

Abstract
A series of 1,3,4-oxadiazol-2-ones was synthesized and tested for activity as antagonists at GPR55 in cellular beta-arrestin redistribution assays. The synthesis was designed to be modular in nature so that a sufficient number of analogues could be rapidly accessed to explore initial structure-activity relationships. The design of analogues was guided by the docking of potential compounds into a model of the inactive form of GPR55. The results of the assays were used to learn more about the binding pocket of GPR55. With this oxadiazolone scaffold, it was determined that modification of the aryl group adjacent to the oxadiazolone ring was often detrimental and that the distal cyclopropane was beneficial for activity. These results will guide further exploration of this receptor.
Keywords: GPR55, neuropathic pain, GPCR, antagonist, cancer
Graphical Abstract

GPR55, a recently deorphanized, rhodopsin-like (class A) G protein-coupled receptor (GPCR), is a receptor for L-α-lysophosphatidylinositol (LPI, Figure 1) which serves as the endogenous agonist (GenBank entry NM 005683).1 Initial studies noted that a variety of CB1 and CB2 ligands bind to GPR552-3 and more recent studies have focused on physiological roles for GPR55 in inflammatory pain,2 neuropathic pain,2 bone development,3 and the potential for activation of GPR55 being pro-carcinogenic.4-8 Despite the important potential biological functions of GPR55, the research is limited by the lack of both potent and selective agonists and antagonists.9-10
Figure 1.

LPI and Lead Antagonists of GPR5512
Based on a high-throughput, high-content screen of approximately 300,000 compounds from the Molecular Libraries Probe Production Centers Network initiative,11 a few molecular scaffolds were identified that had relatively good selectivity and potency as antagonists at GPR55. These structures were then docked into the inactive state model of GPR5512 to visualize the key features of the antagonists. Of the compounds that exhibited selective and moderate activity as antagonists at GPR55, three different structural families were identified as illustrated by ML191, ML192, and ML193 (Figure 1).
The docking of the structures in Figure 1 into the inactive state model of GPR55 indicated a few important interactions as we previously reported.12 Briefly, the primary interaction was hydrogen bonding between the lysine at position 2.60(80)13 and the oxadiazolone carbonyl in ML191, the amide carbonyl in ML192, or an oxygen of the sulfonamide in ML193. The hypothesized interactions with K2.60(80) positioned the bottom aryl rings of all three structures, as represented in Figure 1, to maintain the toggle switch interaction between M3.36(105) and F6.48(239). The remaining interactions of the ligands presented in Figure 1 and GPR55 are primarily aromatic stacking with various residues. Specifically for ML191, the toluene ring attached to the cyclopropane stacks with F169 and the phenyl group attached to the oxadiazolone stacks with F6.55(246) and F3.33 (102; Figure 2). In addition to these interactions, moderate beneficial van der Waals interactions were identified between the oxadiazolone and both M7.39(274) and Y3.32(101). Since the interactions between ML191 and GPR55 centered on the three aromatic rings of ML191, compounds were desired that modified the electronics and sterics of these areas. Hence, the ML191 synthetic studies reported herein were undertaken to explore the SAR of this oxadiazolone class of compounds. ML191 was also chosen as the lead antagonist since there are very few structurally related compounds that could be purchased and screened compared to the available compounds for ML192 and ML193.
Figure 2.

A. Docking and Key Interactions Between ML191 and GPR55. ML191 (green) has a key H-bond interaction with K2.60 (pink). ML191 also has π-stacking or other van der Waals inter-actions with F169, F3.33, F6.55, M7.39, and Y3.32 (all mustard). The interactions with M7.39 and F6.55 appear to hinder the rotation of M3.36 and F6.48 (both purple) which are considered the toggle switch for GPR55. B. Electrostatic potential map of ML191. [This figure is adapted from previously published work, see ref. 12].
Our synthetic approach to GPR55 antagonists was designed so that many different structures could be accessed to rapidly explore initial SAR, along with validating or modifying our current model (Figure 2).11 The synthesis begins with the coupling of a carboxylic acid to 4-piperidone by first forming the acid chloride (Scheme 1). The different acids chosen, based on the initial hit, modify the electronics and sterics of this section of the molecule. Relative to ML191, compound 2a reduces the steric impact, 2b increases the electron-density in the aromatic ring, and compounds 2c and 2d decrease the electron-density. Compounds 2e and 2f were selected to examine the influence of steric bulk at the position of the cyclopropane ring. The largest change in overall structure relates to the 1-naphthoic acid derivative (2f). Although the naphthalene ring is structurally different, this analogue can position the distal aromatic ring in a similar position as the phenyl rings of the other analogues since the bond angle for the Cα will be similar to that of the cyclopropane analogues, however, this structure is much flatter.
Scheme 1.

Synthesis of Acylated Piperidones
With a handful of acylated piperidones prepared, the final two steps first involved a reductive coupling of aryl hydrazides (3t-z) with the previously synthesized piperidones (2a-f) to yield hydrazides 4 (Scheme 2).14 These compounds were then cyclocarbonylated using triphosgene to yield oxadiazolones 5.15 The reductive coupling reactions proceeded smoothly but the products of that step were often unstable to silica gel chromatography. Therefore, the unpurified products were treated with triphosgene without further purification. This modification of the synthesis typically improved the yields of the final compounds (see supplementary data for individual yields).
Scheme 2.

Synthesis of GPR55 Antagonists
Similar to the cyclopropane starting materials (1a-f), the hydrazides (3t-z) were selected to probe the electronic and steric opportunities in the binding site. Based on the current model (Figure 2), the aromatic ring adjacent to the oxadiazolone is involved in an interaction with M3.36(105) and F6.48(239). Additionally, the oxadiazolone contributes as the key interaction between the basic carbonyl oxygen with the ammonium of K2.60(80). Thus, electron rich aromatic rings adjacent to the oxadiazolone should make the carbonyl oxygen more basic and strengthen this interaction.
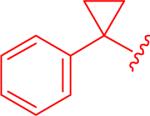
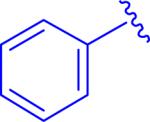
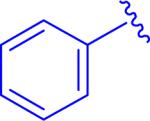
A targeted exploration of the SAR of all six acids (1a-f) with hydrazide 3t and all seven hydrazides (3t-z) with acid 1a (Figure 3 and Table 1) was performed instead of synthesizing and exploring the biological activity of all 42 permutations of the six acids and seven hydrazides. Acid 1a and hydrazide 3t were chosen as the constants since these were the most simplified pieces consisting of an unsubstituted phenyl ring. Unfortunately, there were solubility issues with some of the compounds (e.g. 5bt and 5bv), so additional combinations were required to elucidate the effect of the different areas of the scaffold.
Figure 3.

Analogues with Poor Activity (>15 μM)
Table 1.
GPR55 Antagonist Activity of Compounds.
| Entry, Compound | R | Ar | IC50a (95% CI) |
|---|---|---|---|
| 1,5at |
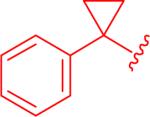
|
|
12 μM (3.9 - 36) |
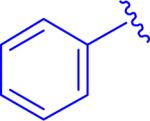
| 2, 5au |
|
|
0.42 μM (0.075 - 2.4) |
| 3, 5av |
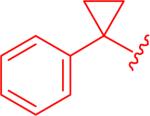
|

|
7.0 μM (0.47 - 100) |
| 4, 5aw |
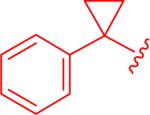
|
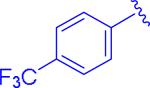
|
insol.b |
| 5, 5bt |
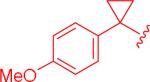
|
|
insol.b |
| 6, 5bv |
|
|
insol.b |
| 7, 5bw |
|
|
1.8 μM (0.74 – 4.3) |
| 8, 5ct |

|
|
2.5 μM (1.3 – 5.0) |
| 9, 5dt |
|
|
0.64 μM (0.33 - 1.2) |
| 10, 5et |
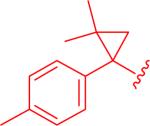
|
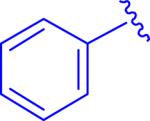
|
0.77 μM (0.39 - 1.5) |
IC50 values and 95% confidence intervals (CI) were determined by running the sample in triplicate versus a 6 μM concentration of LPI
compound was not completely soluble at the concentrations of the assay.
Compounds were initially screened via an image-based cell assay to identify antagonist activity. The rationale for using the β-arrestin recruitment assay was to provide a fair comparison of IC50 values since our initial report employed this assay.11-12 Briefly, U2OS cells overexpressing GPR55 and βarr2-GFP were exposed to LPI (6 μM; EC80) resulting in the recruitment of β-arrestin. Antagonist activity was evaluated by ligand-mediated inhibition of LPI-induced receptor activation. This strategy quickly identified the compounds that had IC50 values higher than 15 μM which were excluded from further analysis (Figure 3).
Concentration response curves were generated for compounds that were active at concentrations below 15 μM employing both the image-based β-arrestin recruitment assay and the DiscoveRx PathHunter® chemiluminescent β-arrestin complementation assay. In the DiscoveRx PathHunter® system, CHO-K1 cells stably expressing GPR55 (fused with a β-galactosidase enzyme fragment), and β-arrestin (fused to an N-terminal deletion mutant of β-galactosidase) were used to quantitate the inhibition of LPI-induced β-arrestin activity. (Figure 4, Table 1). Hence, antagonist activity was evaluated through the use of two differential means of β-arrestin quantitation, in two different cellular backgrounds (see Supplemental Information, Biological Assay). IC50 values were similar in both methodologies.
Figure 4.

Representative Images of Antagonist Screening. A) 0.1 μM 5dt + LPI; B) 1.0 μM 5dt + LPI; C)10 μM 5dt + LPI; D) 3 μM LPI; E) 10 μM 5dt; F) DMSO.
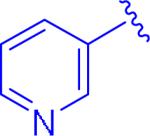
Screening of the compounds allowed for a number of interesting SAR observations. First of all, pyridyl analogue 5au demonstrated that a pyridine ring would be beneficial for both clogP as well as increasing the potency (entry 2 versus entry 1). Electron-poor aryl groups next to the oxadiazolone were detrimental (Figure 3), but electron-rich groups were also not beneficial (entry 3). As discussed earlier, it was anticipated that the more electron-rich aryl groups next to the oxadiazolone would be beneficial electronically, but the results obtained could be validated by the electron-donating groups being larger and creating some detrimental steric interactions.
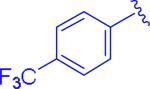
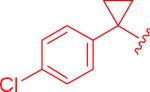
It was found that electron-rich cyclopropylaryl groups had relatively good activities (entry 7), but also typically had solubility problems (entries 5 and 6). Fortunately, analogue 5bw was soluble, but the moderate activity illustrates that the p-methoxy group is detrimental since the most closely related analogue (ML191, Figure 1) was more active and the electron-withdrawing p-chloro analogue (5dt) was even more active. Dichlorophenyl analogue 5ct had good activity, but was not as potent as compared to the monochloroaryl compound (entries 8 and 9) which could be justified based on the larger steric bulk of the second chlorine atom. Structure 5et with the dimethylcyclopropyl group was similarly active (entry 10) as compared to the parent compound, ML191 (Figure 1) which is interesting since this compound adds more steric bulk to the cyclopropyl aryl section of the molecule, albeit in a slightly different location than analogue 5ct. It should be noted that analogue 5et is the only structure analyzed that is chiral. The synthesis of 5et was racemic and the model indicates that there are no major anticipated differences in activities between the two enantiomers.
In conclusion, this letter presents initial SAR for piperidine-substituted oxadiazolone antagonists at GPR55, a recently deorphanized G protein-coupled receptor that lacks a potent and selective ligand of nanomolar potency. These data help to better define areas for improvement of this family of GPR55 antagonists since both halves of the molecule were independently modified. The activities spanned about two orders of magnitude and will be used as a guide for future efforts which will be published in due time.
Supplementary Material
Acknowledgments
This research was supported by National Institutes of Health Grants R21NS077347 and R01DA023204, T32DA007237, P30DA013429. The authors thank Dr. Franklin J. Moy (UNCG) for assisting with analysis of NMR data and Dr. Brandie M. Ehrmann (UNCG) for acquisition of the high resolution mass spectrometry data at the Triad Mass Spectrometry Laboratory at the University of North Carolina at Greensboro.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/XXXX.
References and notes
- 1.Henstridge CM, Balenga NAB, Kargl J, Andradas C, Brown AJ, Irving A, Sanchez C, Waldhoer M. Mol. Endocrinol. 2011;25:1835. doi: 10.1210/me.2011-1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Staton PC, Hatcher JP, Walker DJ, Morrison AD, Shapland EM, Hughes JP, Chong E, Mander PK, Green PJ, Billinton A, Fulleylove M, Lancaster HC, Smith JC, Bailey LT, Wise A, Brown AJ, Richardson JC, Chessell IP. PAIN. 2008;139:225. doi: 10.1016/j.pain.2008.04.006. [DOI] [PubMed] [Google Scholar]
- 3.Whyte LS, Ryberg E, Sims NA, Ridge SA, Mackie K, Greasley PJ, Ross RA, Rogers MJ. Proc. Natl. Acad. Sci. USA. 2009;106:16511. doi: 10.1073/pnas.0902743106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ford LA, Roelofs AJ, Anavi-Goffer S, Mowat L, Simpson DG, Irving AJ, Rogers MJ, Rajnicek AM, Ross RA. Br. J. Pharmacol. 2010;160:762. doi: 10.1111/j.1476-5381.2010.00743.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Andradas C, Caffarel MM, Pérez-Gómez E, Salazar M, Lorente M, Velasco G, Guzmán M, Sánchez C. Oncogene. 2011;30:245. doi: 10.1038/onc.2010.402. [DOI] [PubMed] [Google Scholar]
- 6.Pineiro R, Maffucci T, Falasca M. Oncogene. 2011;30:142. doi: 10.1038/onc.2010.417. [DOI] [PubMed] [Google Scholar]
- 7.Pérez-Gómez E, Andradas C, Flores JM, Quintanilla M, Paramio JM, Guzmán M, Sánchez C. Oncogene. 2013;32:2534. doi: 10.1038/onc.2012.278. [DOI] [PubMed] [Google Scholar]
- 8.Sharir H, Abood ME. Pharmacol. Ther. 2010;126:301. doi: 10.1016/j.pharmthera.2010.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brown AJ, Daniels DA, Kassim M, Brown S, Haslam CP, Terrell VR, Brown J, Nichols PL, Staton PC, Wise A, Dowell SJ. J. Pharmacol. Exp. Ther. 2011;337:236. doi: 10.1124/jpet.110.172650. [DOI] [PubMed] [Google Scholar]
- 10.Yin H, Chu A, Li W, Wang B, Shelton F, Otero F, Nguyen DG, Caldwell JS, Chen YA. J. Biol. Chem. 2009;284:12328. doi: 10.1074/jbc.M806516200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heynen-Genel S, Dahl R, Shi S, Milan L, Hariharan S, Sergienko E, Hendrick M, Dad S, Stonich D, Su Y, Vicchiarelli M, Mangravita-Novo A, Smith LH, Chung TDY, Sharir H, Caron MG, Barak LS, Abood ME. Screening for Selective Ligands for GPR55: Antagonists. National Institutes of Health; Bethesda, MD: 2010. [PubMed] [Google Scholar]
- 12.Kotsikorou E, Sharir H, Shore DM, Hurst DP, Lynch DL, Madrigal KE, Heynen-Genel S, Milan LB, Chung TDY, Seltzman HH, Bai Y, Caron MG, Barak LS, Croatt MP, Abood ME, Reggio PH. Biochem. 2013;52:9456. doi: 10.1021/bi4008885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kapur A, Zhao P, Sharir H, Bai Y, Caron MG, Barak LS, Abood ME. J. Biol. Chem. 2009;284:29817. doi: 10.1074/jbc.M109.050187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Verheijen JC, Richard DJ, Curran K, Kaplan J, Lefever M, Nowak P, Malwitz DJ, Brooijmans N, Toral-Barza L, Zhang W-G, Lucas J, Hollander I, Ayral-Kaloustian S, Mansour TS, Yu K, Zask A. J. Med. Chem. 2009;52:8010. doi: 10.1021/jm9013828. [DOI] [PubMed] [Google Scholar]
- 15.Jiang L-L, Tan Y, Zhu X-L, Wang Z-F, Zuo Y, Chen Q, Xi Z, Yang G-F. J. Ag. Food Chem. 2009;58:2643. doi: 10.1021/jf9026298. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.