

ABSTRACT
Mitochondrial fragmentation due to imbalanced fission and fusion of mitochondria is a prerequisite for mitophagy, however, the exact “coupling” of mitochondrial dynamics and mitophagy remains unclear. We have previously identified that FUNDC1 recruits MAP1LC3B/LC3B (LC3) through its LC3-interacting region (LIR) motif to initiate mitophagy in mammalian cells. Here, we show that FUNDC1 interacts with both DNM1L/DRP1 and OPA1 to coordinate mitochondrial fission or fusion and mitophagy. OPA1 interacted with FUNDC1 via its Lys70 (K70) residue, and mutation of K70 to Ala (A), but not to Arg (R), abolished the interaction and promoted mitochondrial fission and mitophagy. Mitochondrial stress such as selenite or FCCP treatment caused the disassembly of the FUNDC1-OPA1 complex while enhancing DNM1L recruitment to the mitochondria. Furthermore, we observed that dephosphorylation of FUNDC1 under stress conditions promotes the dissociation of FUNDC1 from OPA1 and association with DNM1L. Our data suggest that FUNDC1 regulates both mitochondrial fission or fusion and mitophagy and mediates the “coupling” across the double membrane for mitochondrial dynamics and quality control.
KEYWORDS: DNM1L/DRP1, mitochondrial dynamics, mitophagy, mitophagy receptor, OPA1
Introduction
Mitochondria are essential and multitasking organelles for cellular ATP production, cellular metabolic activities, reactive oxygen species generation and programmed cell death.1,2 To fulfill such diverse, and at times opposite, cellular functions, mitochondria must behave properly and their quality control must be closely monitored to ensure normal cellular activities.3,4 One major mechanism for mitochondrial quality control is mitophagy,5,6 which enables cells to remove damaged or redundant mitochondria. Mitophagy is a specific and selective form of autophagy that requires crosstalk between mitochondria and the autophagy machinery.4,7,8 Mitophagy receptors that have conserved MAP1LC3/LC3-interacting regions (LIRs) mark damaged mitochondria for the recruitment of the autophagy machinery through direct interaction with LC3 and other ATG proteins.4,9 BNIP3L/Nix10-12 and FUNDC113 have been described as mitophagy receptors in mammalian cells, and Atg32 is a mitophagy receptor in yeast.7,8,14,15 Upon mitochondrial stress, such as bioenergetic and oxidative stresses, these receptors that are localized at the outer membrane of mitochondria become phosphorylated or dephosphorylated to enhance the interaction with LC3 or other autophagy genes for the initiation of mitophagy. How these mitophagy receptors sense the mitochondrial stresses within mitochondria to activate mitophagy remains largely unknown.
Mitochondria are highly dynamic organelles that undergo constant fusion and fission. Mitochondrial dynamics are regulated by dynamin family GTPases. DNM1L is a cytosolic molecule recruited to mitochondria for mitochondrial fission, and mitofusins (MFN1 and MFN2) are required for the fusion of mitochondrial outer membrane. OPA1 is another GTPase that localizes at the inner membrane and intermembrane space and plays an essential role in both fusion and fission of mitochondrial inner membrane, maintaining the cristae structure. Mutation of OPA1 and increased susceptibility of cells toward apoptosis causes dominant optic atrophy.16 There are 8 different isoforms of OPA1 due to the alternative splicing of OPA1 pre-mRNA. Mature OPA1 undergoes further processing by YME1L1/YmeL1 and OMA1, leading to the accumulation of long uncleaved OPA1 and short OPA1 forms. The oligomerization of long and short forms of OPA1 determines the fusion or fission of the mitochondrial inner membrane, although this underlying mechanism also remains unclear. Mitochondrial stresses including mitophagy and apoptotic stimulation disrupt these complexes, leading to altered mitochondrial fission or fusion, which is a prerequisite for the mitophagic or apoptotic response. It has been suggested that mitochondrial fission and fusion cycles enable a cell to segregate damaged mitochondria from its network. The segregated mitochondria that have lower membrane potential can regain their membrane potential and refuse to the mitochondria network. Mitophagy occurs when the segregated mitochondria fail to retain their membrane potential. So far, mitophagy in mammalian cells is known to occur through a PARK2 (parkin RBR E3 ubiquitin protein ligase)-PINK1 (PTEN-induced putative kinase 1) pathway17,18 or a mitophagy receptor-dependent pathway.19 Therefore, mitochondrial fission or fusion cycling and mitophagy are integral components of mitochondrial quality control.20
We have previously found that FUNDC1 is a mammalian mitophagy receptor that interacts with and recruits LC3 to mitochondria for mitophagy.13 We have also found that FUNDC1 is phosphorylated at tyrosine 18 (Y18)13 and serine 13 (S13)19 by SRC kinase and CK2, respectively. The phosphorylation prevents the interaction between FUNDC1 and LC3 for subsequent mitophagy in a mammalian system. We sought to understand how mitochondrial dynamics contribute to receptor-mediated mitophagy and we were interested to find if FUNDC1 interacts with both DNM1L and OPA1 for mitochondrial dynamics and mitophagy. Our results reveal a novel function of FUNDC1 and suggest that its interactions may serve as a platform for coordinating mitochondrial fission of both inner and outer membrane of mitochondria and mitophagy.
Results
DNM1L is required for FUNDC1-induced mitochondrial fragmentation and mitophagy
We have previously shown that overexpression of FUNDC1 induced mitochondrial fragmentation in addition to its fundamental role in mitophagy. We thus sought to address the question of how FUNDC1 affects mitochondrial fragmentation and how mitochondrial fragmentation contributes to mitophagy. Knockdown of DNM1L blocked FUNDC1-induced mitochondrial fragmentation and LC3 aggregation (Fig. 1A, B, and C). DNM1L knockdown also blocked FCCP or selenite-induced mitochondrial fragmentation and mitophagy (Fig. S1A, S1B, S1C, S1D). Biochemical analysis also revealed that DNM1L knockdown attenuated the degradation of mitochondrial proteins such as TOMM20 (a mitochondrial outer membrane protein) and TIMM23 (a mitochondrial inner membrane protein) that were induced by FUNDC1 overexpression (Figs. 1D, right panel, S1E). The dominant negative DNM1L mutant (DNM1LK38A) was used to cause loss of function of DNM1L and prevent mitochondrial fragmentation. Coexpression of DNM1LK38A and FUNDC1 blocked mitochondrial fragmentation (Fig. 1E, F, S2A), LC3 aggregation (Fig. 1G, H) and mitochondrial protein degradation (Figs. 1I, right panel, S2B, S2C).
Figure 1.

DNM1L is required for FUNDC1-induced mitochondrial fragmentation and mitophagy. (A) Scrambled shRNA-treated and DNM1L knockdown cells were transfected with FUNDC1-MYC and GFP-LC3 for 24 h. The cells were then fixed and immunostained to detect HSP60 (red) and MYC (purple). Scale bar: 10 µm. (B) Cells treated as in (A), mitochondria fragmentation was quantified by counting numbers of cells with fragmented mitochondria versus all counted cells (mean+/−SEM; n = 100 cells from 3 independent experiments; **, P < 0 .01). (C) The GFP-LC3 aggregates in cells treated as in (A) were quantified with imageJ. The GFP-LC3 aggregation area vs. whole cell area was used to indicate the GFP-LC3 aggregation ratio (mean+/−SEM; n = 100 cells from 3 independent experiments; *, P < 0 .05). (D) Scrambled shRNA-treated and DNM1L knockdown cells were transfected with vector or FUNDC1-MYC for 24 h. The cells were then analyzed via western blotting. (E) HeLa cells were transfected with DNM1LK38A-Flag, FUNDC1-MYC and Mito-DsRed for 24 h. The cells were then fixed and immunostained to detect Flag (Green) and MYC (purple). Scale bar: 10 µm. (F) Cells treated as in (E), % mitochondria fragmentation was quantified as in (B) (mean+/−SEM; n = 100 cells from 3 independent experiments; *, P < 0 .05). (G) HeLa cells were transfected with DNM1LK38A-Flag, FUNDC1-MYC and GFP-LC3 for 24 h. The cells were then fixed and immunostained to detect MYC (purple) and Flag (red). Scale bar: 10 µm. (H) The GFP-LC3 aggregates in cells treated as in (G) were quantified with imageJ. The GFP-LC3 aggregation area versus whole cell area was used to indicate the GFP-LC3 aggregation ratio (mean+/−SEM; n = 100 cells from 3 independent experiments; *, P < 0 .05). (I) Scrambled shRNA-treated and DNM1L knockdown cells were transfected with DNM1LK38A and FUNDC1-MYC for 24 h. The cells were then analyzed via western blotting.
We next detected whether DNM1L interacts with FUNDC1 and found that endogenous FUNDC1 interacted with ectopically expressed DNM1L (Fig. 2A). Endogenous DNM1L also interacted with ectopically expressed FUNDC1 (Fig. 2B). Next, we determined the interaction domains of FUNDC1 and DNM1L and found that the cytosolic domain (1 to ˜50 amino acids [aa]) of FUNDC1 interacted with DNM1L even in the absence of the LIR motif (Fig. 2D). Overexpression of FUNDC1 or its LIR mutants recruited DNM1L to mitochondria except in the 1 to 50 deletion-mutants (Fig. 2C). Affinity isolation assays showed that DNM1L directly interacted with FUNDC1 but not the mutant lacking a cytosolic domain (1 to 50 aa) (Fig. 2E). This interaction could be physiologically relevant, as knockdown of FUNDC1 attenuates DNM1L mitochondrial translocation under selenite treatment, which has been shown to induce mitochondrial stress.23 A subcellular fractionation assay and immunostain assay further confirmed that FUNDC1 or its LIR mutants recruited DNM1L to mitochondria and induced translocation, while mutants lacking a cytosolic domain (deletion of 1 to 50 aa) failed to do so (Fig. 2F, G, H). Immunostain and subcellular fractionation assays also confirmed that FUNDC1 knockdown blocked DNM1L mitochondria translocation after treatment with selenite (Fig. 2I, J, K). These data demonstrate that mitochondrial fission is coupled with mitophagy through the direct interaction between FUNDC1 and DNM1L, the critical mitochondrial fission molecule.
Figure 2.

FUNDC1 interacts with, and recruits DNM1L to mitochondria. (A and B) HeLa cells were transfected with FUNDC1-MYC or DNM1L-MYC (JOSD2-MYC was used as negative control) for 24 h, harvested and lysed, and they were then subjected to immunoprecipitation (IP) with an anti-DNM1L antibody (A) or anti-FUNDC1 antibody (B). CoIP with FUNDC1 (A) or DNM1L (B) was detected by western blotting using an anti-MYC antibody. ((C)and D) HeLa cells were transfected with FUNDC1-MYC or the indicated mutants for 24 h and then were subjected to immunoprecipitation (IP) with an anti-DNM1L antibody. wild-type FUNDC1 and FUNDC1 mutants were detected through western blotting using an anti-MYC antibody. (E) 0.5 μg DNM1L protein was incubated with 1 μg anti-DNM1L antibody and 30 μl protein G-Sepharose beads in 500 μl 1% NP-40 lysis buffer (pH 7.4) following incubating for 4 h and washing 3 times with PBS. HeLa cells were transfected with FUNDC1-MYC or the indicated mutant for 24 h and then were lysed in 1% NP-40 lysis buffer for 30 min. Cell lysis and protein G-Sepharose beads were incubated for 6 h following washing 5 times. The affinity isolation of FUNDC1-MYC (wild type or its mutant) was detected by western blotting using an anti-MYC-antibody. (F) HeLa cells were transfected with Mito-DsRed and FUNDC1 or its mutants for 24 h. The cells were then fixed and immunostained to detect MYC (green) and DNM1L (purple). Scale bar: 10 µm. (G) The proportion of mitochondria translocated DNM1L in (F) were quantified with imageJ by measuring DNM1L (purple) mitochondria (red) merged area and mitochondria area (red), merged area vs. mitochondria area was used to indicate the translocated DNM1L. The ratio was normalized to cells transfected with scrambled shRNA vector plasmids (mean+/−SEM; n = 100 cells from 3 independent experiments; *, P < 0 .05). (H) HeLa cells were subfractionated to detect DNM1L translocation from the cytosol fraction to mitochondrial pellets in the transfection in (F). (I) Scrambled shRNA-treated and FUNDC1 knockdown cells were then treated with 10 µM selenite for 12 h and then fixed and immunostained to detect DNM1L (green) and HSP60 (red). Scale bar: 10 µm. (J) The proportion of mitochondria-translocated DNM1L in (I) was quantified with imageJ by measuring DNM1L (green) mitochondria (red) merged area (yellow) and mitochondria area (red), the ratio of translocated DNM1L was analyzed as in (G) (mean+/−SEM; n = 100 cells from 3 independent experiments; *, P < 0 .05). (K) HeLa cells were subfractionated to detect DNM1L translocation from the cytosol fraction to mitochondrial pellets in the treatment in (I).
Knockdown of FUNDC1 promotes mitochondrial fusion
As mitochondrial fragmentation is the result of the imbalance between mitochondrial fission and fusion, we were also interested in whether defective mitochondrial fusion also contributes to FUNDC1-mediated mitophagy. We thus measured mitochondrial fusion activity using the photoactivation technique22 that directly visualizes and quantitatively measures mitochondrial fusion ability by expressing a mitochondrial matrix-targeted photoactivatable GFP (PAGFP). Knockdown of FUNDC1 resulted in a faster decrease of mito-PAGFP fluorescence intensity of activated mitochondria, indicating a higher fusion ability of mitochondria compared to the parental cells. In contrast, overexpression of wild-type FUNDC1 or the Deletion-LIR (D-LIR) mutant reduced the mitochondrial fusion ability (Fig. 3A to C). Knockdown of FUNDC1 led to accumulation of mitochondrial proteins, such as TOMM20 and TIMM23, due to the inhibition of mitophagy, and overexpression of the FUNDC1-MYC mutant of FUNDC1 decreased the same mitochondrial proteins, which the D-LIR mutant of FUNDC1 did not (Fig. 3D). Additionally, we detected fusion ability after FUNDC1 knockdown and selenite and FCCP treatments. The results showed that knockdown of FUNDC1 enhances mitochondrial fusion upon selenite (Fig. 3E to G) and FCCP treatments (Fig. S3), compared to their corresponding controls. FUNDC1-KD also attenuated the degradation of mitochondrial proteins, such as TOMM20 and TIMM23, under selenite treatment (Figs. 3H, S4A, S4B), suggesting that enhanced fusion is negatively correlated with mitophagy.
Figure 3.

FUNDC1-KD promotes mitochondrial fusion. (A) FUNDC1 knockdown cells were transfected with FUNDC1-MYC and the D-LIR mutant. Then, scrambled shRNA-treated and FUNDC1 knockdown cells were transfected with mito-DsRed and mito-PAGFP and were subsequently photoactivated. Images were collected every 15 min over a 30-min time course. Representative images are projections of z-series to show changes in the fluorescence intensities. Scale bar: 10 µm. (B) Normalized fluorescence intensity of mito-PAGFP in scrambled shRNA-transfected cells and FUNDC1 knockdown or mutant FUNDC1-transfected cells was measured and plotted. Each experimental group contains at least 20 cells. (C) Values of mito-PAGFP percent fluorescence in scrambled shRNA-transfected cells and FUNDC1 knockdown or mutant FUNDC1-transfected cells decrease after 15 and 30 min in each experimental group, and were plotted (Mean+/−SEM; n> = 20 cells from 3 independent experiments; ***, P < 0 .001). (D) Scrambled shRNA-treated and FUNDC1 knockdown cells were transfected with FUNDC1-MYC or D-LIR mutants for 24 h. The cells were then analyzed via western blotting. FUNDC1-MYC or D-LIR mutants were mutated to be RNAi-resistant. (E) Scrambled shRNA-treated and FUNDC1 knockdown cells were transfected with mito-DsRed and mito-PAGFP, after 10 µM selenite treatment, followed by photoactivation. Images were collected every 15 min over 30 min. Representative images are projections of z-series to show changes in the fluorescence intensities. Scale bar: 10 µm. (F) Normalized fluorescence intensity of mito-PAGFP in scrambled shRNA-treated and FUNDC1 knockdown cells was measured and plotted. Each experimental group contains at least 20 cells. (G) Values of mito-PAGFP percent fluorescence in scrambled shRNA-treated and FUNDC1 knockdown cells decrease after 15 and 30 min in each experimental group, and they are plotted (mean+/−SEM; n> = 20 cells from 3 independent experiments; *, P < 0 .05; ***, P < 0.001). (H) Scrambled shRNA-treated and FUNDC1 knockdown cells were treated with 10 µM selenite for 4, 8, and 12 h. The cells were then analyzed via western blotting.
FUNDC1 interacts with OPA1 at the K70A site
We next addressed the question of how FUNDC1 affects mitochondrial fusion. It is possible that FUNDC1 regulates mitochondrial fusion through its interaction with mitochondrial fusion proteins. Indeed, we found that endogenous FUNDC1 interacts with both DNM1L and OPA1, while its interaction with mitofusins is rarely detectable under experimental conditions (Fig. 4A). Selenite treatment reduced its interaction with OPA1 while enhancing its interaction with DNM1L (Fig. 4B). Affinity isolation assays showed that FUNDC1 directly interacts with both Long-OPA1 (L-OPA1) and Short-OPA1 (S-OPA1) (Fig. 4C, D). Additionally, endogenous OPA1 interacted with ectopically expressed FUNDC1 even in the absence of its cytosolic domains (mutant of D1 to 50), and transmembrane domain (mutant D139 to 155) (Fig. 4E). However, mutations or reversal of the order of residues in FUNDC1 between 69 to 73 aa, which are predicted to face the intermembrane space, decrease the interaction between OPA1 and FUNDC1 (Fig. 4F). Single amino acid mutant analysis showed that the FUNDC1K70A mutant has reduced interaction between OPA1 and FUNDC1, suggesting that lysine 70 (K70) of FUNDC1 is important for OPA1 FUNDC1 interaction (Fig. 4G). Interestingly, if lysine 70 was replaced with positively charged arginine, but not alanine, the interaction between OPA1 and FUNDC1 was retained (Fig. 4H). The affinity isolation assay showed that FUNDC1K70A abolished its interaction with OPA1, while the K70R mutant retained this interaction (Fig. 4I).
Figure 4.

FUNDC1 interacts with OPA1 at the K70A site. (A) HeLa cells were subject to immunoprecipitation (IP) with an anti- FUNDC1 antibody. The immunoprecipitation complex was probed via western blotting using an anti-OPA1, MFN1 or MFN2 antibody. (B) HeLa cells were treated with 10 µM selenite for 6 h and were then subjected to immunoprecipitation (IP) with an anti-FUNDC1 antibody. CoIP of OPA1 or DNM1L was detected by western blotting using an anti-OPA1 antibody or DNM1L antibody, (C and D) 0.5 μg L-OPA1 (C) or 0.5 μg S-OPA1 (D) were incubated with 0.5 μg FUNDC1 proteins in 500 μl 1% NP-40 lysis buffer (pH 7.4), and affinity isolation was performed by addition of 1 μg an anti-FUNDC1 or 1 μg IgG antibody and 30 μl protein A-Sepharose beads following incubation for 4 h. After washing for 5 times, the affinity-isolated complexes were detected by western blotting. (E, F, G and H) HeLa cells were transfected with FUNDC1-MYC or FUNDC1 mutants for 24 h. The cells were then subject to immunoprecipitation. FUNDC1 (wild type and mutants) were detected by western blotting using and anti-MYC antibody. (I) 0.5 μg wild-type FUNDC1 or the K70A and K70R mutant proteins were incubated with 0.5 μg S-OPA1 proteins in 500 μl 1% NP40 lysis buffer (pH 7.4), and affinity isolation was performed by addition of 1 μg an anti-OPA1 or 1 μg IgG antibody and 30 μl protein G-Sepharose beads following incubation for 4 h. After washing for 5 times, the affinity-isolated complexes were detected by western blotting.
Loss of interaction between OPA1 and FUNDC1 is important for mitophagy
We next determined the functional consequence of the interaction between FUNDC1 and OPA1. We compared the effect of the wild-type FUNDC1 and the K70A or K70R mutants on mitochondrial fragmentation and LC3 aggregation. The results showed that the FUNDC1K70A mutant that lacks the interaction with OPA1 has reduced LC3 aggregation as compared to wild-type FUNDC1 and the K70R mutant (Fig. 5A, B). Biochemical analysis further confirmed that K70A mutants strongly enhanced the degradation of mitochondrial proteins such as TOMM20 and TIMM23 compared to wild-type FUNDC1 (Fig. 5C). These data suggest that the interaction between FUNDC1 and OPA1 through K70 is important for mitophagy, and mutants that fail to interact with OPA1 have higher capacity for mitophagy. Knockdown of OPA1 enhanced mitophagy, as revealed by increased LC3 aggregation (Fig. 5D, E) and mitochondrial protein degradation induced by FUNDC1 expression (Fig. 5F). These data suggest that OPA1 and its interaction with FUNDC1 are involved in mitophagy.
Figure 5.

Interaction between OPA1 and FUNDC1 is important for mitophagy. (A) HeLa cells were transfected with FUNDC1-MYC or mutants and GFP-LC3 for 24 h. The cells were then fixed and immunostained to detect MYC (red) and HSP60 (purple). Scale bar: 10 µm. (B) The GFP-LC3 aggregates in cells treated as in (A) were quantified with imageJ. The GFP-LC3 aggregation area versus whole cell area was used to indicate the GFP-LC3 aggregation ratio (mean+/−SEM; n = 100 cells from 3 independent experiments; *, P < 0 .05, NS: no significance). (C) HeLa cells were transfected with FUNDC1 and mutants for 24 h, and then subjected to western blotting. (D) HeLa cells were transfected with scrambled or OPA1 knockdown shRNA vectors for 24 h and were then transfected with FUNDC1-MYC or FUNDC1 mutants for 24 h. The cells were fixed and immunostained to detect MYC (red) and HSP60 (purple). Scale bar: 10 µm. (E) The GFP-LC3 aggregates in cells treated as in (D) were quantified with imageJ. The GFP-LC3 aggregation area vs. whole cell area was used to indicate the GFP-LC3 aggregation ratio (mean+/−SEM; n = 100 cells from 3 independent experiments; *, P < 0 .05). (F) Scrambled shRNA-treated or DNM1L-KD cells were transfected by FUNDC1 for 24 h and then subjected to western blotting.
FUNDC1 cooperates with OPA1 and DNM1L for mitochondrial fission and mitophagy
As both OPA1 and DNM1L interact with FUNDC1, we investigated how OPA1 and DNM1L cooperate to regulate FUNDC1-induced mitophagy and mitochondrial fragmentation. Using an immunoprecipitation (IP) assay, we found that mitochondrial stress conditions such as selenite or FCCP treatments decreased the interaction between FUNDC1 and OPA1 and enhanced the interaction between FUNDC1 and DNM1L (Fig. 6A). This was further confirmed by crosslinking-IP assay (Fig. S5A). Oligomycin or antimycin treatment also induced mitochondrial fission and mitophagy (Fig. S6A, S6B) and decreased OPA1-FUNDC1 interaction while increased DNM1L-FUNDC1 interaction (Fig. 6B). We also checked whether knockdown of DNM1L could block mitophagy and mitochondrial fragmentation induced by the K70A mutant that does not interact with OPA1. The results showed that knockdown of DNM1L inhibited wild-type FUNDC1- or the FUNDC1K70A mutant-induced mitochondrial fragmentation and mitophagy (Fig. 6C to E, S6C). The K70A mutant did enhance DNM1L mitochondria translocalization (Fig. 6F to H). These data suggest that FUNDC1 cooperates with OPA1 and DNM1L for mitochondrial fission and mitophagy.
Figure 6.
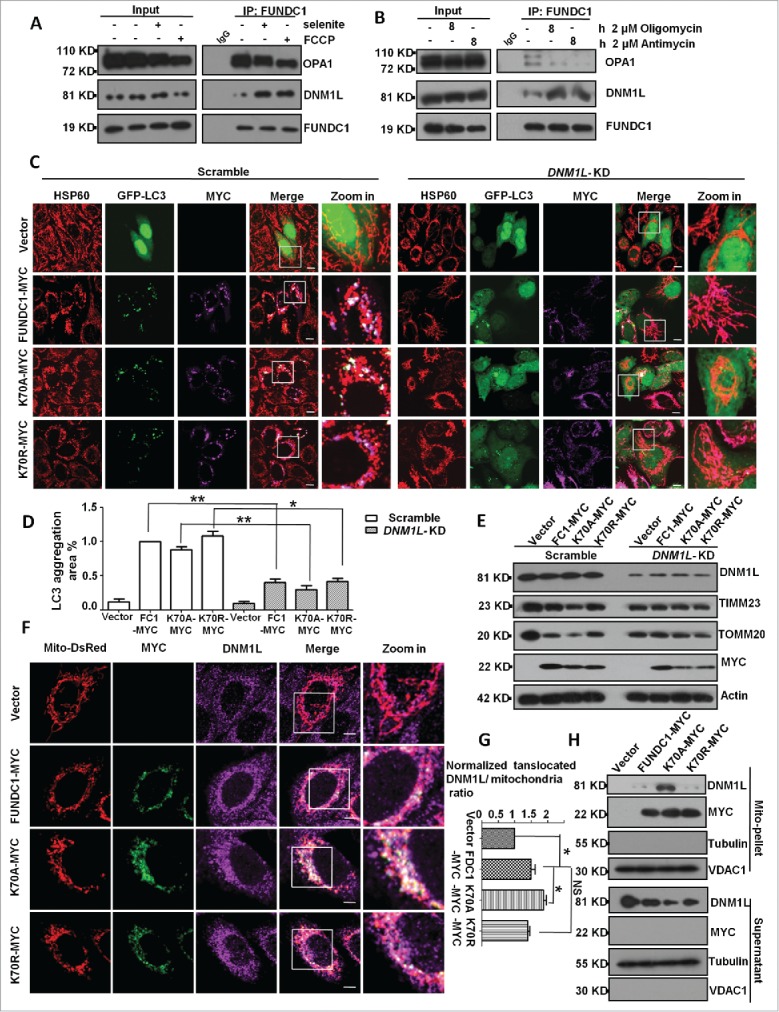
FUNDC1 cooperates with OPA1 and DNM1L for mitochondrial fragmentation and mitophagy. (A) HeLa cells were treated with 10 µM selenite or 10 µM FCCP for 3 h. The cells were then subjected to immunoprecipitation with an anti-FUNDC1 antibody. The immune complex was analyzed by western blotting. (B) Hela cells were treated with 2 µM oligomycin or 2 µM antimycin for 8 h. The cells were then subjected to CoIP with an anti-FUNDC1 antibody. The immune complexes were detected by western blotting. (C) Scrambled shRNA-treated and DNM1L knockdown cells were transfected with FUNDC1 or FUNDC1 mutants and GFP-LC3, and then fixed and immunostained for HSP60 (red) and MYC (purple). Scale bar: 10 µm. (D) The GFP-LC3 aggregates in cells treated as in (C) were quantified with imageJ. The GFP-LC3 aggregation area vs. whole cell area was used to indicate the GFP-LC3 aggregation ratio (mean+/−SEM; n = 100 cells from 3 independent experiments; *, P < 0 .05; **, P < 0 .01). (E) Cells were transfected by FUNDC1 (wild type and mutants), and then subjected to western blotting. (F) HeLa cells were transfected with FUNDC1 or FUNDC1 mutants and mito-DsRed for 24 h and then fixed and immunostained to detect MYC (green) and DNM1L (purple). Scale bar: 10 µm. (G) The proportion of translocated DNM1L in (F) was quantified with imageJ by measuring DNM1L (purple) mitochondria (red) merged area and mitochondria area (red), the merged area vs. whole mitochondrial area was used to indicate the translocated DNM1L. The ratio was normalized to cells transfected with shRNA vector plasmids (mean+/−SEM; n = 100 cells from 3 independent experiments; *, P < 0 .05). (H) HeLa cells were subfractionated to detect DNM1L translocation from the cytosol fraction to mitochondrial pellets in the treatment in (F).
FUNDC1 phosphorylation status is important for FUNDC1-induced mitophagy and mitochondrial fragmentation
FUNDC1 is phosphorylated at serine 13 by CK2 and tyrosine 18 by SRC kinase, and dephosphorylation of FUNDC1 is important for the initiation of mitophagy.19 We asked whether FUNDC1 phosphorylation status affects its interactions with DNM1L and OPA1 for subsequent mitochondrial fragmentation and mitophagy. Using an immunoprecipitation (IP) assay, we found that FUNDC1S13A, a Ser13 dephosphorylation mimic mutant, has a reduced interaction with OPA1 and an increased interaction with DNM1L (Fig. 7A to C). After treatment with CK2 inhibitor TBB in HeLa cells, mitochondria fragmented (Fig. S7B), and the interaction between FUNDC1 and OPA1 decreased while the interaction between FUNDC1 and DNM1L increased (Fig. 7D). Knockdown of the CSNK2A1 subunit increased the DNM1L-FUNDC1 interaction but decreased the OPA1-FUNDC1 interaction under selenite or FCCP treatment (Fig. 7E). Furthermore, knockdown of the CSNK2A1 subunit induced pronounced mitochondrial fragmentation under selenite or FCCP treatments (Fig. S7A). PGAM5 is a phosphatase which dephosphorylates FUNDC1 at Ser13.19 We found that under normal and stress conditions (selenite or FCCP), depletion of PGAM5 enhanced the OPA1-FUNDC1 interaction and decreased the DNM1L-FUNDC1 interaction (Fig. S8A). Besides, we used the S13D phosphomimetic mutant to mimic the FUNDC1 Ser13 phosphorylated-status by CK2. We detected the interaction between FUNDC1S13D and OPA1 or DNM1L, the results showed that the S13D phosphomimetic mutant of FUNDC1 had a lower interaction with DNM1L and a stronger interaction with OPA1 (Fig. S8B, S8C).
Figure 7.

CK2 phosphorylates FUNDC1 Ser13 to inhibit mitochondrial fragmentation and mitophagy. (A) HeLa cells were transfected with FUNDC1-MYC (wild type or mutants) for 24 h and then subjected to immunoprecipitation (IP) with an anti-MYC antibody. CoIP of OPA1 or DNM1L was detected by western blotting using an anti-OPA1 antibody or DNM1L antibody. FUNDC1 (wild type and the Y18W or S13A mutants) were used to mimic the dephosphorylation status of FUNDC1. (B and C) HeLa cells were transfected with FUNDC1-MYC or FUNDC1 mutants for 24 h and were then subjected to immunoprecipitation (IP) with an anti-OPA1 antibody (B) or an anti-DNM1L antibody (C). MYC was detected through western blotting using an anti-MYC antibody. (D) HeLa cells were treated with 10 µM TBB or 10 µM PP2 for 12 h and were then collected and subjected to CoIP using an anti-FUNDC1 antibody. Immunoprecipitated OPA1 and DNM1L were detected by western blotting using anti-OPA1 and anti-DNM1L antibodies. PP2 is an inhibitor of SRC and TBB is an inhibitor of CK2. (E) Scrambled shRNA-treated and CSNK2A1 knockdown HeLa cells were collected for CoIP using an anti-FUNDC1 antibody. CoIP of OPA1 or DNM1L was detected by western blotting. (F) Model of the OPA1-DNM1L interaction axis associated with mitophagy.
Discussion
Both mitochondrial fission or fusion and mitophagy are integral components of mitochondrial quality control. The intriguing issue is how these events are coordinated. Our current work presents the “coupling” mechanism between mitochondrial dynamics and receptor-mediated mitophagy at molecular level in mammalian cells. We found that DNM1L, the mitochondrial fission factor, and OPA1, a mitochondrial inner membrane fission or fusion protein, both interact with FUNDC1 for receptor-induced mitophagy. It appears that there is a “see-saw model” of interaction between DNM1L and OPA1 with FUNDC1. Specifically, FUNDC1 can interact with OPA1 under normal conditions, and the interaction is reduced under mitochondrial stress conditions. In contrast, FUNDC1 can also recruit DNM1L toward mitochondria under stress conditions from its normal cytosolic localization (Fig. 7F). Interestingly, HSP60 levels were reduced under FUNDC1 overexpression or stress (FCCP or selenite) treatment, suggesting that mitoUPR were deactivated during these treatments (Fig. S9A, S9B).
Previous reports suggest that DNM1L-mediated mitochondrial fission of outer membrane is a prerequisite for receptor-mediated mitophagy in yeast.21 We extend this finding in FUNDC1-mediated mitophagy in mammalian cells, and, importantly, we have found that the interaction between FUNDC1 and OPA1 is also important for the fission and fusion of mitochondrial inner membrane and mitophagy. We have identified that K70 of FUNDC1 is important for its interaction with OPA1, and mutation of this lysine residue to A, but not R, abolished the interaction, suggesting that the charged residue is important for this interaction. An overwhelming number of studies have already shown that OPA1 forms an oligomerized complex between long and short forms to determine fusion and fission of the mitochondrial inner membrane,24,25 although the exact stoichiometry and mode of action remain subjects of debate.25 Under normal conditions, FUNDC1 is able to anchor OPA1 through its charged lysine residue toward the inner surface of mitochondrial outer membrane. In response to mitochondrial stresses, OPA1 becomes cleaved or even degraded, thus promoting mitochondrial fission, which is required for mitophagy (see Fig. 3H). Recent studies have shown that mitochondrial stresses from the respiration chain may affect OPA cleavage by YME1L1 and OMA1 for mitochondrial fission or fusion.26 We thus speculate that the interaction between OPA1 and FUNDC1 may serve as an “inside-out” mechanism for mitochondrial stress sensing for mitophagy. Mitochondrial bioenergetic or oxidative stresses could affect YME1L1-dependent cleavage of OPA1 for mitochondrial fission and subsequent mitophagy,22 thus coupling the stress response with the protein and mitochondrial quality control. Indeed, we have observed that knockout of both YME1L1 and OMA1 strongly blocks FUNDC1-mediated mitophagy (unpublished observation). It is also interesting to note that the phosphorylation status of FUNDC1 can regulate the interaction between FUNDC1 and DNM1L or OPA1, which mirrors the finding that the phosphorylation status of FUNDC1 regulates mitophagy.13,19 Our results thus suggest the reciprocal regulation of mitochondrial fission or fusion and mitophagy and the bidirectional signaling across the double membrane of mitochondrial for mitochondrial fission or fusion and mitophagy.
Our data argue that there exists an “interactome” and FUNDC1 lies at the center of this interactome for mitochondrial quality control. We have previously found that kinases such as SRC kinase and CK2 and phosphatases such as PGAM5 interact with FUNDC1 and determine its phosphorylation status in a reversible fashion.19 The phosphorylation status of FUNDC1 dictates the interaction between FUNDC1 with DNM1L and OPA1 (this paper) and with LC3 as we have previously shown.13 Furthermore, we have also shown that BCL2L1, the master regulator of apoptosis, regulates FUNDC1-mediated mitophagy through its interaction with PGAM5 for the dephosphorylation of FUNDC1. This “interactome” thus coordinates these distinct mechanisms for mitochondrial dynamics, mitochondrial quality control and apoptosis in response to different signaling pathways and mitochondrial stresses. Further understanding the molecular details of FUNDC1 interactome will offer mechanistic insights for these critical processes governing mitochondrial and cellular activities.
Materials and methods
Reagents and antibodies
Mitochondrial proteins such as TIMM23, TOMM20, MFN1, MFN2 and FUNDC1 were examined by western blotting. The following antibodies were used: anti-TIMM23 (1:1,000; BD Biosciences, 611223), anti-TOMM20 (1:1,000; BD Biosciences, 612278), anti-HSP60, (1:1,000; Cell Signaling Technology, 12165S); anti-OPA1, (1:1,000; BD Biosciences, 612607), anti-DNM1L/DRP1, (1:1,000; BD Biosciences, 611113); anti-MFN1 monoclonal antibody (1:1,000; Abnova, H000556691-M04), anti-MFN2 monoclonal antibody (1:1,000; Abnova, H00009927-M03); anti-FUNDC1 polyclonal antibody (1:1,000; Abgent, AP17377a); anti-FLAG polyclonal antibody (1:2,000; Sigma, F-1804), anti-PGAM5 polyclonal antibody (1:1,000; Abcam, ab126534), anti-CSNK2A1/CK2α1 casein kinase 2, α 1 polypeptide (1:1,000; Santa Cruz Biotechnology, sc-6479), HRP-conjugated secondary antibodies were purchased from Jackson ImmunoResearch Laboratories (31160).
For the immunoprecipitation assay, an anti-DNM1L monoclonal antibody 1:1,000 (BD, 611113), anti-OPA1 monoclonal antibody 1:1,000 (BD, 612607), anti-FUNDC1 1:1,000 (Abgent, AP17377a), and anti-MYC 1:1000 (Sigma, M4439) were used.
For immunofluorescence, the concentrations used were as follows: anti-HSP60 1:300 (Cell Signaling Technology, 12165S), anti-DNM1L 1:100, (BD, 611113), anti-FLAG 1:200 (Sigma, F-1804), anti-MYC 1:400 (Sigma, M4439), anti-c-MYC polyclonal antibody, 1:1,00 (Santa Cruz Biotechnology, sc-789).
The following fluorescent secondary antibodies were used: goat anti-mouse IgG FITC 1:200 (Life Technologies, 626312); goat anti-rabbit IgG FITC (Life Technologies, 656111) 1:200; goat anti-mouse IgG Cy3 (Life Technologies, 1575605) 1:200; goat anti-rabbit IgG Cy3 (Life Technologies, 167203) 1:200; goat anti-mouse Cy5 (Life Technologies, 1511347) 1:200; goat anti-rabbit Cy5 1:200 (Life Technologies, 1322326).
Cell culture and transfection
Dulbecco's modified Eagle's medium (GIBCO, 11965–118) containing 10% fetal bovine serum (Hyclone, SV30087.02) and 0.1 mg/ml penicillin-streptomycin was used to culture HeLa cells at 37°C under 5% CO2. The target sequence in FUNDC1 for RNA interference is 5-GCAGCACCTGAAATCAACA-3; the scrambled RNA interference sequence is 3-GACATTTGTAACGGGATTC-5; for OPA1, the target sequence is 5′-GATGAAGTTATCAGTCTGAGCCAGGTTAC-3′; for DNM1L, the target sequence is 5′-TTCAATCCGTGATGAGTATGCT TTTCTTC-3; and the control shRNA for DNM1L is 5-TCGTACTCATAATCAGCTCTGCAT ACATC-3. FUNDC1 RNAi resistant sequence is: 5-GCCGCCCCTGAGATAAACA-3. DNA transfections were performed using PEI (Polysciences, 23966) according to the manufacturer's instructions. PGAM5-KO HeLa cell was created by Cas9/CRISPR system.
SDS-PAGE and western blotting
Lysis buffer (20 mM Tris, pH 7.4, 137 mM NaCl, 2 mM EDTA, 10% glycerol (Sigma, 49770), 1% NP-40 (NEW industry, 728601), and protease inhibitors (Roche Applied Science, 04693132001) was used to lyse cells or membrane fractions. Equivalent protein quantities (20 µg) were subjected to SDS-PAGE and transferred to nitrocellulose membranes. Membranes were probed with the indicated primary antibodies, followed by the appropriate HRP-conjugated secondary antibodies. A chemiluminescence kit (ThermoFisher, 32109) was used to visualize immunoreactive bands.
Immunoprecipitation
HeLa cells were transiently transfected using the PEI method for 24 h. The cells were then lysed with 0.5 ml of lysis buffer plus protease inhibitors (Roche Applied Science, 04693132001) for 30 min on ice and centrifuged at 12,000 g for 15 min. The lysates were immunoprecipitated with a specific antibody and protein A-Sepharose (Life Technology, 10–1041) or protein G-Sepharose (Life Technology, 10–1243) for 8 h at 4°C. Thereafter, the precipitants were washed 3 times with phosphate-buffered saline (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4), and the immune complexes were eluted with sample buffer containing 1% SDS for 3 min at 95°C and analyzed by SDS-PAGE.
Immunofluorescence microscopy
Cells were grown on a coverslip to 60% confluence. After treatment, cells were fixed with freshly prepared 3.7% formaldehyde at 37°C for 15 min and washed 3 times with PBS (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 2 mM KH2PO4). 0.2% Triton X-100 (ROTH, 3010244) was used to increase antigen accessibility. Cells were incubated with primary antibodies for 2 h, washed 2 times with PBS, and then stained with a secondary antibody for 45 min. Cell images were captured using an LSM 510 Zeiss confocal microscope (Carl Zeiss Jena, Germany).
Affinity isolation assay
E. coli BL21 (DE3) (Transgen Biotech, CD601–01) was used to express GST tagged proteins. GST fusion proteins were purified on glutathione -Sepharose 4 Fast Flow beads (Amersham Biosciences, 17–5132–01) following thrombin cleavage to remove GST tag. For GST affinity isolation with purified FUNDC1 protein, 0.5 µg of DNM1L or OPA1 protein was incubated with 0.5 µg of FUNDC1 protein in 500 µl of 1% NP-40 lysis buffer (20 mM Tris-HCl, 137 mM NaCl, 1% NP-40, 2 mM EDTA, 10% glycerol, adjust pH to 7.4) for 2 h at 4°C with 1 µg of FUNDC1 antibody and then washed 5 times with 1 ml of PBS buffer. The precipitate complex was boiled with sample buffer containing 1% SDS for 5 min at 95°C and analyzed by SDS-PAGE.
Photoactivation assay
Cells were grown in 2-well chambers for confocal microscopy. a microscope (model LSM 510; Carl Zeiss MicroImaging) and a 63×1 .4 NA Apochromat objective (Carl Zeiss MicroImaging, Thornwood, NY) were used to capture images. 405-nm wavelength light was used to photoactivate the PAGFP. After selecting the regions of interest, a series of z-sections from the cell top to the bottom with 1-μm intervals were irradiated with 405-nm wavelength light. The same intervals between optical sections were used for imaging. Red color indicates mito-DsRed 22 to mark mitochondria and green color indicates mito-PAGFP 22 to detect mitochondria fusion ability.
Statistical analysis
For quantitative analysis, values were obtained from 3 independent experiments and shown as the mean ± SEM. Statistical analyses were performed using the Student t test, with P values < 0.05 being considered significant. Significance levels *, P < 0.05 and **, P < 0.01 were considered as relatively significant levels according to the controls. All statistical data were calculated using GraphPad Prism software.
Supplementary Material
Abbreviations
- aa
amino acids
- ACTB
actin, β
- BCL2L1/Bcl-xL
BCL2-like 1
- CK2
casein kinase 2
- CSNK2A1/CK2α1
casein kinase 2, α 1 polypeptide
- D
deletion
- DNM1L/DRP1
dynamin 1-like
- FCCP
carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone
- FUNDC1
FUN-14 domain containing protein 1
- GFP
green fluorescent protein
- GST
glutathione S-transferase
- IP
immunoprecipitation
- JOSD2
Josephin domain containing 2
- L-OPA1
long OPA1
- MAP1LC3B/LC3B
microtubule associated protein 1 light chain 3 β
- MFN1/2
mitofusin 1/2
- OPA1
optic atrophy 1 (autosomal dominant)
- PGAM5
PGAM family member 5, serine/threonine protein phosphatase, mitochondrial
- RFP
red fluorescent protein
- SC
scrambled
- S-OPA1
short OPA1
- SRC
SRC proto-oncogene, non-receptor tyrosine kinase
- TIMM23
translocase of inner mitochondrial membrane 23 homolog (yeast)
- TM
transmembrane
- TOMM20
translocase of outer mitochondrial membrane 20 homolog (yeast)
- VDAC1
voltage-dependent anion channel 1.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We'd like to thank Richard J. Youle from National Institute of Health for OPA1, DNM1L knockdown and control plasmids. We also want to thank Fei Sun from Institute of Biophysics, Chinese Academy of Sciences for OPA1 and DNM1L proteins.
Funding
This research was supported by the 973 program project (No. 2015CB856300 and NO. 2013CB531200) from the MOST and Natural Science Foundation of China (31520103904, 31471306, 81130045, 31201042).
References
- [1].Galluzzi L, Kepp O, Trojel-Hansen C, Kroemer G. Mitochondrial control of cellular life, stress, and death. Circ Res 2012; 111:1198-207; PMID:23065343; http://dx.doi.org/ 10.1161/CIRCRESAHA.112.268946 [DOI] [PubMed] [Google Scholar]
- [2].Wallace DC, Brown MD, Melov S, Graham B, Lott M. Mitochondrial biology, degenerative diseases and aging. Biofactors 1998; 7:187-90; PMID:9568243; http://dx.doi.org/ 10.1002/biof.5520070303 [DOI] [PubMed] [Google Scholar]
- [3].Chen H, Chan DC. Mitochondrial dynamics—fusion, fission, movement, and mitophagy—in neurodegenerative diseases. Hum Mol Genet 2009; 18 (R2):R169-76; PMID:19808793; http://dx.doi.org/ 10.1093/hmg/ddp326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol 2011; 12:9-14; PMID:21179058; http://dx.doi.org/ 10.1038/nrm3028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Lemasters JJ. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res 2005; 8:3-5; PMID:15798367; http://dx.doi.org/ 10.1089/rej.2005.8.3 [DOI] [PubMed] [Google Scholar]
- [6].Batlevi Y, La Spada AR. Mitochondrial autophagy in neural function, neurodegenerative disease, neuron cell death, and aging. Neurobiol Dis 2011; 43:46-51; PMID:20887789; http://dx.doi.org/ 10.1016/j.nbd.2010.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kanki T, Wang K, Cao Y, Baba M, Klionsky DJ. Atg32 is a mitochondrial protein that confers selectivity during mitophagy. Dev Cell 2011; 17:98-109; http://dx.doi.org/ 10.1016/j.devcel.2009.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Okamoto K, Kondo-Okamoto N, Ohsumi Y. Mitochondria -anchored receptor Atg32 mediates degradation of mitochondria via selective autophagy. Dev Cell 2009; 17:87-97; PMID:19619494; http://dx.doi.org/ 10.1016/j.devcel.2009.06.013 [DOI] [PubMed] [Google Scholar]
- [9].Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 2011; 7:279-96; PMID:21189453; http://dx.doi.org/ 10.4161/auto.7.3.14487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A, Rogov V, Löhr F, Popovic D, Occhipinti A, et al.. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep 2010; 11:45-51; PMID:20010802; http://dx.doi.org/ 10.1038/embor.2009.256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, Wang J. Essential role for Nix in autophagic maturation of erythroid cells. Nature 2008; 454:232-5; PMID:18454133; http://dx.doi.org/ 10.1038/nature07006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Schweers RL, Zhang J, Randall MS, Loyd MR., Li W, Dorsey FC, Kundu M, Opferman JT, Cleveland JL, Miller JL, et al.. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci USA 2007; 104:19500-5; http://dx.doi.org/ 10.1073/pnas.0708818104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, Ma Q, Zhu C, Wang R, Qi W, et al.. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol 2012; 14:177-85; PMID:22267086; http://dx.doi.org/ 10.1038/ncb2422 [DOI] [PubMed] [Google Scholar]
- [14].Aoki Y, Kanki T, Hirota Y, Kurihara Y, Saigusa T, Uchiumi T, Kang D. Phosphorylation of Serine 114 on Atg32 mediates mitophagy. Mol Biol Cell 2011; 22:3206-17; PMID:21757540; http://dx.doi.org/ 10.1091/mbc.E11-02-0145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Liu L, Sakakibara K, Chen Q, Okamoto K. Receptor-mediated mitophagy in yeast and mammalian systems. Cell Res 2014; 24:787-95; PMID:24903109; http://dx.doi.org/ 10.1038/cr.2014.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science 2012; 337:1062-5; PMID:22936770; http://dx.doi.org/ 10.1126/science.1219855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, et al.. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J 2008; 27:433-46; PMID:18200046; http://dx.doi.org/ 10.1038/sj.emboj.7601963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Twig G, Shirihai OS. The interplay between mitochondrial dynamics and mitophagy. Antioxid. Redox Signal 2011; 14:1939-51; http://dx.doi.org/ 10.1089/ars.2010.3779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Chen G, Han Z, Feng D, Chen Y, Chen L, Wu H, Huang L, Zhou C, Cai X, Fu C, et al.. A Regulatory Signaling Loop Comprising the PGAM5 Phosphatase and CK2 Controls Receptor-Mediated Mitophagy. Mol Cell 2014; 54:362-77; PMID:24746696; http://dx.doi.org/ 10.1016/j.molcel.2014.02.034 [DOI] [PubMed] [Google Scholar]
- [20].Mao K, Wang K, Liu X, Klionsky DJ. The scaffold protein Atg11 recruits fission machinery to drive selective mitochondria degradation by autophagy. Dev Cell 2013; Jul 15; 26(1):9-18; PMID:23810512; http://dx.doi.org/ 10.1016/j.devcel.2013.05.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Nieto-Jacobo F, Pasch D, Bassea CW. The mitochondrial Dnm1-like fission component is required for lga2-Induced mitophagy but dispensable for starvation-induced mitophagy in Ustilago maydis. Eukaryotic Cell 2012; 11(9):p:1154-66; PMID:22843561; http://dx.doi.org/ 10.1128/EC.00115-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].MacVicar TD, Lane JD. Impaired OMA1-dependent cleavage of OPA1 and reduced DRP1 fission activity combine to prevent mitophagy in cells that are dependent on oxidative phosphorylation. J Cell Sci 2014; May 15; 127(Pt 10):2313-25; PMID:24634514; http://dx.doi.org/ 10.1242/jcs.144337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Karbowski M, Arnoult D, Chen H, Chan DC, Smith CL, Youle RJ. Quantitation of mitochondrial dynamics by photo labeling of individual organelles shows that mitochondrial fusion is blocked during the Bax activation phase of apoptosis. JCB, 2004; 164 (4):493-9; PMID:14769861; http://dx.doi.org/ 10.1083/jcb.200309082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Wojewoda M, Duszyński J. Effect of selenite on basic mitochondrial function in human osteosarcoma cells with chronic mitochondrial stress. Mitochondrion 2012; 12(1):149-55; PMID:21742063; http://dx.doi.org/ 10.1016/j.mito.2011.06.010 [DOI] [PubMed] [Google Scholar]
- [24].Yamaguchi R, Lartigue L, Perkins G, Scott RT, Dixit A, Kushnareva Y, Kuwana T, Ellisman MH, Newmeyer DD. Opa1-mediated cristae opening is Bax/Bak- and BH3-dependent, required for apoptosis, and independent of Bak oligomerization. Mol Cell 2008; Aug 22; 31(4):557-69; http://dx.doi.org/ 10.1016/j.molcel.2008.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Anand R, Wai T, Baker MJ, Kladt N, Schauss AC, Rugarli E, Langer T. The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J Cell Biol 2014; 204(6):919-29; PMID:24616225; http://dx.doi.org/ 10.1083/jcb.201308006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].MacVicar TD, Lane JD. Impaired OMA1-dependent cleavage of OPA1 and reduced DRP1 fission activity combine to prevent mitophagy in cells that are dependent on oxidative phosphorylation. J Cell Sci 2014; 127(Pt 10):2313-25; PMID:24634514 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.