

ABSTRACT
Activation of the immune system is metabolically costly, yet a hallmark of an infection is a reduction in appetite with a subsequent reduction in metabolite provision. What is the functional value of decreasing nutrient intake when an infection imposes large demands on metabolic parameters? Here, we propose that sickness-associated anorexia (SAA) upregulates the ancient process of autophagy systemically, thereby profoundly controlling not only immune- but also nonimmune-competent cells. This allows an advanced impact on the resolution of an infection through direct pathogen killing, enhancement of epitope presentation and the contribution toward the clearance of noxious factors. By rendering a ‘free meal,’ autophagy is thus most fundamentally harnessed during an anorexic response in order to promote both host tolerance and resistance. These findings strongly suggest a reassessment of numerous SAA-related clinical applications and a re-evaluation of current efforts in patient care.
KEYWORDS: anorexia, autophagy, infection, sickness behavior, xenophagy
Introduction
… she drank only half a cup and swallowed a tiny piece of bread. The veal she refused with disgust and irritation. “You are ill, Marie, all this is a sign of illness,” Shatov remarked timidly as he waited upon her. - Fyodor Dostoyevsky (Demons, 1872)
A decrease in appetite is a universal and cardinal manifestation of an established infection.1 Since both vertebrates and invertebrates, exhibit anorexia during an infection, this response likely plays a vital function, which remained conserved across species. However, considering the high metabolic cost of mobilizing an immune response, it may seem counterproductive to undergo an anorexic response during an infection. This is exacerbated by the febrile response, where for every 1°C increase in body temperature, metabolic output may be upregulated by 10–15%, translating into a 30–60% increase in basal metabolic rate.2 What could be the functional value of decreasing nutritional intake at a time where extreme metabolic demands are imposed on the host?
Here we provide intriguing evidence suggesting that sickness-associated anorexia (SAA) represents an evolutionarily conserved response dedicated toward a systemic upregulation of autophagic activity. Substantiating this perspective, we mechanistically relate the physiological response observed during anorexia and an infection to an upregulation of autophagic flux, i.e. the rate of protein degradation through macroautophagy, leading to enhanced hepatic autophagy, an altered amino acid profile as well as a profound impact on not only immune- but also nonimmune-competent tissues. In doing so, we assess possible underlying design principles of SAA-induced autophagy, that may point toward powerful implications to exploit autophagy to advance the resolution of infections.
Anorexia enhances hepatic autophagy and has an impact on disease trajectory
SAA may play a critical role in sustaining hepatic autophagy with major impact on disease trajectory. It has recently been demonstrated that NR1H4/FXR, nuclear receptor subfamily 1 group H member 4 and PPARA/PPARα (peroxisome proliferator-activated receptor α) are prominent transcriptional regulators of hepatic autophagy.3 Both NR1H4 and PPARA competitively bind to gene promoters with antagonistic transcriptional regulation of autophagy genes. In a fasted state, PPARA is activated, leading to an upregulation of hepatic autophagy. In contrast, NR1H4 is triggered postprandial by bile acids, leading to an inhibition of autophagy.3 Thus, fasting and feeding, particularly high-fat foods, would transcriptionally control autophagy in hepatocytes, indicating that anorexia may be a specific behavioral response to maintain high levels of hepatic autophagic activity during an infection.
Upregulating autophagy in the liver may have a critical impact on the disease trajectory in a number of ways. The liver plays a major role in lipopolysaccharide (LPS) clearance during sepsis,4 although the process by which LPS is neutralized remains poorly understood. However, recent evidence indicates that the uptake of LPS by SCARB1 (scavenger receptor class B member I), ubiquitously expressed in the liver, may provide a mechanism for LPS4 as well as bacteria5 uptake. Indeed, earlier studies identified LPS in “phagocytic vacuoles” of Kupffer cells,6 suggesting that autophagy may play a role in degrading LPS. Supporting this view, Kupffer cells from mice fed a high-fat diet display a significant decrease in autophagic markers with a concomitant increased sensitivity toward LPS treatment ex vivo.7 Similarly, in a cecum-ligation-and-puncture mouse model system, it was illustrated that pharmacological induction of autophagy is protective in cardiomyocytes8 as well as in hepatocytes,9 whereas inhibition of autophagy has detrimental consequences. These results indicate that upregulation of hepatic autophagy may provide survival benefits during sepsis. Thus, SAA may represent a mechanism to promote autophagic flux in the liver.
Finally, it has been demonstrated that NR1H4 similarly antagonizes the transcriptional activity of CREB (cAMP response element-binding protein) after feeding and that this “NR1H4-CREB axis” likely modulates autophagy in various other tissue types during feeding/fasting cycles.10 This finding suggests that fasting may also play a role that widely affects peripheral tissue. However, chronic activation of hepatic autophagy adversely affects liver homeostasis. In patients suffering from anorexia nervosa, a psychiatric disorder that is distinct from SAA, prolonged activation of hepatic autophagy has detrimental consequences.11 Indeed, it has been observed that starvation-induced autophagy in patients suffering from anorexia nervosa may lead to hepatic cell death.12 However, these pathologies manifest in severely undernourished individuals as a consequence of prolonged upregulation of autophagy. Since infections resolve within weeks, anorexia nervosa is not only distinct from SAA in terms of etiology but also in terms of the time frame in which SAA would invoke autophagic processes.
Anorexia induces autophagy by altering the profile of plasma amino acids
During an infection, energy-rich molecules are mobilized in order to sustain metabolic activity of immune effectors. Mechanistically, LPS binding to TLR4 (toll-like receptor 4) on 3T3-L1 adipocytes induces insulin resistance,13 while inducing lipolysis in adipocytes14 and promoting triglyceride production in hepatocytes.15 Similarly, glucose is preferentially shunted toward immune cells during an infection. Cytokines such as TNF and IL1B/IL-1β induce a state of insulin resistance in peripheral tissues, particularly by inhibiting glucose uptake in the 2 major ‘glucose-sinks’—myocytes16 and adipocytes,17 thereby providing more glucose for aerobic glycolysis (Warburg effect), and driving the anabolic metabolism of activated immune cells.18
However, in sharp contrast to glucose and fatty acids, various circulating amino acids (AA), including branched-chain AA (BCAA), progressively decrease during an infection.19 Such a decrease occurs in the context of rapid muscle catabolism, suggesting the prime recipients of liberated amino acids may be the liver where amino acids are used for gluconeogenesis, and immune cells for rapid cell division or anabolism. Decreased circulating AA could promote autophagy in 2 ways: first, cellular efflux of glutamine coupled with an influx of BCAA is the rate-limiting step in the induction of the autophagy inhibitor MTOR.20 Thus, decreased serum levels of BCAA may promote autophagic flux by inhibiting the action of MTOR. Second, AAs also influence the expression of growth factors and hormones that may affect autophagy.21 For example, arginine promotes insulin secretion,22 which in turn is a potent inhibitor of autophagy, independent of the effect of amino acids.23 Of note, serum arginine levels are often decreased during sepsis,24 suggesting that a decline in arginine may be mechanistically related to a systemic induction of autophagic flux. Collectively, these observations argue that SAA may represent a strategy to maintain autophagic turnover in cells by modulating circulating AA levels (Fig. 1).
Figure 1.

The autophagic machinery deployed in housekeeping functions is fundamentally implicated in pathogen clearance and subsequent loading and expression of pathogen epitopes onto MHC II complexes. Pathogens have evolved numerous mechanisms to avoid autophagic degradation. Note, in addition to being phagocytosed, viruses and bacteria have evolved numerous strategies to gain entry into nonphagocytic cells via endocytosis.30 Endosomes can be targeted by autophagy or fuse with autophagosomes to form amphisomes. Regardless, similar autophagic components are implicated in these processes.
Anorexia-mediated autophagy in immune cells
Short-term starvation induced by 4-h amino acid and growth factor withdrawal upregulates autophagy, protecting macrophages against intracellular pathogens such as Mycobacterium tuberculosis (TB).25 This is intriguing since TB manipulates the host autophagic machinery by preventing fusion of lysosomes with TB-containing vesicles (phagosomes),26 thus suggesting redundancy or synergy between starvation- and immunologically-induced autophagy. The same starvation protocol has also been used to mediate “starvation-induced autophagy killing” of TB by immune cells.27,28 These studies thus establish that both starvation-induced macroautophagy as well as xenophagy implement the same mechanistic pathways in degradation of both pathogens and cellular content (Fig. 1).
An additional novel immunological mechanism involving the autophagy machinery has recently been described.27 It was observed that SQSTM1/p62, a receptor molecule that targets ubiquitinated cytoplasmic components for autophagic degradation, is essential for “starvation-induced autophagy killing.” Surprisingly, however, elimination of bacteria is not mediated by direct lysosomal degradation of bacteria. Instead, it was shown that SQSTM1 shuttles FAU/RPS30 to autolysosomes where it is processed by lysosomal CTSL (cathepsin L) into peptides with antimicrobial properties against Mycobacteria. Other components required in this process include LC3 and the recognition of ubiquitin moieties, illustrating that this immune function indeed involves an autophagic response. Thus, in addition to the established role of xenophagy, autophagy may also aid in controlling an infection by processing cytoplasmic components into peptides with antimicrobial activity.
Autophagic pathways are also utilized to secrete protein products,29 thus raising the question of whether an autophagic immune response is restricted to intracellular defenses, or if such processed cytoplasmic peptides may be liberated into the local extracellular environment. It is also of interest to consider whether nonimmune cells could similarly process ribosomes into antimicrobial peptides.
Anorexia-induced autophagy in nonimmune cells
Pathogens have also evolved a number of mechanisms by which to gain entry into nonphagocytic cells.30 Pathogens entering a cell have to subvert autophagic processes rapidly in order to avoid being degraded. Therefore, when an increase in autophagic flux is maintained, the pathogen would be confronted with a narrower window of opportunity for subversion.31 Thus, SAA enhances cell-autonomous defenses by increasing autophagic flux, thereby restricting the pathogen's infectiousness.
In this regard, autophagic processes have been observed to contribute toward cell-autonomous defense in nonimmune cells. This is well exemplified by infections with group A Streptococcus (GAS) where it has been shown that autophagic processes in nonimmune cells play a vital role in managing infection by GAS.32 Following entry into the host cell, GAS is rapidly trapped in LC3-positive vacuoles exhibiting morphological features distinct from starvation-induced autophagosomes.32 However, these bacteria are killed via a lysosomal-dependent mechanism, suggesting that autophagic processes are indeed involved. Furthermore, ATG5-deficient cells are unable to sequester and kill GAS in similar autophagosome-like vesicles, illustrating an underlying role of autophagic processes in mediating cell-autonomous defense.
Furthermore, autophagy is implicated in mobilizing an adaptive immune response by processing and presenting epitopes in both immune and nonimmune cells. For example, both muscle33 and endothelial34 cells can be induced to express epitopes processed by autophagy on major histocompatibility complex (MHC) class II molecules. Adipocytes derived from obese individuals similarly demonstrate an ability to express MHC II at levels comparable to macrophages35 which, considering the immune function of (pre-) adipocytes,36 may also be intact in the adipocytes of nonobese individuals. Furthermore, since the autophagic machinery is also used to mobilize fat stores (lipophagy), these cells could conceivably recruit the same autophagic machinery for epitope expression by MHC II.
Interestingly, inhibition of MTOR has recently been shown to not only enchase survival in mice during primary infection with influenza virus, but also to provide enhanced protection to secondary infection by different substrains through altering the antibody repertoire and enhanced crossreactivity.37 We speculate that superior epitope presentation as a result of enhanced autophagy may contribute to the heterosubtypic immunity to influenza substrains. SAA may thus represent a mechanism by which enhanced epitope presentation augments an adaptive immune response.
The position of the autophagic pathway at the intersection between cell death and survival may also play an important role during an infection. Besides the documented prosurvival effect of autophagy, autophagic proteins such as BECN1, ATG5, ATG7, and ATG12 are directly or indirectly involved in apoptosis.38 Moreover, autophagy may play a pivotal role in immune function by regulating cell death in virally infected cells. Virus-infected cells often commit a form of programmed cell death, apoptosis or programmed necrosis (necroptosis), in an effort to deny virus access to the cellular replication machinery. In turn, viruses have evolved various mechanisms for preventing programmed cell death.39 In this regard it has been shown that cells can undergo ‘autophagy-mediated’ cell death, autosis, which is distinct both molecularly and morphologically from necrosis and apoptosis.40 In particular, autosis is inducible through starvation,40 indicating that this response may provide an alternative cell-death pathway. Thus, starvation may ‘prime’ autophagy-mediated cell death as a failsafe response when subversive signaling by a virus compromises canonical cell-death pathways.
In addition to an immune function, autophagy may also modulate the cell's responsiveness to the developing disease state. Clearance of apoptotic bodies is crucial in preventing secondary necrosis and a subsequent upregulation of the inflammatory response. Within a developmental context, autophagy plays a pivotal role in the removal of apoptotic bodies by providing energy-dependent engulfment signals.41 Thus, autophagy may play a critical role in processing the phagocytosed cellular corpses. Autophagy has also been implicated in the selective degradation of the inflammasome complex, thus limiting the inflammatory response.42 In addition, hyperthermia, associated with a febrile-response, induces the expression of heat-shock proteins in a number of tissue types,43 demonstrating that a febrile response may result in the accumulation of misfolded proteins. In this regard, autophagy may promote cell survival by providing additional means for removing protein aggregates systemically, irrespective of immune competence. Taken together, these mechanisms demonstrate a formative role for autophagy in mediating cell survival and modulating the ‘inflammatory tone’ of cells.
Anorexia-induced autophagy: Underlying design principles
Forgoing feeding during an infection is truly an energetically expensive immunological strategy. Why induce autophagy by fasting and not by some less costly mechanism? We suggest that dual activation of autophagy by starvation via the MTOR pathway in combination with immunological activation via LPS-activated autophagy via immune activators such as TLR4 or interferon signaling may enhance killing efficacy in 2 ways. First, activation of different autophagic signaling circuits may converge synergistically, giving rise to a robust and rapid increase in autophagic flux. Second, activating autophagy through multiple signaling circuits may render cells more resilient against attacks by pathogens attempting to subvert or undermine recruitment of autophagic effectors. Indeed, various pathogens, including certain viruses, bacteria and protists have evolved mechanisms for modulating the autophagic apparatus for their own purposes.44 Consequently we suggest that SAA represents an evolutionarily conserved strategy to robustly increase autophagy systemically thereby providing added ‘signaling security’ against pathogenic subversive strategies.
It is well appreciated that animals are capable of “self-medicating” by selecting food sources with specific properties in order to rid themselves of various diseases.45 We suspect that this paradigm also applies for macronutrient manipulation, as indicated by meat aversion and other alterations in food perception. Indeed, as indicated at the start of the Introduction, Marie's “disgust” at the veal is reflective of meat aversion often present during an infection and well recognized in cancer patients.46 It is tempting to speculate that meat aversion may be a direct strategy to maintain suppressed AA levels in an attempt to ensure upregulated autophagy systemically. Indeed, even in the presence of growth factors, deprivation of AAs, particularly BCAAs such as leucine and isoleucine, are potent inhibitors of MTORC1 and subsequently upregulate autophagy.47 As argued here, an upregulation of autophagy may provide a number of benefits (Table 1, Fig. 2).
Table 1.
Sickness-associated anorexia may aid in the systemic upregulation of autophagy, thereby providing a number of survival benefits and aids in the pathogen clearance process.
| PROCESS OF SAA-INDUCED AUTOPHAGY | UNDERLYING MECHANISM | REFERENCE |
|---|---|---|
| Responsiveness to developing infection | Autophagic degradation of inflammasome | 42 |
| Inflammation resulting from secondary necrosis of apoptotic corpses | Phagocytosis and autophagic degradation of apoptotic bodies by neighboring cells | 41,59 |
| Clearance of damaged organelles such as mitochondria that would result in oxidative stress | Selective targeting of compromised mitochondria | 60 |
| Degrading missfolded and aggregating proteins damaged by biocidal chemicals and heat (febrile response) | Lysosomal degradation of large protein aggregates | 61 |
| Clearance of circulating LPS | Increased LPS clearance | 6 |
| Direct targeting of intracellular pathogens | Lysosomal degradation of pathogens | 50,62 |
| AMP-mediated targeting of pathogens | Processing of cellular components into AMP | 27 |
| Augmented adaptive immune system | Autophagic processing of pathogen components into epitopes | 33,34 |
| Co-activation of converging signaling cascades and mechanistic redundancy | Convergence/redundancy between immune- and nutrition-induced autophagic signaling cascades protecting cells against ‘signaling attacks’ | 44,63 |
AMP, antimicrobial peptide; LPS, lipopolysaccharides; SAA, sickness-associated anorexia.
Figure 2.
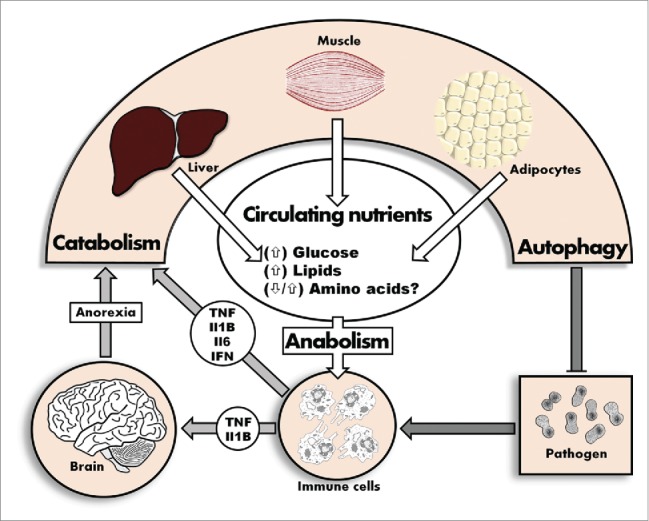
During an infection, immune effectors remodel the nutrient context in order to induce and maintain heightened autophagic flux.
Yet, it should also be taken into account that therapeutic benefit may arise from attenuating SAA because aspects of SAA are activated prophylactically where infection is not an eminent threat. This is illustrated by the fact that almost half of all clinically observed febrile responses are of noninfectious origin,48 indicating that, even in the absence of any infection, trauma may induce aspects of sickness behavior. Indeed, ‘danger signals’ such as HMGB1 can bind to TLR4, the same innate sensor for LPS.49 Since excessive tissue damage is associated with a breach of barrier defense, such a pre-emptive response might have afforded protection in an environment with a higher pathogen load. In a clinical setting, however, such a response to sterile tissue damage would be excessive. Similarly, tissue disruption associated with metastatic cancer, in conjunction with cell death may underlie an anorexic response in cancer patients.
Implications
It has long been speculated that sickness-induced anorexia contributes to the resolution of an infection.1 Correspondingly, the immunological function of autophagy has resulted in speculation on how fasting or pharmacological simulation thereof could be introduced as a novel therapeutic strategy in treating various diseases.50 The hypothesis presented here unifies these 2 concepts by providing evidence that implicates SAA as a mechanism to upregulate autophagy systemically (Fig. 1). It thus seems likely that pharmacological recapitulation of fasting-induced autophagy may hold therapeutic value. Furthermore, the argument presented here provides mechanistic insight into the benefits occasionally observed with permissive underfeeding. Indeed, it was found that patients receiving early parenteral nutrition had a small but significantly higher incidence of infection51 while it was demonstrated in another study that permissive underfeeding reduced mortality rates.52
The observation that BCAAs are often decreased in the muscles of septic patients, coupled with the ability of leucine to inhibit muscle degradation ex vivo, has initially generated much excitement for the use of AA as a potential supplement in these patients. However, results from studies applying supplementation with these or other amino acids have been disappointing.53 Similarly, there is currently no evidence indicating the existence of an optimum protein provision in critically ill patients.54 It is likely that at least some of the negative results obtained from nutritional studies may relate to the fact that such supplementation may hamper autophagic turnover in cells by supplying BCAAs, thereby potently inhibiting autophagy. Indeed, a comparison between patients receiving early as opposed to late parenteral nutrition demonstrated that autophagic processes are inhibited by feeding regimes.55 Interestingly, an increase in LC3-II protein levels is associated with better recovery and a significant decrease in muscle weakness,55 suggesting that upregulation of autophagy may hold additional benefits within the clinical setting. Collectively, these observations highlight the need for evaluating the specific context in which nutritional support is provided, as derailing autophagic processes in the context of an infection may hamper patient recovery.
Likewise, the fact that pathogens are able to subvert autophagy44 (Fig. 2) warrants careful further investigation. On the one hand, certain pathogens might have evolved special mechanisms to cope well with anorexia-induced autophagy, rendering permissive underfeeding futile or even detrimental. On the other hand, permissive underfeeding may recapitulate the effects of SAA as a means to recruit additional autophagic processes, rendering the autophagic response more robust against pathogen subversion. We suspect that a re-evaluation of different supplementation regimes on autophagic flux47 and patient outcome could prove insightful: evaluating patients according to pathogen-type may indicate infections in which permissive underfeeding as opposed to aggressive supplementation proves most effective.
However, although it is well established that autophagy is induced during starvation, and as evidence presented here indicates, that this activation of autophagy indeed relates mechanistically to an enhanced immune response, evaluating the effect of fasting-induced autophagy on pathogen clearance is required. Here, accurately measuring autophagic flux in vivo is a prerequisite, since the autophagic machinery is often hijacked by pathogens and as such, markers that demonstrate an increase in autophagic activity (e.g. accumulation of autophagosomes) may not relate to an increase in autophagic flux, but rather indicate infectious status.
The hypothesis presented here, involving a systemic upregulation of autophagic flux may also have implications for vaccine protocols. Inhibition of MTOR during primary infection with influenza enhances cross-reactivity to different substrains during a secondary challenge.37 If the upregulation of autophagy results in enhanced presentation of epitopes, a more robust response to vaccination is to be expected after fasting. Such an approach may be of particular value in the context of vaccines that induce only modest protection, or in situations where a compromised immune system may render the vaccine less effective. For example, HIV patients are notoriously poor responders to the hepatitis B vaccine, likely as a result of depleted immune effectors.48 Fasting-induced autophagy may augment vaccine efficacy by incorporating non-immunological cells to assist in epitope presentation. Clearly, future work addressing the interplay between the type of pathogen, infection and autophagic flux will enhance our understanding of SAA-stimulated autophagy.
In addition to facilitating epitope expression, inhibition of MTOR with rapamycin could increase both the number of memory CD8+ T cells during the expansion phase, as well as increase the number of memory cells during the contraction phase.56 This suggests that SAA could be directed at inhibiting MTOR, and, in doing so, upregulate autophagy in order to enhance T cell expansion and memory formation. However, recent findings from carefully designed experiments have challenged the role of autophagy in the initial response to viral infection in CD8 T cells. It has been shown that, although autophagy plays a critical role in mediating T cell survival during the contraction phase, autophagy is expendable during the generation of effector cells.57 Others obtained similar results, demonstrating that T cell attrition resulting from conditional knockout of key autophagic proteins manifests more than one wk after the viral challenge.58 The fact that an intact autophagic machinery only becomes critical late in the disease process, or after pathogen clearance, would suggest that SAA may not play a pivotal role in mediating CD8+ T cells during the initial phase of an infection. Elucidating the defining role that autophagy plays during specific phases of the developing disease trajectory may aid in establishing the potential role of SAA in T cell function.
Conclusion
SAA and the concomitant disturbed AA profile observed during an infection are typically viewed as manifestations of pathology—a condition in need of being remedied. Yet, the hypothesis presented here outlines a different immunological narrative. Decreasing food intake may represent an evolutionarily conserved response pursuant to a systemic upregulation of autophagic processes, which performs a multifaceted role in host defense and cell survival. The comparative contribution of the mechanisms discussed here is unclear and in urgent need of empirical evaluation. Similarly, an understanding of how different autophagic ‘triggers’ such as signaling cascades and nutrient availability converge on autophagic signaling pathways is required to develop new and better therapeutic interventions. Finally, we argue for an urgent re-evaluation of nutritional support in the context of controlled permissive underfeeding and vaccination regimen, where enhanced autophagy may provide superior support.
Abbreviations
- AA
amino acids
- AMP
antimicrobial peptide
- BCAA
branched-chain AA
- CREB
cAMP response element-binding protein
- GAS,
group A Streptococcus
- LPS
lipopolysaccharide
- MHC
major histocompatibility complex
- NR1H4/FXR
nuclear receptor subfamily 1 group H member 4
- TB
Mycobacterium tuberculosis
- PPARA/PPARα
peroxisome proliferator-activated receptor
- SAA
sickness-associated anorexia
- TLR
toll-like receptor
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by the Cancer Association of South Africa (CANSA) and the National Research Foundation (NRF).
References
- [1].Hart BL. Biological basis of the behavior of sick animals. Neuroscience & Biobehavioral Reviews 1988; 12:123-137; PMID:3050629; http://dx.doi.org/ 10.1016/S0149-7634(88)80004-6 [DOI] [PubMed] [Google Scholar]
- [2].Ashley NT, Weil ZM, Nelson RJ. Inflammation: Mechanisms, costs, and natural variation. Annual Review of Ecology, Evolution, and Systematics 2012; 43:385-406; http://dx.doi.org/ 10.1146/annurev-ecolsys-040212-092530 [DOI] [Google Scholar]
- [3].Lee JM, Wagner M, Xiao R, Kim KH, Feng D, Lazar MA, Moore DD. Nutrient-sensing nuclear receptors coordinate autophagy. Nature 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Guo L, Zheng Z, Ai J, Huang B, Li XA. Hepatic scavenger receptor BI protects against polymicrobial-induced sepsis through promoting LPS clearance in mice. J Biol Chem 2014; 289:14666-14673; PMID:24719333; http://dx.doi.org/ 10.1074/jbc.M113.537258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Schäfer G, Guler R, Murray G, Brombacher F, Brown GD. The role of scavenger receptor B1 in infection with mycobacterium tuberculosis in a murine model. PloS one 2009; 4:e8448; http://dx.doi.org/ 10.1371/journal.pone.0008448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Mathison JC, Ulevitch RJ. The clearance, tissue distribution, and cellular localization of intravenously injected lipopolysaccharide in rabbits. J Immunol 1979; 123:2133-2143; PMID:489976 [PubMed] [Google Scholar]
- [7].Fukada H, Yamashina S, Izumi K, Komatsu M, Tanaka K, Ikejima K, Watanabe S. Suppression of autophagy sensitizes kupffer cells to endotoxin. Hepatol Res 2012; 42:1112-1118; PMID:22583683; http://dx.doi.org/ 10.1111/j.1872-034X.2012.01024.x [DOI] [PubMed] [Google Scholar]
- [8].Hsieh CH, Pai PY, Hsueh HW, Yuan SS, Hsieh YC. Complete induction of autophagy is essential for cardioprotection in sepsis. Ann Surg 2011; 253:1190-1200; PMID:21412148; http://dx.doi.org/ 10.1097/SLA.0b013e318214b67e [DOI] [PubMed] [Google Scholar]
- [9].Lin CW, Lo S, Perng DS, Wu DB, Lee PH, Chang YF, Kuo PL, Yu ML, Yuan SS, Hsieh YC. Complete activation of autophagic process attenuates liver injury and improves survival in septic mice. Shock 2014; 41:241-249; PMID:24365881; http://dx.doi.org/ 10.1097/SHK.0000000000000111 [DOI] [PubMed] [Google Scholar]
- [10].Seok S, Fu T, Choi S, Li Y, Zhu R, Kumar S, Sun X, Yoon G, Kang Y, Zhong W. Transcriptional regulation of autophagy by an FXR-CREB axis. Nature 2014; 516:108-111; PMID:25383523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kheloufi M, Boulanger CM, Durand F, Rautou P. Liver autophagy in anorexia nervosa and acute liver injury. BioMed research international 2014; 2014:701064; PMID:25250330; http://dx.doi.org/ 10.1155/2014/701064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Rautou P, Cazals–Hatem D, Moreau R, Francoz C, Feldmann G, Lebrec D, Ogier–Denis É, Bedossa P, Valla D, Durand F. Acute liver cell damage in patients with anorexia nervosa: A possible role of starvation-induced hepatocyte autophagy. Gastroenterology 2008; 135:840-848. e3; PMID:18644371; http://dx.doi.org/ 10.1053/j.gastro.2008.05.055 [DOI] [PubMed] [Google Scholar]
- [13].Song MJ, Kim KH, Yoon JM, Kim JB. Activation of toll-like receptor 4 is associated with insulin resistance in adipocytes. Biochem Biophys Res Commun 2006; 346:739-745; PMID:16781673; http://dx.doi.org/ 10.1016/j.bbrc.2006.05.170 [DOI] [PubMed] [Google Scholar]
- [14].Zu L, He J, Jiang H, Xu C, Pu S, Xu G. Bacterial endotoxin stimulates adipose lipolysis via toll-like receptor 4 and extracellular signal-regulated kinase pathway. J Biol Chem 2009; 284:5915-5926; PMID:19122198; http://dx.doi.org/ 10.1074/jbc.M807852200 [DOI] [PubMed] [Google Scholar]
- [15].Feingold KR, Staprans I, Memon RA, Moser AH, Shigenaga JK, Doerrler W, Dinarello CA, Grunfeld C. Endotoxin rapidly induces changes in lipid metabolism that produce hypertriglyceridemia: Low doses stimulate hepatic triglyceride production while high doses inhibit clearance. J Lipid Res 1992; 33:1765-1776; PMID:1479286 [PubMed] [Google Scholar]
- [16].Wei Y, Chen K, Whaley-Connell AT, Stump CS, Ibdah JA, Sowers JR. Skeletal muscle insulin resistance: Role of inflammatory cytokines and reactive oxygen species. Am J Physiol Regul Integr Comp Physiol 2008; 294:R673-80; PMID:18094066; http://dx.doi.org/ 10.1152/ajpregu.00561.2007 [DOI] [PubMed] [Google Scholar]
- [17].Grant RW, Dixit VD. Adipose tissue as an immunological organ. Obesity 2015; 23(3):512-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Vander Heiden MG, Cantley LC, Thompson CB. Understanding the warburg effect: The metabolic requirements of cell proliferation. Science 2009; 324:1029-1033; PMID:19460998; http://dx.doi.org/ 10.1126/science.1160809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Su L, Li H, Xie A, Liu D, Rao W, Lan L, Li X, Li F, Xiao K, Wang H, Yan P, Li X, Xie L. Dynamic changes in amino acid concentration profiles in patients with sepsis. PLoS One 2015; 10:e0121933; PMID:25849571; http://dx.doi.org/ 10.1371/journal.pone.0121933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, Yang H, Hild M, Kung C, Wilson C. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 2009; 136:521-534; PMID:19203585; http://dx.doi.org/ 10.1016/j.cell.2008.11.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kuhara T, Ikeda S, Ohneda A, Sasaki Y. Effects of intravenous infusion of 17 amino acids on the secretion of GH, glucagon, and insulin in sheep. Am J Physiol 1991; 260:E21-6; PMID:1987790 [DOI] [PubMed] [Google Scholar]
- [22].Newsholme P, Brennan L, Rubi B, Maechlen P. New insights into amino acid metabolism, b-cell function and diabetes. Clin Sci 2005; 108:185-194; PMID:15544573; http://dx.doi.org/ 10.1042/CS20040290 [DOI] [PubMed] [Google Scholar]
- [23].Kanazawa T, Taneike I, Akaishi R, Yoshizawa F, Furuya N, Fujimura S, Kadowaki M. Amino acids and insulin control autophagic proteolysis through different signaling pathways in relation to mTOR in isolated rat hepatocytes. J Biol Chem 2004; 279:8452-8459; PMID:14610086; http://dx.doi.org/ 10.1074/jbc.M306337200 [DOI] [PubMed] [Google Scholar]
- [24].Luiking YC, Poeze M, Dejong CH, Ramsay G, Deutz NE. Sepsis: An arginine deficiency state? Crit Care Med 2004; 32:2135-2145; PMID:15483426; http://dx.doi.org/ 10.1097/01.CCM.0000142939.81045.A0 [DOI] [PubMed] [Google Scholar]
- [25].Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and< i>mycobacterium tuberculosis survival in infected macrophages. Cell 2004; 119:753-766; PMID:15607973; http://dx.doi.org/ 10.1016/j.cell.2004.11.038 [DOI] [PubMed] [Google Scholar]
- [26].Hestvik ALK, Hmama Z, Av-Gay Y. Mycobacterial manipulation of the host cell. FEMS Microbiol Rev 2005; 29:1041-1050; PMID:16040149; http://dx.doi.org/ 10.1016/j.femsre.2005.04.013 [DOI] [PubMed] [Google Scholar]
- [27].Ponpuak M, Davis AS, Roberts EA, Delgado MA, Dinkins C, Zhao Z, Virgin IV HW, Kyei GB, Johansen T, Vergne I. Delivery of cytosolic components by autophagic adaptor protein p62 endows autophagosomes with unique antimicrobial properties. Immunity 2010; 32:329-341; PMID:20206555; http://dx.doi.org/ 10.1016/j.immuni.2010.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Pilli M, Arko-Mensah J, Ponpuak M, Roberts E, Master S, Mandell MA, Dupont N, Ornatowski W, Jiang S, Bradfute SB. TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity 2012; 37:223-234; PMID:22921120; http://dx.doi.org/ 10.1016/j.immuni.2012.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Manjithaya R, Subramani S. Role of autophagy in unconventional protein secretion. Autophagy 2010; 6:650-651; PMID:20473033; http://dx.doi.org/ 10.4161/auto.6.5.12066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Cossart P, Helenius A. Endocytosis of viruses and bacteria. Cold Spring Harb Perspect Biol 2014; 6; pii: a016972; PMID:25085912; http://dx.doi.org/ 10.1101/cshperspect.a016972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Baxt LA, Garza-Mayers AC, Goldberg MB. Bacterial subversion of host innate immune pathways. Science 2013; 340:697-701; PMID:23661751; http://dx.doi.org/ 10.1126/science.1235771 [DOI] [PubMed] [Google Scholar]
- [32].Nakagawa I, Amano A, Mizushima N, Yamamoto A, Yamaguchi H, Kamimoto T, Nara A, Funao J, Nakata M, Tsuda K, Hamada S, Yoshimori T. Autophagy defends cells against invading group A streptococcus. Science 2004; 306:1037-1040; PMID:15528445; http://dx.doi.org/ 10.1126/science.1103966 [DOI] [PubMed] [Google Scholar]
- [33].Keller CW, Fokken C, Turville SG, Lunemann A, Schmidt J, Munz C, Lunemann JD. TNF-alpha induces macroautophagy and regulates MHC class II expression in human skeletal muscle cells. J Biol Chem 2011; 286:3970-3980; PMID:20980264; http://dx.doi.org/ 10.1074/jbc.M110.159392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Mai J, Virtue A, Shen J, Wang H, Yang X. An evolving new paradigm: Endothelial cells–conditional innate immune cells. J Hematol Oncol 2013; 6:61; PMID:23965413; http://dx.doi.org/ 10.1186/1756-8722-6-61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Deng T, Lyon CJ, Minze LJ, Lin J, Zou J, Liu JZ, Ren Y, Yin Z, Hamilton DJ, Reardon PR, Sherman V, Wang HY, Phillips KJ, Webb P, Wong ST, Wang RF, Hsueh WA. Class II major histocompatibility complex plays an essential role in obesity-induced adipose inflammation. Cell Metab 2013; 17:411-422; PMID:23473035; http://dx.doi.org/ 10.1016/j.cmet.2013.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Zhang L, Guerrero-Juarez CF, Hata T, Bapat SP, Ramos R, Plikus MV, Gallo RL. Dermal adipocytes protect against invasive staphylococcus aureus skin infection. Science 2015; 347:67-71; PMID:25554785; http://dx.doi.org/ 10.1126/science.1260972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Keating R, Hertz T, Wehenkel M, Harris TL, Edwards BA, McClaren JL, Brown SA, Surman S, Wilson ZS, Bradley P. The kinase mTOR modulates the antibody response to provide cross-protective immunity to lethal infection with influenza virus. Nat Immunol 2013; 14:1266-1276; PMID:24141387; http://dx.doi.org/ 10.1038/ni.2741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Subramani S, Malhotra V. Non-autophagic roles of autophagy-related proteins. EMBO Rep 2013; 14:143-151; PMID:23337627; http://dx.doi.org/ 10.1038/embor.2012.220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Mocarski ES, Upton JW, Kaiser WJ. Viral infection and the evolution of caspase 8-regulated apoptotic and necrotic death pathways. Nature Reviews Immunology 2012; 12:79-88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Liu Y, Shoji-Kawata S, Sumpter RM Jr, Wei Y, Ginet V, Zhang L, Posner B, Tran KA, Green DR, Xavier RJ, Shaw SY, Clarke PG, Puyal J, Levine B. Autosis is a na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc Natl Acad Sci U S A 2013; 110:20364-20371; PMID:24277826; http://dx.doi.org/ 10.1073/pnas.1319661110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Qu X, Zou Z, Sun Q, Luby-Phelps K, Cheng P, Hogan RN, Gilpin C, Levine B. Autophagy gene-dependent clearance of apoptotic cells during embryonic development. Cell 2007; 128:931-946; PMID:17350577; http://dx.doi.org/ 10.1016/j.cell.2006.12.044 [DOI] [PubMed] [Google Scholar]
- [42].Shi C, Shenderov K, Huang N, Kabat J, Abu-Asab M, Fitzgerald KA, Sher A, Kehrl JH. Activation of autophagy by inflammatory signals limits IL-1 [beta] production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol 2012; 13:255-263; PMID:22286270; http://dx.doi.org/ 10.1038/ni.2215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Ostberg J, Kaplan K, Repasky E. Induction of stress proteins in a panel of mouse tissues by fever-range whole body hyperthermia. Int J Hyperthermia 2002; 18:552-562; PMID:12537754; http://dx.doi.org/ 10.1080/02656730210166168 [DOI] [PubMed] [Google Scholar]
- [44].Ko Y, Choi JH, Ha NY, Kim IS, Cho NH, Choi MS. Active escape of orientia tsutsugamushi from cellular autophagy. Infect Immun 2013; 81:552-559; PMID:23230293; http://dx.doi.org/ 10.1128/IAI.00861-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].de Roode JC, Lefevre T, Hunter MD. Ecology. self-medication in animals. Science 2013; 340:150-151; PMID:23580516; http://dx.doi.org/ 10.1126/science.1235824 [DOI] [PubMed] [Google Scholar]
- [46].Fanelli FR, Cangiano C, Ceci F, Cellerino R, Franchi F, Menichetti E, Muscaritoli M, Cascino A. Plasma tryptophan and anorexia in human cancer. Eur J Cancer Clin Oncol 1986; 22:89-95; PMID:3456893; http://dx.doi.org/ 10.1016/0277-5379(86)90346-9 [DOI] [PubMed] [Google Scholar]
- [47].Efeyan A, Zoncu R, Sabatini DM. Amino acids and mTORC1: From lysosomes to disease. Trends Mol Med 2012; 18:524-533; PMID:22749019; http://dx.doi.org/ 10.1016/j.molmed.2012.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Launey Y, Nesseler N, Mallédant Y, Seguin P. Clinical review: Fever in septic ICU patients—friend or foe. Crit Care 2011; 15:01-07; http://dx.doi.org/ 10.1186/cc10097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Andersson U, Tracey KJ. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu Rev Immunol 2011; 29:139-162; PMID:21219181; http://dx.doi.org/ 10.1146/annurev-immunol-030409-101323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Ma Y, Galluzzi L, Zitvogel L, Kroemer G. Autophagy and cellular immune responses. Immunity 2013; 39:211-227; PMID:23973220; http://dx.doi.org/ 10.1016/j.immuni.2013.07.017 [DOI] [PubMed] [Google Scholar]
- [51].Casaer MP, Mesotten D, Hermans G, Wouters PJ, Schetz M, Meyfroidt G, Van Cromphaut S, Ingels C, Meersseman P, Muller J. Early versus late parenteral nutrition in critically ill adults. N Engl J Med 2011; 365:506-517; PMID:21714640; http://dx.doi.org/ 10.1056/NEJMoa1102662 [DOI] [PubMed] [Google Scholar]
- [52].Arabi YM, Tamim HM, Dhar GS, Al-Dawood A, Al-Sultan M, Sakkijha MH, Kahoul SH, Brits R. Permissive underfeeding and intensive insulin therapy in critically ill patients: A randomized controlled trial. Am J Clin Nutr 2011; 93:569-577; PMID:21270385; http://dx.doi.org/ 10.3945/ajcn.110.005074 [DOI] [PubMed] [Google Scholar]
- [53].De Bandt JP, Cynober L. Therapeutic use of branched-chain amino acids in burn, trauma, and sepsis. J Nutr 2006; 136:308S-13S; PMID:16365104 [DOI] [PubMed] [Google Scholar]
- [54].Hoffer LJ, Bistrian BR. Appropriate protein provision in critical illness: A systematic and narrative review. Am J Clin Nutr 2012; 96:591-600; PMID:22811443; http://dx.doi.org/ 10.3945/ajcn.111.032078 [DOI] [PubMed] [Google Scholar]
- [55].Hermans G, Casaer MP, Clerckx B, Güiza F, Vanhullebusch T, Derde S, Meersseman P, Derese I, Mesotten D, Wouters PJ. Effect of tolerating macronutrient deficit on the development of intensive-care unit acquired weakness: A subanalysis of the EPaNIC trial. The lancet Respiratory medicine 2013; 1:621-629; PMID:24461665; http://dx.doi.org/ 10.1016/S2213-2600(13)70183-8 [DOI] [PubMed] [Google Scholar]
- [56].Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, Larsen CP, Ahmed R. mTOR regulates memory CD8 T-cell differentiation. Nature 2009; 460:108-112; PMID:19543266; http://dx.doi.org/ 10.1038/nature08155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Xu X, Araki K, Li S, Han J, Ye L, Tan WG, Konieczny BT, Bruinsma MW, Martinez J, Pearce EL. Autophagy is essential for effector CD8 T cell survival and memory formation. Nat Immunol 2014; 15:1152-1161; PMID:25362489; http://dx.doi.org/ 10.1038/ni.3025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Schlie K, Westerback A, DeVorkin L, Hughson LR, Brandon JM, MacPherson S, Gadawski I, Townsend KN, Poon VI, Elrick MA, et al.. Survival of effector CD8+ T cells during influenza infection is dependent on autophagy. J Immunol 2015; 194:4277-4286; PMID:25833396; http://dx.doi.org/ 10.4049/jimmunol.1402571 [DOI] [PubMed] [Google Scholar]
- [59].Mellén M, De La Rosa E, Boya P. The autophagic machinery is necessary for removal of cell corpses from the developing retinal neuroepithelium. Cell Death & Differentiation 2008; 15:1279-1290; http://dx.doi.org/ 10.1038/cdd.2008.40 [DOI] [PubMed] [Google Scholar]
- [60].Chu CT, Ji J, Dagda RK, Jiang JF, Tyurina YY, Kapralov AA, Tyurin VA, Yanamala N, Shrivastava IH, Mohammadyani D. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol 2013; 15:1197-1205; PMID:24036476; http://dx.doi.org/ 10.1038/ncb2837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007; 282:24131-24145; PMID:17580304; http://dx.doi.org/ 10.1074/jbc.M702824200 [DOI] [PubMed] [Google Scholar]
- [62].Crotzer VL, Blum JS. Autophagy and adaptive immunity. Immunology 2010; 131:9-17; PMID:20586810; http://dx.doi.org/ 10.1111/j.1365-2567.2010.03309.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Wing EJ, Barczynski LK, Boehmer SM. Effect of acute nutritional deprivation on immune function in mice. I. macrophages. Immunology 1983; 48:543-550; PMID:6402445 [PMC free article] [PubMed] [Google Scholar]