs1. We first confirmed their interaction by pulldown experiment (Fig. 7D), then compared the expression level of Acs1 at the early meiosis stage, and found that with the induction of meiosis the expression level of Acs1 also increased (Fig. 7B and E), correspondingly, more Acs1 could be pulled down by Ego4-TAP (Fig. 7F). In addition, we found that Acs1 was stabilized in ego4Δ cells during the prophase of meiosis I (Fig. 6E). These results suggest that Ego4 might negatively regulate meiosis by destabilization Acs1.
Figure 7.
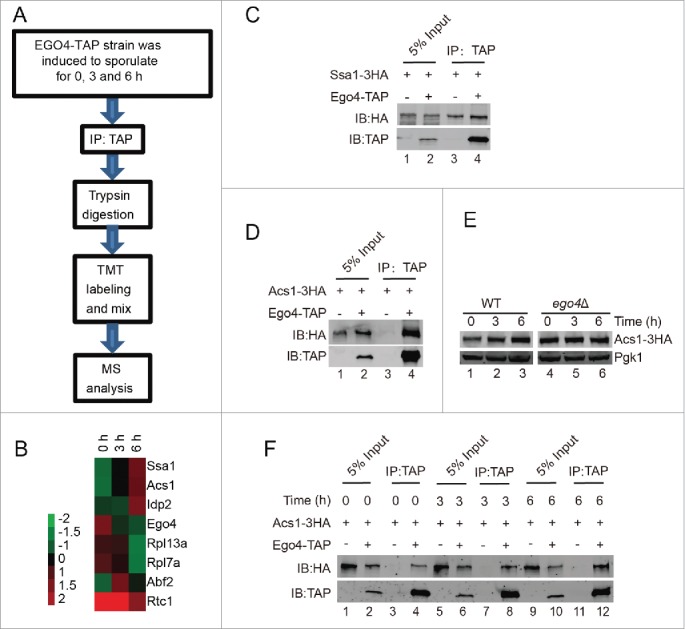
Identification of Ego4 interacting proteins at the prophase of meiosis. (A) Workflow for Ego4 interacting protein identification. (B) Cluster analysis of the identified proteins. The expression levels of proteins were normalized and clustered by Cluster 3.0 and visualized by TreeView software. (C, D) Interactions between Ego4 and Ssa1 (C) or Acs1 (D). Cells were sporulated and collected from 0, 3 and 6 h after being transferred into sporulation medium. Proteins were pooled together for affinity isolation experiments; proteins were purified from cell lysates using calmodulin beads. (E) The expression of Acs1 during sporulation induction. WT and ego4Δ ACS1-3HA cells were induced for sporulation, and samples were collected at 0, 3 and 6 h after induction. Acs1 was detected by anti-HA antibody. (F) Ego4 interacts with Acs1 at the early stages of meiosis. Cells were collected at the indicated time points, Ego4 was then affinity isolated with calmodulin beads, and the proteins were then detected by western blotting. To compare the amount of the co-purified Acs1, the Ego4-TAP proteins from each time points were adjusted to equal amounts.
Finally, to test whether Ego4 is a major meiotic regulator or not, we created the ego4Δ atg12Δ double mutant strain, then tested its sporulation efficiency. Even after 2 d induction, the sporulation efficiency of this double mutant was not rescued compared to the atg12Δ strain (Fig. S11) suggesting that Ego4 is one of the negative regulator of meiosis that needs to be degraded by autophagic pathway, other negative regulators that must be degraded by the autophagic pathway still need to be identified in the future work.
Metabolic, ribosome and proteasome-related components are major categories of differentially expressed proteins in meiosis
Given that autophagy mainly affects the early stages of meiosis by degrading at least one negative meiotic regulator, the stages at which the ubiquitin proteasome system is involved also need to be defined. The APC/C and other E2s or E3s were identified as differentially expressed proteins (such as Apc4, Cdc16, Ubc1, Ubc5, Dma2 and Saf1). To further understand the cellular functions of these differentially expressed proteins, we performed network analysis using protein interaction information from the search tool for the Retrieval of Interacting Genes/Proteins (STRING) database. The interaction network was visualized using Cytoscape 2.8.2. To our surprise, many metabolism-related proteins, ribosome and proteasome components were found to be closely connected by functional and physical protein-protein interactions (Fig. 8A). These metabolic proteins are involved in fatty acid metabolism, chitin metabolism, aminoglycan metabolism, coenzyme metabolism, carboxylic acid catabolism, polysaccharide metabolism and so on (Table S5). The expression of these metabolic proteins showed different patterns. Proteins involved in coenzyme metabolism (such as Ald6, Tes1, Acs2, Coq5 and Mdh2) were highly expressed in early meiosis. Many proteins participating in fatty acid metabolism (Pox1, Fox2, Cat2, Faa1 and Yat2) were highly expressed during the 2 meiotic divisions, while the expression levels of proteins involved in chitin metabolism, carbohydrate metabolism and polysaccharide metabolism (such as Glc7, Gfa1, Bmh1, Cda1, Cda2, Gal10, Gal7 and Cts2) were higher in later meiosis (Fig. S12). It has been reported that translational control plays an important role during meiosis.2 In support of that result, the expression of ribosomal proteins decreased very quickly after the initiation of meiosis, increased during the cell division process, and peaked during either metaphase II or anaphase II (Fig. 8B). These results suggest that the genes involved in metabolism and the ribosome actively participated in the meiotic renovation process.
Figure 8.
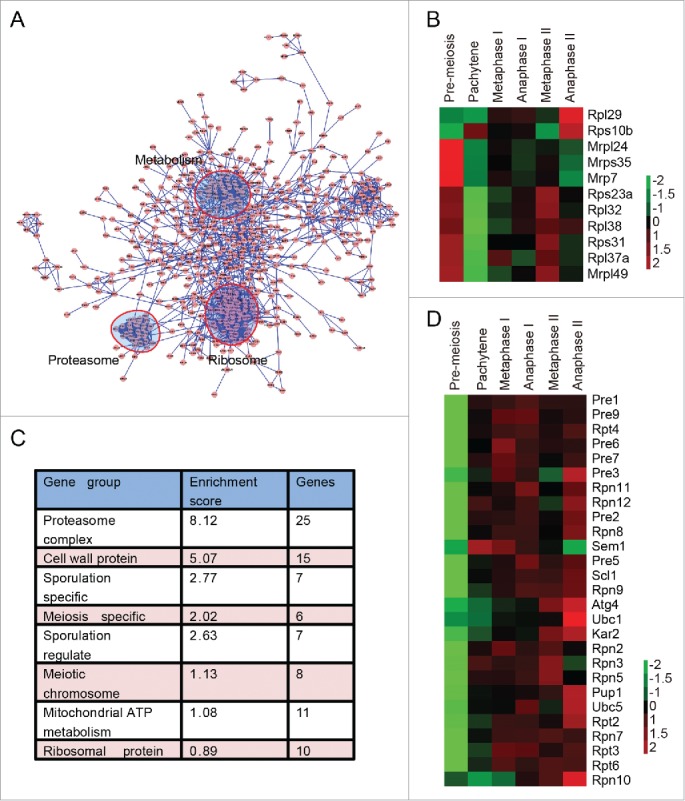
Proteasome and metabolism-related proteins are highly enriched during meiosis. (A) Protein interaction map of differentially expressed proteins during yeast meiosis. The interaction network was calculated by STRING and visualized by Cytoscape 2.8.3. (B) The expression pattern of ribosomal proteins identified by MS data. (C) Gene functional clusters of differentially expressed proteins. Clusters were generated by DAVID 6.7 online. (D) Expression patterns of proteasome members. Almost all the components of the proteasome complex were upregulated after the pachytene stage.
Consistent with the network analysis, Gene Ontology enrichment analysis, which was performed by the functional annotation tool of the DAVID (Database for Annotation, Visualization and Integrated Discovery) bioinformatics resources,45,46 showed that proteasome components were the most enriched proteins during meiosis (Fig. 8C). The enrichment score of the proteasome was even higher than that of sporulation-specific and meiotic chromosome-related proteins (Fig. 8C). The expression levels of almost all components of the proteasome started to increase at the pachytene stage and were highly expressed during the 2 successive meiotic nuclear divisions (Fig. 8D). These results suggest that the upregulation of the proteasome is also needed to facilitate meiotic division, thus providing a new layer of meiotic regulation.
Proteasome activity is essential to meiotic division but not to the initiation of meiosis
To verify the expression level of the proteasome during meiosis, 3HA was fused to the C-terminus of 6 proteasome components genes (RPN9, RPN11, PUP1, SEM1, PRE6 and RPN12). After transfer into SPM and adding β-estradiol to induce sporulation at 6 h, yeast cells were collected at 0, 6, 7.5, 8, 8.5 and 9.5 h, respectively. Consistent with our mass spectrometry data, these 3HA fusion proteins increased after 6 h in SPM (Fig. 9A and Fig. S9). To examine the proteasome activity during meiosis, we then detected the chymotrypsin-like peptidase activity of proteasome at various time points after inducing for sporulation, and found that the chymotrypsin-like peptidase activity of the proteasome increased significantly during meiotic division processes compared with the initial stages of meiosis induction (Fig. 9B). These results confirmed that both the expression level and the activity of proteasome are upregulated during meiotic division and suggest that the proteasome might play a major role during meiotic division rather than initiation.
Figure 9.
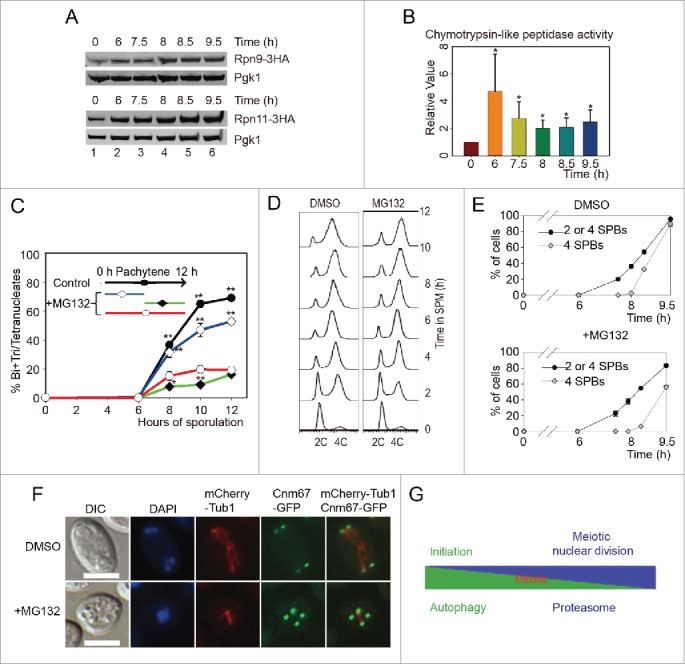
Proteasome activity is essential for meiotic division but not for meiosis initiation. (A) Validation of proteasome upregulation during meiotic division. A 3HA tag was fused at the C termini of RPN9 and RPN11 in the SK1 background yeast strain. Cells were sporulated and released from NDT80 arrest by adding 5 μM β-estradiol at 6 h. Samples were collected and detected by western blotting with anti-HA antibody. (B) Chymotrypsin-like activity of 20S proteasome during meiosis. Total cell lysates from different time points were collected and were used to detect their relative proteasome activity. *, P < 0 .05 vs 0 h sample. (C) Effect of MG132 treatment at different meiotic stages on yeast sporulation. MG132 (100 μM) was added to the SPM at either the time of transfer or 6 h after induction. MG132 was washed out after 6 h, and the effect on sporulation was detected. DMSO was used as the control. Data were presented as the mean ± SD. Asterisk indicates statistically significant difference in comparison with 0 to 12 h treated samples (red line). *, P < 0 .05; **, P < 0 .01. (D) MG132 treatment did not affect premeiotic DNA replication. The WT yeast strain was sporulated in SPM without or with MG132, samples were stained with Sytox Green and analyzed by flow cytometry. (E) Effect of MG132 treatment on yeast nuclear division and SPB duplication. The CNM67-GFP mCherry-TUB1 strain was sporulated in SPM in the absence or presence of MG132, and the numbers of SPBs in either MG132-treated or untreated strains were calculated using microscopy. (F) Samples at 9.5 h were collected and visualized by fluorescence microscopy. Scale bar: 5 μm. DIC, differential interference microscopy. (G) Model for the role of autophagy and the proteasome in yeast meiosis.
Because the proteasome is essential to the cell, it is not possible to directly detect the role of the proteasome in meiotic division by knockout. MG132 is a widely used proteasome inhibitor that can inhibit proteasome-dependent protein degradation, so it was applied to yeast cells during sporulation. After 100 μM MG132 treatment, the sporulation efficiency decreased significantly (Fig. 9C, red line), confirming that the proteasome indeed plays a very important role during meiosis, which is in agreement with many studies show that ubiquitin-proteasome dependent proteolysis of cyclins is essential to meiotic progression.3,47 To test whether the proteasome is essential to the early stage of meiosis, we treated sporulating cells at 0-5 h (early meiosis) with 100 μM MG132, then washed the cells with fresh SPM. The treated cells sporulated with high efficiency, close to that of the control (Fig. 9C, blue line). Thus, we concluded that the proteasome does not play a major role in the initiation of meiosis. To further confirm the role of the proteasome during meiotic division, we treated cells only at the meiotic division stage (5-12 h). To our surprise, the sporulation efficiency of these cells was lower than those treated with MG132 from the beginning, and almost no cells finished the second meiotic nuclear division (Fig. 9C, green line), presumably because of some MG132 have lost their function during the first 5 h incubation.
Further experiments showed that MG132 treatment didn't inhibit premeiotic DNA replication (Fig. 9D), and SPB duplication (Fig. 9E). But the duplicated SPBs couldn't separate; even after 9.5 h induction, 4 GFP dots were still crowded around the nuclear (Fig. 9F). Together, these results suggest that autophagic activity is essential for premeiotic DNA replication, while proteasomal activity plays a very important role during meiotic nuclear divisions but is not essential for meiosis initiation (Fig. 9G).
Discussion
In this article, we confirm that autophagy-related genes are essential to meiosis, and autophagy mainly affects early meiotic phases such as premeiotic DNA replication by degrading some negative meiotic regulators. These results are consistent with previous large-scale screens48 and are further supported by the fact that some cytoplasm-to-vacuole targeting (CVT) and vacuolar protein sorting (VPS) related genes (CCZ1, VPS1, AVT3 and AVT5) are required for the early meiotic phase.49-51 Importantly, our proteomic study also reveals that substrates of the autophagy pathway may include certain negative meiosis regulators, as exemplified by our characterization of a previously uncharacterized ORF (open reading frame), Ego4. We show that endogenous Ego4 is downregulated during sporulation, which is dependent on the selective autophagy pathway. This process appears to contribute to the timely initiation of DNA replication because overexpression of Ego4 is sufficient to slow meiosis initiation. However, because the effect of Ego4 overexpression is moderate, we assume that there might be a host of inhibitors that need to be degraded collectively in order to initiate meiosis. This model is consistent with a previous study that reported Ume6 as an inhibitor of meiosis.52 Our proteomic study further show that Ego4 preferentially interacts with Acs1 during the prophase of meiosis. Acs1 is an acetyl-CoA synthetase, which catalyzes the formation of acetyl-CoA from acetate and CoA. The nuclear acetyl-CoA serves as acetyl donor for histone acetylation, and the cytoplasmic acetyl-CoA is mainly important for the central carbon metabolism.53 Most importantly, the deletion of ACS1 leads to the decreasing of sporulation efficiency.44 So it is possible that Ego4 suppresses meiosis by destabilization Acs1, thus decreasing histone acetylation and finally altering the transcription of some meiotic related genes; alternatively, changing the central carbon metabolism which is specifically required for meiosis. Although additional work is required to identify the full spectrum of meiosis inhibitor, our study raises the possibility that the autophagy-lysosome system can participate in meiosis initiation not only by serving as a dynamic recycling system that produces new building blocks and energy for this cellular renovation process,54 but also by degrading a cohort of negative meiotic regulators.
The development of a new method to synchronize meiotic cells by the Amon lab greatly promoted the application of new ‘omics’ methods to the study of yeast meiosis.16 Recently, ribosome profiling together with RNA sequencing were applied to this system to study meiotic gene expression and translational control.2 Previous microarray and RNA expression profiling studies also provided a global view of the regulation and coordination of gene transcription during yeast meiosis.15 However, a detailed kinetic profile of protein expression during meiosis has not yet been obtained. Proteomic strategies have been applied to investigate yeast mitosis, mitochondria and the difference between haploid and diploid yeast cells.55-57 So far as we know, only 2-D DIGE (2-dimensional fluorescence difference in gel electrophoresis) has been used in the investigation of meiosis. That study found that 142 protein spots were temporally regulated during meiosis but only identified 44 unique proteins by LC-MS/MS.58,59 Here, TMT-based quantitative proteomics was applied to highly synchronized yeast cells at successive intervals following transfer to SPM, and 381 differentially expressed proteins (≥1.5-fold) of 2376 total identified proteins were found, significantly expanding our knowledge of meiosis at the proteome level. According to the studies of Chu et al. and Primig et al., more than 1000 genes among approximately 6200 protein-encoding yeast genes showed significant changes (either induction or repression) in mRNA levels during sporulation.15,60 Considering that only 2376 proteins were identified, the number of differentially expressed proteins should be approximately 1000, which is comparable to the mRNA data.
Although only 44 proteins were identified in the 2-D DIGE studies, 2 functional categories, including carbon metabolism and protein catabolism, were found to be of primary importance. This result is further supported by our network analysis, which shows that metabolism and ribosome-related proteins play central roles in meiosis. It is not surprising that metabolism changes dramatically during meiosis, given that deprivation of glucose and nitrogen trigger autophagy, many long-lived proteins and organelles are degraded to provide building blocks for the next phase of meiosis, which is totally different from mitotic division. In addition, during meiotic division, our quantitative proteomic results showed that ribosome components are significantly upregulated to accommodate the translation of some meiosis-specific mRNAs. Thus our studies provide additional evidence for the translational control of yeast meiosis.
Another intriguing finding of our quantitative proteomic study is that proteasome components are highly regulated compared to genes in other functional categories during starvation-induced sporulation in yeast. This is consistent with our proteomic studies during human and mouse spermatogenesis.61,62 Many reports have investigated the role of the ubiquitin-proteasome system in meiosis and gametogenesis. For example, many E3s such as APC/C, SCF and other UPS elements were found to be involved in the regulation of the meiotic cell cycle.4,47,63,64 However, most of these studies focused on ubiquitination rather than on the proteasome itself. The only description of proteasome localization during meiosis comes from fission yeast, during the first meiotic division, the proteasome signal is more dispersed throughout the nucleus. In contrast, in meiosis II, the proteasome is restricted to the area between the separating DNA.65 We did not observe this type of dramatic localization change during meiosis in budding yeast, but we did find that the expression levels of proteasome components and proteasome activity significantly increased during meiotic division (Fig. 9A and B, Fig. S9), and proteasome activity is essential for the 2 meiotic nuclear divisions but not for meiosis initiation (Fig. 9C). These results strongly support our recent discovery about spermatoproteasomes, which contain spermatid/sperm-specific subunits in addition to PA200 and are proposed to be involved in the acetylation-dependent degradation of somatic core histones during double-stranded DNA breaks.66 All these results suggest that in addition to canonical ubiquitination-dependent protein degradation, the proteasome itself also undergoes noncanonical regulation to fit the specific requirements of meiosis during sexual reproduction.
Materials and methods
Strains and plasmids
All strains are SK1 derivatives and are described in Table S6. GAL-NDT80 and GAL4.ER constructs were obtained from Angelika Amon,16 and construction methods were described in a previous study.67 NDT80-Myc, CDC14-GFP, CNM67-GFP, EGO4-GFP and all other GFP knock-in strains were constructed by using a PCR-based method.68 Yeast deletion strains were constructed by the PCR-mediated gene replacement method as described previously.69 Diploid deletion strains were obtained by genetic crosses of appropriate MATa and MATα haploids. Overexpression plasmids were constructed by inserting genes between the HindIII and BamH1 loci of the pYC2/NTC vector (Addgene, 631903) using the In-Fusion™ Advantage PCR Cloning Kit according to the user's manual.
Sporulation conditions
Strains were grown 24 h in YPD, diluted in YPA (1% yeast extract, 2% peptone, 2% potassium acetate) to OD600 = 0.3, and grown for 14 h for A14201-derived strains and 10 h for A14200-derived strains. Cells were washed 3 times and resuspended in sporulation medium (2% potassium acetate) to OD600 = 1.9 and sporulated at 30°C. GAL-NDT80 GAL4.ER strains were released from the pachytene arrest by the addition of 1μM β-estradiol (5 mM stock in ethanol; Sigma, E2758-1G) at 6 h as described by Carlile and Amon.16 For overexpression, strains were grown in SC for 12 h, washed 3 times and resuspended in SG (SC medium without sugar, add 1% galactose) to OD600 = 0.6 and grown for 40 h for induction. Cells were washed and resuspended in sporulation medium (1% KAc, containing 0.5% galactose).
Western blotting analysis
Cells were subjected to mild alkali treatment and then boiled in a standard electrophoresis loading buffer as described by Kushnirov.70 The samples were run on SDS-PAGE (sodium dodecyl sulfate PAGE) gels, and electrophoretic protein transfer was performed with a Bio-Rad Trans-Blot® SD Semi-Dry Transfer Cell. For GFP-Atg8–PE formation assay, we used 12% SDS PAGE gels containing 6% urea as described by Klionsky et al.17 The antibodies were used at 1:2000, except for anti-Pgk1 (generated in rabbit), which was used at 1:5000. GFP tagged proteins were detected by western blotting with GFP-tag (7G9) mouse monoclonal antibody (mAb; Abmart, M20004). The Rec8 antibody was generated from a rabbit. Ime1-MYC was blotted by MYC-tag (19C2) mouse mAb (Abmart, M20002). 3HA fusion proteins were detected by western blotting with HA-tag (26D11) Mouse mAb (Abmart, M20003).
Proteasome activity assay
Cells were collected at indicated sporulation time-points, then washed 2 times with H2O, and resuspended in a buffer containing 50 mmol/L HEPES, pH 7.5, 150 mmol/L NaCl, 5 mmol/L EDTA, and protease inhibitors (Sigma-Aldrich, E9884).71 The resuspended cells were disrupted by glass beads (Sigma-Aldrich, G1145) using a Genie vortex. The chymotrypsin-like activities of proteasomes were determined with protein extracts and the fluorogenic substrates Suc-LLVY-AMC (Boston Biochem, S-280). All measurement were performed in triplicate and further replicated in independent experiments. One-way ANOVA was used to determine significant differences among sample groups.
Protein extraction, reduction, alkylation and digestion
Yeast cells were finely ground using liquid nitrogen and lysed in protein extraction buffer consisting of 7 M urea (Sigma-Aldrich, 51456), 2 M thiourea (GE Healthcare, RPN6301), 65 mM DTT and 1% (v/v) protease inhibitor cocktail.60 Protein content was measured using a Bradford assay; cysteine residues were reduced by incubating lysates with 200 mM DTT for 1 h at 56°C followed by alkylation in 375 mM iodoacetamide for 45 min at room temperature in the dark. Protein lysates were cleaned by acetone precipitation and digested with trypsin overnight at 37°C at a 1:50 enzyme:protein ratio.
TMT labeling
TMT labeling was performed according to the manufacturer's instructions. Briefly, TMT Label Reagents (Thermo Scientific, 90064) were equilibrated at room temperature, each aliquot was resuspended in 41 μL of anhydrous acetonitrile, and 42 μL of the TMT Label Reagent was added to 100 μg peptides were dissolved in 200 mM triethylammonium bicarbonate. After 60 min of reaction at room temperature, 8μL hydroxylamine 5% (w:v; Thermo Scientific, 90115) was added to each tube, which was incubated for 15 min. The aliquots were then combined, and the pooled samples were evaporated under vacuum.
Strong cation exchange chromatography sample fractionation
The peptide mixture was resuspended in buffer A (10 mM NH4COOH, 5% acetonitrile [ACN], pH 2.7) and loaded onto a strong cation ion exchange column (1 mm ID × 10 cm packed with Poros 10 S, DIONEX) with the UltiMate® 3000 HPLC system at a flow rate of 50 μL/min. The following linear gradient was used: 0% to 56% buffer B (800 mM NH4COOH, 5% ACN, pH 2.7) in 20 min; 56% to 100% B for 1 min; 100% B for 5 min; 100% to 0% B in 1 min; 0% B for 20 min before the next run. Effluents were monitored at 214 nm based on the UV-light trace, and a total of 17 fractions were collected in 2 min intervals during the SCX gradient.
Mass spectrometry analysis
SCX fractions were sequentially loaded onto a μ-precolumn™ cartridge (0.3×5 mm, 5 μm, 100 Å; DIONEX,) at a flow rate of 20 μL/min. The trap column effluent was then transferred to a reverse-phase microcapillary column (0.075×150 mm, Acclaim® PepMap100 C18 column, 3 μm, 100 Å; DIONEX) with a flow rate of 300 nL/min. The reverse-phase separation of peptides was performed using the following buffers: 2% ACN, 0.5% acetic acid (buffer A) and 80% ACN, 0.5% acetic acid (buffer B); a 120-min gradient was used (4% to 7% buffer B for 3 min, 7% to 33% buffer B for 102 min, 33% to 50% buffer B for 10 min, 50% to 100% buffer B for 1 min, 100% buffer B for 3 min, 100% to 4% buffer B for 1 min).
Peptide analysis was performed using a LTQ Orbitrap Velos (Thermo Scientific, Waltham, MA) coupled directly to a LC column. An MS survey scan was obtained for the m/z range 400-1800, and CID MS/MS spectra were acquired from the survey scan for the 8 most intense ions (as determined by X-calibur mass spectrometer software in real time). After each CID MS/MS, the most intense fragment ion in an m/z range between 110-160% of the precursor m/z was selected for HCD-MS3, adapted from Ting et al.'s method.72 Dynamic mass exclusion windows of 60 s were used, and siloxane (m/z 445.120025) was used as the lock mass.
Protein identification and quantification
RAW files for MS/MS were identified using MaxQuant (version: 1.2.2.5) with fixed TMT 6-plex modification. The sequence database for identification was downloaded from the Saccharomyces Genome Database (www.yeastgenome.org), which contains proteins translated from ORFs and pseudogenes. The default settings were used for the other parameters. The false discovery rate (FDR) for peptide and protein identification were both set to 0.01 as evaluated by MaxQuant based on reverse sequences. For each identified peptide, quantification signals were extracted from the corresponding HCD-MS3 spectra, and the relative protein expression levels were calculated according to the Libra algorithm of TPP73,74 using in-house developed scripts. In brief, each channel of reporter ion intensity was normalized by the sum of the signals in the corresponding channels. For each peptide, spectra with intensities that deviated from the mean by more than 2 folds of sigma were removed. Each peptide channel was then re-normalized by the sum across channels. The protein intensity was calculated as the median of normalized intensity of the corresponding peptides. For the identification of peptides and proteins, the FDR was estimated by searching against the protein database with the reversed protein sequences using MaxQuant. For statistical comparison of protein expression levels, one-way analysis of variance (ANOVA) was used to calculate significant differences in abundance among groups. A permutation-based FDR value less than 0.05, and a fold change greater than 1.5 was considered significant using Perseus software75 for protein quantification. Thus spectra with abnormal intensity were already removed. And proteins with only a single unique peptide were removed to increase the confidence of quantification.
Clustering and bioinfomatic analysis
Before clustering analysis, the expression values of each protein were normalized across time points to have an average value of 0 and a standard deviation of 1, to better show the expression trend, and they were showed in column 23-30 of supplementary Table S1. We then performed clustering analysis by Cluster 3.0 software and visualization using TreeView software. Protein interaction network was generated by STRING and visualized by cytoscape 2.8.3. Gene functional clusters were performed by DAVID 6.7 online tools.
FM 4-64 staining
Cells were collected after pre-sporulation and incubated with 5μg/ml FM 4-64 (from 100μg/ml stock solution in Me2SO; Invitrogen, T13320) for 5 min at room temperature. Then cells were diluted in 1% KAc for sporulation.
Flow cytometry
For DNA content analysis, 1×107 cells were fixed with 1 ml cold 70% ethanol overnight, then resuspended in 1 ml 50 mM sodium citrate. The samples were centrifuged at 376 x g for 5 min, and the supernatant fraction was removed. Samples were digested with RNase A (Sigma Aldrich, R6513) for 2 h at 37°C and then sonicated for 2 s at 20% power, stained with 1 μM Sytox Green (Molecular Probes, S-7020), and analyzed by BD FACSvantage SE Flow Cytometry System. For GFP detection, samples were collected and washed once with cold phosphate-buffered saline (PBS; HyClone, SH30256.01B), then immediately subjected to BD FACSvantage SE Flow Cytometry.
Transmission electron microscopy analysis
Yeast cells were sporulated and collected at different time points for TEM analysis. Sample preparation was conducted according to a previously described protocol.76 Briefly, sporulated samples were collected and wash 3 times with PBS (pH 6.5), then fixed by 4% K2MnO4 for 4h at 4°C. After pre-embedding with 1% argarose, samples were dehydrated and embedded using spur's low viscosity embedding kit (EMS, 14300) following the manuscript. Sections were cut to about 90 nm and examined in a JEM-1400 electron microscope.
Autophagic body size estimation
The estimation of autophagic bodies was performed exactly as described by Backues et al.77 Briefly, photos taken by TEM analysis was outlined by photoshop and measured using ImageJ, the measured particle sizes were input in the “Size_Estimation.xlsx” offered by Backues et al. Then adjusted the empirical parameters, the section thickness was set to 90 nm. After that, we adjusted the values of mu (μ) and sigma (σ) to fit the actual data, at last we got the best-fit values of μ and σ in order to calculate the mean and standard deviation of the radii and the mean volumes of the original autophagic bodies.
Identification of Ego4 interacting proteins
For purification of Ego4-TAP interacting proteins, yeast cells collected at different time points were ground to powder using liquid nitrogen. The lysate was purified by IgG Sepharose 6 Fast Flow (GE Healthcare, 17-0969-01) and calmodulin Sepharose 4B (GE Healthcare, 17-0529-01) beads according to a tandem affinity purification method.78 After purification, the proteins were digested using the previous on-beads digestion method.79 In brief, after reduction and alkylation, the beads were suspended in 200 μL 25mM NH4HCO3 with 250 ng trypsin, and incubated overnight at 37°C with shaking. The reaction was stopped by adding formic acid to 5% final concentration, and the peptides were purified with the OASIS HLB C18 cartridges (Waters, Milford, MA) before TMT labeling. The TMT labeling, LC separation, CID MS/MS identification and HCD-MS3 quantification were the same as described for whole yeast proteome quantification. Wild-type strain was used as negative control. All proteins identified in the negative control were removed as nonspecific binding.
Supplementary Material
Abbreviations
- Abs
autophagic bodies
- ACN
acetonitrile
- APC
anaphase-promoting complex
- ATG
autophagy related gene
- CID
collision-induced dissociation
- CMA
chaperone-mediated autophagy
- CQ
chloroquine
- Cvt
cytoplasm-to-vacuole targeting
- DAVID
Database for Annotation, Visualization and Integrated Discovery
- DTT
dithiothreitol
- FDR
false discovery rate
- GAL4.ER
Gal4-estrogen receptor fusion protein
- GFP
green fluorescent protein
- HCD
higher energy collision dissociation
- LC
liquid chromatography
- MEN
mitotic exit network
- MS
mass spectrometry
- ORF
open reading frame
- PAGE
polyacrylamide gel electrophoresis
- PE
phosphatidylethanolamine
- SCF
Skp Cullin, F-box containing complex
- SCX
strong cation exchange chromatography
- SDS
sodium dodecyl sulfate
- SPBs
spindle pole bodies
- SPM
sporulation medium
- STRING
Search Tool for the Retrieval of Interacting Genes/Proteins
- TEM
transmission electron microscopy
- TMT
tandem mass tag
- UPS
ubiquitin-proteasome system
- VPS
vacuolar protein sorting
- WT
wild type
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Lilin Du, Yihong Ye and Haining Du for their critical reading of the manuscript. We are grateful to Angelika Amon and Hongguo Yu for providing SK1 background strains, Steven I. Reed for pRS406 pHIS3-mCherry-TUB1 plasmid. We also thank Jingnan Liang (from Institute of Microbiology, Chinese Academy of Science) for technical assistance in TEM analysis.
Funding
This work is supported by the National Basic Research Program of China (Grant No.: 2011CB944303, 2013CB911400), the National Natural Science Foundation of China (31171374, 81222006), a Knowledge Innovation Program (Grant No. : KSCX2-YW-N-071) and One Hundred Talents Program of Chinese Academy of Sciences.
References
- [1].Neiman AM. Sporulation in the budding yeast Saccharomyces cerevisiae. Genetics 2011; 189:737-65; PMID:22084423; http://dx.doi.org/ 10.1534/genetics.111.127126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Brar GA, Yassour M, Friedman N, Regev A, Ingolia NT, Weissman JS. High-resolution view of the yeast meiotic program revealed by ribosome profiling. Science 2012; 335:552-7; PMID:22194413; http://dx.doi.org/ 10.1126/science.1215110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Pesin JA, Orr-Weaver TL. Regulation of APC/C activators in mitosis and meiosis. Annual Rev Cell Dev Biol 2008; 24:475-99; http://dx.doi.org/ 10.1146/annurev.cellbio.041408.115949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Peters JM. SCF and APC: the Yin and Yang of cell cycle regulated proteolysis. Curr Opin Cell Biol 1998; 10:759-68; PMID:9914180; http://dx.doi.org/ 10.1016/S0955-0674(98)80119-1 [DOI] [PubMed] [Google Scholar]
- [5].Cheng CH, Lo YH, Liang SS, Ti SC, Lin FM, Yeh CH, Huang HY, Wang TF. SUMO modifications control assembly of synaptonemal complex and polycomplex in meiosis of Saccharomyces cerevisiae. Genes Dev 2006; 20:2067-81; PMID:16847351; http://dx.doi.org/ 10.1101/gad.1430406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Kurz T, Pintard L, Willis JH, Hamill DR, Gönczy P, Peter M, Bowerman B. Cytoskeletal regulation by the Nedd8 ubiquitin-like protein modification pathway. Science 2002; 295:1294-8; PMID:11847342; http://dx.doi.org/ 10.1126/science.1067765 [DOI] [PubMed] [Google Scholar]
- [7].Komatsu M, Ichimura Y. Selective autophagy regulates various cellular functions. Genes Cells 2010; 15:923-33; PMID:20670274; http://dx.doi.org/ 10.1111/j.1365-2443.2010.01433.x [DOI] [PubMed] [Google Scholar]
- [8].Abeliovich H, Klionsky DJ. Autophagy in yeast: mechanistic insights and physiological function. Microbiol Mol Biol Rev 2001; 65:463-79, Table of contents; PMID:11528006; http://dx.doi.org/ 10.1128/MMBR.65.3.463-479.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Maria Cuervo A. Autophagy: in sickness and in health. Trends Cell Biol 2004; 14:70-7; PMID:15102438; http://dx.doi.org/ 10.1016/j.tcb.2003.12.002 [DOI] [PubMed] [Google Scholar]
- [10].Nakatogawa H, Suzuki K, Kamada Y, Ohsumi Y. Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat Rev Mol Cell Biol 2009; 10:458-67; PMID:19491929; http://dx.doi.org/ 10.1038/nrm2708 [DOI] [PubMed] [Google Scholar]
- [11].Enyenihi AH, Saunders WS. Large-scale functional genomic analysis of sporulation and meiosis in Saccharomyces cerevisiae. Genetics 2003; 163:47-54; PMID:12586695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Deutschbauer AM, Williams RM, Chu AM, Davis RW. Parallel phenotypic analysis of sporulation and postgermination growth in Saccharomyces cerevisiae. Proc Natl Acad Sci 2002; 99:15530-5; PMID:12432101; http://dx.doi.org/ 10.1073/pnas.202604399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Zubenko GS, Jones EW. Protein Degradation, Meiosis and Sporulation in Proteinase-Deficient mutants of saccharomyces cerevisiae. Genetics 1981; 97:45-64; PMID:7021321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Tsukada M, Ohsumi Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett 1993; 333:169-74; PMID:8224160; http://dx.doi.org/ 10.1016/0014-5793(93)80398-E [DOI] [PubMed] [Google Scholar]
- [15].Chu S, DeRisi J, Eisen M, Mulholland J, Botstein D, Brown PO, Herskowitz I. The transcriptional program of sporulation in budding yeast. Science 1998; 282:699-705; PMID:9784122; http://dx.doi.org/ 10.1126/science.282.5389.699 [DOI] [PubMed] [Google Scholar]
- [16].Carlile TM, Amon A. Meiosis I is established through division-specific translational control of a cyclin. Cell 2008; 133:280-91; PMID:18423199; http://dx.doi.org/ 10.1016/j.cell.2008.02.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Klionsky DJ, Cuervo AM, Seglen PO. Methods for monitoring autophagy from yeast to human. Autophagy 2007; 3:181-206; PMID:17224625; http://dx.doi.org/ 10.4161/auto.3678 [DOI] [PubMed] [Google Scholar]
- [18].Nakamura N, Matsuura A, Wada Y, Ohsumit Y. Acidification of vacuoles is required for autophagic degradation in the yeast Saccharomyces cerevisiae. J Biochem 1997; 121:338-44; PMID:9089409; http://dx.doi.org/ 10.1093/oxfordjournals.jbchem.a021592 [DOI] [PubMed] [Google Scholar]
- [19].Ohsumi Y. Molecular dissection of autophagy: two ubiquitin-like systems. Nat Rev Mol Cell Biol 2001; 2:211-6; PMID:11265251; http://dx.doi.org/ 10.1038/35056522 [DOI] [PubMed] [Google Scholar]
- [20].San-Segundo PA, Roeder GS. Pch2 links chromatin silencing to meiotic checkpoint control. Cell 1999; 97:313-24; PMID:10319812; http://dx.doi.org/ 10.1016/S0092-8674(00)80741-2 [DOI] [PubMed] [Google Scholar]
- [21].Watanabe Y, Nurse P. Cohesin Rec8 is required for reductional chromosome segregation at meiosis. Nature 1999; 400:461-4; PMID:10440376; http://dx.doi.org/ 10.1038/22774 [DOI] [PubMed] [Google Scholar]
- [22].Klein F, Mahr P, Galova M, Buonomo SB, Michaelis C, Nairz K, Nasmyth K. A central role for cohesins in sister chromatid cohesion, formation of axial elements, and recombination during yeast meiosis. Cell 1999; 98:91-103; PMID:10412984; http://dx.doi.org/ 10.1016/S0092-8674(00)80609-1 [DOI] [PubMed] [Google Scholar]
- [23].Shou W, Seol JH, Shevchenko A, Baskerville C, Moazed D, Chen Z, Jang J, Shevchenko A, Charbonneau H, Deshaies RJ. Exit from mitosis is triggered by Tem1-dependent release of the protein phosphatase Cdc14 from nucleolar RENT complex. Cell 1999; 97:233-44; PMID:10219244; http://dx.doi.org/ 10.1016/S0092-8674(00)80733-3 [DOI] [PubMed] [Google Scholar]
- [24].van Werven FJ, Amon A. Regulation of entry into gametogenesis. Philosophical Transactions Royal Society B 2011; 366:3521-31; http://dx.doi.org/ 10.1098/rstb.2011.0081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Simchen G. Commitment to meiosis: what determines the mode of division in budding yeast? Bioessays 2009; 31:169-77; PMID:19204989; http://dx.doi.org/ 10.1002/bies.200800124 [DOI] [PubMed] [Google Scholar]
- [26].Farmer S, Hong EJE, Leung WK, Argunhan B, Terentyev Y, Humphryes N, Toyoizumi H, Tsubouchi H. Budding yeast Pch2, a widely conserved meiotic protein, is involved in the initiation of meiotic recombination. PloS One 2012; 7:e39724; PMID:22745819; http://dx.doi.org/ 10.1371/journal.pone.0039724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Rockmill B, Roeder GS. RED1: a yeast gene required for the segregation of chromosomes during the reductional division of meiosis. Proc Natl Acad Sci 1988; 85:6057-61; PMID:3413075; http://dx.doi.org/ 10.1073/pnas.85.16.6057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Woltering D, Baumgartner B, Bagchi S, Larkin B, Loidl J, de los Santos T, Hollingsworth NM. Meiotic segregation, synapsis, and recombination checkpoint functions require physical interaction between the chromosomal proteins Red1p and Hop1p. Mol Cell Biol 2000; 20:6646-58; PMID:10958662; http://dx.doi.org/ 10.1128/MCB.20.18.6646-6658.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Iwamoto MA, Fairclough SR, Rudge SA, Engebrecht J. Saccharomyces cerevisiae Sps1p regulates trafficking of enzymes required for spore wall synthesis. Eukaryotic Cell 2005; 4:536-44; PMID:15755916; http://dx.doi.org/ 10.1128/EC.4.3.536-544.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].van der Vaart JM, Caro L, Chapman JW, Klis FM, Verrips CT. Identification of three mannoproteins in the cell wall of Saccharomyces cerevisiae. J Bacteriol 1995; 177:3104-10; PMID:7768807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].De Virgilio C, DeMarini DJ, Pringle JR. SPR28, a sixth member of the septin gene family in Saccharomyces cerevisiae that is expressed specifically in sporulating cells. Microbiol 1996; 142:2897-905; http://dx.doi.org/ 10.1099/13500872-142-10-2897 [DOI] [PubMed] [Google Scholar]
- [32].Christodoulidou A, Bouriotis V, Thireos G. Two sporulation-specific chitin deacetylase-encoding genes are required for the ascospore wall rigidity of Saccharomyces cerevisiae. J Biol Chem 1996; 271:31420-5; PMID:8940152; http://dx.doi.org/ 10.1074/jbc.271.49.31420 [DOI] [PubMed] [Google Scholar]
- [33].Percival-Smith A, Segall J. Characterization and mutational analysis of a cluster of three genes expressed preferentially during sporulation of Saccharomyces cerevisiae. Mol Cell Biol 1986; 6:2443-51; PMID:3023934; http://dx.doi.org/ 10.1128/MCB.6.7.2443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Krisak L, Strich R, Winters RS, Hall JP, Mallory MJ, Kreitzer D, Tuan RS, Winter E. SMK1, a developmentally regulated MAP kinase, is required for spore wall assembly in Saccharomyces cerevisiae. Genes Dev 1994; 8:2151-61; PMID:7958885; http://dx.doi.org/ 10.1101/gad.8.18.2151 [DOI] [PubMed] [Google Scholar]
- [35].Onodera J, Ohsumi Y. Ald6p is a preferred target for autophagy in yeast, Saccharomyces cerevisiae. J Biol Chem 2004; 279:16071-6; PMID:14761979; http://dx.doi.org/ 10.1074/jbc.M312706200 [DOI] [PubMed] [Google Scholar]
- [36].Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al.. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445-544; PMID:22966490; http://dx.doi.org/ 10.4161/auto.19496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].PA C, AP M. Repression by the yeast meiotic inhibitor RME1. Genes Dev 1993; 7:11. [DOI] [PubMed] [Google Scholar]
- [38].Yorimitsu T, Klionsky DJ. Atg11 links cargo to the vesicle-forming machinery in the cytoplasm to vacuole targeting pathway. Mol Biol Cell 2005; 16:1593-605; PMID:15659643; http://dx.doi.org/ 10.1091/mbc.E04-11-1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].SV SJG, MU HJK, DJ K. Cvt19 is a receptor for the cytoplasm-to-vacuole targeting pathway. Mol Cell 2001; 7:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].EH H, MJ L, MW B, HR P. Retromer and the sorting nexins Snx4/41/42 mediate distinct retrieval pathways from yeast endosomes. EMBO J 2003; 22:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Kanki T, Wang K, Baba M, Bartholomew CR, Lynch-Day MA, Du Z, Geng J, Mao K, Yang Z, Yen WL, et al.. A genomic screen for yeast mutants defective in selective mitochondria autophagy. Mol Biol Cell 2009; 20:4730-8; PMID:19793921; http://dx.doi.org/ 10.1091/mbc.E09-03-0225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Lu K, Psakhye I, Jentsch S. Autophagic clearance of polyQ proteins mediated by ubiquitin-Atg8 adaptors of the conserved CUET protein family. Cell 2014; 158:549-63; PMID:25042851; http://dx.doi.org/ 10.1016/j.cell.2014.05.048 [DOI] [PubMed] [Google Scholar]
- [43].Welter E, Thumm M, Krick R. Quantification of nonselective bulk autophagy inS. cerevisiaeusing Pgk1-GFP. Autophagy 2014; 6:794-7; http://dx.doi.org/ 10.4161/auto.6.6.12348 [DOI] [PubMed] [Google Scholar]
- [44].Deutschbauer AM, Williams RM, Chu AM, Davis RW. Parallel phenotypic analysis of sporulation and postgermination growth in Saccharomyces cerevisiae. Proc Natl Acad Sci 2002; 99:15530-5; PMID:12432101; http://dx.doi.org/ 10.1073/pnas.202604399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protocols 2008; 4:44-57; http://dx.doi.org/ 10.1038/nprot.2008.211 [DOI] [PubMed] [Google Scholar]
- [46].Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 2009; 37:1-13; PMID:19033363; http://dx.doi.org/ 10.1093/nar/gkn923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].LB-Y Josefsberg, Galiani D, Dantes A, Amsterdam A, Dekel N. The proteasome is involved in the first metaphase-to-anaphase transition of meiosis in rat oocytes. Biol Repro 2000; 62:1270-7; http://dx.doi.org/ 10.1095/biolreprod62.5.1270 [DOI] [PubMed] [Google Scholar]
- [48].Rabitsch KP, Tóth A, Gálová M, Schleiffer A, Schaffner G, Aigner E, Rupp C, Penkner AM, Moreno-Borchart AC, Primig M, et al.. A screen for genes required for meiosis and spore formation based on whole-genome expression. Curr Biol 2001; 11:1001-9; PMID:11470404; http://dx.doi.org/ 10.1016/S0960-9822(01)00274-3 [DOI] [PubMed] [Google Scholar]
- [49].Meiling-Wesse K, Barth H, Thumm M. Ccz1p/Aut11p/Cvt16p is essential for autophagy and the cvt pathway. FEBS Lett 2002; 526:71-6; http://dx.doi.org/ 10.1016/S0014-5793(02)03119-8 [DOI] [PubMed] [Google Scholar]
- [50].Yu X, Cai M. The yeast dynamin-related GTPase Vps1p functions in the organization of the actin cytoskeleton via interaction with Sla1p. J Cell Sci 2004; 117:3839-53; PMID:15265985; http://dx.doi.org/ 10.1242/jcs.01239 [DOI] [PubMed] [Google Scholar]
- [51].Yeh E, Driscoll R, Coltrerai M, Olinsi A, Bloom K. A dynamin-like protein encoded by the yeast sporulation. Nature 1991; 349:21; http://dx.doi.org/ 10.1038/349713a0 [DOI] [PubMed] [Google Scholar]
- [52].Anderson SF, Steber CM, Esposito RE, Coleman JE. UME6, a negative regulator of meiosis in Saccharomyces cerevisiae, contains a C-terminal Zn2Cys6 binuclear cluster that binds the URS1 DNA sequence in a zinc-dependent manner. Protein Sci 1995; 4:1832-43; PMID:8528081; http://dx.doi.org/ 10.1002/pro.5560040918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Takahashi H, McCaffery JM, Irizarry RA, Boeke JD. Nucleocytosolic acetyl-coenzyme a synthetase is required for histone acetylation and global transcription. Mol Cell 2006; 23:207-17; PMID:16857587; http://dx.doi.org/ 10.1016/j.molcel.2006.05.040 [DOI] [PubMed] [Google Scholar]
- [54].Mukaiyama H, Kajiwara S, Hosomi A, Giga-Hama Y, Tanaka N, Nakamura T, Takegawa K. Autophagy-deficient Schizosaccharomyces pombe mutants undergo partial sporulation during nitrogen starvation. Microbiol 2009; 155:3816-26; http://dx.doi.org/ 10.1099/mic.0.034389-0 [DOI] [PubMed] [Google Scholar]
- [55].Koch A, Krug K, Pengelley S, Macek B, Hauf S. Mitotic substrates of the kinase aurora with roles in chromatin regulation identified through quantitative phosphoproteomics of fission yeast. Sci Signal 2011; 4:rs6; PMID:21712547; http://dx.doi.org/ 10.1126/scisignal.2001588 [DOI] [PubMed] [Google Scholar]
- [56].Reinders J, Zahedi RP, Pfanner N, Meisinger C, Sickmann A. Toward the complete yeast mitochondrial proteome: multidimensional separation techniques for mitochondrial proteomics. J Proteome Res 2006; 5:1543-54; PMID:16823961; http://dx.doi.org/ 10.1021/pr050477f [DOI] [PubMed] [Google Scholar]
- [57].de Godoy LM, Olsen JV, Cox J, Nielsen ML, Hubner NC, Fröhlich F, Walther TC, Mann M. Comprehensive mass-spectrometry-based proteome quantification of haploid versus diploid yeast. Nature 2008; 455:1251-4; PMID:18820680; http://dx.doi.org/ 10.1038/nature07341 [DOI] [PubMed] [Google Scholar]
- [58].Scaife C, Mowlds P, Grassl J, Polden J, Daly CN, Wynne K, Dunn MJ, Clyne RK. Two-D DIGE analysis of the budding yeast pH 6-11 proteome in meiosis. Proteomics 2010; 10:4401-14; PMID:21136594; http://dx.doi.org/ 10.1002/pmic.201000376 [DOI] [PubMed] [Google Scholar]
- [59].Grassl J, Scaife C, Polden J, Daly CN, Iacovella MG, Dunn MJ, Clyne RK. Analysis of the budding yeast pH 4-7 proteome in meiosis. Proteomics 2010; 10:506-19; PMID:20029842; http://dx.doi.org/ 10.1002/pmic.200900561 [DOI] [PubMed] [Google Scholar]
- [60].Primig M, Williams RM, Winzeler EA, Tevzadze GG, Conway AR, Hwang SY, Davis RW, Esposito RE. The core meiotic transcriptome in budding yeasts. Nat Genetics 2000; 26:415-23; PMID:11101837; http://dx.doi.org/ 10.1038/82539 [DOI] [PubMed] [Google Scholar]
- [61].Guo X, Zhang P, Huo R, Zhou Z, Sha J. Analysis of the human testis proteome by mass spectrometry and bioinformatics. Proteomics-Clin Applications 2008; 2:1651-7; http://dx.doi.org/ 10.1002/prca.200780120 [DOI] [PubMed] [Google Scholar]
- [62].Guo X, Shen J, Xia Z, Zhang R, Zhang P, Zhao C, Xing J, Chen L, Chen W, Lin M, et al.. Proteomic analysis of proteins involved in spermiogenesis in mouse. J Proteome Res 2010; 9:1246-56; PMID:20099899; http://dx.doi.org/ 10.1021/pr900735k [DOI] [PubMed] [Google Scholar]
- [63].Peter M, Castro A, Lorca T, Le Peuch C, Magnaghi-Jaulin L, Dorée M, Labbé JC. The APC is dispensable for first meiotic anaphase in Xenopus oocytes. Nat Cell Biol 2000; 3:83-7 [DOI] [PubMed] [Google Scholar]
- [64].Guardavaccaro D, Kudo Y, Boulaire J, Barchi M, Busino L, Donzelli M, Margottin-Goguet F, Jackson PK, Yamasaki L, Pagano M. Control of meiotic and mitotic progression by the F box protein β-Trcp1 in vivo. Dev Cell 2003; 4:799-812; PMID:12791266; http://dx.doi.org/ 10.1016/S1534-5807(03)00154-0 [DOI] [PubMed] [Google Scholar]
- [65].Wilkinson CR, Wallace M, Morphew M, Perry P, Allshire R, Javerzat JP, McIntosh JR, Gordon C. Localization of the 26S proteasome during mitosis and meiosis in fission yeast. EMBO J 1998; 17:6465-76; PMID:9822592; http://dx.doi.org/ 10.1093/emboj/17.22.6465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Qian MX, Pang Y, Liu CH, Haratake K, Du BY, Ji DY, Wang GF, Zhu QQ, Song W, Yu Y, et al.. Acetylation-Mediated Proteasomal Degradation of Core Histones during DNA Repair and Spermatogenesis. Cell 2013; 153:1012-24; PMID:23706739; http://dx.doi.org/ 10.1016/j.cell.2013.04.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Benjamin KR, Zhang C, Shokat KM, Herskowitz I. Control of landmark events in meiosis by the CDK Cdc28 and the meiosis-specific kinase Ime2. Genes Dev 2003; 17:1524-39; PMID:12783856; http://dx.doi.org/ 10.1101/gad.1101503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Longtine MS, Mc Kenzie A III, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 1998; 14:953-61; PMID:9717241; http://dx.doi.org/ 10.1002/(SICI)1097-0061(199807)14:10%3c953::AID-YEA293%3e3.0.CO;2-U [DOI] [PubMed] [Google Scholar]
- [69].Wach A, Brachat A, Pöhlmann R, Philippsen P. New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast 1994; 10:1793-808; PMID:7747518; http://dx.doi.org/ 10.1002/yea.320101310 [DOI] [PubMed] [Google Scholar]
- [70].Kushnirov VV. Rapid and reliable protein extraction from yeast. Yeast 2000; 16:857-60; PMID:10861908; http://dx.doi.org/ 10.1002/1097-0061(20000630)16:9%3c857::AID-YEA561%3e3.0.CO;2-B [DOI] [PubMed] [Google Scholar]
- [71].Depre C, Wang Q, Yan L, Hedhli N, Peter P, Chen L, Hong C, Hittinger L, Ghaleh B, Sadoshima J, et al.. Activation of the cardiac proteasome during pressure overload promotes ventricular hypertrophy. Circulation 2006; 114:1821-8; PMID:17043166; http://dx.doi.org/ 10.1161/CIRCULATIONAHA.106.637827 [DOI] [PubMed] [Google Scholar]
- [72].Ting L, Rad R, Gygi SP, Haas W. MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat Meth 2011; 8:937-40; http://dx.doi.org/ 10.1038/nmeth.1714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Keller A, Eng J, Zhang N, Li XJ, Aebersold R. A uniform proteomics MS/MS analysis platform utilizing open XML file formats. Mol Sys Biol 2005; 1:2005.0017; http://dx.doi.org/ 10.1038/msb4100024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Witze ES, Old WM, Resing KA, Ahn NG. Mapping protein post-translational modifications with mass spectrometry. Nat Meth 2007; 4:798-806; http://dx.doi.org/ 10.1038/nmeth1100 [DOI] [PubMed] [Google Scholar]
- [75].Cox J, Mann M. 1D and 2D annotation enrichment: a statistical method integrating quantitative proteomics with complementary high-throughput data. BMC Bioinformatics 2012; 13 Suppl 16:S12; PMID:23176165; http://dx.doi.org/ 10.1186/1471-2105-13-S16-S12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Kaiser CA, Schekman R. Distinct sets of SEC genes govern transport vesicle formation and fusion early in the secretory pathway. Cell 1990; 61:723-33; PMID:2188733; http://dx.doi.org/ 10.1016/0092-8674(90)90483-U [DOI] [PubMed] [Google Scholar]
- [77].Backues SK, Chen D, Ruan J, Xie Z, Klionsky DJ. Estimating the size and number of autophagic bodies by electron microscopy. Autophagy 2014; 10:155-64; PMID:24270884; http://dx.doi.org/ 10.4161/auto.26856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Passmore LA, Barford D, Wade Harper J. Purification and Assay of the Budding Yeast Anaphase-Promoting Complex In: Raymond JD, ed. Methods Enzymol 2005:195-219 [DOI] [PubMed] [Google Scholar]
- [79].Fukuyama H, Verdier Y, Guan Y, Makino-Okamura C, Shilova V, Liu X, Maksoud E, Matsubayashi J, Haddad I, Spirohn K, et al.. Landscape of protein–protein interactions in Drosophila immune deficiency signaling during bacterial challenge. Proc Natl Acad Sci 2013; 110:10717-22; PMID:23749869; http://dx.doi.org/ 10.1073/pnas.1304380110 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.