

ABSTRACT
LAMP2A is the key protein of chaperone-mediated autophagy (CMA), downregulation of LAMP2A leads to CMA blockade. CMA activation has been implicated in cancer growth, but the exact mechanisms are unclear. Elevated expression of LAMP2A was found in 8 kinds of tumors (n=747), suggesting that LAMP2A may have an important role in cancer progression. Unsurprisingly, LAMP2A knockdown in gastric cancer (GC) cells hindered proliferation, accompanied with altered expression of cell cycle-related proteins and accumulation of RND3/RhoE. Interactomic and KEGG analysis revealed that RND3 was a putative CMA substrate. Further study demonstrated that RND3 silencing could partly rescue the proliferation arrest induced by LAMP2A knockdown; RND3 was increased upon lysosome inhibition via both chemicals and LAMP2A-shRNA; Furthermore, RND3 could interact with CMA components HSPA8 and LAMP2A, and be engulfed by isolated lysosomes. Thus, constant degradation of RND3 by CMA is required to sustain rapid proliferation of GC cells. At last, the clinical significance of LAMP2A was explored in 593 gastric noncancerous lesions and 173 GC tissues, the results revealed that LAMP2A is a promising biomarker for GC early warning and prognosis of female GC patients.
KEYWORDS: CMA, gastric cancer, LAMP2A, proliferation, RND3
Introduction
Autophagy mediates the degradation of cellular components such as proteins and organelles in lysosomes,1 and contributes to cellular homeostasis, quality control, maintenance of energetic balance.2,3 Macroautophagy and chaperone-mediated autophagy (CMA) are 2 well-characterized autophagic pathways in mammalian cells.4,5 CMA is a selective process mediated in particular by HSPA8 and LAMP2A; HSPA8 can recognize and interact with the substrates bearing a “KFERQ”-like motif in their amino acid sequence,6,7 and LAMP2A serves as a receptor on the lysosome membrane for the HSPA8-substrate complex.8 After unfolding,9 the substrate is translocated into the lysosome lumen through a multimeric protein complex mainly composed of LAMP2A.10-12 During this process, LAMP2A is the rate-limiting protein of CMA and is widely used as a target to block CMA activity.13-16 CMA plays an important role in multiple cellular processes by degrading target proteins. For example, it contributes to maintain genome stability and cell survival in fibroblasts through regulated degradation of CHEK1/Chk1 after the genotoxic insult,16 CMA regulates T cell response through the targeted degradation of negative regulators of T cell activation.17
CMA deficiency has been linked with the pathogenesis of certain diseases such as neurodegenerative disorders, Danon disease, lysosomal storage disorders,18-23 and also with liver metabolic dysfunction24 and aging.25,26 In contrast, high activation of CMA is observed in tumors and is required for tumor growth and survival.13,15 In lung cancer, LAMP2A knockdown increases wild-type TP53, which suppresses the transcription of the genes encoding the glycolysis enzymes GAPDH, and PGK and results in reduced glycolysis and growth arrest,15 but this mechanism may not apply to cancer cells with mutant TP53, and CMA substrates responsible for proliferation arrest need to be identified to fully understand the link between CMA and cancer proliferation.
Here we identified MG5 as a specific antibody against LAMP2A. MG5 was produced in our lab by immunizing the BALB/C mice directly with gastric cancer cells and then by monoclonal hybridoma techniques. Although it had a higher affinity to cancer tissue than to normal mucosa, its antigen was previously unclear. We used an interactomic approach and KEGG analysis to screen for putative CMA substrates responsible for proliferation and survival in gastric cancer (GC). Furthermore, rigorous experiments were performed to validate the CMA substrate RND3 and its function in GC. Finally, the early warning and prognostic values of LAMP2A were evaluated in 593 cases of gastric noncancerous lesions and 173 cases of gastric adenocarcinoma.
Results
Identification of MG5 as a specific antibody against LAMP2A
Western blot (WB) analysis showed that MG5 could specifically recognize a band at about 120 kDa in several gastric cancer cell lines, but did not crossreact with other proteins ranging from 10 to 250 kDa (Fig. 1A). Confocal immunofluorescence demonstrated that MG5 staining predominantly merged with LysoTracker Red (Fig. 1B), but not with the Golgi maker GOLGB1/Giantin (golgin B1) (Fig. 1C), suggesting that it is mainly located in lysosomes. Immunoprecipitates of MG5 and control IgG from the BGC823 gastric cancer cells were subject to western blot (WB) analysis (MG5 was used as a detecting antibody), and to Coomassie Blue staining (CBS). A band at 120 kDa could be seen in both WB and CBS (Fig. 1D, as indicated in red rectangle). Then mass spectrum (MALDI-TOF-TOF) was used to identify the protein at 120 kDa. By matching with MS database, several proteins were selected as putative MG5 targets (Fig. 1E, indicated in blue), but only LAMP2 is a lysosome protein with an apparent relative molecular mass of about 120 kDa (Fig. 1E, indicated in red). The fragment spectrum of MS revealed that the peptide specific to LAMP2 is “IPLNDLFR” (Fig. 1F). These data obtained from MS analysis suggest that LAMP2 is the antigen of MG5. Consistently, the commercial LAMP2 antibody (Santa Cruz biotechnology) could recognize the MG5 immunoprecipitate at 120 kDa (Fig. 1G). Because LAMP2 has 3 different isoforms, we transiently overexpressed LAMP2A, LAMP2B and LAMP2C in 293T cells, WB results showed that the commercial LAMP2 antibody (anti-L2) could recognize all 3 isoforms, while MG5 could only recognize LAMP2A (Fig. 1H). Then LAMP2A mRNA was overexpressed or silenced via lentivirus-mediated infection in BGC823 cells, RT-PCR assay showed that the manipulation of LAMP2A mRNA was successful (Fig. 1I; *, P < 0.001), and then WB showed that the changes of MG5 immunoblot were consistent with LAMP2A mRNA (Fig. 1J). All these data proved that MG5 is specific antibody against LAMP2A, and does not crossreact with other proteins. Thus, we used MG5 to detect or immunoprecipitate LAMP2A in this study.
Figure 1.

Identification of MG5 as a specific antibody against LAMP2A. (A) MG5 immunoblot in gastric cancer cell lines MKN45, BGC823, SGC7901 and AGS, the entire blot (from 10 to 250 kDa) is shown. ((B)and C) coimmunofluorescence of MG5 and LysoTracker Red (DND99) in BGC823 cells, GOLGB1 was used as a negative control, DAPI was used to stain the cell nucleus. Scale bar (in red) was 10 µm. (D) Immunoprecipitates of MG5 (MG5-IP) and control IgG (IgG-IP) form BGC823 cells were subject to western blot (WB) and Coomassie Blue staining. The bands in red rectangles of (D) were excised and prepared for mass spectrometry (MS). (E) Putative antigens for MG5 (in blue or red) were identified by MS and database matching. (F) MS peptide profile matching showed that LAMP2 is a candidate antigen of MG5. (G) WB assay of MG5-IP and IgG-IP using anti-LAMP2 (Santa Cruz Biotechnology). (H) 293T cells were transfected with LAMP2A, LAMP2B, LAMP2C or empty vector (control), and then were subjected to WB with either the MG5 or LAMP2 antibodies Anti-2L (Santa Cruz Biotechnology), ACTB was used as an internal loading control. (I) BGC823 cells were infected with lentivirus expressing LAMP2A, empty vector (Ctr), LAMP2A shRNA and control shRNA (NC), and were subjected to RT-PCR to confirm LAMP2A mRNA changes (I), then WB was performed to examine MG5 immunoreactivity (J), ACTB was used as an internal loading control.
CMA is required for rapid proliferation of gastric cancer cells
A tissue microarray (TMA) was immunostained with MG5 to screen the expression of LAMP2A in 10 different tumors (Fig. 2A, N = 60). Compared with normal tissues, LAMP2A expression was significantly increased in GA, CA, RA, PDC, LSCC, LA, BDC, and ESCC (Fig. 2A; P < 0.05); Moreover, 8 TMAs with more tissues (N = 747) were immunostained with MG5; the results showed that the positive rates in ESCC (77.8%), GA (53.8%), CA (51.2%), LSCC (59.7%), LA (73.8%), BDC (47.7%), RA (51.9%) and PDC (81.5%) were all significantly higher as compared with adjacent normal tissues (Fig. 2A, right; *, P < 0.05, **, P < 0.001). These data indicate that LAMP2A is overexpressed in many cancers, and CMA might play an important role in cancer. In order to block CMA, we silenced LAMP2A in both BGC823 and AGS cell lines by using lentivirus expressing targeted shRNA, and established 4 stable cell lines: BGC823-L2A−, BGC823-NC, AGS-L2A−, AGS-NC. WB confirmed that the LAMP2A level was successfully knocked down in both BGC823-L2A− and AGS-L2A- as compared with their control cell lines BGC823-NC and AGS-NC (Fig. 2B, C, P < 0.05). MTT assays showed that the growth rates of BGC823-L2A− and AGS-L2A− were both slowed down as compared with their controls (Fig. 2D and E; *, P < 0.05); FACS showed that the apoptotic rates of BGC823-NC were significantly increased at d 5 as compared with BGC823-L2A− (Fig. 2F; *, P < 0.05) , and this phenomenon could also be noticed in AGS cells at d 4 (Fig. 2G; *, P < 0.05), supporting the notion that increased apoptosis often appears as a secondary result of increased proliferation. These data implied that CMA is required for rapid proliferation of GC cells. Besides, the colony formation number of BGC823-L2A− was significantly reduced compared with BGC823-NC (Fig. 2H; P < 0.05). Consistently, pro-proliferation proteins such as E2F3 and PCNA in BGC823-L2A− were downregulated compared with BGC823-NC, whereas antiproliferation proteins CDKN1B/p27 and RND3 were upregulated (Fig. 2I; *, P < 0.05). All these data demonstrate that CMA blockade impedes GC cell proliferation.
Figure 2.

CMA blockade impedes gastric cancer cell proliferation. (A, left) Immunostaining of LAMP2A using MG5 in 10 kinds of tumors and normal tissues. Abbreviations: GA, gastric adenocarcinoma; CA, colon adenocarcinoma; RA, rectal adenocarcinoma; PDC, pancreatic ductal carcinoma; LSCC-lung squamous cell carcinoma; LA, lung adenocarcinoma; BDC, breast ductal carcinoma; RCCC, renal clear cell carcinoma; the adjacent normal tissues were immunostained as controls. (Manifestation: X 200). (A, Right) LAMP2A positivity was examined by MG5 staining in 8 kinds of tumors and normal tissues (*, P < 0.05; **, P < 0.001; Chi-square test). ((B)and C) Four stable cell lines (BGC823-NC, BGC823-L2A−, AGS-NC, and AGS-L2A−) were established by infecting BGC823 with lentiviral vectors containing LAMP2A shRNA or control shRNA (NC), and LAMP2A expression was validated by WB, ACTB was used as an internal control. ((D)and E) Growth of the 4 stable cell lines were assayed by MTT (values are means ± SEM of 3 independent experiments; *, P < 0.05; t test). ((F)and G) Apoptosis of the 4 stable cell lines were assayed by FACS (values are means ± SEM of 3 independent experiments). (H) This panel shows the clones formed by BGC823-NC and BGC823-L2A− cell lines (left), and the bar diagram of colony formation numbers of the 2 cell lines (right), the values are means ± SEM of 3 independent experiments; *, P < 0.05; t test. (I) Expression of LAMP2A and cell cycle-related proteins were examined by WB, ACTB was used as an internal control (left). The densitometric analyses of the indicated proteins with respect to ACTB from 3 independent experiments are shown as bar diagram (right).
Screening for candidate substrates of CMA that can regulate proliferation through an interactomic approach and KEGG analysis
Wild-type TP53/p53 has been reported to mediate CMA deficiency-induced proliferation arrest by downregulating GAPDH and PGK at transcription levels in lung cancer.15 Thus we tested this mechanism in gastric cancer. WB showed that although GAPDH and PGK were downregulated in AGS cells with wild-type TP53, upon LAMP2A silencing, they were actually upregulated in BGC823 cells with mutant TP53, despite the increase of TP53 in both cell lines (Fig. 3A; P < 0.05). And IHC results indicated that the correlation between LAMP2A and TP53 in GC tissues was very weak (Fig. 3B, R= −0.24, P < 0.05). These results suggest that wild-type TP53-mediated downregulation of glycolytic enzymes cannot fully account for CMA blockade induced proliferation arrest, especially when TP53 is mutated. Thus we have speculated that TP53-independent mechanisms may exist in GC and certain CMA substrates can play an important role in proliferation. In order to find some cues, we adopted interactomic approach combined with KEGG analysis to screen for the functional CMA targets. Firstly, we used MG5 and anti-HSPA8 to coimmunoprecipitate the binding proteins of LAMP2A and HSPA8 in BGC823, WB showed that LAMP2A and HSPA8 were markedly immunoprecipitated (Fig. 3C), suggesting that the immunoprecipitation (IP) assay was successful. Then the IP products were subject to SDS-PAGE and mass spectrometry (MS) analysis, the results revealed that 624 proteins could interact with both LAMP2A and HSPA8 (Fig. 3D), these proteins were candidates for CMA substrates or cochaperones. Then we adopted WebGestalt (an online tool) to perform KEGG analysis, and found that these proteins were significantly enriched in 16 biological pathways (Fig. 3E; P < 0.01), including reported pathways such as metabolic pathways, Parkinson disease, Alzheimer disease, Huntington disease, and antigen presentation; as well as unreported pathways, organelles and structures such as ribosomes, regulation of cytoskeleton, spliceosome, tight junction, RNA transport, protein processing in the ER, aminoacyl-tRNA biosynthesis, protein export and base excision repair. The related proteins were listed here (Fig. 3E), among which GAPDH, and PKM are well characterized CMA substrates, HSPA8 is the classic CMA chaperone, and PARK7/DJ-1 has been predicted to be a CMA substrate.27 These proteins may help us to better understand the mechanisms through which CMA can regulate a range of biological pathways. We thoroughly examined our interactomic results, and found that RND3 was selected by KEGG analysis as a key regulator of the cytoskeleton, and its antiproliferative action has also been well characterized. Additionally, the RND3 level was increased in both AGS and BGC823 cells upon LAMP2A silencing (Fig. 3A; P < 0.05), and the correlation between RND3 and LAMP2A expression in GC tissues was strong (Fig. 3D; R = −0.71, P < 0.001), suggesting that it is a CMA substrate that may exert an antiproliferative function independent of TP53 status.
Figure 3.

Screening for candidate substrates of CMA that can regulate proliferation, through an interactomic approach and KEGG analysis. (A) LAMP2A, TP53, GAPDG, PGK and RND3 levels were evaluated by WB in BGC823-NC, BGC823-L2A−, AGS-NC, and AGS-L2A− cell lines, ACTB was used as an internal control. (B) Expression correlation among LAMP2A, TP53 and RND3 based on IHC positivity in consecutive gastric cancer tissue slides, (n = 180, Spearman test). Scale bar was 100 µm. (C) HSPA8 and LAMP2A were immunoprecipitated from BGC823 cells and the IP efficacy was evaluated by WB. (D) Immunoprecipitates of (C) were subjected to SDS-PAGE and Coomassie Blue staining, and then the 2 lanes were excised and prepared for mass spectrum analysis, 624 proteins were selected as candidates for CMA substrates or cochaperones, and were subjected to KEGG analysis. The enriched KEGG pathways and related proteins are listed in (E).
Identification of RND3 as a novel CMA substrate that mediates CMA blockade-induced proliferation arrest
As mentioned above, the antiproliferative protein RND3 was selected as a putative CMA substrate and its level was increased upon LAMP2A silencing. Here we decided to prove that CMA can regulate GC cell proliferation at least partly through RND3 and examine whether RND3 can be degraded via CMA. RND3 was upregulated in BGC823 cells by plasmid transfection, and WB showed that the transfection was successful (Fig. 4A), and RND3 transfection significantly impeded cell growth as compared with control plasmid (Fig. 4B; *P < 0.05), but the apoptotic rate was unchanged at 90% cell confluency (Fig. 4C; P > 0.05), indicating that RND3 upregulation inhibits proliferation in GC cells. Next, we downregulated RND3 in the BGC823-L2A− cell line by lentiviral vectors expressing specific shRNA, WB showed that RND3 level was dramatically reduced by RND3 shRNA compared with nonspecific control (NC; Fig. 4D), and RND3 shRNA significantly rescued the impeded growth observed in BGC823-L2A− as compared with control shRNA (Fig. 4E; *, P < 0.05), whereas apoptosis was unchanged at 90% cell confluency (Fig. 4F; P > 0.05). Consistently, FACS analysis of cell cycle distribution revealed that more cells were arrested in G1 phase and S phase (Fig. 4G and H; *, P < 0.05) in BGC823-L2A− as compared with BGC823-NC, and RND3 shRNA could partly rescue BGC823-L2A− from G1 arrest (Fig. 4G and H; *, P < 0.05). In vivo experiments also showed that the subcutaneous xenografts of BGC823-L2A− grew markedly slower than xenografts of BGC-NC (Fig. 4I and J; P < 0.05), and this phenomenon could be significantly reversed upon RND3 knockdown (Fig. 4I and J; P < 0.05). Thus, RND3 accumulation is partly responsible for the reduced proliferation caused by impaired CMA.
Figure 4.

RND3 mediates CMA blockade-induced proliferation arrest. The BGC823 cell line was transected with both RND3 or control plasmid (A), and the effect of RND3 overexpression on cell growth was assayed by MTT (B), additionally, the effect of RND3 overexpression on cell apoptosis was assayed by FACS (C). (D) Four stable cell lines (BGC823-NC, BGC823-L2A−, BGC823-L2A-/NC and BGC823-L2A− RND3− were established by infecting BGC823 with lentivirus expressing LAMP2A shRNA (L2A−), RND3 shRNA (RND3−), and control shRNA (NC), and the silencing efficacy was evaluated by WB. (E) Growth of the 4 cell lines was examined by MTT assay. (F) Apoptosis of the 4 cell lines was assayed by FACS. ((G)and H) Cell cycle distribution of the 4 cell lines was assayed by FACS. ((I)and J) Subcutaneous growth of the 4 cell lines in nude mice was evaluated by measuring the final volume at Day 10. The values are means ± SEM of 3 independent experiments (B, C, E, F, H, (J)*, P < 0.05; t test).
Next, we investigated whether RND3 was a substrate of CMA. By examining the amino acid sequence of RND3, we found the pentapeptide “QRIEL” (Fig. 5A), which was a putative selection signal for CMA degradation. WB showed that RND3 was increased by the lysosome inhibitor chloroquine (CQ), bafilomycin A1 (Baf) and by the proteasome inhibitor MG132 (Fig. 5B, C). And as observed previously, RND3 was increased upon CMA blockade by LAMP2A shRNA (Fig. 5D). Besides, RND3 could be coimmunoprecipitated by both HSPA8 and LAMP2A, and vice versa (Fig. 5E). To test whether the “QRIEL” motif is essential for HSPA8 binding, the RND3 amino acid “Q” was mutated to “A.” CoIP assay showed that the RND3 mutant (Q54A), but not wild-type RND3, lost the ability to interact with HSPA8 (Fig. 5F), Furthermore, coimmunofluorescence assay revealed that RND3 markedly colocalized with LAMP2A when lysosome degradation was inhibited by Baf (Fig. 5G). Consistently, WB showed that RND3 levels in both whole cell lysate and purified lysosomes were markedly increased by Baf treatment (Fig. 5H). Furthermore, lysosome binding and uptake assay demonstrated that both RND3 and GAPDH (positive control) could be detected in the presence (protein bound to the lysosomal membranes and taken by lysosomes) and absence (protein bound to the lysosomal membranes) of protease inhibitors (PtdIns) (Fig. 5I), and the percentages (lysosomes/input) of both RND3 and GAPDH in the presence of PI were higher than in the absence of PtdIns (Fig. 5J, P < 0.05), demonstrating that they can be engulfed by lysosomes. Competition assays showed that binding and uptake of RND3 by lysosomes was significantly suppressed by the purified CMA substrate GAPDH (Fig. 5K, L). All these data point to that RND3 is a CMA substrate, and its accumulation caused by impaired CMA impedes proliferation in GC cells.
Figure 5.
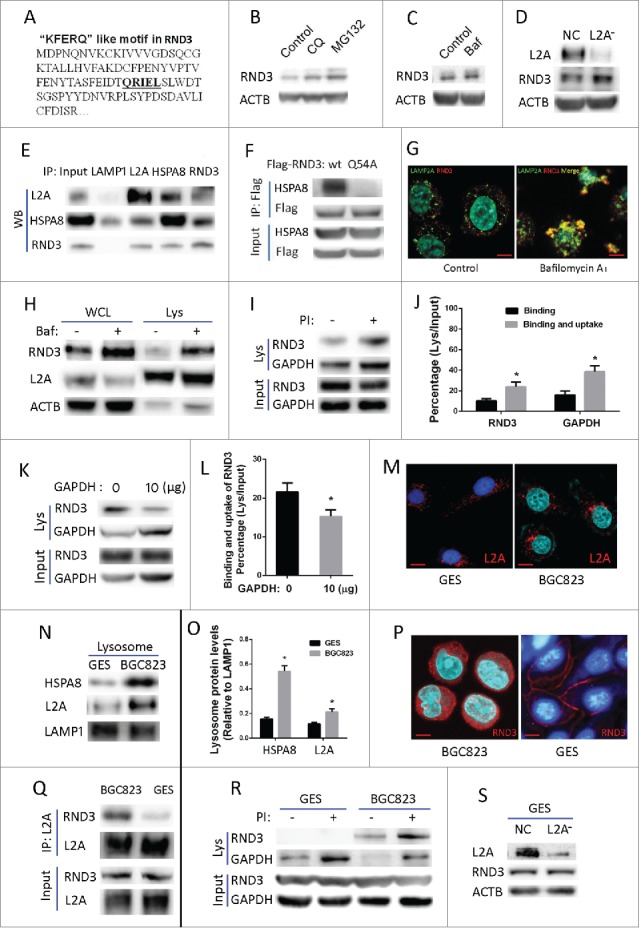
Identification of RND3 as a novel CMA substrate. (A) One “KFERQ”-like motif (QRIEL) was found in the amino sequence of RND3. ((B)and C) RND3 expression was examined in BGC823 cells after exposure to the lysosome inhibitor chloroquine (CQ, 50 µM), Baf, (10 nM), and the proteasome inhibitor MG132 (20 µM) for 12 h. ACTB was used as an internal control. (D) RND3 expression was examined by WB in BGC823-NC and BGC823-L2A−, ACTB was used as an internal control. (E) Interaction among LAMP2A, HSPA8 and RND3 was validated by reciprocal IP assay, LAMP1 was used as a negative control. (F) Interaction between HSPA8 and wild-type RND3 or the Q54A mutant was assayed by CoIP. (G) Coimmunofluorescence was performed to observe the colocalization of LAMP2A and RND3 in BGC823 cell upon inhibition of lysosome degradation by Baf, (10 nM, 12 h). Scale bar (in red) was 10 µm. (H) RND3 levels in whole cell lysates (WCL) and purified lysosomes (Lys) of BGC823 cells were assayed by WB following treatment with Baf (10 nM, 12 h); LAMP2A (lysosome marker) and ACTB (cytosol marker) were examined to verify the enrichment of lysosomes. ((I)and J) Binding and uptake assay was performed for RND3, with a bar diagram that shows percentage of RND3 with regard to the assay (* P < 0.05). GAPDH was used as a positive control. PI indicates protease inhibitors. ((K)and L) Competition assay with bona fide CMA substrate GAPDH for binding and uptake of RND3, bar diagram shows percentage of RND3 with regard to the assay (* P < 0.05). (M) Location of LAMP2A-positive lysosomes relative to nuclei in GES and BGC823 were assayed by IF, Red puncta indicate LAMP2A-positive lysosomes, Blue signal indicates cell nucleus. Scale bar (in red) was 10 µm. ((N)and O) HSPA8 and LAMP2A levels in isolated lysosomes of GES and BGC823 were examined by WB, bar diagram indicates densitometric analyses of the indicated proteins from 3 independent experiments. LAMP1 was used as an internal control for lysosomes (*, P < 0.05). (P) Location of RND3 in GES and BGC823 was assayed by IF; red signal indicates RND3, blue signal indicates cell nucleus. Scale bar (in red): 10 µm. (Q) Interaction between LAMP2A and RND3 in GES and BGC823 was assayed by CoIP. (R) Lysosomal binding and uptake assay was performed for endogenous RND3 in GES and BGC823, GAPDH was used as a positive control and PtdIns indicates protease inhibitors. (S) RND3 level in GES was examined by WB following LAMP2A knockdown. ACTB was used as an internal control.
Finally, we wondered whether CMA activity is higher in GC cells than in normal cells and whether CMA's regulation of RND3 for proliferation is specific only to cancer cells. Immunofluorescence (IF) assay showed that the perinuclear relocalization of LAMP2A-positive lysosomes, which has been considered as a marker of CMA activation,28 was more evident in BGC823 as compared with normal gastric cell line GES (Fig. 5M); besides, WB assay revealed that the HSPA8 and LAMP2A levels in purified lysosomes were both significantly increased in BGC823 as compared with GES (Fig. 5N, O), suggesting that CMA activity is higher in cancer cell as lysosomal HSPA8 (Lys-HSPA8) and lysosomal LAMP2A (lys-LAMP2A) are 2 essential components required for CMA activation.26,29 These data indicate that CMA activity in GC cells is elevated as compared with normal cells. Additionally, IF assay showed that RND3 was primarily located in the cell membrane in GES cells; whereas in BGC823, it was mainly located in the cytosol (Fig. 5P). It seems that in GES, RND3 would not be readily accessible to CMA degradation because it is not in the cytosol. As expected, CoIP assays demonstrated that the interaction between LAMP2A and RND3 in GES was markedly decreased as compared with BGC823 (Fig. 5Q). Besides, lysosomal binding and uptake of RND3 was not observed in GES, but was evident in BGC823 (Fig. 5R). In line with those findings, RND3 level was not changed upon LAMP2A knockdown in GES (Fig. 5S). These data suggest that CMA's regulation of RND3 is absent or very weak in normal gastric cells, whereas it is predominant in cancer cells.
Prognostic and early warning values of LAMP2A in gastric cancer
As demonstrated above, LAMP2A expression was elevated in 8 kinds of malignancies, and its higher expression is important to maintain rapid proliferation and survival in gastric cancer. But its clinical value is poorly understood. Here we evaluated its prognostic values in 173 patients with gastric adenocarcinoma (GA) by Log-rank test, and no significant correlation was noticed between LAMP2A expression and the median survival time (MST) (Fig. 6A; P < 0.05), after stratification by gender, we found that in male patients, LAMP2A did not correlate with MST either (Fig. 6A; P < 0.05), whereas in female patients, the MST of LAMP2A-negative patients was more than 60 mo, while the MST of LAMP2A-positive patients was only 23 mo (Fig. 6A; P < 0.05). Additionally, multivariate analysis revealed that LAMP2A was an independent prognostic indicator for female GA patients, regardless of age, pathological grade, TNM, and clinical stage (Table 1; HR = 2.711, P < 0.05). And this is not likely to be a by-chance discovery, because we have found a consistent result in a cohort of esophageal cancer patients (data not shown). Next, we investigated its early warning ability for gastric cancer. LAMP2A expression in 593 cases of gastric noncancerous lesions was examined by IHC, and the representative IHC images of precancerous tissues and corresponding GC tissues were presented here (Fig. 6B). The correlation between LAMP2A and GC development was analyzed by unconditional logistic regression. For patients with chronic atrophic gastritis, 2 out of 182 patients developed GC in the LAMP2A-negative group and 3 out of 154 patients developed GC in the LAMP2A -positive group, and there was no significant difference in GC risk (Fig. 6C; P > 0.05); For patients with intestinal metaplasia (IM) and dysplasia (DYS), 5 out of 151 patients developed GC in the LAMP2A-negative group and24 out of 106 patients developed GC in the LAMP2A-positive group. The GC risk of LAMP2A-positive patients is 5.52 times higher than that of the LAMP2A-negative group (Fig. 6C; P < 0.05), suggesting that LAMP2A is a promising classifier for IM/DYS patients with high risk of GC development.
Figure 6.
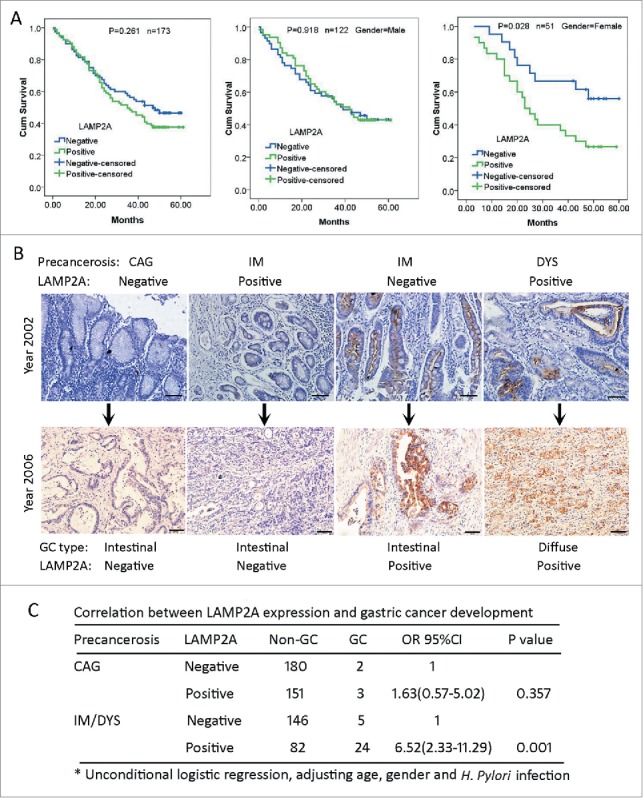
LAMP2A expression in GC and noncancerous tissues with regard to prognosis and GC development. (A) Kaplan-Meier curves for postoperative survival of all the GC patients (left), of male GC patients (middle) and of female GC patients (right), according to LAMP2A positivity. The medium survival times of LAMP2A-negative and positive patients were compared by log-rank test, P < 0.05 was considered to be statistically significant. (B) Immunostaining of LAMP2A in gastric precancerous tissues and the corresponding GC tissues. Scale bar (in black): 200 µm. (X 200). (C) Correlation between LAMP2A positivity and GC development risk of gastric precancerosis was analyzed by unconditional logistic regression, P < 0.05 was considered to be statistically significant.
Table 1.
Multivariate analysis for female gastric cancer patients.
| HR (95% CI) | P | B | SE | Wald | |
|---|---|---|---|---|---|
| Distant metastasis | 6.329 (3.451–8.254) | 0.023 | 1.845 | .814 | 5.138 |
| Clinical stage | 2.321 (1.902–3.215) | 0.028 | .842 | .383 | 4.825 |
| LAMP2A | 2.711 (2.012–4.064) | 0.015 | .997 | .409 | 5.958 |
Note: Cox regression test done by SPSS17.0; age, gender, node metastasis, invasion depth, pathological grade, clinical stage and LAMP2A expression were examined in this analysis. P values less than 0.05 were considered to be statistically significant. HR, hazard ratio; CI, confidence interval; P, P value; B, β value; SE, standard error; Wald, Wald chi-square test.
Discussion
CMA has been linked with cancer progression in recent years, but the mechanisms are not thoroughly explored. In this study, we mainly investigated the mechanisms underlying the regulatory function of CMA in proliferation of gastric cancer. A putative CMA substrate, RND3 was selected by an interactomic approach combined with KEGG analysis. Further study revealed that RND3 is a novel substrate of CMA, and its accumulation could partly account for the impaired proliferation caused by CMA blockade.
One of the unique features of CMA is that substrates that undergo degradation are specifically selected through a pentapeptide motif.6 This specific selectivity permits CMA to play regulatory roles in various cellular processes by modulating levels of enzymes, transcription factors and cell maintenance proteins.29 CMA has been associated with the proliferation and survival of cancer cells.13-15 CMA activity in several cancer cell lines and lung tumor tissues is upregulated,15 mostly due to an elevation in LAMP2A level. In this study, we examined the expression of LAMP2A in ten kinds of tumors, and found that LAMP2A was significantly upregulated in 8 kinds of human tumors, including lung cancer, breast cancer and gastric cancer, whereas, no significant increase was noticed in hepatic cell carcinoma and renal clear cell carcinoma, as its expression in normal liver cell and renal tubular cell was also very high. This is consistent with a previous finding,27 supporting the notion that CMA is required for hepatic metabolism24 and for proteostasis in renal tubular cells.27 Nevertheless, our results strongly suggest that high CMA activity is a hallmark of malignancies. The mechanism behind CMA overactivity in tumor is still unclear, and it is tempting to speculate that deregulation of certain miRNAs may account for LAMP2A overexpression as seen in neurodegeneration.30
Genetic knockdown of LAMP2A in lung cancer cells revealed that CMA is required for lung cancer cell proliferation.15 The elevated CMA has been proven to be necessary to sustain enhanced glycolysis to meet the bioenergetic demand of rapid proliferation, as blockage of CMA by silencing LAMP2A leads to wild-type TP53-mediated transcriptional suppression of 2 glycolytic enzymes, GAPDH and PGK, and the subsequent reduction in glycolysis and ATP production.15 But this mechanism may not fully apply to gastric cancer, because IHC results indicate that the correlation between LAMP2A and TP53 is weak, and although GAPDH and PGK are decreased in ATG cells with wild-type TP53 upon LAMP2A silencing, they are actually upregulated in BGC823 cells with mutant TP53. This is normal, because they are well-characterized CMA substrates, and surely will be upregulated by CMA blockade without the transcriptional suppression by wild-type TP53. In this study, we found that RND3 was increased in both AGS and BGC823 cells upon LAMP2A silencing; supporting that it is a CMA substrate that exerts an antiproliferative function independent of TP53 status. Thus our study provides a novel mechanism to explain the impaired proliferation caused by CMA deficiency from the point of its modulation on substrate levels. Besides, we found that high expression of LAMP2A is closely correlated with low expression of RND3 in plenty of GC tissues, suggesting that this novel mechanism may still work in vivo.
RND3 belongs to the RND subfamily of the RHO family. Unlike other RHO proteins, it can only associate with GTP, and become constitutively activated.31,32 Several studies have proven that it is an antiproliferation protein. Tang et al. have reported that RND3 is downregulated in lung cancer cell lines, the reintroduction of RND3 can block the proliferation of cancer cells; Mechanistically, NOTCH intracellular domain (NICD) protein abundance is regulated by RND3-mediated NICD proteasome degradation, indicating that Rnd3 regulates cell proliferation through a NOTCH1 NICD-HES1 signaling axis.33 Zhu et al. have demonstrated that wild-type TP53 can strongly increase RND3 expression, and enhanced RND3 expression can markedly inhibit proliferation, suggesting that RND3 is a TP53-regulated tumor suppressor.34 Additionally, RND3 downregulation in ESCC cells promotes cell proliferation, as well as cell cycle progression, and RND3 upregulation inhibits cell proliferation and arrests the cell cycle at the G0/G1 phase. Also, RND3 overexpression increases PTEN and CDKN1B/p27, and decreases pAKT, as well as CCND1 (cyclin D1).35 Villalonga et al. have reported that RND3 prevents the release of EIF4E from EIF4EBP1/4E-BP1, inhibiting cap-dependent translation, accordingly, RND3 also inhibits the expression and the transcriptional activity of the EIF4E target MYC/c-Myc, and EIF4E can rescue cell cycle progression in RND3-expressing cells, indicating that the inhibition of EIF4E function is critical to mediate the antiproliferative effects of RND3.36 Poch et al. have proven that RND3 inhibits ERK activation, thereby decreasing CCND1 expression and leading to a reduction in RB1/Retinoblastoma 1 inactivation, and this mechanism is involved in the RND3-induced growth inhibition in glioblastoma cells.37 RND3 induction inhibits fibroblast cell proliferation and serum-induced S-phase entry; additionally, human papillomavirus E7, adenovirus E1A, and CCNE/cyclin E, can rescue cell cycle progression in RND3-expressing cells, indicating that RND3 can inhibit cell cycle progression upstream of the phosphorylated-RB1checkpoint.38 Thus, the mechanisms underlying the antiproliferation function of RND3 are context-dependent. Furthermore, here we have found that RND3 upregulation caused by CMA knockdown can also inhibit the proliferation of GC cells, but whether RND3 exerts its suppressive action on proliferation through the above-mentioned mechanisms is worthy of further investigation.
RND3 always remains bound to GTP in a constitutively active state, suggesting that its function is regulated by other mechanisms such as gene expression, protein stability and protein interactions. Lonjedo et al. have proposed a mechanism according to which proteasomal degradation of RND3 by SKP2 regulates its protein levels to control cellular proliferation.39 In agreement with those findings, here we have found that RND3 level is increased by proteasome inhibitor MG132, but we have also indentified CMA as alternative pathway for RND3 degradation. In fact, several proteins are degraded by both the proteasome and CMA pathways, including NFKBIA/IκBα, FOS/c-fos, ALDO/aldolase, SNCA/α-synuclein and HIF1A/HIF1-a,40-43 suggesting that UPS and CMA can coordinate with each other to realize a robust modulation of key substrates. One important concern is whether CMA's regulation of RND3 for proliferation is specific only to cancer cells or if this also occurs in normal cells. Our study has proven that, compared with normal gastric cell line GES, CMA activity in the GC cell line BGC823 is boosted; CMA's regulation of RND3 was undetectable in GES, but is evident in BGC823. As phosphorylation of RND3 translocates RND3 from membrane to cytosol,44 and CMA can only target cytosolic proteins,6 it is tempting to speculate that phosphorylation may promote its interaction with CMA components and the subsequent degradation. Our data suggest that CMA's regulation of RND3 is absent or very weak in normal gastric cells. And we propose that 2 factors may prevent RND3 degradation in normal cells: plasma membrane localization of RND3 and low basal CMA activity. In contrast, the CMA-RND3-proliferation pathway may be predominant and specific in gastric cancer cells.
Last, we have evaluated the clinical relevance of LAMP2A in plenty of precancerous tissues and GC tissues. All the GC patients were treated postoperatively with standard adjuvant chemotherapy. And we have proven that LAMP2A is a prognostic marker for female GC patients that could be used to predict the survival time of female patients. We do not know exactly why it does not associate with prognosis in male patients, but this is not likely to be a by-chance discovery, because we have found a consistent result in a cohort of esophageal cancer patients (data not shown). Based on the fact that LAMP2A is required to resist chemotherapy,13 and that estrogen can promote drug resistance,45 we hypothesize that in female patients estrogen may coordinate with LAMP2A to aggravate chemotherapy resistance and lead to worse prognosis. It has been convincingly shown by Cuervo's group that CMA has important implications for aging.26 But we didn't find any influence of patient age on LAMP2A's prognostic effect. The reason may be that, the ages of GC patients are all relatively old (mostly older than 50), which may attenuate the influence of aging on prognosis due to a lack of enough young patients. Besides, although our study suggests a prooncogenic function for CMA, the effect of CMA in normal cells seems to be quite the opposite, as it protects cells from the damage caused by extracellular and intracellular injuries, which, if allowed to accumulate could facilitate oncogenesis,26 could partly account for the increased cancer risk associated with aging. Although we have found that LAMP2A was an unfavorable factor for GC patient survival, we cannot conclusively ascertain CMA's significance in patient prognosis, because LAMP2A can also be important for other functions beyond CMA. For example, recently it has been shown that LAMP2A levels regulate surface exposure of calreticulin, which is a crucial danger signal for immunogenic cell death, but the link between CMA and calreticulin surface exposure has not been validated.46 Furthermore, for patients with gastric precancerous lesions (IM/DYS), GC risk of LAMP2A+ patients is 5.52 times higher than that of the LAMP2A− patients, suggesting that LAMP2A is a promising early warning marker for gastric precancerous lesions that can predict the risk of GC occurrence. And we propose that IM and DYS with positive LAMP2A expression should be excised to prevent GC development.
In summary, we have screened and identified a novel CMA substrate that plays an important role in regulating GC cell proliferation. Constant turnover of RND3 via CMA is crucial to sustaining rapid proliferation in GC, highlighting the important role of CMA in controlling cancer proliferation. Our study also suggests that, as a key component of CMA, LAMP2A is not only a promising therapeutic target but also a useful early warning marker for GC development in gastric precancerous lesions and an independent prognostic indicator for female GC patients.
Materials and methods
Cells, animals, tissue micromicroarrays and specimens
Gastric cancer cell lines BGC823, AGS, MKN45, SGC7901 and the immortalized normal gastric epithelial cell line GES were used. Nu/Nu athymic male mice were used. The following tissue micromicroarrays were used: Horg-C120PG-01 (10 kinds of tumors, n = 120), HLug-Squ150Sur-01 (LSCC, n = 67), HLug-Ade150Sur-01 (LA, n = 61), HCol-Ade180Sur-04 (CA, n = 86), HBre-Duc170Sur-01 (BDC, n = 107), HEso-Squ172Sur-02 (ESCC, n = 90), HStm-Ade-Sur-01 (GA, n = 173), HPan-Ade180Sur-01 (PDC, n = 81) and HRec-Ade180Sur-01 (RA, n = 82). All the TMAs were purchased from Shanghai Outdo Biotech Company. Tissues from 593 cases of gastric precancerosis were obtained from our hospital and this study was approved by the Ethics Committee of the Fourth Military Medical University.
Primary antibodies, chemicals and proteins
MG5 (a specific antibody against LAMP2A) was developed by hybridoma techniques and was preserved in our laboratory, Monoclonal antibody sequencing demonstrated that the isotype of MG5 hybridoma was IgG1 heavy chain and κ light chain (Report from: GenScript, Piscataway, NJ, USA). Additionally, the following antibodies were used: anti-LAMP2 (Santa Cruz Biotechnology, sc-18822), anti-E2F3 (Santa Cruz Biotechnology, sc-878), anti-PCNA (Santa Cruz Biotechnology, sc-7909), anti-CDKN1B (Santa Cruz Biotechnology, sc-528), anti-TP53 (Santa Cruz Biotechnology, sc-126), anti-HSPA8 (Santa Cruz Biotechnology, sc-33575), anti-GAPDH (Santa Cruz Biotechnology, sc-365062), anti-CASP3 (Santa Cruz Biotechnology, sc-7272), anti-cleaved CASP3 (Santa Cruz Biotechnology, sc-22171), anti-BAX (Santa Cruz Biotechnology, sc-20067), anti-BCL2 (Santa Cruz Biotechnology, sc-492), anti-LAMP2 (Abcam, ab-37024), anti-ACTB (Sigma Aldrich, A1978), and anti-RND3 (Millipore, 05-723). The following chemicals were used: Chloroquine (Sigma Aldrich, C6628), MG132 (Sigma Aldrich, M7449), Bafilomycin A1/Baf (Gene Operation, IIC2501), 6-aminonicotinamide (Sigma Aldrich, A68203). Recombinant protein of human GAPDH (OriGene, TP302309).
Transfection and infection
RND3 plasmid and control plasmid were transfected into cells as described previously.47 LAMP2A, LAMP2B and LAMP2C plasmids (OriGene, SC118738, SC127527 and SC318864) and their control plasmids (OriGene, PCMV6XL5 and PCMV6XL6) were used. The lentiviral vectors with shRNAs against LAMP2A were purchased from GeneChem Company, Shanghai, China (PIEL248064052). Cells were infected with lentivirus according to the manufacturer's manual.
Cell growth and apoptosis
Rates of cellular proliferation and viability were determined by MTT assay as described previously.48 100 cells were cultured in 6-well plates for 12 d to assess the colony formation ability of tumor cells. Xenografts of gastric cancer cells in nude mice were generated by subcutaneous injection of 1 × 106 cells into their left flank 10 d later, and tumor volumes were measured. Cell apoptosis was examined by FACS analysis after labeling with 7-ADD and ANXA5/annexin V-PE (Beyotime, C1065).
Quantitative real-time PCR
RT-PCR was performed as described previously.49 Briefly, total RNA was isolated by using TRIzol, and was purified using a TURBO DNA-free™ kit (Thermo Fisher Scientific, AM1907) to remove potential genomic DNA. Reverse transcription was performed using the RETROscript® Reverse Transcription Kit (Thermo Fisher Scientific, AM1710). Quantitative PCR was performed using a Stratagene Mx 3000P System (Stratagene,La Jolla, USA) with the SuperScript® III Platinum® SYBR® Green One-Step qRT-PCR Kit (Thermo Fisher Scientific, 11736-059). Primer sequences were as follows:
LAMP2A forward: 5′-GTCTCAAGCGCCATCATACT-3′;
LAMP2A reverse: 5′-TCCAAGGAGTCTGTCTTAAGTAGC-3′.
ACTB forward: 5′- ATTGGCAATGAGCGGTTC-3′;
ACTBreverse: 5′- GGATGCCACAGGACTCCAT-3′.
RND3 mutagenesis
pCMV6-Entry-RND3 (OriGene, RC233531) was used. RND3 was cloned into the pET28 vector for bacterial expression. Flag-tagged versions of RND3 were generated using the pCMV6-Entry vector (OriGene, PS100001). Site-directed mutagenesis on RND3 was performed using Stratagene QuikChange-II kit.
Immunoprecipitation
IP was done with the Pierce Classic IP Kit (Pierce Biotechnology, 26146): Briefly, Cells were lysed with IP Lysis/Wash Buffer on ice for 5 min, lysate was centrifugated at 13000 × g for 10 min, and then the supernatant fraction was precleared using Control Agarose Resin for 1 h, adding 10 µg primary antibodies and 40 µl Protein A/G Agarose Resin to 600 µl precleared lysate, and incubated overnight at 4°C. Normal IgG (10 µg) was used as a control antibody. The resin was washed twice with 200 µl Lysis/Wash Buffer and once with 100 µl 1x conditioning buffer. A volume of 50 µl 2x sample buffer with 20 mM DDT was added and incubated at 100°C for 10 min. Then it was centrifuged at 1000 × g for 3 min to collect the eluate. For CoIP, the IP eluate was examined by western blot with another primary antibody against the protein of interest.
Mass spectrum analysis
After Coomassie Blue staining, the bands of interest were manually excised. Then the slices were destained by washing twice with 25 mM NH4HCO3/30% acetonitrile. After shrinking with 100% acetonitrile and drying, reduction was performed with 20 mM DTT at 56°C for 1 h, followed by alkylation with 50 mM iodoacetamide at room temperature in the dark for 20 min. After shrinking with 100% acetonitrile, the gel pieces were dried in a SpeedVac Vacuum. Protein digestions were performed with modified sequencing-grade trypsin at 37°C overnight. Peptides were extracted with 60% acetonitrile/0.1% trifluoroacetic acid (TFA) twice in a sonication bath. The combined supernatant fractions were dried in the SpeedVac Vacuum apparatus (Thermo Fisher Scientific, UVS400A-230, Waltham, MA, USA.), desalted with Ultramicrospin and MicroSpin columns (Thermo Fisher Scientific, 29924) and redissolved with 2% acetonitrile/98% H2O/0.1% TFA for mass spectrometry analysis. For matrix-assisted laser desorption and ionization time-of-flight a MALDI-TOF-TOF 5800 instrument was used (AB Sciex, Framingham, MA, USA) analysis, peptide samples were loaded onto a C18 column (UF-Column-52/2150, 200A, 0.2*150 mm, ULTRA-FAST, Zhejiang, China), mixed with a matrix solution (4-hydroxy–cyanocinnamic acid in 70% acetonitrile/0.1% TFA) and spotted onto MALDI-TOF-TOF plates. The droplet was dried at room temperature. Once the liquid was completely evaporated, the sample was loaded into the mass spectrometer and analyzed. Data analysis was performed using Protein Pilot software (AB Sciex, USA). The following searching parameters were used: trypsin specificity for protein cleavage; minimum number of matched peptides 1; detected Protein Threshold [Unused ProtScore (Conf)]: >0 .05 (10.0 %). The spectrums were searched against the database uniprot_sprot_can+iso_20100622+Contams.
Lysosome isolation
Lysosomes were isolated as described previously.50 Briefly, all steps were performed at 4°C. Cells were washed with phosphate-buffered saline (PBS; Beyotime Co., C0221A), and suspended in homogenization buffer (0.25 M sucrose [Sigma, S9378], 2 mM EDTA, 10 mM HEPES, pH 7.4), and were homogenized with a glass homogenizer. The homogenate was centrifuged at 800 × g for 15 min to remove cells debris. The supernatant fraction was centrifuged at 6800 × g for 15 min to remove the large organelles. The supernatant fraction was then centrifuged at 25000 × g for 10 min to obtain the light organelles. The precipitate was layered over 10 ml of a 27% Percoll solution (Pharmacia Inc., 17-0891-01) in homogenization buffer, under-layered with 0.5 ml of a 2.5 M sucrose solution. Centrifugation was done in a rotor for 2 h at 35000 × g. The layer of crude lysosomes of about 1.2 ml was collected at the bottom, and then was centrifuged at 100000 × g for 1 h to remove the other light organelles. The lysosomal solution was washed with PBS and was centrifuged at 18000 × g for 30 min to remove the sucrose at the bottom of the tube. Following isolation, the purity of lysosomes was verified by detecting LAMP2A and ACTB levels. Lysosomal integrity was verified by measuring the activity of HEX/β-hexosaminidase. Preparations with more than 10% of broken lysosomes after isolation or more than 20% at the end of the incubation were discarded.
Lysosome binding and uptake assay
Lysosome binding and uptake assay were performed as described previously.50 Briefly, GBC823 cells were transfected with RND3 plasmid for 48 h, and then the cell lysate was incubated with freshly isolated lysosomes in MOPS buffer at 37°C for 5 and 10 min. Where indicated, lysosomes were pretreated with a cocktail of protease inhibitors for 15 min at 0°C. Lysosomes were collected by centrifugation at 35000 × g for 2 h, washed with PBS buffer, and subjected to SDS-PAGE and western blot for RND3 and GAPDH. Uptake was evaluated by subtracting the level of RND3 associated with lysosomes in the presence (protein bound to the lysosomal membranes and taken by lysosomes) and absence (protein bound to the lysosomal membranes) of protease inhibitors. Additionally, lysosome binding and uptake assays of endogenous RND3 in GES and BGC823 cells were also performed where indicated.
General methods
Immunohistochemistry was performed as described previously,47 except that different primary antibodies were used. For evaluation of cell staining, sections were examined by 2 independent observers without prior knowledge of the clinical or clinicopathological status of the specimens. The expression was stratified and scored as negative (−) or positive (+) according to the intensity of stained cancer cells by optical evaluation. Confocal immunofluorescence was performed as described previously,47 except that different primary antibodies were used. Western blots were performed as described previously,47 except that different primary antibodies and dilutions were used, the densitometric analyses of the indicated proteins with respect to ACTB from 3 independent experiments were performed.
Statistical analysis
Statistical analyses were performed using IBM SPSS 17.0 software. Measurement data were analyzed using the Student t test, while categorical data were analyzed by χ2 test. Log-rank test and Cox proportional hazards regression model were performed for survival analysis. Statistical significance was set at P < 0.05.
Funding Statement
This work was supported by the National Natural Science Foundation of China (81430072, 81120108005, 81372609, 81421003, 81272652, 81225003, 81227901), Project from Ministry of Science and Technology of China 2015BAI13B07, 2012AA02A504, 2012AA02A203-A03, 2012ZX09303011.
Abbreviations
- BDC
breast ductal carcinoma;
- CMA
chaperone-mediated autophagy;
- CA
colon adenocarcinoma;
- CoIP
coimmunoprecipitation;
- CQ
chloroquine;
- DYS
dysplasia;
- GA
gastric adenocarcinoma;
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase;
- GC
gastric cancer;
- HCC
hepatic cell carcinoma;
- HSPA8
heat shock protein family A (Hsp70) member 8;
- IF
immunofluorescence;
- IHC
immunohistochemistry;
- IM
intestinal metaplasia;
- IP
immunoprecipitation;
- LA
lung adenocarcinoma;
- LAMP2A
lysosomal-associated membrane protein 2A;
- LSCC
lung squamous cell carcinoma;
- MST
median survival time;
- MTT
4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide;
- NICD
NOTCH intracellular domain;
- PBS
phosphate-buffered saline;
- PDC
pancreatic ductal carcinoma;
- PI
protease inhibitors;
- RA
rectal adenocarcinoma;
- RND3
Rho Family GTPase 3;
- SEM
standard error of the mean;
- TFA
trifluoroacetic acid;
- TMA
tissue microarray;
- WB
western blot.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We gratefully acknowledge Prof. Qian Yang, Prof. Xiaoyong Yang, Dr. Xiqiang Cai, Zheng Chen, Guangbo Tang, Jianhua Dou, Miaomiao Tian, Zuhong Tian, Xiaofang, Yi, Rui Wang, Gang Liu and the other members of our laboratory for valuable suggestions and comments.
References
- [1].Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol 2010; 12:814-22; PMID:20811353; http://dx.doi.org/ 10.1038/ncb0910-814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nat 2008; 451:1069-75; PMID:18305538; http://dx.doi.org/ 10.1038/nature06639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Mizushima N. The pleiotropic role of autophagy: from protein metabolism to bactericide. Cell Death Differentiation 2005; 12 Suppl 2:1535-41; http://dx.doi.org/ 10.1038/sj.cdd.4401728 [DOI] [PubMed] [Google Scholar]
- [4].Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol 2010; 22:124-31; PMID:20034776; http://dx.doi.org/ 10.1016/j.ceb.2009.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Cuervo AM. Chaperone-mediated autophagy: selectivity pays off. Trends Endocrinol Metab: TEM 2010; 21:142-50; PMID:19857975; http://dx.doi.org/ 10.1016/j.tem.2009.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Dice JF. Peptide sequences that target cytosolic proteins for lysosomal proteolysis. Trends Biochem Sci 1990; 15:305-9; PMID:2204156; http://dx.doi.org/ 10.1016/0968-0004(90)90019-8 [DOI] [PubMed] [Google Scholar]
- [7].Chiang HL, Terlecky SR, Plant CP, Dice JF. A role for a 70-kgdalton heat shock protein in lysosomal degradation of intracellular proteins. Science 1989; 246:382-5; PMID:2799391; http://dx.doi.org/ 10.1126/science.2799391 [DOI] [PubMed] [Google Scholar]
- [8].Cuervo AM, Dice JF. A receptor for the selective uptake and degradation of proteins by lysosomes. Science 1996; 273:501-3; PMID:8662539; http://dx.doi.org/ 10.1126/science.273.5274.501 [DOI] [PubMed] [Google Scholar]
- [9].Salvador N, Aguado C, Horst M, Knecht E. Import of a cytosolic protein into lysosomes by chaperone-mediated autophagy depends on its folding state. J Biol Chem 2000; 275:27447-56; PMID:10862611 [DOI] [PubMed] [Google Scholar]
- [10].Bandyopadhyay U, Sridhar S, Kaushik S, Kiffin R, Cuervo AM. Identification of regulators of chaperone-mediated autophagy. Mol Cell 2010; 39:535-47; PMID:20797626; http://dx.doi.org/ 10.1016/j.molcel.2010.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Bandyopadhyay U, Kaushik S, Varticovski L, Cuervo AM. The chaperone-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol Cell Biol 2008; 28:5747-63; PMID:18644871; http://dx.doi.org/ 10.1128/MCB.02070-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Agarraberes FA, Dice JF. A molecular chaperone complex at the lysosomal membrane is required for protein translocation. J Cell Sci 2001; 114:2491-9; PMID:11559757 [DOI] [PubMed] [Google Scholar]
- [13].Saha T. LAMP2A overexpression in breast tumors promotes cancer cell survival via chaperone-mediated autophagy. Autophagy 2012; 8:1643-56; PMID:22874552; http://dx.doi.org/ 10.4161/auto.21654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Lv L, Li D, Zhao D, Lin R, Chu Y, Zhang H, Zha Z, Liu Y, Li Z, Xu Y, et al.. Acetylation targets the M2 isoform of pyruvate kinase for degradation through chaperone-mediated autophagy and promotes tumor growth. Mol Cell 2011; 42:719-30; PMID:21700219; http://dx.doi.org/ 10.1016/j.molcel.2011.04.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kon M, Kiffin R, Koga H, Chapochnick J, Macian F, Varticovski L, Cuervo AM. Chaperone-mediated autophagy is required for tumor growth. Sci Transl Med 2011; 3:109ra17; http://dx.doi.org/ 10.1126/scitranslmed.3003182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Park C, Suh Y, Cuervo AM. Regulated degradation of Chk1 by chaperone-mediated autophagy in response to DNA damage. Nat Commun 2015; 6:6823; PMID:25880015; http://dx.doi.org/ 10.1038/ncomms7823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Valdor R, Mocholi E, Botbol Y, Guerrero-Ros I, Chandra D, Koga H, Gravekamp C, Cuervo AM, Macian F. Chaperone-mediated autophagy regulates T cell responses through targeted degradation of negative regulators of T cell activation. Nat Immunol 2014; 15:1046-54; PMID:25263126; http://dx.doi.org/ 10.1038/ni.3003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Venugopal B, Mesires NT, Kennedy JC, Curcio-Morelli C, Laplante JM, Dice JF, Slaugenhaupt SA. Chaperone-mediated autophagy is defective in mucolipidosis type IV. J Cell Physiol 2009; 219:344-53; PMID:19117012; http://dx.doi.org/ 10.1002/jcp.21676 [DOI] [PubMed] [Google Scholar]
- [19].Di Blasi C, Jarre L, Blasevich F, Dassi P, Mora M. Danon disease: a novel LAMP2 mutation affecting the pre-mRNA splicing and causing aberrant transcripts and partial protein expression. Neuromuscular Disorders 2008; 18:962-6; PMID:18990578; http://dx.doi.org/ 10.1016/j.nmd.2008.09.008 [DOI] [PubMed] [Google Scholar]
- [20].Nixon RA. Autophagy in neurodegenerative disease: friend, foe or turncoat? Trends Neurosci 2006; 29:528-35; PMID:16859759; http://dx.doi.org/ 10.1016/j.tins.2006.07.003 [DOI] [PubMed] [Google Scholar]
- [21].Massey AC, Zhang C, Cuervo AM. Chaperone-mediated autophagy in aging and disease. Curr Topics Dev Biol 2006; 73:205-35; PMID:16782460; http://dx.doi.org/ 10.1016/S0070-2153(05)73007-6 [DOI] [PubMed] [Google Scholar]
- [22].Saftig P, Tanaka Y, Lullmann-Rauch R, von Figura K. Disease model: LAMP-2 enlightens Danon disease. Trends Mol Med 2001; 7:37-9; PMID:11427988; http://dx.doi.org/ 10.1016/S1471-4914(00)01868-2 [DOI] [PubMed] [Google Scholar]
- [23].Napolitano G, Johnson JL, He J, Rocca CJ, Monfregola J, Pestonjamasp K, Cherqui S, Catz SD. Impairment of chaperone-mediated autophagy leads to selective lysosomal degradation defects in the lysosomal storage disease cystinosis. EMBO Mol Med 2015; 7:158-74; PMID:25586965; http://dx.doi.org/ 10.15252/emmm.201404223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Schneider JL, Suh Y, Cuervo AM. Deficient chaperone-mediated autophagy in liver leads to metabolic dysregulation. Cell Metab 2014; 20:417-32; PMID:25043815; http://dx.doi.org/ 10.1016/j.cmet.2014.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Schneider JL, Villarroya J, Diaz-Carretero A, Patel B, Urbanska AM, Thi MM, Villarroya F, Santambrogio L, Cuervo AM. Loss of hepatic chaperone-mediated autophagy accelerates proteostasis failure in aging. Aging Cell 2015; 14:249-64; PMID:25620427; http://dx.doi.org/ 10.1111/acel.12310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Cuervo AM, Wong E. Chaperone-mediated autophagy: roles in disease and aging. Cell Res 2014; 24:92-104; PMID:24281265; http://dx.doi.org/ 10.1038/cr.2013.153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Franch HA. Chaperone-mediated autophagy in the kidney: the road more traveled. Semin Nephrol 2014; 34:72-83; PMID:24485032; http://dx.doi.org/ 10.1016/j.semnephrol.2013.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Patel B, Cuervo AM. Methods to study chaperone-mediated autophagy. Methods 2015; 75:133-40; PMID:25595300; http://dx.doi.org/ 10.1016/j.ymeth.2015.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kaushik S, Cuervo AM. Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends Cell Biol 2012; 22:407-17; PMID:22748206; http://dx.doi.org/ 10.1016/j.tcb.2012.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Alvarez-Erviti L, Seow Y, Schapira AH, Rodriguez-Oroz MC, Obeso JA, Cooper JM. Influence of microRNA deregulation on chaperone-mediated autophagy and α-synuclein pathology in Parkinson disease. Cell Death Dis 2013; 4:e545; PMID:23492776; http://dx.doi.org/ 10.1038/cddis.2013.73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Chardin P. Function and regulation of Rnd proteins. Nat Rev Mol Cell Biol 2006; 7:54-62; PMID:16493413; http://dx.doi.org/ 10.1038/nrm1788 [DOI] [PubMed] [Google Scholar]
- [32].Vega FM, Ridley AJ. Rho GTPases in cancer cell biology. FEBS Lett 2008; 582:2093-101; PMID:18460342; http://dx.doi.org/ 10.1016/j.febslet.2008.04.039 [DOI] [PubMed] [Google Scholar]
- [33].Tang Y, Hu C, Yang H, Cao L, Li Y, Deng P, Huang L. Rnd3 regulates lung cancer cell proliferation through notch signaling. PloS One 2014; 9:e111897; PMID:25372032; http://dx.doi.org/ 10.1371/journal.pone.0111897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Zhu Y, Zhou J, Xia H, Chen X, Qiu M, Huang J, Liu S, Tang Q, Lang N, Liu Z, et al.. The Rho GTPase RhoE is a p53-regulated candidate tumor suppressor in cancer cells. Int J Oncol 2014; 44:896-904; PMID:24399089 [DOI] [PubMed] [Google Scholar]
- [35].Zhao H, Yang J, Fan T, Li S, Ren X. RhoE functions as a tumor suppressor in esophageal squamous cell carcinoma and modulates the PTEN/PI3K/Akt signaling pathway. Tumour Biol 2012; 33:1363-74; http://dx.doi.org/ 10.1007/s13277-012-0384-5 [DOI] [PubMed] [Google Scholar]
- [36].Villalonga P, Fernandez de Mattos S, Ridley AJ. RhoE inhibits 4E-BP1 phosphorylation and eIF4E function impairing cap-dependent translation. J Biol Chem 2009; 284:35287-96; PMID:19850923; http://dx.doi.org/ 10.1074/jbc.M109.050120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Poch E, Minambres R, Mocholi E, Ivorra C, Perez-Arago A, Guerri C, Pérez-Roger I, Guasch RM. RhoE interferes with Rb inactivation and regulates the proliferation and survival of the U87 human glioblastoma cell line. Exp Cell Res 2007; 313:719-31; PMID:17182035; http://dx.doi.org/ 10.1016/j.yexcr.2006.11.006 [DOI] [PubMed] [Google Scholar]
- [38].Villalonga P, Guasch RM, Riento K, Ridley AJ. RhoE inhibits cell cycle progression and Ras-induced transformation. Mol Cell Biol 2004; 24:7829-40; PMID:15340047; http://dx.doi.org/ 10.1128/MCB.24.18.7829-7840.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Lonjedo M, Poch E, Mocholi E, Hernandez-Sanchez M, Ivorra C, Franke TF, Guasch RM, Pérez-Roger I. The Rho family member RhoE interacts with Skp2 and is degraded at the proteasome during cell cycle progression. J Biol Chem 2013; 288:30872-82; PMID:24045951; http://dx.doi.org/ 10.1074/jbc.M113.511105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Hubbi ME, Hu H, Kshitiz, Ahmed I, Levchenko A, Semenza GL. Chaperone-mediated autophagy targets hypoxia-inducible factor-1alpha (HIF-1alpha) for lysosomal degradation. J Biol Chem 2013; 288:10703-14; PMID:23457305; http://dx.doi.org/ 10.1074/jbc.M112.414771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant α-synuclein by chaperone-mediated autophagy. Science 2004; 305:1292-5; PMID:15333840; http://dx.doi.org/ 10.1126/science.1101738 [DOI] [PubMed] [Google Scholar]
- [42].Cuervo AM, Hu W, Lim B, Dice JF. IkappaB is a substrate for a selective pathway of lysosomal proteolysis. Mol Biol Cell 1998; 9:1995-2010; PMID:9693362; http://dx.doi.org/ 10.1091/mbc.9.8.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Aniento F, Papavassiliou AG, Knecht E, Roche E. Selective uptake and degradation of c-Fos and v-Fos by rat liver lysosomes. FEBS Letters 1996; 390:47-52; PMID:8706827; http://dx.doi.org/ 10.1016/0014-5793(96)00625-4 [DOI] [PubMed] [Google Scholar]
- [44].Riento K, Totty N, Villalonga P, Garg R, Guasch R, Ridley AJ. RhoE function is regulated by ROCK I-mediated phosphorylation. EMBO J 2005; 24:1170-80; PMID:15775972; http://dx.doi.org/ 10.1038/sj.emboj.7600612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Teixeira C, Reed JC, Pratt MA. Estrogen promotes chemotherapeutic drug resistance by a mechanism involving Bcl-2 proto-oncogene expression in human breast cancer cells. Cancer Res 1995; 55:3902-7; PMID:7641210 [PubMed] [Google Scholar]
- [46].Garg AD, Dudek AM, Agostinis P. Calreticulin surface exposure is abrogated in cells lacking, chaperone-mediated autophagy-essential gene, LAMP2A. Cell Death Dis 2013; 4:e826; PMID:24091669; http://dx.doi.org/ 10.1038/cddis.2013.372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Zhou J, Li K, Gu Y, Feng B, Ren G, Zhang L, Wang Y, Nie Y, Fan D. Transcriptional up-regulation of RhoE by hypoxia-inducible factor (HIF)-1 promotes epithelial to mesenchymal transition of gastric cancer cells during hypoxia. Biochem Biophys Res Commun 2011; 415:348-54; PMID:22037464; http://dx.doi.org/ 10.1016/j.bbrc.2011.10.065 [DOI] [PubMed] [Google Scholar]
- [48].Zheng X, Dong J, Gong T, Zhang Z, Wang Y, Li Y, Shang Y, Li K, Ren G, Feng B, et al.. MicroRNA library-based functional screening identified miR-137 as a suppresser of gastric cancer cell proliferation. J Cancer Res Clin Oncol 2015; 141:785-95; PMID:25342326; http://dx.doi.org/ 10.1007/s00432-014-1847-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Gao L, She H, Li W, Zeng J, Zhu J, Jones DP, Mao Z, Gao G, Yang Q. Oxidation of survival factor MEF2D in neuronal death and Parkinson disease. Antioxidants Redox Signal 2014; 20:2936-48; PMID:24219011; http://dx.doi.org/ 10.1089/ars.2013.5399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Qi L, Zhang XD, Wu JC, Lin F, Wang J, DiFiglia M, Qin ZH. The role of chaperone-mediated autophagy in huntingtin degradation. PloS One 2012; 7:e46834; PMID:23071649; http://dx.doi.org/ 10.1371/journal.pone.0046834 [DOI] [PMC free article] [PubMed] [Google Scholar]