

Abstract
Hsp90 and its co-chaperones are known to be important for cancer cell survival. The N-terminal inhibitors of Hsp90 that are in ongoing clinical trials as anti-tumor agents have unfortunately shown disappointing efficacies in the clinic. Thus, novel inhibitors of the Hsp90 machine with different mechanism of action are urgently needed. We report here the development of a novel high-throughput drug-screening (HTS) assay platform to identify small molecule inhibitors of Hsp90 and its co-chaperones. This assay quantitatively measures the ability of Hsp90 and its co-chaperones to refold/protect the progesterone receptor (PR), a physiological client of Hsp90, in 96-well plate format. We screened the NIH clinical collection drug library and identified capsaicin as a hit molecule. Capsaicin is an FDA-approved drug for topical use in pain management. Cell survival assays showed that capsaicin selectively kills cancer cells and destabilizes several Hsp90 client proteins. Thus, our data may explain the seemingly pleotropic effect of capsaicin.
Keywords: Molecular Chaperones, Hsp90 machine, High-throughput screening assay, Progesterone receptor, Capsaicin
INTRODUCTION
Hsp90 has been implicated in a multitude of cellular functions and connects multiple signaling networks. It drives the folding, assembly, translocation, and degradation of many regulatory proteins that contribute to the hallmarks of cancer. Many Hsp90 client proteins are primary oncology targets, especially protein kinases (ErbB2, BCR-Abl, Cdk4, PKB/Akt, Raf-1, B-Raf, and FAK) and steroid receptors1.
Inhibitors of Hsp90, such as 17-AAG and other geldanamycin derivatives, or purine-scaffold derivatives, are effective in different tumor types including melanoma, multiple myeloma, breast and prostate cancers2. Although there are no FDA-approved Hsp90 inhibitors yet, 17 different Hsp90 inhibitors have shown promise in clinical trials for cancer of various origins. The majority of these drugs inactivate the ATPase activity of Hsp90, causing proteasomal degradation of its “client” proteins3. Unfortunately, these Hsp90 ATPase inhibitors also induce overexpression of Hsp70 and Hsp27, which are apoptosis inhibitors4 and may contribute to the low efficacy of the drugs. This highlights the need for novel strategies to inactivate the Hsp90 machine. Great effort has been made to develop compounds with reduced stress response induction or to target other domains of Hsp90, such as the C-terminus ATP binding pocket5. However, alternative strategies to inactivate the Hsp90 machine, including inactivating its co-chaperones, need to be pursued more aggressively.
The proper functioning of steroid receptors, such as PR and glucocorticoid receptor (GR), is strictly dependent on the Hsp90 chaperoning machine6. Thus, they have been used as model systems to dissect the Hsp90 chaperoning pathway. Most of these studies used cell-free systems, such as rabbit reticulocyte lysate (RL). This knowledge has been harnessed to develop a PR reconstitution assay using not only RL as a source of molecular chaperones but also purified chaperones6. Because of the robustness and reliability of the PR reconstitution assay we sought to develop it into a high throughput screen (HTS) format and make it suitable for screening small molecule libraries to identify new modulators of the core components of the Hsp90 chaperoning machine.
MATERIAL AND METHODS
PR reconstitution assay using RL
This assay is based on the reconstitution assays previously published7. Untreated RL is purchased from Green Hectares (Oregon, WI). Lysates are supplemented with 3 mM MgCl2, 1 mM ATP, and an ATP-regenerating system consisting of phosphocreatinine and creatine phosphokinase. Chicken PR-A was over-expressed in SF9 cells. A PR22 antibody was used to adsorb PR complexes to Protein A resin. Protein A-PR22-PR resin that has been stripped of endogenous associated chaperones with 400 mM KCl are suspended in 250 μl of RL and incubated for 30 min at 30 °C. Resin is re-suspended every 5 min. After incubation at 30 °C for 30 min, the samples are chilled and supplemented with 5 nM [1,2-3H]progesterone (1.9 TBq/mmol, NEN Life Science Products) plus 100 nM unlabeled progesterone. The samples are incubated for 1 h on ice and then washed with 4 × 1 ml of incubation buffer by centrifugation and suspension. At the last wash, 100 μl are removed for measurement of [3H]progesterone using a Microbeta scintillation counter (PekinElmer). The remainding 900 μl are centrifuged. The buffer is removed and the resin is re-suspended in sample buffer and analyzed by SDS-PAGE.
High-throughput PR reconstitution assay
Chicken PR-A was over-expressed in SF9 cells. A PR22 antibody was used to adsorb PR complexes on the well floor of a high-binding 96-well plate (Greiner Bio-One, catalog no. 655094). Briefly, 96-well plates were loaded with 50 μl per well of protein A solution (20 μg/ml) in 1X PBS and incubated overnight at 4°C. Plates were then washed twice with reaction buffer (200 μl per well; 20 mM Tris-base, pH 7.8, 100 mM KCl, 1 mM DTT, 0.03% NP40) and blocked with 200 μl of 5% BSA in reaction buffer for 1.5 h at 4°C. Next, 50 μl of 20 μg/ml purified PR22 antibody in 5% BSA was added, and plates were incubated for 1.5 h at 4°C. Excess PR22 was washed three times with reaction buffer (200 μl per well). SF9-PR lysate (50 μl) in high salt stripping buffer (20 mM Tris-base, pH 7.8, 300 mM KCl, 1 mM DTT, 5 mM MgCl2, 5 mM ATP, 0.03% NP40) was then added, and plates were incubated for 1 h at 4°C. This was followed by 2 washes with stripping buffer and 3 washes with reaction buffer (200 μl each) to remove excess PR and its associated proteins. We then add 100 μl of a RL mixture consisting of 50 μl of pure RL and 50 μl of RL recipe buffer (40 mM Tris-base, pH 7.8, 200 mM KCl, 2 mM DTT, 0.25 mM MgCl2, 0.06% NP40, 0.25 mM ATP, 5% glycerol, supplemented with 178 nM creatine phosphokinase and 1.11 mM phospho creatine as ATP regeneration system). DMSO (1%) or a drug at 10 μM final concentration was added, and plates were incubated for 30 min (shaking every 5 min) at 30°C in a humid environment. After the incubation, [3H]-progesterone (American Radiolabelled Chemicals, Inc., catalog no. ART 0063) was added. Plates were incubated for 3 h at 4°C. The assembled PR complexes were washed five times with 200 μl per well reaction buffer. Bound [3H]-progesterone was monitored using scintillation liquid and a Microbeta plate reader (PerkinElmer Life Sciences). Each sample is duplicated on another 96-well plate in the same experimental conditions.
MTT cell survival assay
Cells were grown in 96-well tissue culture plates (Corning, catalog no. 3599) to 60% confluence and treated with indicated concentrations of 17-AAG, capsaicin or DMSO control (2% total DMSO concentration) for the indicated times. Cells were incubated with 10 μl of The CellTiter 96® AQueous One Solution Cell Proliferation Assay reagent (Promega, catalog no. G3580) and 90 μl of culture media/well for 1 h at 37°C. Absorbance at 495 nm was measured using a SAFIRE-TECAN plate reader.
Colony formation assay
Cells were grown in 6-well tissue culture plates to 60% confluence and treated with 200 μM capsaicin or DMSO control for 24 h. Cells were then collected and 1000 of these cells were re-plated per 10 cm tissue culture dish (Falcon, catalog no. 353003) in triplicate experiments. Cells were grown for 15 days in MEM 1X media supplemented with 10% FBS. Cells were fixed with 6% glutaraldehyde and 0.5% Crystal Violet, and colonies that contained 50 cells were counted.
RESULTS AND DISCUSSION
Parent assay: reconstitution of the PR complexes using RL
PR is a physiological client of the Hsp90 chaperoning machine in cells. Seminal work from the laboratories of David Toft, William Pratt, David F. Smith, and other researchers has lead to the reconstitution of PR chaperone complexes in vitro using either rabbit RL, which serves as a complete source of molecular chaperones7. Properly folded PR binds progesterone with high affinity. The assay is a protein A-Sepharose resin-based immuno-precipitation assay, where recombinant avian PR multi-chaperone complexes are isolated from SF9 cells using the specific monoclonal antibody PR22. PR is then stripped from SF9 endogenous associated proteins with a high salt buffer. Incubation of the naked PR at 30°C leads to total loss of PR hormone-binding activity in less than five min. However, adding RL in the presence of an ATP regeneration system reconstitutes PR multi-chaperone complexes and preserves the hormone-binding activity for as long as 30 min at 30°C (not shown). This assay therefore reflects the ability of Hsp90 and its co-chaperones to protect/refold partially heat-denatured PR to its ligand-binding conformation. As seen in Fig. 1, RL efficiently refolded PR to its hormone binding state. Geldanamycin (10 μM) inhibited the folding process to about 40% (Fig. 1A). As expected, analysis of protein complexes using SDS-PAGE followed by Coomassie blue staining (Fig. 1B) showed that the active, mature PR complex, contained mainly PR, Hsp90, the immunophilin cyclophilin 40, and p23, which is know to stabilize the Hsp90-client protein complexes. Addition of geldanamycin blocked the chaperoning process in intermediate state where substantial Hsp70, Hop, and Hsp40 are detected, and the level of Hsp90 was lower than in samples with no drug added. No p23 or immunophilins were detected in geldanamycin-containing samples.
Figure 1.
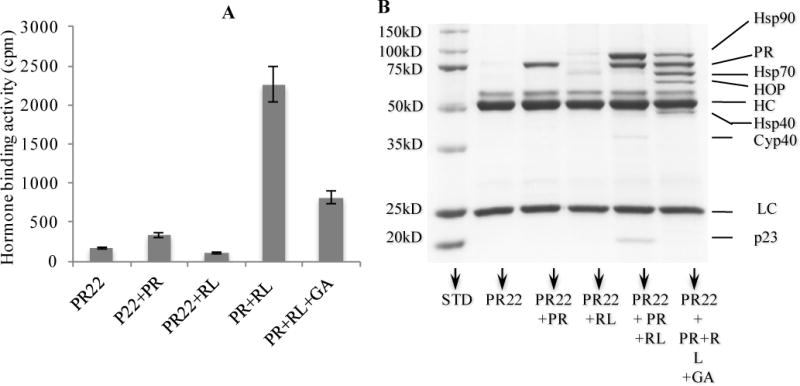
Parent assay: Reconstitution of PR complexes using protein A Sepharose beads, PR antibody, and RL. PR was incubated in presence of RL alone or with geldanamycin (GA; 10 μM) as described in Material and Methods. A. Hormone binding activity is expressed in Cpm. Lane PR22 indicates the resin blank containing the PR antibody PR22 alone. Lane PR22+PR shows the PR bound to PR22 with no RL added. Lane PR22+RL shows the antibody PR22 incubated with RL. Lane PR+RL shows PR bound to PR22 incubated with RL. Lane PR+RL+GA is the same as lane 5 but with 10 μM GA. B. Samples from A were isolated and analyzed for protein content by SDS-PAGE and Coomassie blue staining. All samples were incubated for 30 min at 30°C. HC, heavy chain; LC, light chain of the PR22 antibody.
Transformation of the PR reconstitution assay to 96-well platform screen
Transforming the PR reconstitution assay from an immunoprecipitation-based assay on protein A beads to a microtitre 96-well plate required optimization at every step of the process (Fig. 2A). We tested several kinds of plates and optimized the concentrations of protein A and PR antibody, the amount of PR, the stripping conditions, the volume of RL, and the assay buffer additives (glycerol, ATP regeneration system, MgCl2, glycerol, NP40, and DMSO). Our goal was to obtain an optimal hormone binding activity that was greater than at least six times the background.
Figure 2.
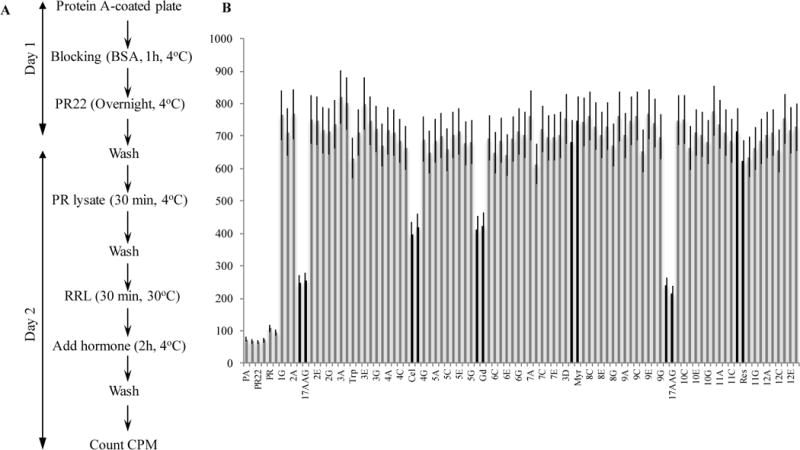
A. Schematic representation of PR reconstitution assay. B. Validation of the assay in the 96-well plate format using known Hsp90 machine inhibitors. All controls are done in duplicates. Two identical plates were tested simultaneously, and the experiment was repeated three times. PA indicates protein A alone. PR22 indicates blank containing the antibody PR22 alone incubated with RL. PR shows the PR bound to PR22 with no RL added. Wells from 1G to 12E contain RL. Black wells contain 17-AAG, celastrol (Cel), gedunin (Gd), Myrecitin (Myr) or resveratrol (Res). 17-AAG was tested at 1 μM. All the other compounds were used at 10 μM concentration.
To evaluate the suitability of the PR reconstitution assay for HTS, we designed the incubation conditions at 30°C to minimize the variation between the central and peripheral wells. We investigated the intra-plate variability using RL with no inhibitors on a full plate (Supplementary Fig. S1). RL efficiently refolds the PR immunoadsorbed onto the 96-well plate to its hormone binding state while BSA does not. The reconstitution is homogeneous throughout the plate with less than 10% variability. The average hormone binding activity for the 86 wells was 734 cpm with a standard deviation of 70.1. The signal/background ratio was about 7 and inter-day variability was around 10%.
The usefulness of the PR reconstitution assay in 96-well plates to detect the Hsp90 machine inhibitors was validated using well-known inhibitors of the ATPase activity of Hsp90, such as geldanamycin derivative 17-AAG (Fig. 2B). We also used celastrol and gedunin, which inactivate the Hsp90 machine through interaction with the co-chaperone p238. As negative controls, we used myricetin, resveratrol, and triptolide, which do not show any effect on the Hsp90 chaperoning of steroid receptors.
We further tested the robustness of the assay using multiple plates. We calculated the Z′-factor using the previously published formula9. The positive controls are wells with RL without inhibitors or with myricetin, which does not affect the chaperoning of PR; negative controls are wells with RL and 17-AAG or gedunin that inhibit the system to less than 40% or 55% activity, respectively. We found the Z′ factor to be 0.723 for four different plates in two-day measurements with inhibitors dispersed into end wells, which demonstrated the excellent quality of the assay. The assay is thus robust and can be used as a reliable tool for high-throughput drug screening.
Validation of the PR reconstitution assay in 96-well plate using NIH small molecule library
We further tested the usefulness of this assay using a small chemical library consisting of 446 compounds called ‘NIH clinical collection’ (Cat# NCC-002). This library consists of FDA-approved drugs or compounds that are in Phase I, II, or III of human clinical trials for various diseases and have been evaluated for toxicity, safety and bioavailability. This library thus offers an attractive set of drugs or drug-like molecules that have shown promising results in human patients. Each compound was tested in duplicate at 10 μM final concentration. Because inhibitors of the co-chaperone p23, such as celastrol of gedunin, reduced the hormone binding activity by about 45% (Fig. 2B), we consider a “hit” inhibitor a compound that inhibits the hormone binding activity of PR by approximately 40% or more. Likewise, we consider a “hit” activator a compound that increases the PR hormone binding by at least 40%. The library comes in six 96-well plates. Out of 446 compounds tested, we obtained seven primary hits from different plates (Fig. 3 and data not shown). Inhibitors having chemical structures related to progesterone were not given further consideration, as they compete with 3[H]-progesterone for binding PR. The remaining hits, which are structurally different from progesterone, were rescreened in triplicate at 20 μM final concentration (not shown). Among these, only capsaicin showed a reproducible effect on blocking the hormone-binding activity of PR by about 40%, which seems to be the maximum inhibition that capsaicin causes in this assay. The hit rate is thus 0.22%, well within 1%, the maximum acceptable hit rate by NIH guidelines for a robust high-throughput drug-screening assay.
Figure 3. Screening of NIH Clinical Collection drug library.
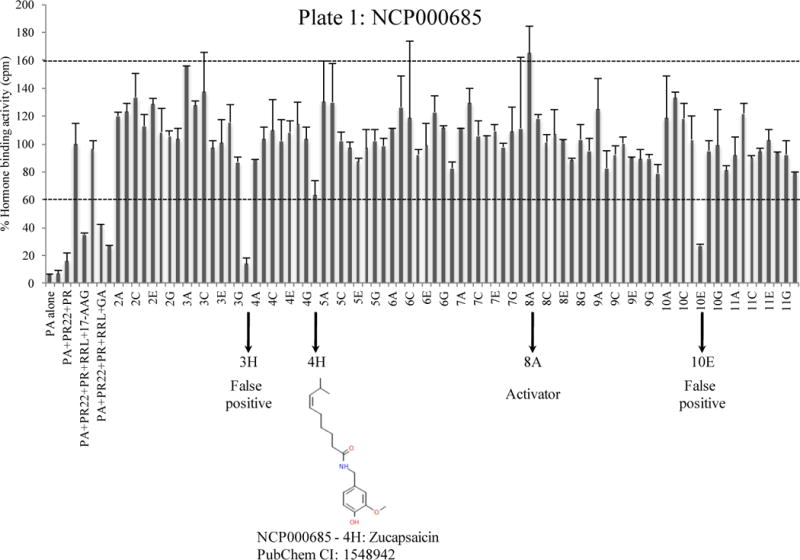
PR complexes were reconstituted on a 96-well plate using RL as the source of molecular chaperones and accessory proteins. Each bar represents percent hormone-binding activity of PR in the presence or absence of chemical compounds. The first samples represent the following internal controls: 1. Protein A alone, 2. Protein A + PR22, 3. Protein A + PR22 + PR, 4. Protein A + PR22 + PR + RL, 5. Protein A + PR22 + PR + RL + 17-AAG (2 μM), 6. Protein A + PR22 + PR + RL + Myrecetin (20 μM), 7. Protein A + PR22 + PR + RL + geldanamycin (20 μM), 8. Protein A + PR22 + PR + RL + gedunin (20 μM). The remaining samples contain compounds from the NCP000685 plate used at 10 μM final concentration. Primary hits are represented with arrows on the x-axis. The standard deviation of duplicate samples is shown as error bars.
Capsaicin selectively kills cancer cells of various origins
Since capsaicin consistently and reproducibly blocked PR chaperoning in vitro, we decided to test the effect of capsaicin in killing cancer cells. Using an MTT assay, the cytotoxic effect of capsaicin was assessed with concentrations ranging from 1 μM to 200 μM, and at three time points using MCF7 breast cancer cells and LNCaP prostate cancer cells (Fig. 4A). These data suggest that capsaicin starts to cause significant death of LNCaP cells at concentrations higher than 50 μM, confirming the range of doses reported by other investigators. We compared capsaicin’s effect on immortalized normal breast epithelial cells (Hs578Bst) versus breast cancer cells (MCF7) with doses of 50, 100, and 200 μM for 24 or 96 hours. As shown in Figure 4A, capsaicin selectively kills MCF7 breast cancer cells in a time- and dose-dependent manner, without affecting Hs578Bst normal breast cells. These results confirm previous report showing that another non-transformed mammary cell line MCF10A is less sensitive to capsaicin treatment10. We then assessed the effect of capsaicin on the colony formation efficiency of LNCaP, MCF7, HeLa, and HepG2 cancer cell lines. As shown in Fig. 4B, capsaicin efficiently reduces the ability of cancer cells to form colonies.
Figure 4. Capsaicin selectively kills cancer cells.
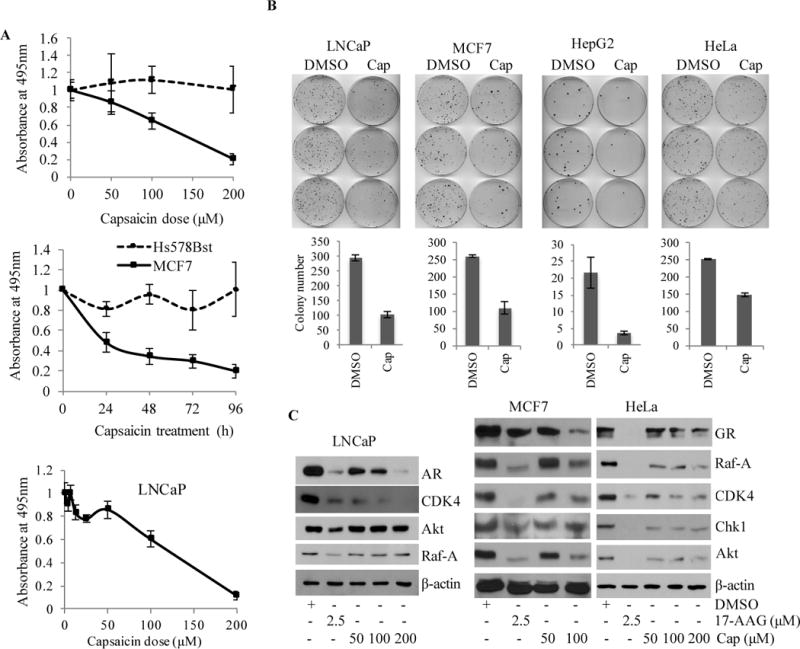
(A) MTT cell survival assays showing that capsaicin kills MCF7 breast cancer cells in a concentration- (upper panel) and time-(lower panel) dependent manner. Hs578Bst normal mammary epithelial cells were unaffected after capsaicin treatment. In the upper panel, cells were treated with 50 to 200 μM capsaicin for 96 h. In the lower panel, cells were treated with 200 μM capsaicin for 24 to 96 h. The standard deviation of quadruplet samples is shown as error bars. (B) Capsaicin blocks colony formation of LNCaP, MCF7, HepG2, and HeLa cells. Cells were treated with 200 μM capsaicin for 24 h. Adherent cells were then collected and plated (1000 cells per 10 cm dish) for further incubation. Colonies that formed after 15 days are shown (upper panel). Colonies containing 50 or more cells were quantified (lower panel). The standard deviation of three dishes is shown as error bars. The experiment was repeated three times. C. Capsaicin blocks Hsp90 chaperoning machine in LNCaP, MCF7 and HeLa cells. Cells were treated with 50, 100, and 200 μM capsaicin for 24 h. Cell lysates were made and Western blotted for indicated Hsp90 client proteins. DMSO was used as a negative control; 17-AAG (2.5 μM) was used as a positive control; and β-actin used as a loading control. For each cell line, the experiment was repeated three times.
Capsaicin (8-methyl-N-vanillyl-6-nonenamide), a homovanillic acid derivative, is the main pungent ingredient of red pepper, genus Capsicum. Capsaicin was reported to bind to the transient receptor potential vanilloid (TRPV1) receptor, leading to desensitization of peripheral nerves to noxious stimuli11. Numerous recent research reports have convincingly shown that capsaicin induces cell cycle arrest and apoptotic cancer cell death in a variety of cancer cell lines including colon adenocarcinoma, pancreatic cancer, hepatocellular carcinoma, prostate cancer, breast cancer, and many others12. Interestingly, many of the pathways inhibited by capsaicin treatment involve Hsp90 client proteins. These include EGFR/HER-2 pathway10, cyclin-dependent kinase13 and Src kinase14 among others.
Capsaicin Inactivates the Hsp90 chaperoning machine in cancer cells
To verify whether capsaicin targets the Hsp90 machine in cells, we treated LNCaP, MCF7, and HeLa cells with increasing doses of capsaicin and studied the effect of capsaicin on the expression of Hsp90 client proteins. The majority of Hsp90 client proteins such as nuclear receptors (GR and AR), and other cell signaling protein kinases, including CDK4, Chk1, Raf-A, and Akt, were down-regulated upon capsaicin treatment (Fig. 4C). There is, however, some cell-specific effects of capsaicin. Akt and Raf-A, for example, are destabilized in MCF7 and HeLa cells but are insensitive to capsaicin treatment in LNCaP cells. Taken together, these data indicate that capsaicin blocks the Hsp90 chaperone system as reflected by destabilization of protein clients of Hsp90 machine. Previous repots have shown that capsaicin blocks the activity of many other oncogenic signaling proteins known to be clients of Hsp90 such as NF-κB, ER, EGFR/HER2, Src, VEGF, and PI3K/Akt, among many others. We therefore propose that the seemingly pleiotropic effects of capsaicin15 may be due to inhibition of the central Hsp90 chaperoning machine.
In conclusion, we have developed a new high-throughput drug-screening platform that is sensitive and robust enough to identify small molecule modulators of the Hsp90 chaperoning machine in vitro. This assay is uniquely qualified to identify compounds that inhibit or activate the Hsp90 chaperoning machine. We have designed a robot capable of executing all the steps of this assay (Fig 2A). automatically. This provides the ability to screen small compound libraries at a rate of 1000 compounds a day.
Supplementary Material
Evaluation of PR reconstitution assay in 96-well plate format using RL as a source of molecular chaperones. The whole plate was loaded with protein A, PR22, and PR as indicated in Material and Methods. Eight wells were incubated with BSA as controls and RL was added to all remaining wells.
Acknowledgments
We thank Dr. Lisa Middleton for the critical editing of the manuscript.
Footnotes
This work was supported by NIH R01 grant GM102443-01 for Ahmed Chadli.
References
- 1.Neckers L, Workman P. Hsp90 molecular chaperone inhibitors: are we there yet? Clin Cancer Res. 2012;18(1):64–76. doi: 10.1158/1078-0432.CCR-11-1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Taldone T, Gozman A, Maharaj R, et al. Targeting Hsp90: small-molecule inhibitors and their clinical development. Curr Opin Pharmacol. 2008;8(4):370–4. doi: 10.1016/j.coph.2008.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Trepel J, Mollapour M, Giaccone G, et al. Targeting the dynamic HSP90 complex in cancer. Nat Rev Cancer. 2010;10(8):537–49. doi: 10.1038/nrc2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lanneau D, Brunet M, Frisan E, et al. Heat shock proteins: essential proteins for apoptosis regulation. J Cell Mol Med. 2008;12(3):743–61. doi: 10.1111/j.1582-4934.2008.00273.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Duerfeldt AS, Blagg BS. Hsp90 inhibition: elimination of shock and stress. Bioorg Med Chem Lett. 2010;20(17):4983–7. doi: 10.1016/j.bmcl.2010.06.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pratt WB, Toft DO. Regulation of signaling protein function and trafficking by the hsp90/hsp70-based chaperone machinery. Exp Biol Med (Maywood) 2003;228(2):111–33. doi: 10.1177/153537020322800201. [DOI] [PubMed] [Google Scholar]
- 7.Kosano H, Stensgard B, Charlesworth MC, et al. The assembly of progesterone receptor-hsp90 complexes using purified proteins. J Biol Chem. 1998;273(49):32973–9. doi: 10.1074/jbc.273.49.32973. [DOI] [PubMed] [Google Scholar]
- 8.Patwardhan CA, Fauq A, Peterson LB, et al. Gedunin inactivates the co-chaperone p23 protein causing cancer cell death by apoptosis. J Biol Chem. 2013;288(10):7313–25. doi: 10.1074/jbc.M112.427328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang JH, Chung TD, Oldenburg KR. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen. 1999;4(2):67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 10.Thoennissen NH, O’Kelly J, Lu D, et al. Capsaicin causes cell-cycle arrest and apoptosis in ER-positive and -negative breast cancer cells by modulating the EGFR/HER-2 pathway. Oncogene. 2010;29(2):285–96. doi: 10.1038/onc.2009.335. [DOI] [PubMed] [Google Scholar]
- 11.Caterina MJ, Schumacher MA, Tominaga M, et al. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature. 1997;389(6653):816–24. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- 12.Diaz-Laviada I, Rodriguez-Henche N. The potential antitumor effects of capsaicin. Progress in drug research. 2014;68:181–208. doi: 10.1007/978-3-0348-0828-6_8. [DOI] [PubMed] [Google Scholar]
- 13.Chen D, Yang Z, Wang Y, et al. Capsaicin induces cycle arrest by inhibiting cyclin-dependent-kinase in bladder carcinoma cells. Int J Urol. 2012;19(7):662–8. doi: 10.1111/j.1442-2042.2012.02981.x. [DOI] [PubMed] [Google Scholar]
- 14.Pyun BJ, Choi S, Lee Y, et al. Capsiate, a nonpungent capsaicin-like compound, inhibits angiogenesis and vascular permeability via a direct inhibition of Src kinase activity. Cancer Res. 2008;68(1):227–35. doi: 10.1158/0008-5472.CAN-07-2799. [DOI] [PubMed] [Google Scholar]
- 15.Bley K, Boorman G, Mohammad B, et al. A comprehensive review of the carcinogenic and anticarcinogenic potential of capsaicin. Toxicol Pathol. 2012;40(6):847–73. doi: 10.1177/0192623312444471. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Evaluation of PR reconstitution assay in 96-well plate format using RL as a source of molecular chaperones. The whole plate was loaded with protein A, PR22, and PR as indicated in Material and Methods. Eight wells were incubated with BSA as controls and RL was added to all remaining wells.