

abstract
Peroxisomes are autonomously replicating and highly metabolic organelles necessary for β-oxidation of fatty acids, a process that generates large amounts of reactive oxygen species (ROS). Maintaining a balance between biogenesis and degradation of peroxisomes is essential to maintain cellular redox balance, but how cells do this has remained somewhat of a mystery. While it is known that peroxisomes can be degraded via selective autophagy (pexophagy), little is known about how mammalian cells regulate pexophagy to maintain peroxisome homeostasis. We have uncovered a mechanism for regulating pexophagy in mammalian cells that defines a new role for ATM (ATM serine/threonine kinase) kinase as a “first responder” to peroxisomal ROS. ATM is delivered to the peroxisome by the PEX5 import receptor, which recognizes an SRL sequence located at the C terminus of ATM to localize this kinase to peroxisomes. In response to ROS, the ATM kinase is activated and performs 2 functions: i) it signals to AMPK, which activates TSC2 to suppresses MTORC1 and phosphorylates ULK1 to induce autophagy, and ii) targets specific peroxisomes for pexophagy by phosphorylating PEX5 at Ser141, which triggers ubiquitnation of PEX5 at Lys209 and binding of the autophagy receptor protein SQSTM1/p62 to induce pexophagy.
Peroxisomes are highly metabolic, multifunctional, and ubiquitous organelles that participate in β-oxidation of branched and very long chain fatty acids, synthesis of myelin lipids, and turnover of ROS. Peroxisomes are autonomously replicating, and maintaining the balance between peroxisome biogenesis and degradation is crucial for normal cellular homeostasis. Too few peroxisomes can give rise to diseases such as peroxisomal biogenesis disorders, whereas excessive/impaired peroxisomes can increase cellular ROS, causing damage to cells and contributing to aging and other degenerative diseases, as well as cancer.
Autolysis, peroxisomal lon proteases, and pexophagy are the 3 known mechanism for peroxisomal degradation, with pexophagy being the major pathway by which excess/impaired peroxisomes are eliminated. However, little is known about how pexophagy is regulated in mammalian cells to maintain the proper balance of peroxisomes (and ROS) in the cell.
ATM kinase is a well-characterized tumor suppressor, which plays a key role in the nucleus in the DNA damage response, acting as a “first responder” to DNA damage. We have focused on exploring the cytosolic functions of ATM, and were the first to discover that ATM plays a key role in the cytoplasm regulating MTORC1. When activated by ROS, ATM signaling via STK11/LKB1-AMPK activates the TSC2 tumor suppressor to suppress MTORC1 and induce autophagy. We also found that the TSC2 signaling node (TSC2, its activation partner TSC1 and its GAP target RHEB) is localized to the peroxisome, and that this localization is required for suppression of MTORC1 in response to peroxisomal ROS. These findings led us to ask whether ATM signaling to TSC2 might also be occurring at this organelle, a hypothesis supported by a previous report from Watters et al., that ATM localizes to peroxisomes.
In our recent paper, we reported that the peroxisomal import receptor PEX5 recognizes the SRL sequence at the C terminus of ATM to deliver this tumor suppressor to the peroxisome. The binding of PEX5 to the SRL sequence was confirmed by site-directed mutagenesis, and an R to Q mutation in this sequence (ATM-RQ) dramatically reduces PEX5 binding to ATM and localization of ATM to peroxisomes.
We and others have previously reported that when activated by ROS, ATM signaling results in suppression of MTORC1 and activation of ULK1 to increase autophagic flux. We have now found that in addition to increasing autophagic flux, ATM plays a key a role in selective autophagy of peroxisomes. ATM kinase activated by ROS phosphorylates PEX5 at Ser141, leading to monoubiquitination of peroxisome-localized PEX5 at Lys209. In the process of autophagosome formation, ubiquitinated proteins and/or organelles bind with a receptor protein, SQSTM1 or NBR1, on one side (through the ubiquitin-binding domain) and with newly formed LC3B on the expanding membrane of the phagophore on the other side. Lys209 monoubiquitinated PEX5 is recognized by SQSTM1, targeting peroxisomes for pexophagy (Figure 1). Interestingly, the phosphomimetic mutation S141E of PEX5 is not sufficient to induce pexophagy, suggesting that activation of ATM and repression of MTORC1 to increase autophagic flux is required to complete pexophagy.
Figure 1.
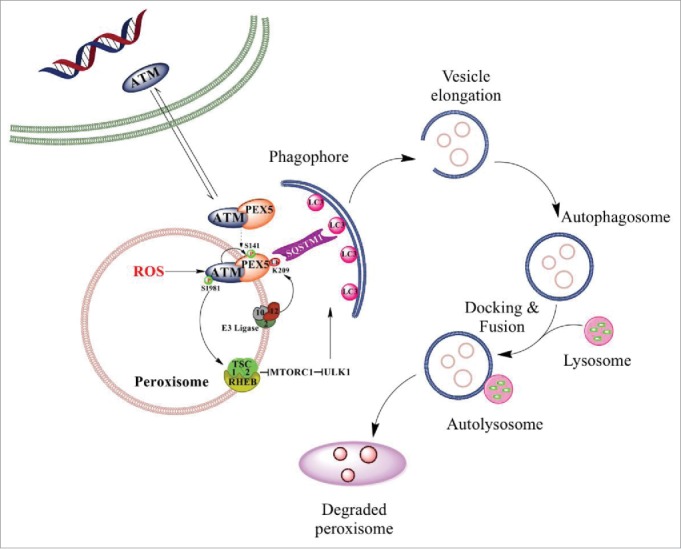
Working model depicting the mechanism of pexophagy in the mammalian system. ATM signaling to the TSC signaling node to repress MTORC1, phosphorylation of PEX5 to induce ubiquitination. by the PEX2-PEX10-PEX.12 E3 ligase, and recognition of Ub-PEX5 by SQSTM1 to induce pexophagy in response to ROS. Phagophores are formed that sequester a .peroxisome; the resulting autophagosomes fuse with a lysosome, and lysosomal enzymes degrade the peroxisome to complete the process of pexophagy.
While this study begins to solve the mystery of how cells regulate peroxisome number by using ROS as a “rheostat,” it also opens other interesting avenues for exploration, such as: i) Can ATM activated in one cellular compartment, such as the nucleus or cytosol, shuttle to and function in another compartment? ii) Is ATM activated as a monomer or dimer at the peroxisome? iii) Since ATM is also activated in response to reactive nitrogen species, do peroxisomal reactive nitrogen species also play a role in pexophagy? Finally, while it is clear ATM functions as a DNA repair protein in the nucleus, we know little about the contribution of ATM signaling in the cytoplasm to its function as tumor suppressor, and whether protecting cells from DNA and oxidative damage are equally important functions for this kinase. These (and other) questions now await further exploration.
Disclosure of potential conflicts of interest
Authors declare there is no conflict of interest.
Funding
National Institutes of Health (NIH) Grant R01 CA143811 to and Robert A. Welch Endowed Chair in Chemistry (BE-0023) funded this work to C.L.W.