

ABSTRACT
Park2/Parkin is a central mediator of selective mitochondrial autophagy for mitochondrial quality control. We showed in mouse hearts that PINK1/Mfn2/Park2 mediated generalized mitophagy is essential to the normal perinatal transition from fetal mitochondria that prefer carbohydrates as metabolic substrates to adult fatty-acid metabolizing mitochondria. Our findings demonstrate how functional interactions between mitophagic mitochondrial removal and biogenic mitochondrial replacement facilitate metabolic maturation of the heart.
KEYWORDS: Heart, metabolomics, mitofusin 2, mitophagy, Parkin, transcriptomics
The major function of mammalian mitochondria is to fuel cellular activity by producing ATP through oxidative phosphorylation of metabolic substrates. Because the mitochondrial electron transport chain generates reactive oxygen species, it is essential that mitochondria be maintained and repaired, thus optimizing ATP production and minimizing cytotoxicity. As cardiomyocytes are the most mitochondria-rich of mammalian cells, it is anticipated that maintaining mitochondrial fitness is a biological imperative in hearts.
A common conceptual view of mitochondrial fitness considers all mitochondria to start out identical, but subsequently suffer from varying degrees of age and damage. A simple metaphor is of many individual cars of the same model, but from different model years, having different mileage and varying accident and repair histories. Just as identifying and culling individual dysfunctional automobiles and replacing them with newer cars can assure the overall fitness of a car pool, damaged or functionally impaired mitochondria are first segregated from those that are functioning optimally, and then selectively removed. The best understood mechanism for mitochondrial triage and elimination involves proteins encoded by 2 Parkinson disease genes, the mitochondrial kinase PINK and a cytosolic ubiquitin ligase PARK2/Parkin, and the mitochondrial dynamism factor MFN2 (mitofusin 2). Because physical mitochondrial removal employs the autophagy apparatus, this process is called mitophagy.
The canonical concept of mitochondrial fitness being maintained through targeted mitophagy of individual organelles seemingly does not consider that mitochondria must also collectively adapt to changing physiological and metabolic environments. In other words, mitochondrial fitness is not simply a matter of individual organelle integrity, but also of appropriateness of the mitochondrial collective to any given bioenergetic context. I view this as a matter of mitochondrial suitability for a given cellular ecology. For example, a mismatch between mitochondrial substrate preference and substrate availability could be functionally disastrous, even though individual mitochondria function properly when provided with their preferred substrate. One example of metabolic transitioning is the normal shift of mammalian hearts that preferentially metabolize carbohydrates in utero to metabolizing fatty acids after birth. This developmental change in mitochondrial substrate preference was commonly considered to be a matter of mitochondrial “programming,” but we were skeptical that mitochondria readily switch back and forth between preferred substrates. Rather, we hypothesized that mitochondria (like automobiles) are hard-wired to optimally metabolize a particular substrate. If this is correct, then switching the mitochondrial pool from one fuel to another is not as simple as reprogramming; it would require physically turning over the mitochondrial collective, which is analogous to replacing all gasoline cars in the automobile pool with diesel versions of the model.
We used 2 complementary approaches to examine the hypothesis that mitochondrial replacement is required for perinatal cardiac metabolic transitioning: First, we deleted the gene encoding the central mitophagic factor PARK2 in cardiomyocytes; and second, we interrupted PARK2 translocation to cardiomyocyte mitochondria by expressing a nonfunctioning PARK2 receptor, MFN2 in which 2 PINK1-phosphorylatable amino acids are mutated to nonphosphorylatable alanine (MFN2 AA). The former approach abrogates all PARK2 functionality in cardiomyocytes, whereas the latter approach leaves PARK2 intact, but specifically inhibits PARK2-mediated mitophagy. In both cases the consequence of suppressing the PARK2 mitophagy pathway is that the normal perinatal transition from embryonic to adult cardiomyocyte mitochondria does not occur, with lethal consequences. Detailed studies of MFN2 AA mice (that lived up to 6 wk) revealed that cardiomyocyte mitochondria in young adult Park2-interrupted hearts exhibit a morphometry characteristic of embryonic rather than adult mitochondria. Moreover, mitochondrial capacity to metabolize fatty acids is impaired, fatty acid metabolic gene expression is suppressed, and myocardial fatty acid intermediates are inappropriately low, compared to normal young adult hearts. Each of these metrics recapitulates features of normal embryonic mitochondria. Our stated interpretation of these findings is “Persistence of fetal carbohydrate-metabolizing mitochondria in adult Mfn2 AA hearts revealed a requirement for organelle removal through the PINK1-Mfn2-Parkin mitophagy mechanism before mitochondrial transitioning to normal adult fatty acid metabolism.” We concluded that, “the Parkin-Mfn2 interaction drives general mitophagic turnover of fetal mitochondria in the perinatal heart, enabling their replacement with mitochondria incorporating biogenically derived metabolic systems optimized for the high energetic demands of contracting adult hearts” (Fig. 1).
Figure 1.
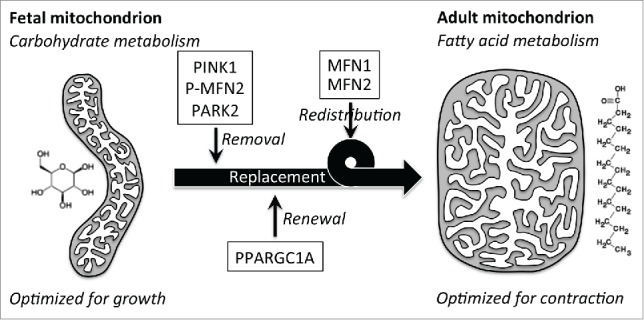
Schematic depiction of putative roles for the PINK1 -MFN2-PARK2 mitophagy pathway, PPARGC1A-directed biogenesis, and mitochondrial fusion mediated by MFN1 and MFN2 in the perinatal transition from fetal to adult heart metabolism. Our recent study defined the PARK2 pathway's mechanism. The role of PPARGC1A and biogenesis was inferred from mRNA expression profiling results. We hypothesize that mitochondrial fusion can be a mechanism for distributing biogenically synthesized adult components throughout the mitochondrial pool.
One consideration is whether interruption of perinatal cardiomyocyte PARK2 mitophagy signaling actually causes individual fetal mitochondria to be retained, vs. the fetal heart mitochondria simply being replaced by phenotypically similar (i.e., fetal) mitochondria instead of by adult mitochondria. In other words, do fetal mitochondria actually “persist” as stated? We think this is a semantic distinction without a biological difference. Indeed, we do not accept that any mitochondria, whether fetal or adult, are immortal or immutable. As with other cell components, mitochondria will wear out or sustain damage and require replacement, i.e., replenishment with newly synthesized proteins (in large part transcribed in the nucleus, translated in the cytosol, and then imported: i.e., mitochondrial biogenesis). Thus, it is not that defective PARK2-mediated mitophagy causes every individual cardiomyocyte mitochondrion in the fetal heart to be retained until adulthood, but that normal maturation of mitochondria is interrupted and the mitochondrial pool retains seminal characteristics of a fetal heart. Under normal conditions, we envision that mitochondrial renewal via PARK2-mediated mitophagy and biogenic synthesis of fresh components is accelerated and developmentally modified in the perinatal myocardium so that organelles with adult morphology and functional profiles are substituted for existing fetal-type mitochondria. Our data reveal that (in hearts at least) a generalized increase in PARK2-mediated mitophagy is a prerequisite for mitochondrial maturation. Thus, cardiomyocyte mitochondria must be removed before they can be developmentally replaced. Whether this is also true in the reverse metabolic remodeling observed in diseased adult hearts is currently unknown.
We emphasize that the MFN2 AA separation-of-function mutant, which is fully capable of promoting mitochondrial fusion but dominantly inhibits mitochondrial PARK2 translocation, was essential to these insights and is likely to prove useful as an investigative tool in other systems. Moreover, our observation that mitochondrial fusion and PARK2 recruitment are mutually exclusive functions of MFN2, determined by PINK1 phosphorylation, establishes a plausible mechanism for immediate and selective sequestration (by disrupting mitochondrial fusion) of mitochondria targeted for mitophagy.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.