

ABSTRACT
Macroautophagy (hereafter autophagy) is a cellular catabolic process that is essential for maintaining tissue homeostasis and regulating various normal and pathologic processes in human diseases including cancer. One cancer-driving process is accumulation of genetic mutations due to impaired DNA damage repair, including nucleotide excision repair. Here we show that autophagy positively regulates nucleotide excision repair through enhancing DNA damage recognition by the DNA damage sensor proteins XPC and DDB2 via 2 pathways. First, autophagy deficiency downregulates the transcription of XPC through TWIST1-dependent activation of the transcription repressor complex E2F4-RBL2. Second, autophagy deficiency impairs the recruitment of DDB2 to ultraviolet radiation (UV)-induced DNA damage sites through TWIST1-mediated inhibition of EP300. In mice, the pharmacological autophagy inhibitor Spautin-1 promotes UVB-induced tumorigenesis, whereas the autophagy inducer rapamycin reduces UVB-induced tumorigenesis. These findings demonstrate the crucial role of autophagy in maintaining proper nucleotide excision repair in mammalian cells and suggest a previously unrecognized tumor-suppressive mechanism of autophagy in cancer.
Keywords: autophagy, DDB2, nucleotide excision repair, UV, XPC
Introduction
Occurring at a low basal level, macroautophagy (hereafter autophagy) can be induced to maintain tissue homeostasis in response to a variety of physiological and pathological stresses,1 including solar UV B radiation (UVB).2,3 Autophagy dysfunction is associated with multiple human diseases, such as neurodegeneration, infectious diseases, metabolic diseases, cardiovascular diseases, aging, and cancer.4-7
In carcinogenesis and cancer progression, autophagy can be both oncogenic and tumor suppressive.6-9 On the one hand, autophagy promotes tumorigenesis and tumor progression at least in part through promoting cell survival and invasion.1-3,10,11 On the other hand, autophagy suppresses tumorigenesis and tumor progression through selectively degrading SQSTM1/p62, an autophagy receptor protein and signaling adaptor that promotes inflammation, cell proliferation and migration,12-16 through removing damaged proteins and organelles to prevent genomic damage,4,6,15,17,18 or by promoting homologous recombination DNA repair in response to ionizing radiation-induced DNA double-strand breaks in mammalian cells.19,20
In addition, autophagy may suppress tumor initiation by regulating another essential DNA repair pathway: nucleotide excision repair (NER). NER is a versatile DNA repair pathway that eliminates a wide variety of helix-distorting base lesions induced by environmental carcinogenic sources, including 2 products induced by solar UVB radiation, namely, cyclobutane pyrimidine dimers (CPD) and (6-4) photoproducts (6-4PP), as well as other bulky adducts induced by air pollutants.21-27 Defects in the global genome NER (GG-NER) subpathway cause xeroderma pigmentosum (XP), an autosomal recessive disorder predisposing affected individuals to cancer development not only in the skin, but also in the brain and lungs.24,25,28,29 Seven XP group genes plus one variant have been identified as indispensable NER factors,24,25,29-31 among which XPC and DDB2 are essential for GG-NER-specific damage recognition.32-34
These NER factors are regulated at the transcription and posttranslational levels by a number of endogenous and environmental factors.35,36 Since these XP factors are difficult to target directly, identification of their upstream regulators will provide pharmaceutically accessible tools for enhancing NER and potentially reducing tumorigenesis. Here we show that autophagy positively regulates DNA damage recognition by NER via 2 pathways. First, autophagy deficiency downregulates the transcription of XPC. Second, autophagy deficiency impairs the recruitment of DDB2 to UV-induced CPD sites through TWIST1-mediated inhibition of EP300. In mice, the pharmacological autophagy inducer rapamycin decreases UVB-induced tumorigenesis while the inhibitor Spautin-1 increases it. These findings demonstrate the critical role of autophagy in maintaining proper NER capacity and suggest a new tumor-suppressive mechanism of autophagy in tumor initiation.
Results
Autophagy deficiency inhibits nucleotide excision repair and downregulates XPC
To determine whether autophagy plays a role in NER, we compared NER capacity in mouse embryonic fibroblast (MEF) cells from wild-type (WT) mice with that from mice that were autophagy-deficient through loss of various autophagy-related (Atg) genes. A deficiency in Atg5 significantly reduced the repair of CPDs (Fig. 1A and C), the major DNA damage that drives UV-induced tumorigenesis.37 Similarly, deficiency in the genes Atg7, Atg12, or Atg14 significantly reduced the repair of CPDs (Fig. S1A to F). In addition, Atg5 deficiency significantly reduced the repair of 6-4PPs (Fig. 1B and D), which are repaired much faster than CPDs and do not contribute to UV-induced skin tumorigenesis.37 As compared with the WT group, in both MEF and immortalized baby mouse kidney (iBMK) cells, Atg5 deficiency decreased the levels of XPC (Fig. 1E), an NER protein essential for both CPD and 6-4PP repair.31 However, autophagy deficiency had no effect on the protein levels of other NER factors (Fig. S1G to H). Deficiency of Atg5, Atg7, Atg12, or Atg14 blocked the conversion of LC3-I to LC3-II and induced SQSTM1 accumulation (Fig. S1G, S1J to K),2,16 2 distinctive hallmarks of autophagy,38,39 and Atg5 deficiency blocked basal and induced autophagic flux by either UVB irradiation or rapamycin (Fig. S1L),2 indicating an autophagy deficiency. In iBMK cells, knockdown of either Atg5 or Atg7 inhibited autophagy and decreased the XPC protein level (Fig. 1F). In human keratinocytes (HaCaT cells), the autophagy inhibitor Spautin-1 decreased the XPC protein level (Fig. S1I). Furthermore, in HaCaT cells ATG7 knockdown inhibited autophagy and decreased the basal XPC protein level and UVB-induced XPC ubiquitination (Fig. 1G), a biochemical process critical for DNA damage recognition mediated by the UV-DDB complex.40-42 These results indicate that autophagy deficiency inhibits NER and downregulates XPC and UVB-induced XPC ubiquitination.
Figure 1.
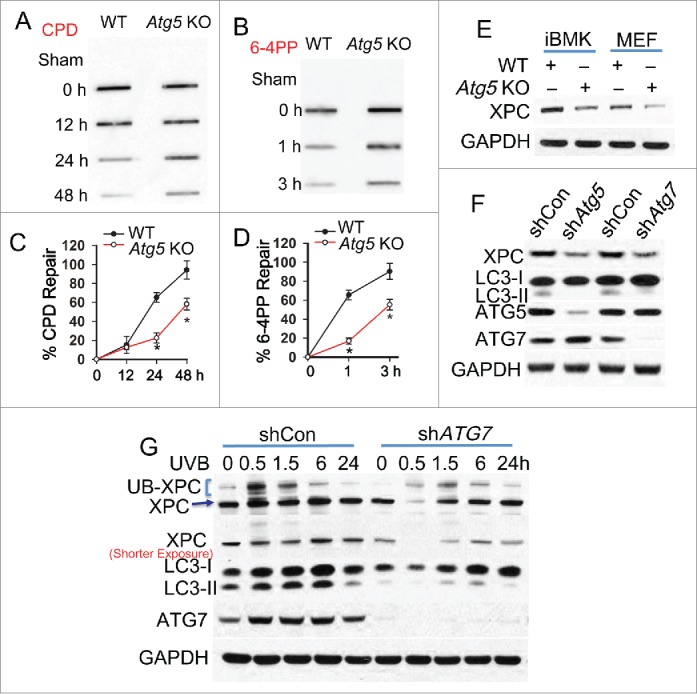
Autophagy deficiency inhibits nucleotide excision repair and downregulates XPC. (A, B) Slot blot analysis of the levels of CPD (A) and 6-4PP (B) in WT and atg5 KO MEF cells at 0, 12, 24 and 48 h post-UVB (10 mJ/cm2) for CPD and 0, 1 and 3 h post-UVB (10 mJ/cm2) for 6-4PP. (C, D) Quantification of percentage (%) of CPD repair (C) from A and 6-4PP repair (D) from B (mean±SD, n=3). *, P < 0.05, compared with WT group, Student t test (C, D). (E) Immunoblot analysis of XPC and GAPDH in WT and atg5 KO MEF and iBMK cells. (F) Immunoblot analysis of XPC, LC3-I/II, ATG5, ATG7 and GAPDH in iBMK cells lentivirally infected with shRNA nontargeted control (shCon) or shRNA targeting Atg5 (shAtg5) or Atg7 (shAtg7). (G) Immunoblot analysis of XPC, UB-XPC (polyubiquitinated XPC), LC3-I, LC3-II, ATG7 and GAPDH in HaCaT cells transfected with shCon or shATG7 at 0, 0.5, 1.5, 6 and 24 h post-UVB (20 mJ/cm2). The results were obtained from 3 independent experiments.
Autophagy deficiency links TWIST1 to suppression of nucleotide excision repair
To determine the mechanism by which autophagy regulates XPC and NER, we assessed the functional significance of TWIST1, an important transcription factor stabilized by autophagy deficiency or SQSTM1 upregulation16,43 (Fig. 2A). In autophagy-deficient cells, knockdown of Twist1 increased the XPC protein level, while it had no effect on the SQSTM1 level (Fig. 2A). In WT and sqstm1 knockout (KO) MEF cells, overexpression of Twist1 decreased the XPC protein level, indicating that TWIST1-induced inhibition of XPC is SQSTM1-independent (Fig. 2B). In autophagy-deficient cells, knockdown of Twist1 restored the repair of both CPD and 6-4PP to a capacity similar to wild-type cells (Fig. 2C to F). These results indicate that autophagy deficiency inhibits NER and downregulates XPC through upregulating TWIST1.
Figure 2.
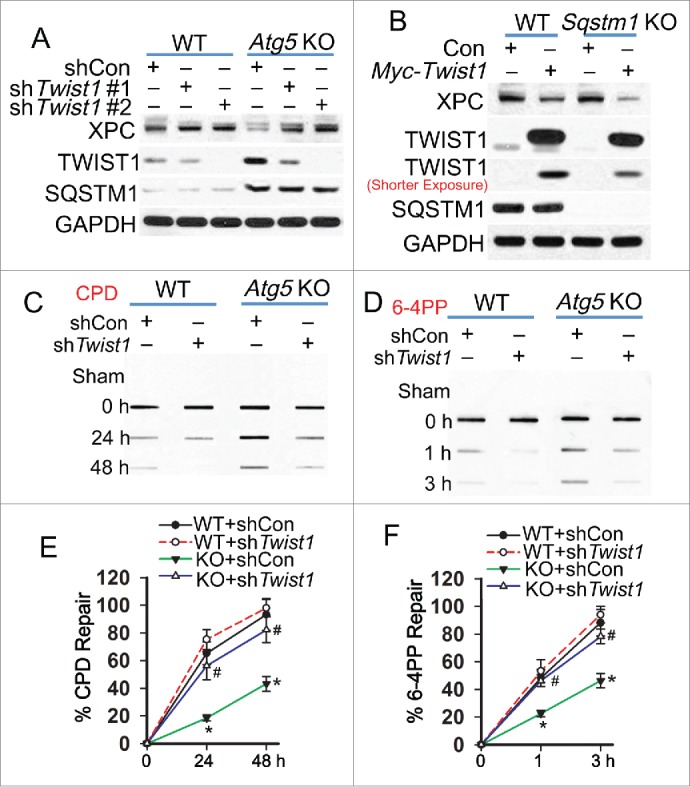
Autophagy deficiency links TWIST1 to suppression of nucleotide excision repair. (A) Immunoblot analysis of XPC, TWIST1, SQSTM1 and GAPDH in WT and atg5 KO MEF cells lentivirally infected with shCon or 2 independent shRNAs targeting Twist1 (shTwist1 #1 or shTwist1 #2). (B) Immunoblot analysis of XPC, TWIST1, SQSTM1 and GAPDH in WT and sqstm1 KO MEF cells lentivirally infected with vector control (Con) or Myc-Twist1 expression vector. (C, D) Slot blot analysis of the levels of CPD (C) and 6-4PP (D) in WT and atg5 KO MEF cells lentivirally infected with shCon or shTwist1. Cells were collected for analysis at 0, 24 and 48 h post-UVB (10 mJ/cm2) for CPD and 0, 1 and 3 h post-UVB (10 mJ/cm2) for 6-4PP. (E, F) Quantification of percentage (%) of CPD repair (E) from C and 6-4PP repair (F) from D (mean±SD, n=3). *, P < 0.05, compared with WT group; #, P < 0.05, compared with KO+shCon group, Student t test. The results were obtained from 3 independent experiments.
Autophagy deficiency inhibits XPC transcription through the TWIST1-AKT pathway
To determine the mechanism by which autophagy deficiency downregulates XPC through TWIST1, we analyzed the mRNA level of Xpc and the transcriptional activity of the Xpc promoter in WT and atg5 KO MEF cells. Autophagy deficiency decreased the Xpc mRNA level and the transcription activity of the Xpc promoter (Fig. 3A-B). Twist1 knockdown abolished the effect of autophagy deficiency (Fig. 3A and B). TWIST1 belongs to a family of basic helix-loop-helix domain-containing transcription factors that regulate gene transcription through interaction with the proximal E-box elements of target gene promoters.44-47 To determine whether E-box elements are involved in TWIST1-mediated inhibition of Xpc transcription, we mutated the putative E-box sites identified in silico individually or in combination (Fig. S2A). Mutation of these E-box sites had no effect on autophagy deficiency-induced inhibition of Xpc transcription (Fig. 3C), indicating an E-box-independent mechanism.
Figure 3.

Autophagy deficiency inhibits XPC transcription through the TWIST1-AKT pathway. (A) RT-PCR analysis of Xpc mRNA levels in WT and atg5 KO MEF cells lentivirally infected with shCon or shTwist1 (mean±SD, n=3). (B) Luciferase reporter assay of the Xpc promoter in WT and atg5 KO MEF cells lentivirally infected with shCon or shTwist1 (mean±SD, n=3). (C) Luciferase reporter assay of the Xpc promoter with wild-type (WT) sequence, deletion of E-Box2/3, mutation of E-Box1, mutation of E-Box4 or deletion of E-Box2/3 in combination with mutation of E-Box1/3 in WT and atg5 KO MEF cells (mean±SD, n=3). *, P < 0.05, compared with WT group; #, P < 0.05, compared with WT or shCon group, Student t test (A-C). (D) Immunoblot analysis of p-AKT (Ser473), total AKT, TWIST1 and GAPDH in WT and atg5 KO MEF cells lentivirally infected with shCon or shTwist1. (E) Immunoblot analysis of RBL2, E2F4, LMNB/LAMIN B and GAPDH in cytosolic and nuclear fraction from WT and atg5 KO MEF cells. (F) Luciferase reporter assay of the Xpc promoter with wild-type (WT) sequence and E2F mutation (E2F mut) in WT and atg5 KO MEF (mean±SD, n=3). (G) Immunoblot analysis of XPC, p-AKT (Ser473), total AKT, TWIST1 and GAPDH in WT and atg5 KO MEF cells treated with or without the PI3K-AKT pathway inhibitor LY294002 (LY, 10 μM). (H, I) Quantification of percentage (%) of CPD repair (H) and 6-4PP repair (I) in WT and atg5 KO MEF cells treated with or without LY294002 (LY, 10 μM) (mean±SD, n=3). *, P < 0.05, compared with WT group; #, P < 0.05, compared with Veh group, Student t test (H, I). The results were obtained from 3 independent experiments.
To determine the underlying mechanism by which TWIST1 suppresses Xpc expression, we assessed the role of AKT activation, since TWIST1 has been demonstrated to activate the AKT pathway,48-50 and AKT activation suppresses Xpc transcription.51 Our results indicated that autophagy deficiency activates the AKT pathway and that Twist1 knockdown blocked the effect of autophagy deficiency on AKT activation (Fig. 3D). We have recently demonstrated that the AKT pathway promotes the nuclear localization of RBL2/p130,51 forming the RBL2-E2F4 complex that binds to the E2F site of the Xpc promoter and inhibits Xpc transcription.52,53 Autophagy deficiency increased the nuclear localization of RBL2, whereas it had no effect on E2F4 localization (Fig. 3E). Mutation of the E2F site in the Xpc promoter abolished autophagy deficiency-mediated inhibition of Xpc transcription (Fig. 3F, Fig. S2B). In autophagy-deficient cells, blocking the AKT pathway with the PI3K inhibitor LY294002 completely restored the XPC protein level and 6-4PP repair but only partially rescued CPD repair (Fig. 3G to I, Fig. S2C and D). These results indicate that autophagy regulates repair of 6-4PP and CPD through promoting Xpc expression, while another mechanism in addition to XPC availability is also critical for autophagy regulation of CPD repair.
Autophagy deficiency inhibits 6-4PP repair via decreasing XPC while it inhibits CPD repair via both decreasing XPC availability and damage recognition by DDB2
To determine the role of XPC in autophagy regulation of NER, XPC was added in both WT and atg5 KO MEF cells. Xpc addition did not affect the TWIST1 protein level (Fig. 4A). In autophagy-deficient cells, Xpc addition completely restored 6-4PP repair to a level similar to WT cells, while it only partially restored CPD repair (Fig. 4B and C, Fig. S3A and B). To determine the role of TWIST1 in CPD repair independent of XPC transcription, we re-expressed XPC driven by the constitutive CMV promoter in XPC-deficient human dermal fibroblasts (XPC-/--CMV-XPC), in which XPC transcription is not affected by transcriptional regulators including TWIST1. In these XPC-/--CMV-XPC cells, TWIST1 addition did not affect XPC expression (Fig. 4D). TWIST1 addition inhibited CPD repair, while it had no effect on 6-4PP repair (Fig. 4E, Fig. S3C to E). It has been demonstrated that unlike 6-4PP repair, CPD repair requires DDB2 for recognizing CPD formed in genomic DNA.54 A micro-irradiation assay demonstrated that TWIST1 addition inhibited the recruitment of both XPC (Fig. 4F and G) and DDB2 to subnuclear UV-induced CPD sites (Fig. 4H and I). These data demonstrated that autophagy deficiency inhibits 6-4PP repair through downregulation of XPC, whereas it inhibits CPD repair through both downregulating XPC and inhibiting CPD damage recognition by DDB2.
Figure 4.

Autophagy deficiency inhibits 6-4PP repair via decreasing XPC while it inhibits CPD repair via both decreasing XPC availability and damage recognition by DDB2. (A) Immunoblot analysis of XPC, TWIST1 and GAPDH in WT and atg5 KO MEF cells transfected with a construct expressing vector control (Con) or XPC. (B, C) Quantification of percentage (%) of CPD repair (B) and 6-4PP repair (C) in WT and atg5 KO MEF cells transfected with Con or XPC (mean±SD, n=3). *, P < 0.05, compared with WT group; #, P < 0.05, compared with KO+Con group, Student t test. (D) Immunoblot analysis of XPC, TWIST1 and GAPDH in XPC reconstituted cells (XPC-/--CMV-XPC) transfected with Con and MYC-TWIST1. (E) Quantification of percentage (%) of CPD repair in XPC-/--CMV-XPC cells transfected with Con and MYC-TWIST1 (mean±SD, n=3). *, P < 0.05, compared with Con group. (F, H) Immunofluorescence assay of the colocalization of XPC (F) and DDB2 (H) with subnuclear CPD in HaCaT cells transfected with Con or MYC-TWIST1 at 0.5 h post-UV (10 mJ/cm2) through a 5 μm micropore filter. Scale bar: 10 μm. (G, I) The relative intensity of XPC (G) and DDB2 (I) foci was calculated by analyzing 100 foci and normalized to that of CPD (n =100, error bar: SD). The results were obtained from 3 independent experiments.
Autophagy deficiency inhibits DDB2 recruitment through TWIST1 binding and inhibition of EP300
To determine the mechanism by which TWIST1 inhibits CPD recognition by DDB2, we assessed the role of EP300, an NER-promoting acetyltransferase55-58 binding with TWIST1.59 EP300 is important for the recruitment of DDB2, an essential step for the recruitment of the DDB1-CUL4A complex and subsequent ubiquitination of XPC.55 We found that TWIST1 binds with EP300, and the TWIST1-EP300 interaction is increased in autophagy-deficient MEF cells (Fig. 5A). In contrast to the partial restoration of CPD repair obtained by adding Xpc alone, in autophagy-deficient cells adding both Xpc and Ep300 completely restored CPD repair to a level similar to WT cells, while it had no effect on the TWIST1 protein level (Fig. 5B and C, Fig. S4A). In XPC-/--CMV-XPC cells, deletion of the EP300-binding domain in TWIST1 completely abolished the inhibitory effect of TWIST1 on CPD repair (Fig. 5D to F, Fig. S4B) and DDB2 recruitment to the CPD sites (Fig. 5G and H). These data indicate that in autophagy-deficient cells, increased TWIST1-EP300 interaction inhibits CPD recognition by DDB2 and thus suppresses CPD repair.
Figure 5.

Autophagy deficiency inhibits DDB2 recruitment through Twist1 binding and inhibition of EP300. (A) Immunoprecipitation was performed in WT and atg5 KO MEF cells with the indicated antibodies, followed by immunoblot analysis of EP300 and TWIST1. (B) Immunoblot analysis of EP300, XPC, TWIST1 and GAPDH in WT and atg5 KO MEF cells transfected with Con or the combination of Xpc and Ep300. (C) Quantification of percentage (%) of CPD repair in WT and atg5 KO MEF cells transfected with Con or the combination of Xpc and Ep300 (mean±SD, n=3). *, P < 0.05, compared with WT group; #, P < 0.05, compared with Con group, Student t test. (D) Immunoblot analysis of XPC, TWIST1 and GAPDH in XPC-/--CMV-XPC cells transfected with Con and MYC-TWIST1 (36 to 72) deletion. (E) Slot blot analysis of the levels of CPD in XPC-/--CMV-XPC cells transfected with Con and MYC-TWIST1 (36 to 72) deletion. (F) Quantification of percentage (%) of CPD repair from (E) (mean±SD, n=3). *, P < 0.05, compared with WT group; #, P < 0.05, compared with Con group, Student t test. (G) Immunofluorescence assay of the colocalization of DDB2 with subnuclear CPD in HaCaT cells transfected with Con or MYC-TWIST1 (36 to 72) deletion at 0.5 h post-UV (10 mJ/cm2) through a 5-μm micropore filter. Scale bar: 10 μm. (H) The relative intensity of DDB2 was calculated by analyzing 100 foci and normalized to that of CPD (n =100, Mean±SD). All results were obtained from 3 independent experiments.
Pharmacological modulators of autophagy regulate UVB-induced skin carcinogenesis
To determine the functional significance of autophagy in skin cancer, we assessed the effect of pharmacological modulators on UVB-induced skin tumorigenesis as a clinically relevant mouse model. We irradiated SKH-1 mice with UVB for 17 wk until they developed 1 to 3 tumors on average that were 1 to 3mm3 in diameter. Then these mice were treated topically with vehicle, the autophagy inducer rapamycin or the autophagy inhibitor Spautin-1, along with real or sham continuing UVB exposure (Fig. 6A). In UVB-irradiated nontumor skin, rapamycin induced autophagy while Spautin-1 inhibited it (Fig. 6B). Autophagy inhibition by Spautin-1 increased the TWIST1 level and decreased the XPC level, while autophagy induction by rapamycin decreased the TWIST1 level and increased the XPC level (Fig. 6B).
Figure 6.

Pharmacological modulators of autophagy regulate UVB-induced skin carcinogenesis. (A) A schematic diagram of the experimental design for B to G, in which mice were treated with UVB irradiation for 17 wk, 3 times per wk, and then with vehicle only or topical rapamycin (Rap, 10 nmol) or Spautin-1 (SP, 25 nmol) 3 h prior to each UVB or sham treatment 3 times a wk for another 10 wk. (B) Immunoblot analysis of LC3-I, LC3-II, SQSTM1, XPC, TWIST1 and GAPDH in mouse skin collected 12 h after the final treatment. (C) Representative histological and immunohistochemical analysis of SQSTM1, TWIST1, and MKI67 protein levels (brown) in UVB-irradiated mouse skin treated with Vehicle (Veh), rapamycin, or Spautin-1. Scale bar: 50 µm. (D, E) Number (#) of new tumors per mouse at different weeks following Rap or Spautin-1 treatment as in (A) (n=5), without (D) or with (E) continuing UVB irradiation. (F, G) Average volume (mm3) of established tumors formed at 17 wk post-UVB at different weeks following treatment as in (A), without (F) or with (G) continuing UVB irradiation. The results were mean±SD (n=5). *, P <0.05; **, P<0.01 compared with the Veh group in D to G). [Change label to Spautin-1.]
Immunohistochemical analysis also showed that Spautin-1 increased while rapamycin decreased the levels of SQSTM1 and TWIST1 and the number of MKI67/Ki67-positive cells (Fig. 6C). Rapamycin alone or in combination with continued UVB exposure significantly reduced new tumor formation and inhibited the growth of established tumors (Fig. 6D to G). Treatment with Spautin-1 alone had no effect on new tumor formation but significantly promoted the growth of established tumors (Fig. 6D and F). In contrast, treatment with Spautin-1 in combination with UVB irradiation significantly increased both new tumor formation and growth of established tumors (Fig. 6E and G). These results indicate that pharmacological autophagy modulators regulate UVB-induced tumorigenesis and tumor growth and suggest a tumor-suppressive role of autophagy in skin tumorigenesis. It is possible that the other targets of Spautin-1 or rapamycin also contribute to their effect on UVB-induced tumorigenesis, including the effect of rapamycin on the autophagy-independent processes regulated by MTOR signaling.60,61 Further investigation will elucidate the specific contribution of autophagy modulation in the effect of these pharmacological autophagy modulators.
Discussion
Autophagy plays critical roles in tissue homeostasis and stress response.1,5 In cancer, the role of autophagy is shown to be dynamic and context-dependent.6-9 Here we show that autophagy is required for efficient repair of UVB-induced DNA damage by nucleotide excision repair (NER). Autophagy deficiency inhibits NER by suppressing transcription of XPC and recruitment of DDB2 to UVB-induced CPD sites. In mice, the pharmacological autophagy inducer rapamycin decreases UVB-induced tumorigenesis while inhibitor Spautin-1 increases it. Our findings elucidate a crucial role of autophagy in NER and provide a vital molecular mechanism linking autophagy and genomic integrity in cancer (Fig. 7).
Figure 7.
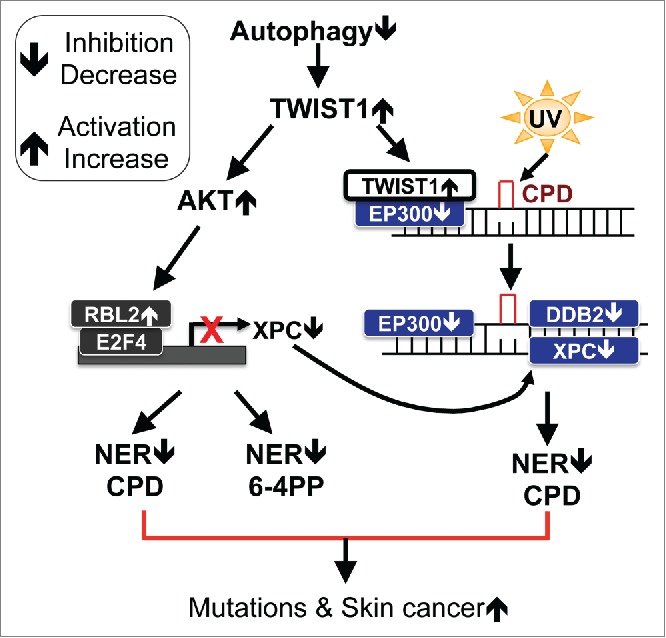
Schematic diagram of autophagy regulation of nucleotide excision repair in skin carcinogenesis. Autophagy positively regulates nucleotide excision repair through decreasing TWIST1 stability, by which autophagy (1) maintains the proper transcription of XPC through inhibiting the transcription repressor complex E2F4-RBL2 and (2) positively regulates the recruitment of DDB2 to UV-induced DNA damage sites through EP300. Thus autophagy may reduce UV-induced accumulation of mutations, thereby reducing skin tumorigenesis.
Our findings have identified a novel role of autophagy in regulating NER in mammalian cells. We demonstrated that autophagy regulates NER via suppressing TWIST1. TWIST1 inhibits NER through at least 2 mechanisms: (1) TWIST1 downregulates XPC transcription through activating the E2F4-RBL2 transcription repressor complex, and (2) TWIST1 inhibits UVB-CPD recognition by DDB2 through binding to EP300 and thus suppressing EP300 activity. Furthermore, in autophagy-deficient cells, inhibition of EP300 activity by TWIST1 upregulation may also impair later steps in NER, since EP300 is important for chromatin remodeling and the function of XPG and PCNA.56-58 Future investigation will demonstrate whether other mechanisms are involved in the regulation of XPC and NER by the autophagy-TWIST1 axis, including RAD23B, which not only binds to XPC to stabilize XPC protein and to stimulate XPC activity in NER62-64 but also binds to TWIST1 in regulating TWIST1 stability by autophagy.16 Additional investigation will also determine whether autophagy also positively regulates NER in lower organisms besides mammalian cells.
In contrast to TWIST1’s suppressive role in NER, other EMT transcription factors, including ZEB1, SNAI1/SNAIL and SNAI2/SLUG, promote DSB repair and cisplatin-DNA damage repair and thus promote radioresistance and cisplatin resistance, respectively.65-67 Thus the role of EMT factors in DNA repair seems to be specific to DNA damage types and the specific repair system required. Our findings demonstrated that TWIST1 mediates NER suppression in autophagy-deficient cells, underscoring the crucial role of TWIST1 regulation by autophagy in cancer initiation.
Our results indicate that TWIST1 suppresses XPC transcription through AKT-mediated activation of the E2F4-RBL2 suppressor complex, but not through the putative E-box elements in the XPC promoter. Future studies will reveal whether E-box elements in the distal region of the XPC gene have active roles in the effect of TWIST1. TWIST1 activates the AKT pathway through upregulating AKT248,49 or downregulating the tumor suppressor PTEN.50 Our findings indicate that inhibiting the PI3K-AKT pathway restored XPC expression, supporting the critical role of the TWIST1-AKT signaling axis in NER suppression.
In summary, our findings demonstrate that autophagy positively regulates UVB-DNA damage recognition by nucleotide excision repair via suppressing TWIST1 and suggest that autophagy is tumor suppressive in the skin. TWIST1 suppresses the transcription of Xpc and the activity of EP300 in DNA damage sensing by DDB2. Our findings may shed light on the previously unrecognized positive role of autophagy in nucleotide excision repair, genomic integrity, and thus tumor suppression.
Materials and methods
Animal treatments
All animal procedures have been approved by the University of Chicago Institutional Animal Care and Use Committee. Hairless SKH-1 mice were obtained from Charles River (477). SKH-1 mice were exposed to UVB (100 mJ/cm2, dose selected to avoid visible sunburn) dorsally or sham-irradiated, 3 times a wk for up to 27 wk, to monitor tumor formation and growth. After 17 wk of irradiation, mice were treated with rapamycin (10 nmol; LC Laboratories, R-5000) or Spautin-1 (25 nmol; Cellagen Technology, C3430-2S) topically. Mouse skin samples were fixed in formalin for histological analysis (HE) or immunohistochemical analysis for TWIST1 (Abcam, 50887), SQSTM1 (PROGEN, GP62) and MKI67 (Abcam, 15580)-positive cells (Immunohistochemistry Core Facility).
Cell culture
WT and atg5 KO MEF cells (obtained from Dr. Noboru Mizushima, University of Tokyo, Japan), WT and atg5 KO iBMK cells (provided by Dr. Eileen White, The Cancer Institute of New Jersey, NJ, USA), WT, sqstm1 KO, atg7 KO, atg12 KO, and Atg14 cKO MEF cells (cKO, derived from Atg14fl/fl MEF cells treated with a Cre-expressing retroviral vector) (obtained from Dr. Seungmin Hwang, University of Chicago, IL, USA), XPC-deficient human fibroblast cells (Coriell Institute, GM15983), HaCaT (human keratinocytes, provided by Dr. Norbert E. Fusenig, German Cancer Research Center, Germany) and HEK-293T (human embryonic kidney cells) were maintained in a monolayer culture in 95% air/5% CO2 at 37°C in Dulbecco’s modified Eagle’s medium (Invitrogen, 11965-092) supplemented with 10% fetal bovine serum (Hyclone, SH30910.03), 100 units per mL penicillin and 100 μg per mL streptomycin (Invitrogen, 15140-122). A pharmacological inhibitor of the AKT pathway (LY294002; Enzo, BML-ST420-0005) was added 12 h before the UVB treatment to a final concentration of 10 μM.
Plasmids transfection
Lentiviral systems were used to reconstitute XPC (CMV-XPC) in XPC-deficient (XPC-/-) cells (Coriell, GM15983) (XPC-/--CMV-XPC), to express Myc-Twist1 in WT and sqstm1 KO MEF cells, and knockdown of mouse Atg5, Atg7, Twist1 and human ATG7, with HEK293T cells used as packaging cells. HEK293T cells were transfected with pLKO.1 vectors, pLKO.1 shAtg5 (mouse), pLKO.1 shAtg7 (mouse), pLKO.1 shTwist1 (mouse), pLKO.1 shATG7 (Human), pLenti con, pLenti-XPC, pLenti-MYC-TWIST1 and packaging mix (psPAX2 and pMG2) using X-tremGENE 9 (Roche, 06366236001) as described as previously.16 The piggyBac transposon system (pBC3) was used to express MYC-TWIST1 or the MYC-TWIST1 (36 to 72) deletion mutant in XPC-/--CMV-XPC cells using X-tremeGENE HP (Roche) according to the manufacturer’s instructions as described previously.16 To stably express MYC-TWIST1 or MYC-TWIST1 (36 to 72) deletion mutants in HaCaT cells, we transfected the cells with pBC3 vector or pBC3- MYC-TWIST1 (36 to 72) using Amaxa Nucleofector according to the manufacturer’s instructions as described previously.16,51 Transient transfection of Xpc and Ep300 in WT and atg5 KO MEF cells was carried out using X-tremeGENE HP (Roche, 06365787001) according to the manufacturer’s instructions.
Plasmids
pCMV6 vector and XPC-pCMV6 were obtained from OriGene Technologies. MYC-TWIST1 pcDNA3.1 was kindly provided by Dr. Anthony Firuli (Indiana University, IN, USA). pLKO.1 shTwist1 (mouse) was obtained from Dharmacon (Dharmacon, RMM4532-EG22160). pLKO.1 shAtg5 (mouse) and pLKO.1 shAtg7 (mouse) were kindly provided by Dr. Seungmin Hwang. Ep300 pcDNA3.1 (Addgene, 30489; deposited by Tso-Pang Yao) and pLenti CMV Dest vectors (Addgene, 17452; deposited by Eric Campeau) were obtained from Addgene. pLKO.1 shATG7 (human) was obtained from Dr. Alec Kimmelman (Harvard Medical School, MA, USA). pGL3 XPC-luc, pGL3 XPC-luc WT and pGL3 XPC-luc with E2F mutation were provided by Dr. Pradip Raychaudhuri (University of Illinois at Chicago, IL, USA).
DNA constructs
We cloned the MYC-TWIST1 (HindIII/XbaI) from the pcDNA3.1 vector to the pBC3 vector (modified piggyBac vector) and the pENTER vector. We also cloned XPC from the pCMV vector to the pENTER vector. TWIST1 deletion (pBC3- MYC-TWIST1-(36 to 72) deletion) was performed using the following primers: primer A 5'- CCATCGATAAGCTTGGTACCGAGC-3', primer B 5'- CTTGCCGCGCTTAGCCCCGCGCTT-3', primer C 5'- AAGCGCGGGGCTAAGCGCGGCAAG-3' and primer D 5'- CGTATCTAGACTAGTGGGACGCGG-3'. Overlap extension PCR was performed to generate pBC3- MYC-TWIST1-(36 to 72) deletion as described previously.68 pENTER MYC-TWIST1 and pENTER XPC were recombined into pLenti CMV Dest vector using Gateway LR Clonase Enzyme Mix (Invitrogen, 11791-019) following the manufacturer’s instructions. All constructs were confirmed by sequencing.
Site-directed mutagenesis
Mutations of the E-Box1/4 binding site and deletion of the E-Box2/3 binding site of wild-type pGL3-XPC plasmids were carried out using a QuikChange XL site-directed mutagenesis kit following the manufacturer’s instructions (Stratagene, 200521) as described previously.16 The amino acid changes were introduced into the pGL3-XPC plasmids by using the following primers. E-Box1 sense 5'- AACTCTAGCTCCAGGGGATAAGATACCTTGTTCTGTCTCAC-3' and E-Box1 antisense 5'- GTGAGACAGAACAAGGTATCTTATCCCCTGGAGCTAGAGTT-3’; E-Box2/3 deletion sense 5'- TGGTGTTTCAATCACTGGTGGTGCTAGTACACAACC-3' and E-Box2/3 deletion antisense 5'- GGTTGTGTACTAGCACCACCAGTGATTGAAACACCA-3’; E-Box4 sense 5'- GCAGGGGCTGTGTGTGAATGTAAGGGTGTGAAAGACAT-3' and E-Box4 antisense 5'- ATGTCTTTCACACCCTTACATTCACACACAGCCCCTGC-3’. All mutants were confirmed by sequencing.
Immunoprecipitation
Immunoprecipitation was performed as described previously by using anti-TWIST1 (Abcam, 50887) antibody.16
Western blotting
Western blotting was performed as described previously.16,51 Antibodies used were as follows: XPC (Santa Cruz Biotechnology, sc30156), GAPDH (Santa Cruz Biotechnology, sc25778), E2F4 (Santa Cruz Biotechnology, sc1082), RBL2 (Santa Cruz Biotechnology, sc317), total AKT (Santa Cruz Biotechnology, sc5298) and EP300 (Santa Cruz Biotechnology, sc585); p-AKT (Ser473) (Cell Signaling Technology, 3789), ATG5 (Cell Signaling Technology, 8540), ATG7 (Cell Signaling Technology, 8558) and LC3-I/II (Cell Signaling Technology, 4108); DDB2 (Abcam, 181136) and TWIST1 (Abcam, 50887) and SQSTM1/p62 (PROGENE GP62). Cytosolic and nuclear proteins were isolated as described previously.51
Luciferase reporter assays
Cells were transfected with pGL3 Xpc -Luc, pGL3 Xpc -Luc WT, pGL3 Xpc -Luc E2F mutation, pGL3 Xpc (E-Box1 Mutation)-Luc, pGL3 Xpc (E-Box2/3-Deletion)-Luc, pGL3 Xpc (E-Box4-Mutation)-Luc and 0.025 μg of pRL-TK (used as a transfection efficiency control; Promega, E2241) using X-tremeGENE HP (Roche, 06366236001) according to the manufacturer’s instructions. Luciferase reporter assays were performed as described previously.16,51
Immunohistochemical analysis
TWIST1 (Abcam, 50887), SQSTM1/p62 (PROGEN, GP62) and MKI67 (Abcam, 15580) were determined using immunohistochemical analysis by the Immunohistochemistry core facility at the University of Chicago. To exclude the contribution of endogenous brown pigmentation due to melanin, we also performed hematoxylin and eosin (H&E) staining.
Real-time PCR
Quantitative real-time PCR assays were performed using a CFX Connect real-time system (Bio-Rad, Hercules, CA) using Bio-Rad iQ SYBR Green Supermix (Bio-Rad, 1708880).51 The threshold cycle number (CQ) for each sample was determined in triplicate. The CQ for values for Xpc were normalized against Gapdh as described previously.51 Amplification primers were 5’- CAAAAAGCAAGGTGGTGGACC-3’ (forward), 5’- TTCATCATCCTCCGCAGGTATG-3’ (reverse) for the mouse Xpc gene; 5’- AGGTCGGTGTGAACGGATTTG-3’ (forward) and 5’- TGTAGACCATGTAGTTGAGGT-3’ (reverse) for the mouse Gapdh gene.
Determination of 2 major forms of UVB-induced DNA damage in genomic DNA by slot blot assay
Slot blot assays of CPD and 6-4PP were performed as described previously.69 Briefly, cells were collected at different time points post-UVB, and DNA was isolated using a QIAamp DNA Mini Kit (Qiagen, 51304). The DNA concentration was calculated from the absorbance at 260 nm using NanoDrop 1000 (NanoDrop products, Wilmington, DE). The CPD and 6-4PP in DNA were quantified by slot blot (Bio-Rad) with antibodies (COSMO BIO Co.,TDM-2 for CPD and 64 M-2 for 6-4PP). For examining repair kinetics, the percentage (%) of repair was calculated by comparing the optical density at the indicated time to that of the corresponding absorbance at time zero when there was no opportunity for repair and 100% of CPDs (or 6-4PPs) were present post-UVB.
Local UV irradiation
Local (micropore) UV irradiation was carried out as described.55 Briefly, an isopore polycarbonate filter (Millipore Co., TMTP02500) containing pores 5 µm in diameter was placed on top of the cell monolayer. The cells were cultured on cover slips and irradiated from above with UVC (254 nm, 10 mJ/cm2). The cells were then permeabilized and incubated with primary rabbit anti-CPD (COSMO BIO Co., TDM-2 for CPD) and 6-4PP (COSMO BIO Co., 64M-2) together with XPC (Santa Cruz Biotechnology, sc30156) or DDB2 (Abcam, 181136). After washing with phosphate-buffered saline (Life Technologies, 20012-050), the cells were incubated with Alexa Fluor 488 F (ab’) 2 fragments of goat anti-mouse IgG antibodies and Alexa Fluor 568 of goat anti-rabbit IgG antibodies (ThermoFisher Scientific, A11017 and A11011, respectively). The cells were then fixed in Prolong Gold Antifade with DAPI (Invitrogen, P-36931) and observed under a fluorescence microscope (Olympus IX71, Olympus, Center Valley, PA).
Statistical analyses
Statistical analyses were performed using Prism 6 (GraphPad software, San Diego, CA, USA). Data were expressed as the mean of at least 3 independent experiments, and analyzed by the Student t test. Error bars indicate the standard deviations of the means (SD). A P value of <0.05 was considered statistically significant.
Supplementary Material
Abbreviations
- 6-4PP
(6-4) photoproducts
- Atg
autophagy related
- CPD
cyclobutane pyrimidine dimers
- DDB2
damage-specific DNA binding protein 2, 48kDa
- EP300/p300
E1A binding protein p300
- iBMK
immortalized baby mouse kidney
- KO
knockout
- MAP1LC3/LC3
microtubule-associated protein 1 light chain 3
- MEF
mouse embryonic fibroblast
- NER
nucleotide excision repair
- RBL2/p130
retinoblastoma-like 2
- shRNA
small hairpin RNA
- SQSTM1/p62
sequestosome 1
- UV
ultraviolet radiation
- UVB
ultraviolet B radiation
- WT
wild type
- XPC
xeroderma pigmentosum, complementation group C
Acknowledgments
We are grateful to Dr. Altaf Wani for helpful suggestions. We thank Dr. Norbert Fusenig for providing the HaCaT cells (human keratinocytes and epithelial cells), Dr. Noboru Mizushima for kindly providing WT and atg5 KO MEFs, Dr. Seungmin Hwang for providing the WT, sqstm1 KO, atg7 KO, atg12 KO, and atg14 KO MEF cells, and pLKO.1 shAtg5 (mouse), and pLKO.1 shAtg7 (mouse) vectors, Dr. Eileen White for providing the WT and atg5 KO iBMK cells, Dr. Tony Firuli for providing MYC-TWIST1 pcDNA3.1 and FLAG-TWIST1 pcDNA3.1, Dr. Kimmelman for providing pLKO.1 shATG7 (human), Dr. Pradip Raychaudhuri for providing the pGL3 XPC-luc, pGL3 Xpc-luc WT and pGL3 Xpc-luc constructs, Terri Li for immunohistochemistry, and Dr. Ann Motten for a critical reading of the manuscript.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by the NIH/NIEHS grant ES024373 and ES016936 (YYH), the American Cancer Society (ACS) grant RSG-13-078-01 (YYH), the University of Chicago Cancer Research Center (P30 CA014599), the CTSA (UL1 TR000430), and the University of Chicago Friends of Dermatology Endowment Fund.
References
- 1.Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell 2010; 40:280-93; PMID:20965422; http://dx.doi.org/ 10.1016/j.molcel.2010.09.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Qiang L, Wu C, Ming M, Viollet B, He YY. Autophagy controls p38 activation to promote cell survival under genotoxic stress. J Biol Chem 2013; 288:1603-11; PMID:23212914; http://dx.doi.org/ 10.1074/jbc.M112.415224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen LH, Chu PM, Lee YJ, Tu PH, Chi CW, Lee HC, Chiou SH. Targeting protective autophagy exacerbates UV-triggered apoptotic cell death. Int J Mol Sci 2012; 13:1209-24; PMID:22312313; http://dx.doi.org/ 10.3390/ijms13011209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature 2008; 451:1069-75; PMID:18305538; http://dx.doi.org/ 10.1038/nature06639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med 2013; 368:1845-6; PMID:23656658; http://dx.doi.org/ 10.1056/NEJMra1205406 [DOI] [PubMed] [Google Scholar]
- 6.White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer 2012; 12:401-10; PMID:22534666; http://dx.doi.org/ 10.1038/nrc3262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Galluzzi L, Pietrocola F, Bravo-San Pedro JM, Amaravadi RK, Baehrecke EH, Cecconi F, Codogno P, Debnath J, Gewirtz DA, Karantza V, et al.. Autophagy in malignant transformation and cancer progression. EMBO J 2015; 34(7):856-80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kimmelman AC. The dynamic nature of autophagy in cancer. Genes Dev 2011; 25:1999-2010; PMID:21979913; http://dx.doi.org/ 10.1101/gad.17558811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhi X, Zhong Q. Autophagy in cancer. F1000Prime Rep 2015; 7:18; PMID:25750736; http://dx.doi.org/ 10.12703/P7-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lock R, Kenific CM, Leidal AM, Salas E, Debnath J. Autophagy-dependent production of secreted factors facilitates oncogenic RAS-driven invasion. Cancer Discov 2014; 4:466-79; PMID:24513958; http://dx.doi.org/ 10.1158/2159-8290.CD-13-0841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maycotte P, Jones KL, Goodall ML, Thorburn J, Thorburn A. Autophagy Supports Breast Cancer Stem Cell Maintenance by Regulating IL6 Secretion. Mol Cancer Res 2015; 13(4):651-8; PMID:25573951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 2005; 171:603-14; PMID:16286508; http://dx.doi.org/ 10.1083/jcb.200507002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007; 282:24131-45; PMID:17580304; http://dx.doi.org/ 10.1074/jbc.M702824200 [DOI] [PubMed] [Google Scholar]
- 14.Duran A, Linares JF, Galvez AS, Wikenheiser K, Flores JM, Diaz-Meco MT, Moscat J. The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer Cell 2008; 13:343-54; PMID:18394557; http://dx.doi.org/ 10.1016/j.ccr.2008.02.001 [DOI] [PubMed] [Google Scholar]
- 15.Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, Bray K, Reddy A, Bhanot G, Gelinas C, et al.. Autophagy suppresses tumorigenesis through elimination of p62. Cell 2009; 137:1062-75; PMID:19524509; http://dx.doi.org/ 10.1016/j.cell.2009.03.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qiang L, Zhao BZ, Ming M, Wang N, He TC, Hwang S, Thorburn A, He YY. Regulation of cell proliferation and migration by p62 through stabilization of Twist1. Proc Natl Acad Sci U S A 2014; 111:9241-6; PMID:24927592; http://dx.doi.org/ 10.1073/pnas.1322913111 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 17.Karantza-Wadsworth V, Patel S, Kravchuk O, Chen G, Mathew R, Jin S, White E. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev 2007; 21:1621-35; PMID:17606641; http://dx.doi.org/ 10.1101/gad.1565707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vessoni AT, Filippi-Chiela EC, Menck CF, Lenz G. Autophagy and genomic integrity. Cell Death Differ 2013; 20:1444-54; PMID:23933813; http://dx.doi.org/ 10.1038/cdd.2013.103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu EY, Xu N, O'Prey J, Lao LY, Joshi S, Long JS, O'Prey M, Croft DR, Beaumatin F, Baudot AD, et al.. Loss of autophagy causes a synthetic lethal deficiency in DNA repair. Proc Natl Acad Sci U S A 2015; 112:773-8; PMID:25568088; http://dx.doi.org/ 10.1073/pnas.1409563112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mo N, Lu YK, Xie WM, Liu Y, Zhou WX, Wang HX, Nong L, Jia YX, Tan AH, Chen Y, et al.. Inhibition of autophagy enhances the radiosensitivity of nasopharyngeal carcinoma by reducing Rad51 expression. Oncol Rep 2014; 32:1905-12; PMID:25175062 [DOI] [PubMed] [Google Scholar]
- 21.Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem 2004; 73:39-85; PMID:15189136; http://dx.doi.org/ 10.1146/annurev.biochem.73.011303.073723 [DOI] [PubMed] [Google Scholar]
- 22.Niggli HJ, Rothlisberger R. Cyclobutane-type pyrimidine photodimer formation and induction of ornithine decarboxylase in human skin fibroblasts after UV irradiation. J Invest Dermatol 1988; 91:579-84; PMID:3192953; http://dx.doi.org/ 10.1111/1523-1747.ep12477095 [DOI] [PubMed] [Google Scholar]
- 23.Vink AA, Berg RJ, de Gruijl FR, Roza L, Baan RA. Induction, repair and accumulation of thymine dimers in the skin of UV-B-irradiated hairless mice. Carcinogenesis 1991; 12:861-4; PMID:2029750; http://dx.doi.org/ 10.1093/carcin/12.5.861 [DOI] [PubMed] [Google Scholar]
- 24.Cleaver JE. Cancer in xeroderma pigmentosum and related disorders of DNA repair. Nat Rev Cancer 2005; 5:564-73; PMID:16069818; http://dx.doi.org/ 10.1038/nrc1652 [DOI] [PubMed] [Google Scholar]
- 25.Cleaver JE, Lam ET, Revet I. Disorders of nucleotide excision repair: the genetic and molecular basis of heterogeneity. Nat Rev Genet 2009; 10:756-68; PMID:19809470; http://dx.doi.org/ 10.1038/nrg2663 [DOI] [PubMed] [Google Scholar]
- 26.Braithwaite E, Wu X, Wang Z. Repair of DNA lesions induced by polycyclic aromatic hydrocarbons in human cell-free extracts: involvement of two excision repair mechanisms in vitro. Carcinogenesis 1998; 19:1239-46; PMID:9683183; http://dx.doi.org/ 10.1093/carcin/19.7.1239 [DOI] [PubMed] [Google Scholar]
- 27.Kad NM, Wang H, Kennedy GG, Warshaw DM, Van Houten B. Collaborative dynamic DNA scanning by nucleotide excision repair proteins investigated by single- molecule imaging of quantum-dot-labeled proteins. Mol Cell 2010; 37:702-13; PMID:20227373; http://dx.doi.org/ 10.1016/j.molcel.2010.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bradford PT, Goldstein AM, Tamura D, Khan SG, Ueda T, Boyle J, Oh KS, Imoto K, Inui H, Moriwaki S, et al.. Cancer and neurologic degeneration in xeroderma pigmentosum: long term follow-up characterises the role of DNA repair. J Med Genet 2011; 48:168-76; PMID:21097776; http://dx.doi.org/ 10.1136/jmg.2010.083022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.DiGiovanna JJ, Kraemer KH. Shining a light on xeroderma pigmentosum. J Invest Dermatol 2012; 132:785-96; PMID:22217736; http://dx.doi.org/ 10.1038/jid.2011.426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature 2001; 411:366-74; PMID:11357144; http://dx.doi.org/ 10.1038/35077232 [DOI] [PubMed] [Google Scholar]
- 31.Sugasawa K. Xeroderma pigmentosum genes: functions inside and outside DNA repair. Carcinogenesis 2008; 29:455-65; PMID:18174245; http://dx.doi.org/ 10.1093/carcin/bgm282 [DOI] [PubMed] [Google Scholar]
- 32.Volker M, Mone MJ, Karmakar P, van Hoffen A, Schul W, Vermeulen W, Hoeijmakers JH, van Driel R, van Zeeland AA, Mullenders LH. Sequential assembly of the nucleotide excision repair factors in vivo. Mol Cell 2001; 8:213-24; PMID:11511374; http://dx.doi.org/ 10.1016/S1097-2765(01)00281-7 [DOI] [PubMed] [Google Scholar]
- 33.Sugasawa K, Ng JM, Masutani C, Iwai S, van der Spek PJ, Eker AP, Hanaoka F, Bootsma D, Hoeijmakers JH. Xeroderma pigmentosum group C protein complex is the initiator of global genome nucleotide excision repair. Mol Cell 1998; 2:223-32; PMID:9734359; http://dx.doi.org/ 10.1016/S1097-2765(00)80132-X [DOI] [PubMed] [Google Scholar]
- 34.Riedl T, Hanaoka F, Egly JM. The comings and goings of nucleotide excision repair factors on damaged DNA. Embo J 2003; 22:5293-303; PMID:14517266; http://dx.doi.org/ 10.1093/emboj/cdg489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim I, He YY. Ultraviolet radiation-induced non-melanoma skin cancer: regulation of DNA damage repair and inflammation. Genes Dis 2014; 1(2):188-198; PMID:25642450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shah P, He YY. Molecular regulation of UV-induced DNA repair. Photochem Photobiol 2015; 91(2):254-64; PMID:25534312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jans J, Schul W, Sert YG, Rijksen Y, Rebel H, Eker AP, Nakajima S, van Steeg H, de Gruijl FR, Yasui A, et al.. Powerful skin cancer protection by a CPD-photolyase transgene. Curr Biol 2005; 15:105-15; PMID:15668165; http://dx.doi.org/ 10.1016/j.cub.2005.01.001 [DOI] [PubMed] [Google Scholar]
- 38.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 2000; 19:5720-8; PMID:11060023; http://dx.doi.org/ 10.1093/emboj/19.21.5720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, et al.. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008; 4:151-75; PMID:18188003; http://dx.doi.org/ 10.4161/auto.5338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sugasawa K, Okuda Y, Saijo M, Nishi R, Matsuda N, Chu G, Mori T, Iwai S, Tanaka K, Tanaka K, et al.. UV-induced ubiquitylation of XPC protein mediated by UV-DDB-ubiquitin ligase complex. Cell 2005; 121:387-400; PMID:15882621; http://dx.doi.org/ 10.1016/j.cell.2005.02.035 [DOI] [PubMed] [Google Scholar]
- 41.Sugasawa K. UV-DDB: a molecular machine linking DNA repair with ubiquitination. DNA Repair (Amst) 2009; 8:969-72; PMID:19493704; http://dx.doi.org/ 10.1016/j.dnarep.2009.05.001 [DOI] [PubMed] [Google Scholar]
- 42.Huang TT, D'Andrea AD. Regulation of DNA repair by ubiquitylation. Nat Rev Mol Cell Biol 2006; 7:323-34; PMID:16633336; http://dx.doi.org/ 10.1038/nrm1908 [DOI] [PubMed] [Google Scholar]
- 43.Bertrand M, Petit V, Jain A, Amsellem R, Johansen T, Larue L, Codogno P, Beau I. SQSTM1/p62 regulates the expression of junctional proteins through epithelial-mesenchymal transition factors. Cell Cycle 2015; 14:364-74; PMID:25496309; http://dx.doi.org/ 10.4161/15384101.2014.987619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell 2008; 14:818-29; PMID:18539112; http://dx.doi.org/ 10.1016/j.devcel.2008.05.009 [DOI] [PubMed] [Google Scholar]
- 45.Eckert MA, Lwin TM, Chang AT, Kim J, Danis E, Ohno-Machado L, Yang J. Twist1-induced invadopodia formation promotes tumor metastasis. Cancer Cell 2011; 19:372-86; PMID:21397860; http://dx.doi.org/ 10.1016/j.ccr.2011.01.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A, Weinberg RA. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004; 117:927-39; PMID:15210113; http://dx.doi.org/ 10.1016/j.cell.2004.06.006 [DOI] [PubMed] [Google Scholar]
- 47.Franco HL, Casasnovas J, Rodriguez-Medina JR, Cadilla CL. Redundant or separate entities?–roles of Twist1 and Twist2 as molecular switches during gene transcription. Nucleic Acids Res 2011; 39:1177-86; PMID:20935057; http://dx.doi.org/ 10.1093/nar/gkq890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cheng GZ, Chan J, Wang Q, Zhang W, Sun CD, Wang LH. Twist transcriptionally up-regulates AKT2 in breast cancer cells leading to increased migration, invasion, and resistance to paclitaxel. Cancer Res 2007; 67:1979-87; PMID:17332325; http://dx.doi.org/ 10.1158/0008-5472.CAN-06-1479 [DOI] [PubMed] [Google Scholar]
- 49.Li J, Zhou BP. Activation of beta-catenin and Akt pathways by Twist are critical for the maintenance of EMT associated cancer stem cell-like characters. BMC Cancer 2011; 11:49; PMID:21284870; http://dx.doi.org/ 10.1186/1471-2407-11-49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yin G, Chen R, Alvero AB, Fu HH, Holmberg J, Glackin C, Rutherford T, Mor G. TWISTing stemness, inflammation and proliferation of epithelial ovarian cancer cells through MIR199A2/214. Oncogene 2010; 29:3545-53; PMID:20400975; http://dx.doi.org/ 10.1038/onc.2010.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ming M, Shea CR, Guo X, Li X, Soltani K, Han W, He YY. Regulation of global genome nucleotide excision repair by SIRT1 through xeroderma pigmentosum C. Proc Natl Acad Sci U S A 2010; 107:22623-8; PMID:21149730; http://dx.doi.org/ 10.1073/pnas.1010377108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cam H, Balciunaite E, Blais A, Spektor A, Scarpulla RC, Young R, Kluger Y, Dynlacht BD. A common set of gene regulatory networks links metabolism and growth inhibition. Mol Cell 2004; 16:399-411; PMID:15525513; http://dx.doi.org/ 10.1016/j.molcel.2004.09.037 [DOI] [PubMed] [Google Scholar]
- 53.Dominguez-Brauer C, Chen YJ, Brauer PM, Pimkina J, Raychaudhuri P. ARF stimulates XPC to trigger nucleotide excision repair by regulating the repressor complex of E2F4. EMBO Rep 2009; 10:1036-42; PMID:19644500; http://dx.doi.org/ 10.1038/embor.2009.139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tang JY, Hwang BJ, Ford JM, Hanawalt PC, Chu G. Xeroderma pigmentosum p48 gene enhances global genomic repair and suppresses UV-induced mutagenesis. Mol Cell 2000; 5:737-44; PMID:10882109; http://dx.doi.org/ 10.1016/S1097-2765(00)80252-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang QE, Han C, Zhao R, Wani G, Zhu Q, Gong L, Battu A, Racoma I, Sharma N, Wani AA. p38 MAPK- and Akt-mediated p300 phosphorylation regulates its degradation to facilitate nucleotide excision repair. Nucleic Acids Res 2013; 41:1722-33; PMID:23275565; http://dx.doi.org/ 10.1093/nar/gks1312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hasan S, Hassa PO, Imhof R, Hottiger MO. Transcription coactivator p300 binds PCNA and may have a role in DNA repair synthesis. Nature 2001; 410:387-91; PMID:11268218; http://dx.doi.org/ 10.1038/35066610 [DOI] [PubMed] [Google Scholar]
- 57.Tillhon M, Cazzalini O, Nardo T, Necchi D, Sommatis S, Stivala LA, Scovassi AI, Prosperi E. p300/CBP acetyl transferases interact with and acetylate the nucleotide excision repair factor XPG. DNA Repair (Amst) 2012; 11:844-52; PMID:22954786; http://dx.doi.org/ 10.1016/j.dnarep.2012.08.001 [DOI] [PubMed] [Google Scholar]
- 58.Cazzalini O, Sommatis S, Tillhon M, Dutto I, Bachi A, Rapp A, Nardo T, Scovassi AI, Necchi D, Cardoso MC, et al.. CBP and p300 acetylate PCNA to link its degradation with nucleotide excision repair synthesis. Nucleic Acids Res 2014; 42:8433-48; PMID:24939902; http://dx.doi.org/ 10.1093/nar/gku533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hamamori Y, Sartorelli V, Ogryzko V, Puri PL, Wu HY, Wang JY, Nakatani Y, Kedes L. Regulation of histone acetyltransferases p300 and PCAF by the bHLH protein twist and adenoviral oncoprotein E1A. Cell 1999; 96:405-13; PMID:10025406; http://dx.doi.org/ 10.1016/S0092-8674(00)80553-X [DOI] [PubMed] [Google Scholar]
- 60.Easton JB, Houghton PJ. mTOR and cancer therapy. Oncogene 2006; 25:6436-46; PMID:17041628; http://dx.doi.org/ 10.1038/sj.onc.1209886 [DOI] [PubMed] [Google Scholar]
- 61.Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell 2007; 12:9-22; PMID:17613433; http://dx.doi.org/ 10.1016/j.ccr.2007.05.008 [DOI] [PubMed] [Google Scholar]
- 62.Masutani C, Sugasawa K, Yanagisawa J, Sonoyama T, Ui M, Enomoto T, Takio K, Tanaka K, van der Spek PJ, Bootsma D, et al.. Purification and cloning of a nucleotide excision repair complex involving the xeroderma pigmentosum group C protein and a human homologue of yeast RAD23. Embo J 1994; 13:1831-43; PMID:8168482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sugasawa K, Masutani C, Uchida A, Maekawa T, van der Spek PJ, Bootsma D, Hoeijmakers JH, Hanaoka F. HHR23B, a human Rad23 homolog, stimulates XPC protein in nucleotide excision repair in vitro. Mol Cell Biol 1996; 16:4852-61; PMID:8756644; http://dx.doi.org/ 10.1128/MCB.16.9.4852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xie Z, Liu S, Zhang Y, Wang Z. Roles of Rad23 protein in yeast nucleotide excision repair. Nucleic Acids Res 2004; 32:5981-90; PMID:15545636; http://dx.doi.org/ 10.1093/nar/gkh934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang P, Wei Y, Wang L, Debeb BG, Yuan Y, Zhang J, Yuan J, Wang M, Chen D, Sun Y, et al.. ATM-mediated stabilization of ZEB1 promotes DNA damage response and radioresistance through CHK1. Nat Cell Biol 2014; 16:864-75; PMID:25086746; http://dx.doi.org/ 10.1038/ncb3013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hsu DS, Lan HY, Huang CH, Tai SK, Chang SY, Tsai TL, Chang CC, Tzeng CH, Wu KJ, Kao JY, et al.. Regulation of excision repair cross-complementation group 1 by Snail contributes to cisplatin resistance in head and neck cancer. Clin Cancer Res 2010; 16:4561-71; PMID:20823140; http://dx.doi.org/ 10.1158/1078-0432.CCR-10-0593 [DOI] [PubMed] [Google Scholar]
- 67.Haslehurst AM, Koti M, Dharsee M, Nuin P, Evans K, Geraci J, Childs T, Chen J, Li J, Weberpals J, et al.. EMT transcription factors snail and slug directly contribute to cisplatin resistance in ovarian cancer. BMC Cancer 2012; 12:91; PMID:22429801; http://dx.doi.org/ 10.1186/1471-2407-12-91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bryksin AV, Matsumura I. Overlap extension PCR cloning: a simple and reliable way to create recombinant plasmids. Biotechniques 2010; 48:463-5; PMID:20569222; http://dx.doi.org/ 10.2144/000113418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ming M, Soltani K, Shea CR, Li X, He YY. Dual role of SIRT1 in UVB-induced skin tumorigenesis. Oncogene 2015; 34:357-63; PMID:24441046; http://dx.doi.org/ 10.1038/onc.2013.583 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.