

abstract
ULK1 (unc-51 like autophagy activating kinase 1), the key mediator of MTORC1 signaling to autophagy, regulates early stages of autophagosome formation in response to starvation or MTORC1 inhibition. How ULK1 regulates the autophagy induction process remains elusive. Here, we identify that ATG13, a binding partner of ULK1, mediates interaction of ULK1 with the ATG14-containing PIK3C3/VPS34 complex, the key machinery for initiation of autophagosome formation. The interaction enables ULK1 to phosphorylate ATG14 in a manner dependent upon autophagy inducing conditions, such as nutrient starvation or MTORC1 inhibition. The ATG14 phosphorylation mimics nutrient deprivation through stimulating the kinase activity of the class III phosphatidylinositol 3-kinase (PtdIns3K) complex and facilitates phagophore and autophagosome formation. By monitoring the ATG14 phosphorylation, we determined that the ULK1 activity requires BECN1/Beclin 1 but not the phosphatidylethanolamine (PE)-conjugation machinery and the PIK3C3 kinase activity. Monitoring the phosphorylation also allowed us to identify that ATG9A is required to suppress the ULK1 activity under nutrient-enriched conditions. Furthermore, we determined that ATG14 phosphorylation depends on ULK1 and dietary conditions in vivo. These results define a key molecular event for the starvation-induced activation of the ATG14-containing PtdIns3K complex by ULK1, and demonstrate hierarchical relations between the ULK1 activation and other autophagy proteins involved in phagophore formation.
Keywords: ATG13, ATG14, BECN1, MTORC1, PIK3C3, ULK1
Introduction
Autophagy is an evolutionarily conserved process through which cytoplasmic components, such as macromolecules and organelles, are captured by double-membrane compartments called phagophores that mature into autophagosomes; these cargoes are subsequently degraded in the lysosomes. Eukaryotic cells can increase their survivability under starvation through nutrient retrieval by autophagic degradation of intracellular components. Autophagy also plays important roles in cellular remodeling during differentiation and animal development, and is responsible for the selective clearance of damaged macromolecules, organelles, and invading microorganisms to maintain the cellular integrity and protection against infection.1-3 Despite recent progress in understanding the fundamental cellular process, the molecular mechanism for autophagosome formation remains unclear.
ULK1 is a serine/threonine kinase that plays a key role in the early processes of autophagy induction. ULK1 is phosphorylated by MTORC1 (mechanistic target of rapamycin [serine/threonine kinase] complex 1) and AMPK (5' AMP-dependent kinase). AMPK-mediated phosphorylations are required for full activation of ULK1,4-6 whereas MTORC1-mediated phosphorylations prevent AMPK-mediated phosphorylations and activation of ULK1.5 ULK1 forms a multiprotein complex by interacting with ATG13, RB1CC1 and ATG101.7-15 ULK1 also interacts with HSP90 and CDC37 to regulate PARK2/PARKIN-dependent mitophagy.16 The ULK1 complex engages in the formation of omegasomes,17,18 the phosphatidylinositol-3-phosphate (PtdIns3P)-enriched membrane platform on which phagophores emerge, as well as in the membrane expansion via ATG9.19,20
ULK1 functionally interacts with PIK3C3/VPS34 (phosphatidylinositol 3-kinase catalytic subunit type 3), a lipid kinase catalyzing the production of PtdIns3P. PIK3C3 interacts with PIK3R4/Vps15 and BECN1 to form a core PtdIns3K complex. The core complex interacts with ATG14, AMBRA1, UVRAG or RUBCN/RUBICON. Each of the interacting proteins exerts specific functions on the PtdIns3K complex at different stages of autophagy. Upon ATG14 binding, the PtdIns3K complex regulates the membrane extension from the endoplasmic reticulum (ER) during the initiation of phagophore formation.17,21-24 A functional linkage between the ULK1 complex and the ATG14-containing PtdIns3K complex was supported by a recent finding in yeast that Atg13 contains a HORMA domain involved in recruiting Atg14 to the ER.25 Another crucial piece of evidence supportive of the ULK1-PtdIns3K link came from the finding that ULK1 phosphorylates BECN1 for starvation-responsive activation of ATG14-containing PtdIns3K.26 Despite these findings, the molecular mechanism for how the ULK1 complex specifically targets the ATG14-containing PtdIns3K complex to trigger autophagy induction has remained unclear.
In this study, we have identified that the ULK1 complex binds and phosphorylates ATG14. The ULK1-mediated phosphorylation of ATG14 is robustly upregulated under cellular conditions that induce autophagy, such as nutrient deprivation or MTORC1 inhibition, and stimulates the kinase activity of the ATG14-containing PtdIns3K complex. We found that the phosphorylation is important for phagophore and autophagosome formation. The phosphorylation allowed us to assess hierachical relations between ULK1 and other autophagy regulators.
Results
The ULK1 complex interacts with the ATG14-containing PtdIns3K complex
ATG14 forms puncta in starved cells in a manner dependent upon ULK1.27 We confirmed the result by monitoring endogenous ATG14 in mouse embryonic fibroblasts (MEFs) using a monoclonal antibody specific to ATG14 (Fig. S1A to C). Because ATG13 binds and stabilizes ULK1, we predicted that ATG13 might also be important for ATG14 puncta formation. Similar to ULK1 deficiency, ATG13 knockdown suppressed the starvation-induced formation of ATG14 puncta in HeLa cells (Fig. 1A). Consistent with the previous report,17 ULK1 puncta colocalized with ATG14 puncta in HCT116 cells and HeLa cells under starvation (Fig. 1B).
Figure 1.

The ULK1 complex interacts with the ATG14-containing PtdIns3K complex. (A) ATG13 knockdown suppresses the formation of ATG14 puncta. shRNA-transduced HeLa cells were incubated in DMEM (full medium) or EBSS (amino acid starvation) for 2 h. Endogenous ATG14 was stained using anti-ATG14 antibody (Cell Signaling Technology, clone D3H2Z). Nuclei were stained by DAPI (blue). Scale bar: 10 μm. (B) ATG13 is important for colocalization of ULK1 and ATG14 puncta. mCherry-ULK1 was transiently expressed in WT ATG13 or ATG13 KO HCT116 cells. Endogenous ATG14 puncta were immunostained and visualized together with mCherry-ULK1 by fluorescence microscopy. (Inset) Higher magnification demonstrates colocalization (yellow) of ULK1 and ATG14 puncta. (C) HA-tagged ULK1 and ATG13 interact with MYC-tagged ATG14. The indicated proteins were expressed in HEK293T cells. The amount of MYC-ATG14 coimmunoprecipitated with HA antibody was analyzed by western blotting (WB). RPS6KB1 was used as a negative control. (D) The amount of MYC-tagged ULK1 recovered with HA immunoprecipitates from HEK293T cells was analyzed by WB. (E) The ATG14-associated PtdIns3K complex is coimmunoprecipitated with ATG13. MYC-ATG13 recovered with HA immunoprecipitates from HEK293T cells was analyzed by WB. RPS6KB1 and PA-PLA were used as negative controls for cytosolic and membrane proteins, respectively. (F) Endogenous ATG14 is coimmunoprecipitated with endogenous ATG13. ATG13 immunoprecipitates were isolated from MEFs using 2 different sources of polyclonal antibodies specific to ATG13 (labeled by Ab1 and Ab2, respectively).9 As a negative control, preimmune serum (IgG) was used. (G) The ATG14-containing PtdIns3K complex is coimmunoprecipitated with endogenous ATG13. Immunoprecipitates obtained from HEK293T cells using ATG13 Ab2 antibody were analyzed for the presence of the indicated proteins by WB. (H) Coimmunoprecipitation of endogenous ULK1 and ATG13 with endogenous ATG14. Immunoprecipitates obtained from HEK293T cells using anti-ATG14 antibody (Santa Cruz Biotechnology, sc-164767) were analyzed for the indicated proteins. Preimmune serum (IgG) and anti-GSK3A goat polyclonal antibody were used as control. (I) Confirmation of the specificity of the ATG13-ATG14 interaction. Anti-ATG13 immunoprecipitates were obtained from WT ATG13 and ATG13 KO HCT116 cells and analyzed by WB.
The similar pattern of induction of ULK1 and ATG14 puncta and their colocalization triggered us to test a possibility of their physical interaction. Using recombinant proteins expressed in HEK293T cells, we found that ATG14 is coimmunoprecipitated with ULK1 and ATG13 but not with RPS6KB1/S6K1 used as a negative control (Fig. 1C). We also found that ULK1 is coimmunoprecipitated with BECN1 (Fig. 1D), indicating that the ULK1 complex might interact with the ATG14-containing PtdIns3K complex. Similar to ULK1, ATG13 was coimmunoprecipitated with ATG14, BECN1, and PIK3C3 but not with a cytosolic RPS6KB1 or a membrane-associated PA-PLA (phosphatidic acid-preferring phospholipase A) used as negative controls (Fig. 1E). Using 2 different sources of polyclonal antibodies made respectively against an N-terminal epitope (amino acids 2 to 15) and an epitope (amino acids 462 to 475) near the C terminus of human ATG13,9 we confirmed that endogenous ATG13 interacts with endogenous ATG14 (Fig. 1F). The ATG13 C-terminal antibody was able to coimmunoprecipitate endogenous PIK3C3 and BECN1 (Fig. 1G). Reciprocally, endogenous ULK1 and ATG13 were coimmunoprecipitated with anti-ATG14 antibody (Fig. 1H). ATG14 was not detected in anti-ATG13 immunoprecipitates isolated from ATG13 KO HCT116 cells, confirming the specificity of the ATG13-ATG14 interaction (Fig. 1I).
ATG13 directly binds to ATG14 and mediates the ULK1-PtdIns3K interaction
Knowing that the ULK1 complex interacts with the ATG14-containing PtdIns3K complex, we investigated whether the interaction is direct, and if then, which protein mediates the interaction. Deficiency of ULK1 in MEFs only marginally reduced the amount of ATG13 coimmunoprecpitated with ATG14 (Fig. 2A), whereas deficiency of ATG13 in HCT116 cells completely disrupted the interaction between ULK1 and ATG14 (Fig. 2B). We also found that deficiency of BECN1 in HCT116 cells did not disrupt the interaction between ATG14 and ATG13 (Fig. 2C), whereas deficiency of ATG14 in HEK293T cells or HCT116 cells disrupted the interaction of ATG13 with PIK3C3 and BECN1 (Fig. 2D and E). This result suggests that ATG13 and ATG14 are important for the interaction between the ULK1 complex and the ATG14-containing PtdIns3K complex.
Figure 2.

ATG13 and ATG14 mediate the interaction between the ULK1 complex and the PtdIns3K complex. (A) ULK1 is not required for the interaction between ATG13, ATG14 and BECN1. Coimmunoprecipitates were obtained from Ulk1+/+ and ulk1−/− MEFs using anti-ATG14 antibody. (B) ATG13 is required for the interaction between ULK1 and ATG14. Anti-ATG14 immunoprecipitates were obtained from WT ATG13 and ATG13 KO HCT116 cells transiently expressing MYC-ULK1. (C) BECN1 is not required for the ATG14-ATG13 interaction. Anti-ATG13 immunoprecipitates were obtained from WT BECN1 and BECN1 KO HCT116 cells transiently expressing MYC-ATG14. (D, E) ATG14 is important for the interaction between ATG13 and PIK3C3. Coimmunoprecipitates were obtained from the indicated shRNA-transduced HEK293T cells using ATG13 Ab1 and Ab2 (D) or from WT ATG14 and ATG14 KO HCT116 cells using ATG13 antibody Ab2. (E). (F, G) ATG13 directly binds to ATG14. Glutathione-S-transferase (GST) affinity isolation assay was conducted using HA-tagged constructs obtained from in vitro translation and GST-tagged ATG13 purified from E. coli (F) or using ATG13 and GST-tagged BECN1 and ATG14 purified from E. coli (G). The proteins bound to glutathione (GSH) resin were analyzed by WB. GST (−) was used as a negative control. (H) ATG14 residues 201 to 395 contain a site important for binding to ATG13. MYC-tagged ATG14 fragments were coexpressed with HA-ATG13 in HEK293T cells. CCD: coiled coil domain; BATS, autophagosome-targeting sequence domain. (I) ATG13 N-terminal region contains a site important for binding to ATG14 and BECN1. MYC-tagged ATG13 fragments were coexpressed with HA-tagged ATG14 or BECN1 in HEK293T cells. (J) Model showing how the ULK1 complex interacts with the ATG14-containing PtdIns3K complex. (K) Chemical crosslinker stabilizes the interaction between ATG13, ULK1 and ATG14. HEK293T cells were treated with DSP (+) or vehicle (−) for 10 min. The proteins crosslinked with ATG14 were isolated by immunoprecipitation using anti-ATG14 antibody. Coimmunoprecipitates were treated with β-mercaptoethanol to reduce crosslinking and analyzed by WB. (L) The interaction between ATG13 and ATG14 is not regulated by MTORC1 inhibition. HEK293T cells were treated with DSP for 10 min following treatment with rapamycin (100 nM) for the indicated periods of time.
To test whether ATG13 and ATG14 directly interact with each other, we prepared glutathione S-transferase (GST)-tagged ATG13 from E. coli and analyzed whether the purified GST-ATG13 can interact with ATG14 or other components of the PtdIns3K complex that were prepared by in vitro translation. The in vitro GST affinity isolation assay revealed that ATG13 binds to ATG14, but not BECN1, VPS15, and PIK3C3 (Fig. 2F; Fig. S2A). We confirmed the direct interaction using ATG13 and GST-tagged ATG14 purified from E. coli (Fig. 2G). Through delimitation analysis, we identified that a region of ATG14 between residues 201 to 395 and a region of ATG13 between residues 1 to 198 are important for the ATG13-ATG14 interaction (Fig. 2H and I). This finding is consistent with the recent report that Atg13 recruits Atg14 to the ER through its N-terminal HORMA domain in yeast.25 In our previous report, we showed that ATG13 binds to ULK1 fragments containing C-terminal residues 829 to 1051.9 Those ULK1 fragments also interacted with ATG14 (Fig. 2I), supporting the role of ATG13 in mediating the interaction between ULK1 and ATG14. Combined, these results suggest that ATG13 and ATG14 directly interact with each other to mediate the interaction between the ULK1 complex and the ATG14-containing PtdIns3K complex (Fig. 2J).
We could not detect any drastic change in the interaction between ATG13 and ATG14 in response to rapamycin, leucine deprivation or the presence of ULK1 (Fig. S2B and C). We considered a possibility that the cell lysis condition might not preserve nutrient-sensitive interactions. As a way to preserve potential nutrient-sensitive interactions, we used a chemical crosslinker DSP before cells were lysed. DSP largely increased the interaction between ATG14, ATG13 and ULK1 (Fig. 2K), indicating that a drastic loss of the ATG13-ATG14 interaction might occur during cell lysis. However, incubation of cells with DSP following rapamycin treatment did still not show any drastic change in the interaction between ATG14 and ATG13 (Fig. 2L). This result suggests that the ATG13-ATG14 interaction might not be regulated by the MTORC1 activity.
ULK1 induces phosphorylation of ATG14 in cells and in vitro
In our interaction assay, we noticed that coexpression of ULK1 induced an upward shift of ATG14 on SDS-PAGE (Fig. 1C). The upshift was seen when ATG14 was coexpressed with the wild-type (WT) ULK1 relative to the kinase-inactivating mutant (KI) ULK1 harboring a point mutation replacing Met92 with alanine (Fig. 3A).9 The mobility shift disappeared when ATG14 was treated with lambda phosphatase, suggesting that the shift might be due to phosphorylation. Supporting the notion, a higher amount of 32P was incorporated into ATG14 in Ulk1+/+ MEFs relative to ulk1−/− MEFs (Fig. 3B). To investigate whether ULK1 can directly phosphorylate ATG14, we expressed MYC-tagged WT or KI ULK1 in HEK293T cells. MYC-immunoprecipitates were obtained and incubated with 32P-ATP and ATG14 purified from E. coli (Fig. S3A). WT ULK1 immunoprecipitates induced a high incorporation of 32P into ATG14, whereas KI ULK1 immunoprecipitates induced only a marginal level of 32P incorporation (Fig. 3C).
Figure 3.

ULK1 and ULK2 phosphorylate ATG14 Ser29. (A) ULK1 induces phosphorylation of ATG14. MTC-tagged ATG14 was coexpressed with HA-tagged WT or KI ULK1 in HEK293T cells. MYC-ATG14 immunoprecipitates were analyzed by WB before and after the treatment with lambda phosphatase. (B) ATG14 phosphorylation depends on ULK1. Ulk1+/+ and ulk1−/− MEFs were incubated in medium containing 32P-orthophosphate. The amount of 32P-labeled ATG14 in ATG14 immunoprecipitates was analyzed by autoradiogram. (C) ULK1 phosphorylates ATG14 in vitro. MYC-tagged WT and KI ULK1 were immunoprecipitated from HEK293T cells and incubated with ATG14 purified from E. coli in the presence of 32P-ATP. Incorporation of 32P into ATG14 was analyzed by autoradiography. (D) ATG14 Ser29 was identified by mass spectrometry. The surrounding sequence of Ser29 was aligned with those of the sites previously reported. (E) ULK1 kinase activity is responsible for ATG14 Ser29 phosphorylation. MYC-tagged WT ATG14 or ATG14S29A was expressed with HA-tagged WT or KI ULK1 in HEK293T cells. The phosphorylation state of Ser29 in immunoprecipitated ATG14 was analyzed by WB. (F) ULK2 induces ATG14 Ser29 phosphorylation. MYC-tagged ATG14 was expressed alone or with HA-tagged ULK2 in HEK293T cells. (G) ULK1 and ULK2 are responsible for the phosphorylation of ATG14 Ser29 in cells. Immunoprecipitates obtained from the indicated MEFs using anti-ATG14 antibody were analyzed for the phosphorylation state of Ser29. (H) ATG14 Ser29 phosphorylation depends on ULK1 in vivo. Liver tissue extract was obtained from Ulk1+/+ and ulk1−/− mice at age 12 weeks old. (I) ULK1 phosphorylates ATG14 Ser29 in vitro. ULK1 kinase fragment containing residues 1 to 649 was incubated with ATG14 purified from E. coli and ATP in vitro. (J) ULK1 and ULK2 kinase activities are responsible for the phosphorylation of ATG14 Ser29 in vitro. In vitro kinase reaction was conducted using MYC-tagged ULK1 or ULK2 immunoprecipitates.
ULK1 and ULK2 phosphorylate ATG14 at Ser29
Knowing that ULK1 induces phosphorylation of ATG14, we attempted to identify ULK1-induced phosphorylation sites of ATG14. We coexpressed MYC-tagged ATG14 with HA-tagged WT or KI ULK1 in HEK293T cells and isolated MYC-ATG14 by immunoprecipitation using anti-MYC antibody. By mass spectrometry, we identified 4 phosphorylation sites (Ser29, Thr48, Ser61, and Ser232) on ATG14 isolated from the cells coexpressing WT ULK1 but not KI ULK1 (Fig. 3D; Figs. S3B, S4, and S5). The sequences surrounding Ser29 and Ser61 showed similarity to that of ATG13 Ser318, one of the the currently known ULK1 target sites (Fig. 3D; Fig. S6A).16,26,28,29 Our attempts to generate polyclonal antibodies for the phosphorylation sites were successful only for ATG14 Ser29 (Fig. S6B and C). We conducted the followup studies only for Ser29 phosphorylation, while leaving other sites for further validation in the future.
Using the polyclonal antibodies, we tested whether ATG14 Ser29 is phosphorylated by ULK1. We found that coexpression of ATG14 with WT ULK1, but not KI ULK1, largely increased ATG14 Ser29 phosphorylation in HEK293T cells (Fig. 3E). Such an increase was not detected with ATG14S29A harboring replacement of Ser29 with alanine. We also found that coexpression of ULK2, which is a homolog of ULK1, induced the Ser29 phosphorylation (Fig. 3F). Ser29 phosphorylation was detected with endogenous ATG14 from Ulk1+/+ MEFs, but only marginally with ATG14 from ulk1−/− MEFs (Fig. 3G). Similar to ULK1 deficiency, ULK2 deficiency also suppressed the ATG14 phosphorylation. The marginal levels of the phosphorylation in ulk1−/− MEFs and ulk2−/− MEFs completely disappeared when both ULK1 and ULK2 were depleted, indicating that the 2 ULK kinases are responsible for ATG14 Ser29 phosphorylation. The ATG14 phosphorylation was decreased by ULK1 deficiency in the mouse liver tissue (Fig. 3H). The residual level of the phosphorylation in ULK1-deficient tissue might be due to ULK2.
To demonstrate that ULK1 phosphorylates ATG14 Ser29, we incubated a catalytically active fragment of ULK1 containing residues 1 to 649 with purified ATG14 in the presence of ATP. The active ULK1 fragment induced Ser29 phosphorylation within 30 min of incubation (Fig. 3I). We also tested MYC-tagged WT and KI ULK1 immunoprecipitated from HEK293T cells for their abilities to phosphorylate ATG14 Ser29. WT ULK1 immunoprecipitates induced a large increase of Ser29 phosphorylation (Fig. 3J). Such an increase was not observed with KI ULK1 immunoprecipitates. Similarly, WT ULK2 immunoprecipitates induced the phosphorylation in vitro.
ATG14 Ser29 phosphorylation is upregulated by starvation or MTORC1 inhibition
Knowing that ULK1 and ULK2 phosphorylate ATG14 Ser29, we predicted that the phosphorylation might be increased by starvation or MTORC1 inhibition. When HEK293T cells were incubated in medium deprived of amino acids, Ser29 phosphorylation was increased reaching the highest level at 30 min (Fig. 4A). The phosphorylation was also greatly increased by rapamycin or Torin1 within 15 min. Serum deprivation also induced a high increase of the phosphorylation (Fig. S7A). An inhibitor for GSK3 (glycogen synthase kinase 3), a kinase previously shown to positively regulate ULK1,30 decreased the ATG14 phosphorylation in serum-deprived culture. The GSK3 inhibitor slightly increased the ATG14 phosphorylation in serum-supplemented full medium. The opposing effects of the GSK3 inhibitor might be related to the serum-dependence of GSK3 functions in the regulation of MTORC1.31 The ATG14 phosphorylation was downregulated in the pancreas of mice in response to refeeding after overnight fasting (Fig. S7B), suggesting that the phosphorylation is regulated by nutritional conditions in vivo. We also observed that glucose starvation increases the Ser29 phosphorylation (Fig. 4A). Since glucose starvation is not a robust inducer of autophagy,32-34 our result suggests that glucose starvation activates ULK1 but it would not induce autophagy because ATP or glucose-derived signals are required for the downstream process of autophagy.
Figure 4.
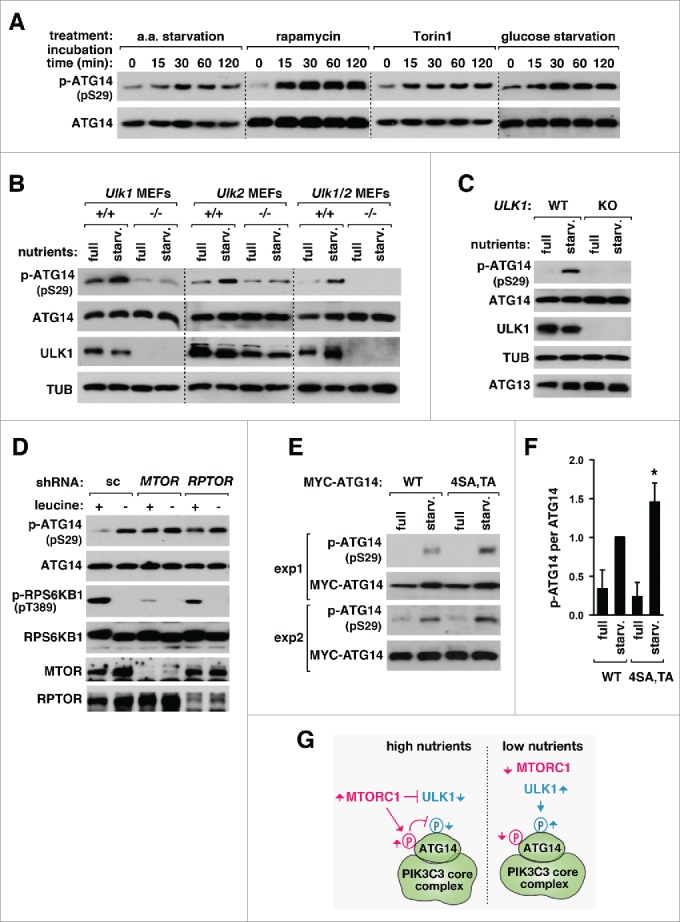
ULK-mediated phosphorylation of ATG14 Ser29 is upregulated under autophagy-inducing conditions. (A) ATG14 Ser29 phosphorylation is induced by nutrient starvation, rapamycin and Torin1. HEK293T cells were incubated in HBSS medium deprived of amino acids or RPMI medium deprived of glucose, or treated with rapamycin (100 nM) or Torin1 (250 nM) in full DMEM medium during the indicated periods of time. The phosphorylation state at Ser29 and the total amount of endogenous ATG14 in cell lysate were analyzed by WB. (B, C) Starvation-induced phosphorylation of ATG14 Ser29 depends on ULK1 and ULK2. MEFs (B) and HCT116 cells (C) with the indicated gene deficiency were incubated in either DMEM (full) or EBSS (starv.) for 30 min. (D) Starvation-induced phosphorylation of ATG14 Ser29 depends on MTORC1. HEK293T cells were stably transduced by the indicated shRNA. The phosphorylation states of ATG14 Ser29 and RPS6KB1 Thr398 and the amounts of the indicated proteins were analyzed by WB. (E) MTORC1-dependent phosphorylation of ATG14 have an effect on ATG14 Ser29 phosphorylation. ATG14 KO HCT116 cells reconstituted with MYC-tagged WT ATG14 or the ATG14[4SA,TA] mutant were incubated with either DMEM or EBSS for 30 min. (F) Quantitative analysis of the effect of the ATG14[4SA,TA] mutation on ATG14 Ser29 phosphorylation. Values are relative to the phosphorylation of WT ATG14 at starvation. Data are represented as mean ± SEM (*, P < 0.05 versus WT; n = 4). (G) Model for the regulation of ATG14 Ser29 phosphorylation.
The starvation-induced phosphorylation of ATG14 Ser29 was drastically suppressed when ULK1 or ULK2 was depleted in MEFs (Fig. 4B). The residual phosphorylation in ULK1- or ULK2-depleted MEFs completely disappeared when both ULK1 and ULK2 were depleted. The ULK1 dependence of the starvation-induced phosphorylation of ATG14 was further confirmed using ULK1-deficient HCT116, which was generated by the TALEN technique (Fig. 4C).35 The ATG14 phosphorylation remained highly even under nutrient-enriched conditions when either MTOR or RPTOR was knocked down (Fig. 4D; Fig. S7C), supporting the dependence of the ULK1 kinase activity on MTORC1.
A previous study showed that MTORC1 phosphorylates ATG14 for suppression of autophagy.36 Replacements of the reported MTORC1 target residues with alanine increased ATG14 Ser29 phosphorylation by about 1.5 fold (Fig. 4E and F; Fig. S7D). The increase implies that MTORC1-mediated phosphorylations of ATG14 might somehow negatively regulate Ser29 phosphorylation (Fig. 4G). We also wondered whether ATG14 Ser29 phosphorylation is affected by BECN1 Ser15 phosphorylation, another target site of ULK1.26 Reconstitution of BECN1S15A in BECN1 KO HCT116 cells did not show any drastic difference in the starvation-induced phosphorylation of ATG14 Ser29 compared to that of WT (Fig. S7E). This result suggests that ATG14 Ser29 phosphorylation is regulated independently of BECN1 Ser15 phosphorylation.
ATG14 Ser29 phosphorylation requires ATG13, RB1CC1 and BECN1 but not PIK3C3 kinase activity
Next, we wondered whether the ULK1-mediated ATG14 phosphorylation relies on the presence of ATG13, RB1CC1 and BECN1. Depletion of ATG13 in HCT116 cells or RB1CC1 in MEFs completely suppressed the induction of the phosphorylation in response to starvation, Torin1, or rapamycin as well as metformin that enhances the AMPK activity (Fig. 5A and B), indicating that the ULK1-binding proteins are important for the ATG14 phosphorylation by ULK1. Depletion of BECN1 in MEFs also abolished the starvation- and the drug-induced phosphorylation of ATG14 (Fig. 5C). BECN1 depletion did not suppress the phosphorylation of ATG13 Ser318 by ULK1 (Fig. S8A), indicating that the ULK1 complex integrity was not perturbed by the depletion. Since we know that BECN1 is not necessary for the ATG14-ATG13 interaction (Fig. 2C), a possible interpretation is that BECN1 plays a role in properly presenting ATG14 Ser29 for the ULK1 catalytic active site for phosphorylation once ATG14 binds to ATG13. Knockdown of UVRAG, which binds to BECN1 and PIK3C3, moderately increased ATG14 phosphorylation in either nutrient-enriched or deprived culture (Fig. S8B). The moderate increase might be due to a higher level of the PtdIns3K core complex available for binding to ATG14 in UVRAG-silenced cells. Knockdown of RUBCN, another binding protein of the PtdIns3K core complex, did not have any drastic effect on ATG14 phosphorylation.
Figure 5.
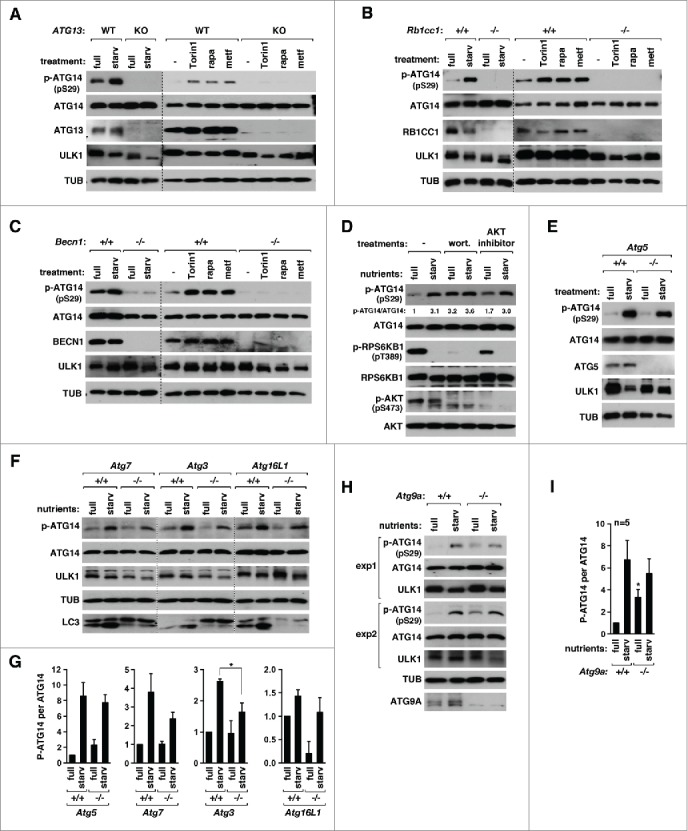
Hierarchical analysis between the ULK1-ATG14 axis and other autophagy regulators. (A) ATG13 is required for ATG14 Ser29 phosphorylation. WT ATG13 and ATG13 KO HCT116 cells were incubated in DMEM (full) or EBSS (starv.) for 30 min or treated with Torin1 (250 nM) or rapamycin (100 nM) for 1 h or with metformin (2 mM) for 2 h. (B, C) RB1CC1 and BECN1 are required for ATG14 Ser29 phosphorylation. The indicated MEFs were treated as indicated in (A). (D) Wortmannin does not suppress the starvation-induced phosphorylation of ATG14 Ser29. HCT116 cells were incubated in DMEM or EBSS in the presence of wortmannin (200 nM), AKT inhibitor VIII (5 µM) or vehicle (−) for 30 min. ((E)and F) ATG5, ATG7, ATG3, and ATG16L1 are not essential but have a positive role for ATG14 Ser29 phosphorylation. The indicated MEFs were incubated in DMEM or EBSS for 30 min. (G) Quantification of the difference in ATG14 phosphorylation between WT and KO cells. Values are mean ± SEM (*, P < 0.05; n = 3). (H) ATG9A is not essential but plays a role in protecting the ULK1 activation under full medium conditions. (I) Quantitative analysis of ATG14 phosphorylation from (H). Values are mean ± SEM (*, P < 0.05 between Atg9a+/+ cells and atg9a−/− cells in full medium; n = 5).
The ATG14 Ser29 phosphorylation allowed us to assess the relation between the ULK1 complex and other autophagy regulators. Wortmannin, an inhibitor of PtdIns3K and class I phosphoinositide 3-kinases (PI3Ks), did not suppress the ATG14 phosphorylation (Fig. 5D), indicating that the PtdIns3K kinase activity might not be necessary for the ULK1 activity. This hierarchical relationship between ULK1 and the PtdIns3K is consistent with the result from the previous cell imaging study.17 The increase of the ATG14 phosphorylation in wortmannin-treated cells under full medium might be due to inhibition of the PIK3C1 (PtdIns3-Kinase Class 1)-AKT-MTORC1 pathway. Supporting this, a chemical inhibition of AKT showed an increase in the ATG14 phosphorylation under the nutrient-enriched full medium (Fig. 5D).
The PE conjugation machinery is not required for ATG14 Ser29 phosphorylation, whereas ATG9A is required to prevent activation of ULK1 under nutrient-enriched conditions
We expanded the analysis to determine whether other autophagy regulators are necessary for the ULK1 activation. The starvation-induced phosphorylation of ATG14 Ser29 was reduced marginally (ATG5), moderately (ATG7 and ATG16L1), or significantly (ATG3) by deficiency of these autophagy genes (Fig. 5E to G; Fig. S8C to E). These results suggest that the PE-conjugation system is not essential but plays a positive role for the ULK1-mediated ATG14 phosphorylation. We also analyzed the effect of deficiency of ATG9A, a protein involved in the phagophore membrane expansion.37-39 ATG9A deficiency in MEFs increased the level of ATG14 phosphorylation in full medium, whereas the deficiency moderately reduced the starvation-induced ATG14 phosphorylation (Fig. 5H and I; Fig. S8F). The change in full medium is unlikely due to an increase of ULK1 level in ATG9A-deficient cells. It is unclear how ATG9A deficiency enhances the basal phosphorylation under the full nutrient condition. It is possible that the ATG9A membrane compartment might retain an unknown positive factor for the ULK1 activation. When ATG9A is absent, the positive factor might be released to increase the activity of ULK1.
ATG14 Ser29 phosphorylation positively regulates the lipid kinase activity of ATG14-associated PIK3C3
To understand the functional consequence of the ATG14 phosphorylation, we analyzed the phosphorylational effect on the lipid kinase activity of ATG14-associated PIK3C3 in converting phosphatidylinositol to PtdIns3P. We assayed the lipid kinase activity by measuring the amount of a p40 phox domain-containing polypeptide that binds to PtdIns3P.40,41 We first validated the specificity of the assay by confirming that the kinase activity of the ATG14-containing PtdIns3K complex was largely increased when ATG14 immunoprecipitates were isolated from nutrient-starved culture compared to nutrient-enriched culture (Fig. 6A and B). PtdIns3P production was not detected with ATG14 immunoprecipitates from ATG14-deficient HCT116 cells, confirming that the assay was specific to the ATG14-containing PtdIns3K complex. Like nutrient starvation, Torin1 or rapamycin also significantly increased the kinase activity of the ATG14 complex (Fig. 6C and D). Wortmannin completely suppressed the kinase activity of the ATG14 complex when it was added to the in vitro reaction, confirming that the assay was specific to PIK3C3. ATG13 deficiency suppressed the kinase activity of the ATG14 complex and desensitized the response of the activity to starvation (Fig. 6E and F). This supports the important role for ATG13 in mediating ULK1 signaling to the activation of the ATG14 complex in response to starvation.
Figure 6.

Figure 6 ATG14 Ser29 phosphorylation is important for starvation-induced activation of ATG14-associated PIK3C3. (A) Validation of our assay to measure the kinase activity of ATG14-associated PIK3C3. WT ATG14 and ATG14 KO HCT116 cells were incubated in DMEM or EBSS for 1 h. ATG14 immunoprecipitates were incubated with PI and ATP for 30 min. The production of PtdIns3P was assayed by dot blot analysis (see Materials and Methods). (B) Quantitative analysis of PtdIns3P production in (A). Values are mean ± SD (**, P < 0.01 vs. full medium). N.D. means “not detected.” (C) The kinase activity of ATG14-associated PIK3C3 is enhanced by Torin1, rapamycin or starvation. HEK293T cells were incubated in DMEM or EBSS or treated with Torin1 (250 nM) or rapamycin (100 nM) for 1 h. The in vitro reaction was conducted as described in (A). Wortmannin (100 nM) was added during the reaction. (D) Quantitative analysis of PtdIns3P production in (C). Values are mean ± SD (*P < 0.05 and **P < 0.01 versus full medium). (E) ATG13 is important for starvation-induced activation of the ATG14-containing PtdIns3K complex. ATG14 immunoprecipitates were isolated from the indicated cells cultured in either DMEM or EBSS for 1 h. The kinase activity of the immunoprecipitates was analyzed as described in (A). (F) Quantitative analysis of PtdIns3P production in (E). (G) ATG14 Ser29 phosphorylation is important for the kinase activity of ATG14-associated PIK3C3. ATG14 immunoprecipitates were obtained from WT ATG14-, ATG14S29A-, or ATG14S29D-reconstituted HCT116 cells. The in vitro reaction was conducted as described in (A). (H) Quantitative analysis of the results from (G). Values are mean ± SD (*P < 0.05; **P < 0.01). (I) ATG14 Ser29 phosphorylation is important for ATG14 puncta formation and their colocalization with 2xFYVE puncta. HCT116 cells reconstituted with WT ATG14, ATG14S29A, or ATG14S29D were transiently transduced by a plasmid encoding RFP-2xFYVE. The cells were incubated in either DMEM or EBSS for 1 h. Endogenous ATG14 was immunostained using anti-ATG14 antibody, and visualized together with RFP-2xFYVE by fluorescence microscope. Nuclei were stained by DAPI (blue). Scale bar: 10 μm. (J to L) Quantitative analysis of the results from (I) (*, P < 0.05; **, P < 0.01; ****, P < 0.0001, n≥35 , the Student t test). Mean and SEM are shown as horizontal bars.
Using the established assay method, we analyzed whether ATG14 Ser29 phosphorylation influences the kinase activity of the ATG14 complex. For this, we stably reconstituted WT ATG14, ATG14S29A or ATG14S29D in ATG14-deficient HCT116 cells. We isolated ATG14-associated PIK3C3 from the reconstituted cells by immunoprecipitation using anti-ATG14 antibody. The ATG14S29A-harboring PtdIns3K complex, compared to the WT complex, showed a significant reduction in the production of PtdIns3P (Fig. 6G and H; Fig. S9A). By contrast, the ATG14S29D-harboring PtdIns3K complex showed a higher production of PtdIns3P compared to the WT complex. The activity of the ATG14S29D complex isolated from full medium was similar to that of the WT complex isolated from nutrient starvation, and the activity was increased only slightly by starvation. A time-course analysis of the kinase activity revealed that the catalytic kinetics of the ATG14S29D complex is much faster than that of the ATG14S29A complex (Fig. S9B and C). This result suggests that the Ser29 phosphorylation might play a major role in the activation of the ATG14 complex in response to starvation.
ATG14 Ser29 phosphorylation is important for starvation-induced activation of ATG14-associated PIK3C3 in cells
Knowing the positive effect of the Ser29 phosphorylation on the activity of ATG14-associated PIK3C3, we analyzed the effects of the phosphorylation on the formation of ATG14 puncta in cells. We used anti-ATG14 antibody for immunostaining that specifically detects ATG14 (Fig. S1A). Under nutrient-enriched conditions, ATG14 puncta were formed in HCT116 cells reconstituted with ATG14S29D but barely with WT or ATG14S29A (Fig. 6I; Figs. S10 to S12). Nutrient starvation increased ATG14 puncta formation in WT and ATG14S29D cells, whereas the increase was significantly suppressed in ATG14S29A cells. The further increase of the ATG14S29D puncta in response to starvation might be due to molecular events other than Ser29 phosphorylation important for phagophore formation.
To determine whether the ATG14 puncta accumulate PtdIns3P, we monitored RFP-tagged 2xFYVE puncta. Although 2xFYVE puncta were formed with similar extents in WT and ATG14S29A cells, the 2xFYVE puncta that colocalized with the ATG14 puncta were significantly reduced in ATG14S29A cells (Fig. 6I to L; Fig. S10 to S12). We still observed that some ATG14S29A puncta localize with 2xFYVE under starvation, which is consistent with a moderate increase of the starvation-induced activation of the PtdIns3K complex associated with ATG14S29A (Fig. 6G and H). We also found that many of the ATG14S29A puncta still colocalize with ULK1 puncta (Fig. S13A to D). Thus, it is likely that ATG14S29A puncta reflect early membrane structures which were arrested and not going through the next processes efficiently without the ATG14 phosphorylation. The colocalization was observed in ATG14S29D cells even under full medium. Although the S29D mutation constitutively upregulated the kinase activity of ATG14-associated PIK3C3, the mutation further increased the accumulation of 2xFYVE on ATG14 puncta in response to starvation. This might be due to factors other than the lipid kinase activity contributing to the production of PtdIns3P and the recruitment of 2xFYVE construct.
ATG14 Ser29 phosphorylation is important for autophagosome formation but not autophagosome maturation
In order to determine whether ATG14 Ser29 phosphorylation is important for the formation of autophagosome, we analyzed MAP1LC3B/LC3B puncta formation. First, we confirmed that ATG14 knockout in HCT116 cells suppresses the starvation-induced formation of LC3 puncta (Fig. S14A). Reconstitution of ATG14 WT in ATG14 KO cells recovered the starvation-induced formation of LC3 puncta (Fig. 7A and B; Fig. S14B). Compared to WT cells, ATG14S29A-reconstituted cells showed a significant reduction in the starvation-induced formation of LC3 puncta. Yet, ATG14S29D-reconstituted cells showed a significant increase in LC3 puncta formation. Furthermore, ATG14S29D cells showed LC3 puncta even under nutrient-enriched conditions, which is consistent with the activation of the PtdIns3K complex at the basal condition (Fig. 6G and H).
Figure 7.

ATG14 Ser29 phosphorylation positively regulates the formation of phagophores and autophagosomes. (A) ATG14 Ser29 phosphorylation positively regulates the formation of LC3 puncta. WT- or mutant ATG14-reconstituted HCT116 cells were incubated in DMEM or EBSS for 2 h. Endogenous LC3B (green) was monitored by immunostaining using anti-LC3B antibody (MBL, PM036). Nuclei were stained by DAPI (blue). Scale bar: 10 μm. (B) Quantitative analysis of the results from (A) (**, P < 0.01; ****, P < 0.0001, n ≥ 20). Mean and SEM are shown as horizontal bars. (C) ATG14 Ser29 phosphorylation moderately affects autophagy flux. WT or mutant ATG14-reconstituted HCT116 cells were incubated in DMEM or EBSS for 1 h in the presence or absence of 100 nM bafilomycin A1 (BAF). (D) ATG14 Ser29 phosphorylation has a drastic effect on SQSTM1 level. Cells were treated as described in (C). (E) ATG14 Ser29 phosphorylation positively regulates the formation of WIPI2 puncta and their colocalization with ATG14 puncta. WT or mutant ATG14-reconstituted HCT116 cells were incubated in DMEM or EBSS for 2 h. Endogenous WIPI2 (red) and ATG14 (green) were monitored by immunostaining. We used anti-WIPI2 antibody from EMD-Millipore (MABC91). (F to H) Quantitative analysis of the results from (F) (****, P < 0.0001, n ≥ 30). Mean and SEM are shown as horizontal bars. (I) Model for the activation of the autophagy initiation machinery.
Knowing the importance of the ATG14 phosphorylation for autophagosome formation, we analyzed whether the phosphorylation is important for autophagy flux. Lysosomal inhibition increased the level of LC3-II in response to starvation to a lesser extent in ATG14S29A cells compared to WT cells and ATG14S29D cells (Figs. 7C; Fig. S15A and B). This indicates that autophagy flux might be lower in ATG14S29A cells compared to WT and ATG14S29D cells. The level of SQSTM1/p62 was higher in ATG14S29A cells compared to that in WT and ATG14S29D cells, and the level was only moderately reduced by starvation (Fig. 7D). The level of SQSTM1 was drastically reduced in ATG14S29D cells, suggesting that the overall activity of autophagy might be higher in the mutant cells. Lysosomal inhibition still increased the levels of LC3-II and SQSTM1 in ATG14S29A cells, indicating that autophagosme maturation might not be disturbed. Consistent with the notion, the S29A or S29D mutation does not alter the affinity of interaction between ATG14 and STX17/syntaxin-17 (Park et al., data not shown), the binding that occurs during autophagosome maturation.42
We found that ATG13-deficient cells showed a similar pattern of changes for LC3-II level as ATG14S29A cells (Fig. S15C). This suggests that ATG13-dependent events, including ATG14 Ser29 phosphorylation, might only moderately influence the LC3 conjugation machinery. We also found that ATG14 deficiency did not prevent the starvation-induced processing of LC3 and the accumulation of LC3-II upon the lysosomal inhibition, although it drastically suppressed LC3 puncta formation (Fig. S14A and S15D). This raises a question of whether the LC3 flux assay could appropriately assess ATG13- and ATG14-dependent autophagy. ATG14 deficiency largely increased the level of SQSTM1, a result consistent with suppression of autophagy. However, lysosomal inhibition could still moderately increase the level of SQSTM1 in nutrient-starved cells, indicating that the starvation-induced autophagy might not be completely suppressed by ATG14 deficiency. Supporting this observation, we found that LC3 puncta formation was not completely suppressed in ATG14 KO cells under starvation (Fig. S14A). This result implies that there might exist a noncanonical type of starvation-induced autophagy that is independent of ATG14.
ATG14 Ser29 phosphorylation is important for phagophore formation
Next, we analyzed whether ATG14 Ser29 phosphorylation is important for phagophore formation by monitoring WIPI2 puncta. ATG14 deficiency suppressed starvation-induced formation of WIPI2 puncta (Fig. S16A). Reconstitution of WT or ATG14S29D enhanced the formation of WIP2 puncta and their colocalization with ATG14 puncta in response to starvation (Fig. 7E to H; Fig. S16B and S17 to S23). By contrast, the enhancement was significantly reduced in ATG14S29A cells. WIPI2 puncta, similar to LC3 puncta, were formed in ATG14S29D cells even in full medium and further increased by starvation. The further increase might reflect molecular events other than Ser29 phosphorylation important for phagophore formation. Lastly, we analyzed the effects of the ATG14 phosphorylation on the formation of ZFYVE1/DFCP1 puncta that accumulate on omegasome.18 Cells reconstituted with ATG14S29A showed a significant reduction in the formation of ZFYVE1 puncta and colocalization with ATG14 puncta in response to starvation compared to WT and ATG14S29D cells (Fig. S24 to S30). ATG14S29D reconstitution induced the formation of ZFYVE1 puncta, many of which colocalized with ATG14 puncta, even under high nutrient conditions. Collectively, these results suggest that ATG14 phosphorylation is important for the formation of the membrane structures developing into phagophore and autophagosome.
It is noteworthy that many ATG14 puncta did not colocalize with 2xFYVE, WIPI2 or ZFYVE1 puncta. It is possible that many ATG14 puncta are not active enough to trigger the accumulation of PtdIns3P. Alternatively, such ATG14 puncta that do not colocalize with PtdIns3P markers might reflect the form involved in membrane tethering and fusion to form endolysosomes, as recently shown.42 Since many of the ATG14 puncta also did not colocalize with ULK1 puncta (Fig. S13A to D), such ATG14 puncta might reflect ULK1-independent autophagy. We also do not exclude the possibility of a technical issue with the use of antibodies (WIPI2 and ATG14) that might be different in the efficiencies of staining or the use of recombinant constructs (2xFYVE, ZFYVE1, and ULK1) for the cell imaging experiments.
Discussion
In this study, we have determined that ULK1 binds ATG14 via ATG13 and phosphorylates ATG14 Ser29 when cells are starved of nutrients or treated with MTOR inhibitors. The phosphorylation positively regulates the kinase activity of the ATG14-containing PtdIns3K complex. Based on our interaction analysis, we propose that the ULK1 complex and the ATG14 complex form the central core for the autophagy initiation machinery (Fig. 7I). As a key element of the machinery, ATG14 may integrate signals from MTORC1 and ULK1 to regulate the kinase activity of PIK3C3. ATG14 Ser29 phosphorylation might be a checkpoint event that monitors the status of MTORC1 and ULK1 to ensure that the cellular status is ready to commit autophagy induction. Such a regulation may enhance the sensitivity of the machinery in response to starvation, making a switch-like response.
The kinase activity of the ATG14-containing PtdIns3K complex was greatly increased by the phosphomimetic mutation S29D of ATG14 (Fig. 6G and H). The activity of the PtdIns3K complex containing ATG14S29D under nutrient-enriched conditions was similar to that of the PtdIns3K complex containing WT ATG14 under nutrient deprivation. This suggests that the Ser29 phosphorylation must be one of the key starvation signals to activate the PtdIns3K complex. It remains to be addressed how the kinase activity of the PtdIns3K complex is increased by the phosphorylation. The ATG14 Ser29 phosphorylation might facilitate other phosphorylations or recruitment of unknown factors important for phagophore formation. Alternatively, it might induce a conformational change of the autophagy initiation machinery, predisposing it for activation. Since we could still observe a moderate increase in the kinase activity of the PtdIns3K complex containing ATG14S29A by starvation, it is likely that mechanisms other than Ser29 phosphorylation exist to activate the PtdIns3K complex. Such mechanisms might involve other phosphorylations of ATG14 (Fig. S6A) or BECN1 Ser15 phosphorylation. Although we found that the phosphorylations of ATG14 and BECN1 by ULK1 are likely regulated independently of each other (Fig. S7E), it is possible that they synergistically contribute to robust responses to starvation by the autophagy initiation machinery. Alternatively, the 2 phosphorylations might activate the ATG14-PtdIns3K complex through different mechanisms relying on crosstalk with other kinases, such as MTORC1 and AMPK. Related to this, we observed that mutations of MTORC1 target sites on ATG14 affected the phosphorylation of ATG14 Ser29 (Fig. 4E). It is possible that BECN1 Ser15 phosphorylation by ULK1 might rely on phosphorylations by different kinases, such as AKT and MAPKs,43,44 for crosstalk.
By monitoring ATG14 Ser29 phosphorylation, we were able to assess the hierachical relation between the ULK1 activation and PtdIns3P. One could speculate that the PtdIns3P-binding capacity of ATG13, which has been recently reported,45 or the ATG13 N-terminal HORMA domain25 might promote the recruitment of the ULK1 complex to the site where ATG14-associated PIK3C3 is activated. The recruited ULK1 may phosphorylate ATG14 and induce a persistent activation of the ATG14-associated PtdIns3K complex. Such a positive-feedback regulation may amplify signals for autophagy induction. Our study does not support this speculated mechanism, since wortmannin, which inhibits the PtdIns3P-producing catalytic activities of the PI3K lipid kinase enzymes, did not suppress the phosphorylation of ATG14 Ser29 by ULK1. However, we do not exclude the possibility that PIK3C3 kinase activity contributes to the proper duration of the ULK1 complex accumulation on omegasomes, which is presumably required prior to the progression to phagophore formation, without affecting the kinase activity of the ULK1 complex.
The hierarchical analysis also allowed us to identify that PE-conjugation machinery is not necessary for the starvation-induced ATG14 phosphorylation. However, we observed a moderate reduction in the ATG14 phosphorylation in cells with defective PE-conjugation machinery, indicating that the lipid-conjugation system might somehow contribute to the ULK1-ATG14 signaling. This could be through the interaction of the PE-conjugation machinery with the ULK1 complex.46 Another interesting observation was that ATG9A deficiency significantly increased ATG14 Ser29 phosphorylation under nutrient-enriched conditions. A possible speculation is that ATG9A compartments might retain a positive factor important for ULK1 activation to suppress abortive induction of autophagy, which might be released from the compartments when ATG9A is absent. Such a potential role of ATG9A warrants further investigation.
Our finding adds another layer of complexity to the regulation of the ATG14-containing PtdIns3K complex that is subject to regulation by MTORC1 and AMPK.36,44,47,48 It would be critical to elucidate the specific functions of ULK1, MTORC1, AMPK and other kinases, such as AKT and MAPKs, in regulating the ATG14-PtdIns3K complex. Our current work enabled us to better understand the hierarchical relationship between ULK1 activation and other autophagy processes. This finding not only advanced the fundamental knowledge of the regulatory mechanism underlying the early stage of autophagy, but also revealed a reliable biomarker for autophagy induction that would be powerful for screening assays and drug development. Further study on the ULK1-ATG14 signaling in the context of interactions and modifications may be central for understanding the canonical autophagy induction process.
Materials and methods
Reagents and antibodies
The sources of antibodies and chemicals used in the experiments are: antibodies for ULK1 (sc-10900 for IP and sc-33182 for WB of human protein), ATG14 (sc-164767 for IP), UVRAG (sc-82115), RUBCN (sc-99955), BECN1 (sc-11427 for WB and sc-10086 for IP), SQSTM1 (sc-28359), GSK3A (sc-1844) and GAPDH (sc-25778) from Santa Cruz Biotechnology; anti-ULK1 antibody (A7481 for WB of mouse protein), anti-WIPI2 antibody (SAB4200400 for immunostaining), EBSS (2888), bafilomycin A1 (B1793–10UG) from Sigma-Aldrich; antibodies for PIK3C3 (3358), ATG9A (13509), ATG14 (rabbit monoclonal clone D3H2Z for immunostaining and WB), MTOR (2972 and 2983 for WB), phospho-AKT Ser473 (4051), AKT (9272), MAP1LC3B/LC3B (2775 for WB), phospho-RPS6KB1 Thr389 (9205), and RPS6KB1 (9202) from Cell Signaling Technology; anti-MYC 9E10 monoclonal antibody (OP10), WIPI2 antibody (MABC91 for immunostaining), and Akt inhibitor VIII (124018) from EMD-Millipore; anti-HA antibody HA.11 from Covance (AFC-101P); anti-GST antibody (27457701), glutathione-sepharose 4B beads (17–0756–01), and PreScission protease (27–0843–01) from GE Healthcare Life Sciences; LC3B antibody (PM036 for immunostaining) from MBL (Woburn, MA); phospho-ATG13 Ser318 (600–401-C49) from Rockland Immunochemicals Inc.; active ULK1 kinase (U01–11G-10) from SignalChem; DMEM (11995), RPMI 1640 (11875093), HBSS (24020), and Lipofectamine 2000 (11668–019) from Life Technologies; Torin1 (4247) and rapamycin(1292/1) from R&D; anti-phosphatidylinositol (840042P) from Avanti Polar Lipids; PtdIns3P Grip (G-0302) from Echelon Biosciences; Protein G-agarose beads (GenDEPOT, P9202); FuGENE™ 6 (PRE2692) and DSP (dithiobis[succinimidylpropionate]) (22586) from (Thermo Fisher Scientific, Inc.). Anti-ATG13 antibodies are described in our previous report.9 We also used anti-ATG13 antibody from Santa Cruz Biotechnology (sc-13468) for IP experiments described in Figures 1I, 2C and 2E. The anti-RPTOR antibody has been described previously.49 The rabbit monoclonal ATG14 antibody (clone D3H2Z) is a custom product prepared by Cell Signaling Technology. The rabbit polyclonal antibody specific to the phosphorylation state of ATG14 Ser29 (13155) was generated by Cell Signaling Technology.
Identification of ATG14 phosphorylation sites
MYC-tagged ATG14 was coexpessed with HA-tagged WT ULK1 or kinase dead ULK1 in HEK293T cells. ATG14 was isolated by immunoprecipitation using anti-MYC antibody. The immunoprecipitated ATG14 was run on SDS-PAGE, and the ATG14 band was isolated from the gel. ATG14 band was subjected to trypsinization followed by enrichment of phosphorylated peptides. Eluted fractions of tryptic digests of peptides were dried and resuspended in solvent (98:2:0.01, water: acetonitrile:formic acid). Aliquots of 1–1.5 ug were run on a Velos Orbitrap mass spectrometer (Thermo Fisher Scientific, Inc., Waltham, MA) as described previously.50 The MS/MS analysis was conducted using Sequest (Thermo Fisher Scientific, Inc., version 27, rev. 12) as well as the PEAKS software (Bioinformatics Solution Inc.). Oxidation of methionine and phosphorylation of serine and threonine were specified in Sequest as variable modifications. Scaffold (version Scaffold 4.2.1, Proteome Software Inc., Portland, OR) was used to validate MS/MS based peptide and protein identification. We also used the PEAKS software (Bioinformatics Solution Inc.) for database search and the spectrum analysis. Peptide identifications were accepted if they could be established at greater than 95% probability by the Peptide Prophet algorithm,51 and contained at least 2 identified peptides. Protein probabilities were assigned by the Protein Prophet algorithm.52 Peptides of interest (phosphorylated candidates) were manually verified for accuracy.
Plasmid constructions and mutagenesis
The human ATG14 gene was amplified from the Kazusa cDNA clone KIAA0831 (Kazusa DNA Research Institute, Japan) by polymerase chain reaction (PCR) and subcloned into pRK5 vector. ATG13, ULK1 and ATG14 DNA fragments used in Figure 2 were generated by PCR amplification and subcloned into pRK5.49 Mutagenesis was performed to generate ATG14S29A, ATG14S29D and BECN1S15A using a site-directed mutagenesis kit (Agilent Technologies, 200522). The sequences of primers used for mutagenesis are: 5'-cgggacctggtggacgccgtggacgatgcggag-3' (ATG14S29A), 5'-gacctggtggacgacgtggacgatgcg-3' (ATG14S29D), and caccatgcaggtggccttcgtgtgccagcgctgc (BECN1S15A). The ATG14 mutant construct harboring 5 alanine substitutions at Ser3, Ser223, Thr233, Ser383 and Ser440 (ATG14[4SA,TA]) was made by the gBlock DNA synthesis service from Integrated DNA Technologies (Coralville, IA) based on the information described by Yuan et al. (2013).36 The RFP-2xFYVE was a generous gift from N. Mizushima. All other constructs used in the experiments have been previously described.9,53,54
Cell culture and transfection
HEK293T, HCT116 and HeLa cells and MEFs were cultured in DMEM supplemented with 10% fetal bovine serum, penicillin and streptomycin at 37°C in 5% CO2. For nutrient starvation, we used either HBSS (Life Technologies, 24020117) or EBSS (Sigma, E2888) supplemented with dialzed fetal bovine serum (Life Technologies, 26400044). For transient expression, cells were transfected with recombinant DNA constructs using FuGENE™ 6 (Thermo Fisher Scientific, PRE2692) following the manufacturer's protocol. Cells were harvested 2 d post-transfection for coimmunoprecipitation assays.
Lentiviral preparation, viral infection, and stable cell-line generation
The lentiviral expression vector CSII-EF-MCS, kindly provided by Dr. Nik Somia, was used to stably transduce HCT116 cells with WT or mutant constructs following the procedure described previously.53 For knockdown of genes, we used the lentiviral vector pLKO.1 system and followed the procedure we previously described.53 Briefly, the pLKO.1 plasmids were introduced into HEK293T together with lentiviral packaging vectors pHR'8.2ΔR and pCMV-VSV-G using Lipofectamine 2000 (Life Technologies, 12566014). Viruses were collected 60 h after transfection, and target cells (HeLa, HEK293T and HCT116) were infected with the collected viruses in the presence of polybrene (Sigma, 107689). Stably transduced cells were selected with puromycin. The target sequences for ATG13, MTOR, RPTOR shRNA and scrambled shRNA are described in our previous report.9,53 The target sequences for ATG14 shRNA are 5′-ccatagaacttggtcatgttt-3′ and 5′-ccactttctttctatgggatt-3′, which are located at 3′-UTR. These shRNAs do not target recombinant ATG14 that lacks the 3′-UTR.
Coimmunoprecipitation and western blotting
For coimmunprecpititation experiments, whole-cell extracts were prepared in buffer containing 40 mM HEPES, pH 7.4, 120 mM NaCl, 1 mM EDTA, 50 mM NaF, 1.5 mM Na3VO4, 10 mM β-glycerophosphate, 0.3% CHAPS (Sigma, C5849), and EDTA-free protease inhibitors (Roche, 11873580001) as described in a previous report.49 We used 2 to 4 μg of antibody and protein G-agarose beads (GenDEPOT, P9202). Immunoprecipitated proteins were washed 4 times using lysis buffer, loaded onto 8% Tris-glycine gels (Life Technologies, EC60185BOX), transferred onto immunoblot polyvinylidene difluoride (PVDF) membrane (Bio-Rad, 620177), and detected with ECL western blotting detection reagents (Advansta, E-1119–50).
In vitro translation
The autophagy genes cloned in the mammalian expression vector pRK5 were expressed using the TNT SP6 Quick Coupled transcription/translation system (Promega, L2080) following the manufacturer's protocol. Briefly, the circular pRK5 vector and methionine (20 μM final concentration) were added to the cell-free expression system and incubated for 90 min at 30°C. The expression of the proteins was confirmed by western blotting using anti-HA antibody (Covance, AFC-101P).
Crosslinking experiments
HEK293T cells growing in 6-cm dishes in DMEM were treated with or without 2 mM DSP (Invitrogen, 22586) for 10 min and reactions were quenched by incubation in 40 mM Tris-HCl, pH7.4 for 10 min. Cell lysate was prepared using 1% Triton-X100 (Sigma, T8787) containing lysis buffer.53 Immunoprecipitation was conducted using anti-ATG14 antibody.
TALE nuclease-mediated gene targeting
To generate cell lines with knockout of ATG14, BECN1, ATG13 or ULK1, we used the Golden Gate TALEN assembly kit.35 The genomic sequences for the genes were scanned for TALEN binding pairs using TALEN-NT (https://tale-nt.cac.cornell.edu/). The binding pairs for the left and right arms were assembled into pc_TALd152+63 provided in the Golden Gate TALEN kit.35 We used HCT116 cells for the application of TALEN targeting approach. HCT116 cells were transfected with 500 ng each piggyBac 7 Transposase (PB7) and pPB-Ef1A-Puro transposon vector in addition to 2 μg of TALEN left and right. Three d post-transfection, cells were seeded into 96-well plates in full media containing puromycin. Wells were screened for single colony formation. Wells with none or 2 or more colonies were discarded. Single colonies were isolated and expanded, and genomic DNA preparations collected. Clones were then analyzed by immunoblotting and DNA sequencing to distinguish clones as true knockouts (with nonsense deletions or insertions) rather than clones with in-frame deletions. The sequences for targeting are 5′-TCCTGGACCGTGTCACCAT ccaggaactcacaggtc AGCGAGACCCTTGGAAAG A-3′ for BECN1; 5′-TCCTCTCAGGCCATCATGGCGT ctcccagtgggaaggg AGCCCGGGCGCTGG A-3′ for ATG14; TCCCTTCTTGCTATAA ctagggtgacaccag CCTATAGGCTCTCCAGGA for ATG13; TGCGCCATGGAGCCCG gccgcggcggcaca GAGACCGTGGGCAAGTTCGA for ULK1.
Assay of 32P incorporation into ATG14 in cells
Ulk1+/+ and ulk1−/− MEFs were incubated in phosphate-free medium (#11971–025, Life Technologies) supplemented with 10% dialyzed fetal bovine serum for 4 h before 0.1 mCi [32P]orthophosphate (Perkin-Elmer, NEX-053) was added for additional 1 h of incubation. Endogenous ATG14 was isolated from MEFs by immunoprecipitation using anti-ATG14 antibody. Isolated proteins were separated by SDS-PAGE, transferred to PVDF membrane, and analyzed by autoradiography.
In vitro kinase assay for ULK1 and ULK2
To determine if ULK1 and ULK2 directly phosphorylate ATG14, we isolated ULK1 and ULK2 immunoprecipitates and analyzed the kinase activity of the isolated immunoprecipitates using purified ATG14 as substrate as described previously.9 Briefly, endogenous or recombinant ULK1 was isolated by immunoprecipitation using anti-ULK1 antibody (Santa Cruz Biotechnology, sc-10900) or anti-MYC antibody (EMD Biosciences, 9E10) from HEK293T cells or MEFs. Recombinant ULK2 was isolated by immunoprecipitation using anti-MYC antibody from HEK293T cells. We also used the active fragment of ULK1 (SignalChem, U01–11G-10) at 100 ng. The reaction buffer contained 25 mM MOPS, pH 7.5, 1 mM EGTA, 0.1 mM Na3VO4, 15 mM MgCl2, and ATP at 100 μM. For 32P incorporation experiments, we added 32P-ATP to the reaction buffer. For each reaction, ATG14, purified from E. coli, was used at 400 ng.
In vitro PIK3C3 activity assay
In vitro PIK3C3 activity assay was performed as described previously with minor modifications.40 In brief, the ATG14-containing PtdIns3K complex was obtained by immunoprecipitation using anti-ATG14 antibody in lysis buffer (50 mM Tris, pH 7.4, 7.5% glycerol, 150 mM NaCl, 1 mM EDTA, protease inhibitors). The immunoprecipitates were washed 3 times with lysis buffer and further washed once in 2.5× substrate buffer (75 mm Tris, pH 7.5, 125 mm NaCl, 12.5 mm MnCl2). The immunoprecipitates were resuspended and incubated in 1x substrate buffer containing PI (250 μg/ml) on ice for 10 min, which were followed by 5 additional min of incubation at room temperature. The PIK3C3 kinase reactions were initiated by adding ATP (10 µM) and incubated for 30 min at room temperature. All the kinase reactions were spotted onto nitrocellulose membrane. The membrane was blocked with 1% fat milk in phosphate-buffered saline (BioExpress, 0780.50L) for 1 h, and incubated with 0.5 μg/ml of GST-p40-PX in PBS containing 3% BSA (EMD-Millipore, 2960–500G) and 0.1% Tween 20 (Sigma, 274348–4L) for 2 h. After multiple times of extensive washing, the amount of GST-p40-PX remaining on the nitrocellulose membrane was analyzed by WB using anti-GST antibody.
Immunostaining and fluorescence microscopy
Immunostaining and fluorescence microscopy experiments were conducted as we described previously.9,55 Briefly, cells were fixed with 4% formaldehyde in PBS for 10 min at room temperature, and permeabilized with 0.3% Triton X-100 for 30 min at room temperature. Permeabilized cells were incubated in PBS containing 1% BSA for 30 min and incubated with antibodies for overnight at 4°C. Endogenous LC3B, ATG14 and WIPI2 were stained by use of anti-LC3B antibody (MBL, PM036), anti-ATG14 antibody (Cell Signaling Technology, clone D3H2Z) and anti-WIPI2 antibody (EMD-Millipore, MABC91 or Sigma, SAB4200400). After the primary antibody labeling, cells were incubated with Alexa Flour 488-conjugated anti-rabbit IgG (Invitrogen, A-21441) and/or Alexa Flour 555-conjugated anti-mouse IgG (Life Technologies, A-31570). Nuclei were stained with DAPI (4′-6-Diamidino-2-phenylindole; Invitrogen, D-1306). Images from stained cells were obtained using a Deltavision PersonelDV microscope (Applied Precision Inc., Issaquah, WA) and analyzed by softWoRx version 6.1.3 (GE Healthcare).
Mouse tissue preparation
Liver tissue was collected from 12-wk-old C57BL6J mice with the genetic background of Ulk1+/+ or ulk1−/−. The mice were originally provided by Dr. Mondira Kundu (St.Jude Children's Research Hospital), and bred with BL6J mice for more than 10 generations. The mice were fed the standard Chow diet after weaning. Tissue was homogenized using a dounce homogenizer with 20 times of up-and-down strokes. Proteins were extracted using a buffer containing 40 mM HEPES, pH 7.4, 120 mM NaCl, 1 mM EDTA, 10 mM NaF, 0.5 mM Na2VO4, 1% Triton X-100 supplemented with protease inhibitor cocktail (Roche, 11873580001). Fifty micrograms of proteins were loaded on SDS-PAGE.
Statistical analysis
Western blot protein band intensities and PtdIns3P dot blot intensities were quantitatively analyzed by ImageJ software. Puncta images were quantitatively analyzed by counting the number in each cell with setting the same threshold for control and experimental groups using ImageJ. Outcomes were summarized by using the mean and the standard error of the mean (SEM) or the standard deviation (SD) as indicated in figure legends. Means were compared by group using the Student t test with Prism 6 (Version 6.0d, GraphPad Software).
Supplementary Material
Funding Statement
This study was supported by the functional proteomics of aging NIH training grant T32AG029796 (to NMO and DG); E0143033654 (to CHJ); HL114662 (to RSN); UL1TR000114 (to CS and DV); P30-DK050456, 7–12-BS-093, GM097057 and AG039758 (to DHK).
Abbreviations
- AMPK
AMP-activated protein kinase
- ATG3
autophag-related 3
- ATG5
autophagy related-5
- ATG7
autophagy-related 7
- ATG9A
autophagy-related 9A
- ATG13
autophagy-related 13
- ATG14:
autophagy-related 14
- ATG16L1
autophagy-related 16-like 1
- BAF
bafilomycin A1
- BAT
brown adipose tissue
- BECN1
Beclin 1, autophagy-related
- DSP
dithiobis(succinimidyl propionate)
- E. coli
Escherichia coli
- ER
endoplasmic reticulum
- FYVE
zinc finger domain found in Fab1, YOTB, Vac1, and EEA1
- HORMA
named after the Hop1p, Rev7p and MAD2 proteins
- GSK3
glycogen synthase kinase 3
- GST
glutathione S-transferase
- KI
kinase-inactivating mutant
- KO
knockout
- MAP1LC3/LC3
microtubule-associated protein 1 light chain 3
- MEFs
mouse embryonic fibroblasts
- MTOR
mechanistic target of rapamycin (serine/threonine kinase)
- MTORC1
MTOR complex 1
- 32P
phosphorus 32
- PE
phosphatidylethanolamine
- PI3K
class 1 phosphoinositide 3-kinase
- PtdIns3K
class 3 phosphatidylinositol 3-kinase
- PtdIns3P
phosphatidylinositol 3-phosphate
- RPS6KB1
ribosomal protein S6 kinase, 70kDa, polypeptide 1
- RPTOR
regulatory-associated protein of MTOR, complex 1
- RB1CC1
RB1 inducible coiled-coil 1
- shRNA
small hairpin RNA
- RUBCN
RUN domain and cysteine-rich domain containing, Beclin 1-interacting protein
- SDS-PAGE
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- SQSTM1
sequestosome 1
- TALEN
transcription activator-like effector nuclease
- TMEM173
transmembrane protein 173
- ULK1
unc-51 like autophagy activating kinase 1
- ULK2
unc-51 like autophagy activating kinase 2
- WIPI2
WD repeat domain, phosphoinositide interacting 2
- WT
wild type
- ZFYVE1
zinc finger, FYVE domain containing 1
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Gary Kasof at Cell Signaling Technology for ATG14 Ser29 phospho-specific antibody production; M. Kundu for Ulk1 and Ulk2 MEFs and ulk1 KO mice; Z. Yue for Becn1 MEFs; M. Komatsu for Atg3 and Atg7 MEFs; N. Mizushima for Atg5 MEFs, 2xFYVE construct and comments on WIPI2 antibodies; T. Saitoh and S. Akira for Atg9a MEFs; S. Hwang for atg16l1 KO MEFs; J.-L. Guan for rb1cc1 KO MEFs; T. Neufeld and Kim lab members for comments; L. Anderson and L. Higgins in the Center for Mass Spectrometry and Proteomics for MS sample prep and analysis; R. Foncea at the Minnesota Obesity Center for lentivirus preparation.
References
- [1].Codogno P, Ogier-Denis E, Houri JJ. Signal transduction pathways in macroautophagy. Cell Signal 1997; 9:125-30; PMID:9113411; http://dx.doi.org/ 10.1016/S0898-6568(96)00130-1 [DOI] [PubMed] [Google Scholar]
- [2].Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell 2008; 132:27-42; PMID:18191218; http://dx.doi.org/ 10.1016/j.cell.2007.12.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nat Cell Biol 2010; 12:823-30; PMID:20811354; http://dx.doi.org/ 10.1038/ncb0910-823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, et al.. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011; 331:456-61; PMID:21205641; http://dx.doi.org/ 10.1126/science.1196371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011; 13:132-41; PMID:21258367; http://dx.doi.org/ 10.1038/ncb2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Lee JW, Park S, Takahashi Y, Wang HG. The association of AMPK with ULK1 regulates autophagy. PLoS One 2010; 5:e15394; PMID:21072212; http://dx.doi.org/ 10.1371/journal.pone.0015394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hara T, Takamura A, Kishi C, Iemura S, Natsume T, Guan JL, Mizushima N. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J Cell Biol 2008; 181:497-510; PMID:18443221; http://dx.doi.org/ 10.1083/jcb.200712064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, Iemura S, Natsume T, Takehana K, Yamada N, et al.. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell 2009; 20:1981-91; PMID:19211835; http://dx.doi.org/ 10.1091/mbc.E08-12-1248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, Kundu M, Kim DH. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell 2009; 20:1992-2003; PMID:19225151; http://dx.doi.org/ 10.1091/mbc.E08-12-1249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ganley IG, Lam du H, Wang J, Ding X, Chen S, Jiang X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem 2009; 284:12297-305; PMID:19258318; http://dx.doi.org/ 10.1074/jbc.M900573200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Chang YY, Neufeld TP. An Atg1/Atg13 complex with multiple roles in TOR-mediated autophagy regulation. Mol Biol Cell 2009; 20:2004-14; PMID:19225150; http://dx.doi.org/ 10.1091/mbc.E08-12-1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hosokawa N, Sasaki T, Iemura S, Natsume T, Hara T, Mizushima N. Atg101, a novel mammalian autophagy protein interacting with Atg13. Autophagy 2009; 5:973-9; PMID:19597335; http://dx.doi.org/ 10.4161/auto.5.7.9296 [DOI] [PubMed] [Google Scholar]
- [13].Mercer CA, Kaliappan A, Dennis PB. A novel, human Atg13 binding protein, Atg101, interacts with ULK1 and is essential for macroautophagy. Autophagy 2009; 5:649-62; PMID:19287211; http://dx.doi.org/ 10.4161/auto.5.5.8249 [DOI] [PubMed] [Google Scholar]
- [14].Kamada Y, Funakoshi T, Shintani T, Nagano K, Ohsumi M, Ohsumi Y. Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J Cell Biol 2000; 150:1507-13; PMID:10995454; http://dx.doi.org/ 10.1083/jcb.150.6.1507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Chan EY, Longatti A, McKnight NC, Tooze SA. Kinase-inactivated ULK proteins inhibit autophagy via their conserved C-terminal domains using an Atg13-independent mechanism. Mol Cell Biol 2009; 29:157-71; PMID:18936157; http://dx.doi.org/ 10.1128/MCB.01082-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Joo JH, Dorsey FC, Joshi A, Hennessy-Walters KM, Rose KL, McCastlain K, Zhang J, Iyengar R, Jung CH, Suen DF, et al.. Hsp90-cdc37 chaperone complex regulates ulk1- and atg13-mediated mitophagy. Mol Cell 2011; 43:572-85; PMID:21855797; http://dx.doi.org/ 10.1016/j.molcel.2011.06.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Itakura E, Mizushima N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy 2010; 6: 764-76; PMID:20639694; http://dx.doi.org/ 10.4161/auto.6.6.12709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, Griffiths G, Ktistakis NT. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol 2008; 182:685-701; PMID:18725538; http://dx.doi.org/ 10.1083/jcb.200803137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Webber JL, Tooze SA. New insights into the function of Atg9. FEBS Lett 2010; 584:1319-26; PMID:20083107; http://dx.doi.org/ 10.1016/j.febslet.2010.01.020 [DOI] [PubMed] [Google Scholar]
- [20].Young AR, Chan EY, Hu XW, Kochl R, Crawshaw SG, High S, Hailey DW, Lippincott-Schwartz J, Tooze SA. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J Cell Sci 2006; 119:3888-900; PMID:16940348; http://dx.doi.org/ 10.1242/jcs.03172 [DOI] [PubMed] [Google Scholar]
- [21].Matsunaga K, Saitoh T, Tabata K, Omori H, Satoh T, Kurotori N, Maejima I, Shirahama-Noda K, Ichimura T, Isobe T, et al.. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol 2009; 11:385-96; PMID:19270696; http://dx.doi.org/ 10.1038/ncb1846 [DOI] [PubMed] [Google Scholar]
- [22].Zhong Y, Wang QJ, Li X, Yan Y, Backer JM, Chait BT, Heintz N, Yue Z. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat Cell Biol 2009; 11:468-76; PMID:19270693; http://dx.doi.org/ 10.1038/ncb1854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Itakura E, Kishi C, Inoue K, Mizushima N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell 2008; 19:5360-72; PMID:18843052; http://dx.doi.org/ 10.1091/mbc.E08-01-0080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Sun Q, Fan W, Chen K, Ding X, Chen S, Zhong Q. Identification of Barkor as a mammalian autophagy-specific factor for Beclin 1 and class III phosphatidylinositol 3-kinase. Proc Natl Acad Sci U S A 2008; 105:19211-6; PMID:19050071; http://dx.doi.org/ 10.1073/pnas.0810452105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Jao CC, Ragusa MJ, Stanley RE, Hurley JH. A HORMA domain in Atg13 mediates PtdIns 3-kinase recruitment in autophagy. Proc Natl Acad Sci U S A 2013; 110:5486-91; PMID:23509291; http://dx.doi.org/ 10.1073/pnas.1220306110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J, Kim H, Neufeld TP, Dillin A, Guan KL. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol 2013; 15:741-50; PMID:23685627; http://dx.doi.org/ 10.1038/ncb2757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Matsunaga K, Morita E, Saitoh T, Akira S, Ktistakis NT, Izumi T, Noda T, Yoshimori T. Autophagy requires endoplasmic reticulum targeting of the PI3-kinase complex via Atg14L. J Cell Biol 2010; 190:511-21; PMID:20713597; http://dx.doi.org/ 10.1083/jcb.200911141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Konno H, Konno K, Barber GN. Cyclic dinucleotides trigger ULK1 (ATG1) phosphorylation of STING to prevent sustained innate immune signaling. Cell 2013; 155:688-98; PMID:24119841; http://dx.doi.org/ 10.1016/j.cell.2013.09.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Egan DF, Chun MG, Vamos M, Zou H, Rong J, Miller CJ, Lou HJ, Raveendra-Panickar D, Yang CC, Sheffler DJ, et al.. Small Molecule Inhibition of the Autophagy Kinase ULK1 and Identification of ULK1 Substrates. Mol Cell 2015; 59:285-97; PMID:26118643; http://dx.doi.org/ 10.1016/j.molcel.2015.05.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lin SY, Li TY, Liu Q, Zhang C, Li X, Chen Y, Zhang SM, Lian G, Liu Q, Ruan K, et al.. GSK3-TIP60-ULK1 signaling pathway links growth factor deprivation to autophagy. Science 2012; 336:477-81; PMID:22539723; http://dx.doi.org/ 10.1126/science.1217032 [DOI] [PubMed] [Google Scholar]
- [31].Shin S, Wolgamott L, Yu Y, Blenis J, Yoon SO. Glycogen synthase kinase (GSK)-3 promotes p70 ribosomal protein S6 kinase (p70S6K) activity and cell proliferation. Proc Natl Acad Sci U S A 2011; 108:E1204-13; PMID:22065737; http://dx.doi.org/ 10.1073/pnas.1110195108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ramirez-Peinado S, Leon-Annicchiarico CL, Galindo-Moreno J, Iurlaro R, Caro-Maldonado A, Prehn JH, Ryan KM, Muñoz-Pinedo C. Glucose-starved cells do not engage in prosurvival autophagy. J Biol Chem 2013; 288:30387-98; PMID:24014036; http://dx.doi.org/ 10.1074/jbc.M113.490581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Moruno-Manchon JF, Perez-Jimenez E, Knecht E. Glucose induces autophagy under starvation conditions by a p38 MAPK-dependent pathway. Biochem J 2013; 449:497-506; PMID:23116132; http://dx.doi.org/ 10.1042/BJ20121122 [DOI] [PubMed] [Google Scholar]
- [34].Lang MJ, Martinez-Marquez JY, Prosser DC, Ganser LR, Buelto D, Wendland B, Duncan MC. Glucose starvation inhibits autophagy via vacuolar hydrolysis and induces plasma membrane internalization by down-regulating recycling. J Biol Chem 2014; 289:16736-47; PMID:24753258; http://dx.doi.org/ 10.1074/jbc.M113.525782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Cermak T, Doyle EL, Christian M, Wang L, Zhang Y, Schmidt C, Baller JA, Somia NV, Bogdanove AJ, Voytas DF. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res 2011; 39:e82; PMID:21493687; http://dx.doi.org/ 10.1093/nar/gkr218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Yuan HX, Russell RC, Guan KL. Regulation of PIK3C3/VPS34 complexes by MTOR in nutrient stress-induced autophagy. Autophagy 2013; 9:1983-95; PMID:24013218; http://dx.doi.org/ 10.4161/auto.26058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Mari M, Griffith J, Rieter E, Krishnappa L, Klionsky DJ, Reggiori F. An Atg9-containing compartment that functions in the early steps of autophagosome biogenesis. J Cell Biol 2010; 190:1005-22; PMID:20855505; http://dx.doi.org/ 10.1083/jcb.200912089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Orsi A, Razi M, Dooley HC, Robinson D, Weston AE, Collinson LM, Tooze SA. Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane integration, are required for autophagy. Mol Biol Cell 2012; 23:1860-73; PMID:22456507; http://dx.doi.org/ 10.1091/mbc.E11-09-0746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Yamamoto H, Kakuta S, Watanabe TM, Kitamura A, Sekito T, Kondo-Kakuta C, Ichikawa R, Kinjo M, Ohsumi Y. Atg9 vesicles are an important membrane source during early steps of autophagosome formation. J Cell Biol 2012; 198:219-33; PMID:22826123; http://dx.doi.org/ 10.1083/jcb.201202061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Farkas T, Daugaard M, Jaattela M. Identification of small molecule inhibitors of phosphatidylinositol 3-kinase and autophagy. J Biol Chem 2011; 286:38904-12; PMID:21930714; http://dx.doi.org/ 10.1074/jbc.M111.269134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Kim YM, Jung CH, Seo M, Kim EK, Park JM, Bae SS, Kim DH. mTORC1 Phosphorylates UVRAG to Negatively Regulate Autophagosome and Endosome Maturation. Mol Cell 2015; 57:207-18; PMID:25533187; http://dx.doi.org/ 10.1016/j.molcel.2014.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Diao J, Liu R, Rong Y, Zhao M, Zhang J, Lai Y, Zhou Q, Wilz LM, Li J, Vivona S, et al.. ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes. Nature 2015; 520:563-6; PMID:25686604; http://dx.doi.org/ 10.1038/nature14147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Wei Y, An Z, Zou Z, Sumpter R, Su M, Zang X, Sinha S, Gaestel M, Levine B. The stress-responsive kinases MAPKAPK2/MAPKAPK3 activate starvation-induced autophagy through Beclin 1 phosphorylation. Elife 2015; 4:e05289; PMID:25693418; http://dx.doi.org/23112296 10.7554/eLife.05289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Wang RC, Wei Y, An Z, Zou Z, Xiao G, Bhagat G, White M, Reichelt J, Levine B. Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science 2012; 338:956-9; PMID:23112296; http://dx.doi.org/ 10.1126/science.1225967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Karanasios E, Stapleton E, Manifava M, Kaizuka T, Mizushima N, Walker SA, Ktistakis NT. Dynamic association of the ULK1 complex with omegasomes during autophagy induction. J Cell Sci 2013; 126:5224-38; PMID:24013547; http://dx.doi.org/ 10.1242/jcs.132415 [DOI] [PubMed] [Google Scholar]
- [46].Nishimura T, Kaizuka T, Cadwell K, Sahani MH, Saitoh T, Akira S, Virgin HW, Mizushima N. FIP200 regulates targeting of Atg16L1 to the isolation membrane. EMBO Rep 2013; 14:284-91; PMID:23392225; http://dx.doi.org/ 10.1038/embor.2013.6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Kim J, Kim YC, Fang C, Russell RC, Kim JH, Fan W, Liu R, Zhong Q, Guan KL. Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell 2013; 152:290-303; PMID:23332761; http://dx.doi.org/ 10.1016/j.cell.2012.12.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Furuya T, Kim M, Lipinski M, Li J, Kim D, Lu T, Shen Y, Rameh L, Yankner B, Tsai LH, et al.. Negative regulation of Vps34 by Cdk mediated phosphorylation. Mol Cell 2010; 38:500-11; PMID:20513426; http://dx.doi.org/ 10.1016/j.molcel.2010.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002; 110:163-75; PMID:12150925; http://dx.doi.org/ 10.1016/S0092-8674(02)00808-5 [DOI] [PubMed] [Google Scholar]
- [50].Lin-Moshier Y, Sebastian PJ, Higgins L, Sampson ND, Hewitt JE, Marchant JS. Re-evaluation of the role of calcium homeostasis endoplasmic reticulum protein (CHERP) in cellular calcium signaling. J Biol Chem 2013; 288:355-67; PMID:23148228; http://dx.doi.org/ 10.1074/jbc.M112.405761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Keller A, Nesvizhskii AI, Kolker E, Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem 2002; 74:5383-92; PMID:12403597; http://dx.doi.org/ 10.1021/ac025747h [DOI] [PubMed] [Google Scholar]
- [52].Nesvizhskii AI, Keller A, Kolker E, Aebersold R. A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem 2003; 75:4646-58; PMID:14632076; http://dx.doi.org/ 10.1021/ac0341261 [DOI] [PubMed] [Google Scholar]
- [53].Vander Haar E, Lee SI, Bandhakavi S, Griffin TJ, Kim DH. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol 2007; 9:316-23; PMID:17277771; http://dx.doi.org/ 10.1038/ncb1547 [DOI] [PubMed] [Google Scholar]
- [54].Woo SY, Kim DH, Jun CB, Kim YM, Haar EV, Lee SI, Hegg JW, Bandhakavi S, Griffin TJ, Kim DH. PRR5, a novel component of mTOR complex 2, regulates platelet-derived growth factor receptor β expression and signaling. J Biol Chem 2007; 282:25604-12; PMID:17599906; http://dx.doi.org/ 10.1074/jbc.M704343200 [DOI] [PubMed] [Google Scholar]
- [55].Kim YM, Stone M, Hwang TH, Kim YG, Dunlevy JR, Griffin TJ, Kim DH. SH3BP4 is a negative regulator of amino acid-Rag GTPase-mTORC1 signaling. Mol Cell 2012; 46:833-46; PMID:22575674; http://dx.doi.org/ 10.1016/j.molcel.2012.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.