

Abstract
In this study we identified the mechanisms underlying the inhibitory effects of NF-κB on the expression of genes encoding multiple liver transport proteins. Well-conserved NF-κB binding sites were found in the promoters of farnesoid X receptor (FXR)-target genes. An electromobility shift assay (EMSA) demonstrated the specific interaction between the NF-κB p65 protein and a 32P-labeled BSEP NF-κB response element (NF-κBE). Chromatin immunoprecipitation (ChIP) analysis confirmed binding of NF-κB p65 to the BSEP locus but not the FXRE in vitro. NF-κB p65 overexpression in Huh-7 cells markedly repressed FXR/RXR transactivation of the BSEP, ABCG5/G8, MRP2, and FXR promoters, which was totally reversed by expression of the IκBα super-repressor. NF-κB interacted directly with FXR on coimmunoprecipitation, suggesting another level for the inhibitory effects of NF-κB on FXR-target genes. In vivo ChIP analysis with liver nuclei obtained from mice after 3 days of common bile duct ligation (BDL) or 6 h post-lipopolysaccharide (LPS) injection showed a markedly increased recruitment of NF-κB p65 to the Bsep promoter compared with controls. There was also increased recruitment of the corepressor silencing mediator of retinoic acid and thyroid hormone receptor (SMRT) and histone deacetylase (HDAC)3 and HDAC2 to the NF-κB sites. We also found that NF-κB p65 was recruited to NF-κB binding sites in the promoters of organic solute transporter, OSTα and OSTβ, and unexpectedly activated rather than repressed gene expression. In mouse liver after BDL NF-κB recruitment to Ostα and Ostβ promoters was associated with increased binding of the potent coactivator cAMP response element binding protein (CREB)-binding protein (CBP)/p300 to the NF-κBE and depletion of CBP/p300 at the FXR element. Overall, these studies demonstrate a novel role for NF-κB in adaptation to obstructive and LPS-induced cholestasis acting through recruitment to specific NF-κB binding sites in the promoters of FXR-target genes and possibly through direct interaction with FXR.
Keywords: NF-κB, FXR, bile acid transporters, gene regulation, cholestasis
several groups including our own have studied the transcriptional regulation of hepatocyte transporters during inflammation-induced and obstructive cholestasis (13, 14, 20, 28). There may be mechanistic overlap between these forms, particularly as absorption of intestinal lipopolysaccharide (LPS) and inflammation assume a greater role in liver injury with increasing duration of bile duct obstruction. Inflammatory signals in hepatocytes induced by LPS directly or indirectly through effector cytokines such as TNFα, IL-1β, or IL-6 reduce the expression levels of the principal transporters for bile acid uptake (NTCP, SLC10a1) and biliary excretion (BSEP, ABCB11) at the gene and protein levels (15). The potentially critical contributions of epigenetic modifications and assembly of coregulators to this process have not been well studied (19).
Transcription factors of NF-κB family play a key role in LPS-induced inflammation (5, 25). NF-κB works in synergy with epigenetic mechanisms including recruitment of histone deacetylases, corepressors, and changes in DNA and histone methylation (4, 17, 23). The different NF-κB dimers exhibit varying affinities for NF-κB binding sites (GGGRNNYYCC; R is purine, Y is pyrimidine, and N is any base) and differ in their ability to activate or repress transcription (24, 25). A heterodimer of RelA (p65) and p50 subunits is the most common combination in the canonical NF-κB signaling pathway, whose activity is tightly controlled by a family of natural inhibitors named IκBs α, β, and ε. In response to cell stimulation, such as by LPS, IκBα is phosphorylated by IκB kinase (IKK) leading to polyubiquitination and 26S proteasome-mediated degradation, allowing NF-κB to translocate into the nucleus (5).
NF-κB is known to downregulate farnesoid X receptor (FxR)-target genes (12, 29, 32), but surprisingly the mechanisms for this effect have not been well defined in the liver. Direct interaction of FXR with NF-κB has been demonstrated by others and will be further examined by us, but the significance and specificity of this mechanism defined by overexpression of FXR and NF-κB in vitro are uncertain (12). In addition, we have discovered well-conserved NF-κB binding sites in the promoters of several genes encoding critical liver transport proteins. The purpose of this study was to determine the significance of these mechanisms as part of an adaptive response to experimental cholestasis.
MATERIALS AND METHODS
Cells and cell culture.
The human hepatoma cell line Huh-7 and the mouse hepatoma cell line Hepa-1 were cultured in RPMI 1640 medium with fetal bovine serum (FBS) and antibiotics. All cells were grown in 5% CO2 in a humidified incubator maintained at 37°C. The cell lines were obtained from the American Tissue Culture Collection.
Chemicals/reagents.
All chemicals were obtained from Sigma-Aldrich (St. Louis, MO) unless stated otherwise. siRNA for human NF-κB p65 (RELA-5970) was obtained from GE Darmacon (Lafayette, CO). Antibodies to NF-κB p65 (ab7970), nuclear receptor corepressor (NCoR; ab3482), and histone deacetylase 1 (HDAC1; ab31263) were from Abcam (Cambridge, MA). Anti-GFP (sc-8334) antibodies was purchased from Santa Cruz Biotechnology (Paso Robles, CA). The anti-HDAC2 and HDAC3 antibodies were purchased from Cell Signaling Technology (Danvers, MA). The anti-silencing mediator of retinoic acid and thyroid hormone receptor (anti-SMRT) antibody was from EMD/Millipore (Billerica, MA).
Animal studies.
All animal studies were approved by the Institutional Animal Care and Use Committee of University of Colorado, Denver.
All animals received humane care according to the criteria outlined in the Guide for the Care and Use of Laboratory Animals prepared by the National Academy of Sciences and published by the National Institutes of Health (NIH publication 86-23, revised 1985).
Common bile duct ligation (BDL) in male C57Bl/6 mice was done at 10–12 wk of age as described by us previously (3, 6). Sham surgery in control mice was accomplished by laparatomy and manipulation of the liver, but the bile duct was not ligated. Livers from sham-operated and bile duct-ligated mice were collected at 3 days postligation.
For the LPS model, LPS (2.0 mg/kg) or saline vehicle was administered to male C57BL6 mice at 10–12 wk of age by intraperitoneal injection, and the mice were killed 6 h later.
Plasmid constructs.
The human BSEP promoter DNA was generated by PCR and subcloned into the luciferase expression vector pSV0AL5Δ(pΔ1445/Luc), as described by us previously (1, 2, 3). The human FXR promoter sequence was amplified by PCR from the genomic DNA and subcloned into the pGL3 luciferase vector into Kpn1/Nhe1 sites. The promoters for the mouse pGL3/Ostα and pGL3/Ostβ were a generous gift from Dr. Paul Dawson (Emory University School of Medicine, Atlanta, GA). Promoter constructs for the human and mouse pGL3/ABCG5/G8 were generously provided by Dr. Tiangang Li (University of Kansas) and Dr. Alan Remaley (National Institutes of Health, Bethesda, MD). Plasmids encoding FXR and RXRα were supplied by Dr. David Mangelsdorf (Dallas, TX). Expression plasmids IκBα super-repressor (IκBα-SR; Addgene-24143), pCMV4 p65 (Addgene-21966), and PCMV4 p50 (Addgene-21965) were obtained from (Addgene, Cambridge, MA). Expression plasmids NCoR and SMRT were obtained from Dr. Philippe Lefebvre (European Genomic Institute for Diabetes, Lille, France). All of the positive clones containing cDNA inserts were verified by restriction enzyme mapping and sequenced using the ABI automated DNA sequencer model 377.
Transient transfections and luciferase assays.
Huh-7 and Hepa-1 cells were plated at a concentration of 1 × 105 cells/well in 24-well plates for 48 h. Cells were transfected at day 0 with the human (mouse) BSEP (Bsep), MRP2 (Mrp2), ABCG5-ABCG8, OSTα/OSTβ, or FXR promoter at 0.5 μg/well (in triplicate/per group) and also cotransfected with 50 ng of FXR/RXRa and various amounts of NF-κB expression plasmids in OPTI-MEM (Invitrogen, Grand Island, NY). Transfections were carried out using TransIT-LT (Mirus Bio, Madison WI) at a DNA:TransIT ratio of 1:3. Following transfection with nuclear receptor plasmids, the cells were suspended in RPMI-1640 medium containing charcoal-stripped 10% FBS. The FXR ligand GW4064 (1 μM) was added at this time and luciferase activities were measured 24 h later using the Promega kit (Promega, Madison, WI). Normalization of transfection efficiencies in the different wells was achieved by cotransfection with pCMV-β galactosidase and the assay of galactosidase activity.
Electrophoretic mobility shift assays.
The electrophoretic mobility shift assay (EMSA) was performed as reported previously by us and others (1, 18). Oligonucleotides for the EMSA are shown in Table 1. In brief, NF-κB p65 cDNA (5 μg/dish) was transfected into Huh-7 cells (5 × 106cells/100-mm dish) in three dishes using Lipofectin at a DNA/Lipofectin ratio of 1:3. Control untransfected cells were left in RPMI 1640 medium. Lipofectin was replaced with RPMI 1640 medium 1 day later, and 3 days later nuclear extracts were prepared using NE-PER from Thermo Fisher Scientific (Grand Island, NY) according to the manufacturer's directions. Nuclear extracts were stored in aliquots at −80°C until used. The BSEP NF-κB p65 oligonucleotide probe for the EMSAs was end labeled with [γ-32P]ATP (3,000 or 6,000 mCi/mmol) by T4 polynucleotide kinase. As a control, the probe was also incubated with the same amount of unprogrammed cell lysate. In competition assays, unlabeled wild-type or mutant oligonucleotides were added to the reaction 15 min before the addition of the probe. DNA-protein complexes were resolved on 4% native polyacrylamide gel electrophoresis containing 0.5× TBE (0.89 M Tris, 0.89 M boric acid, and 0.02 M disodium EDTA for 10× TBE). The gel was dried and exposed to x-ray film for varying lengths of time until a suitable image was obtained.
Table 1.
Oligonucleotides for EMSA, ChIP assays, and primer sets for RT-PCR
| Gene | Sequence 5′-3′ |
|---|---|
| Primers used for qPCR analysis | |
| hBSEP | |
| Forward | aca tgc ttg cga gga cct tta |
| Reverse | gga ggt tcg tgc acc agg ta |
| hFXR | |
| Forward | gac ttt gga cca tga aga ccag |
| Reverse | gcc cag acg gaa gtt tct tatt |
| hOSTα | |
| Forward | ctg ggc tcc att gcc atc tt |
| Reverse | cac ggc ata aaa cga ggt gat |
| hOSTβ | |
| Forward | gca gct gtg gtg gtc att at |
| Reverse | tag gct gtt gtg atc ctt gg |
| hNF-κB p65 | |
| Forward | ccc cac gag ctt gta gga aag |
| Reverse | cca ggt tct gga aac tgt gga t |
| h36B4 | |
| Forward | gca atg ttg cca gtg tct gt |
| Reverse | gcc ttg acc ttt tca gca ag |
| Primers used in ChIP analysis | |
| hBSEP/FXRE site | |
| Forward | ggg ttt ccc aag cac act ctg tgt tt |
| Reverse | gag gaa gcc aga gga aat ggt gg |
| hBSEP/NF-κBE site | |
| Forward | atg ttc ttt tag ggt att tgt ctc |
| Reverse | tac agg cct gta gtt gtg aaa agt tac |
| mBsep/NF-κBE site | |
| Forward | gaa gag tcg ggc ctc tca cca ggc t |
| Reverse | agt cca gat cta gca cag ttc agt g |
| mOstα/NF-κBE site | |
| Forward | agc tct gac act tag atg cta cac |
| Reverse | gcc acc atg cct ggc ttc ta |
| mOstβ/NF-κBE site | |
| Forward | tgg tct ggc ctg cct cga tag |
| Reverse | tgg cga agg tca tga tat gaa c |
| Primers used in control ChIP analysis | |
| hBSEP | |
| Forward | aag cac tgg ccc atc aat tg |
| Reverse | ctc cta agg tgt aac aac t |
| mBsep | |
| Forward | ctc gag att tca cac aag tct aac aac t |
| Reverse | gtt cct gaa atg agg tta gtt |
| mOstα | |
| Forward | tag atg tgg agc ctt gat gag c |
| Reverse | atg gta cag atg gat gga g |
| mOstβ | |
| Forward | tgg gcc tgc ttc ctc ctc |
| Reverse | cag gaa gga gtc aag gct ct |
| Primers used in EMSA analysis | |
| hBSEP wt | |
| Forward | caa cca ggg att ttc caa gag ca |
| Reverse | tgc tct tgg aaa atc cct ggt tg |
| hBSEP mut | |
| Forward | caa cca gcc att ttc caa gag ca |
| Reverse | tgc tct tgg aaa atg gct ggt tg |
| Sequences of siRNAs used in this study | |
| hNF-κB p65 (catalog no. L-003533-00-0005; GE Dharmacon) | |
| siRNA pool 1 | GGAUUGAGGAGAAACGUAA |
| siRNA pool 2 | CCCACGAGCUUGUAGGAAA |
| siRNA pool 3 | GGCUAUAACUCGCCUAGUG |
| siRNA pool 4 | CCACACAACUGAGCCCAUG |
| Nontargeting siRNAs (catalog no. D-001810-01-20; GE Dharmacon) | |
| siRNA pool 1 | UGGUUUACAUGUCGACUAA |
| siRNA pool 2 | UGGUUUACAUGUUGUGUGA |
| siRNA pool 3 | UGGUUUACAUGUUUUCUGA |
| siRNA pool 4 | UGGUUUACAUGUUUUCCUA |
EMSA, electromobility shift assay; ChIP, chromatin immunoprecipitation; qPCR, quantitative polymerase chain reaction.
For in vitro experiments, a cDNA encoding human NF-κB p65 was transcribed and translated using the TNT-coupled reticulocyte lysate system (Promega) according to the manufacturer's instructions. Aliquots of the transcribed/translated extracts were used for EMSAs as described above. A super-shifted band was demonstrated with the addition of an NF-κB p65 antibody.
siRNA-mediated knockdown of NF-κβ p65.
Sequences of siRNAs used in this study are shown in Table 1. Huh-7 cells were plated in six-well plates (1 × 106 cells/well) and incubated 2 days later with 50 nM siRNA using TransIT-TKO (Mirus Bio) at a ratio of 1:1 (μl/μl) according to the manufacturer's instructions. Six hours later, medium was added to the wells, and 24 h later, spent medium was replaced with fresh RPMI 1640. Forty-eight hours later, total RNA was prepared using the TRIzol kit (Thermo-Fisher-Invitrogen), and real-time PCR analysis was done after conversion of mRNA into cDNA.
Chromatin immunoprecipitation analysis of cultured cell lines and mouse liver.
Chromatin immunoprecipitation (ChIP) assays were done by a combination of protocols previously used by us and manufacturer's instructions using EZ ChIP/MagnaChIP G kit from EMD/Millipore (3, 6). Oligonucleotides for ChIP assays are shown in Table 1. Briefly, cells were harvested after fixation with 1% formaldehyde. After cell lysis and sonication, the fragmented DNA was diluted in ChIP dilution buffer and preadsorbed with protein G-Sepharose/salmon sperm DNA (EMD/Millipore) for 1 h at 4°C. Then, 5% of the chromatin was removed and saved as input. It was then incubated overnight at 4°C with 3–5 μg of the appropriate antibodies or normal mouse IgG (control). Antibody-chromatin complexes were captured by incubation with protein G-Sepharose and centrifuged. Reversal of protein cross linking and proteinase K digestion, followed by purification of the DNA, was then achieved. An aliquot of the DNA (2 μl) was used in a PCR (standard and quantitative) reaction using specific primers flanking the FXR element (FXRE) or NF-κB element (NF-κBE) of human BSEP and OSTα and OSTβ promoters. Primers flanking a site distant from the FXRE and NF-κB sites were used as negative controls. PCR products were run in a 2% agarose gel and stained with ethidium bromide to confirm the amplicon size.
The method for in vivo ChIP analysis of liver has been modified from protocols for cells (3, 6, 8). In brief, mouse livers were sliced to small pieces and then incubated with 1% formaldehyde to cross-link proteins to genomic DNA in cells. Following this incubation, excess formaldehyde was quenched by incubation with glycine. Chromatin was prepared from cross-linked livers after isolation of nuclei. Chromatin was sonicated with the appropriate power setting to shear DNA to ∼500-bp fragments for use in ChIP.
Quantitative real-time PCR.
Quantitative real-time PCR was done using the QuantiTect SYBR Green PCR Kit (Qiagen) in combination with primers for BSEP, FXR, NF-κB p65, OSTα (Ostα), or OSTβ (Ostβ) in a Step One Plus Real-time PCR system (ABI), as previously described by us (2, 6). Primer sets for RT- PCR are shown in Table 1.
Immunoprecipitation.
Immunoprecipitation (IP) or coimmunoprecipitations from GFP/FXR and NF-κB p65-transfected Huh-7 cells were done with total cell lysates. Cell lysates prepared in IP buffer (50 mM Tris, pH 8, 150 mM NaCl, 0.5% Nonidet P-40, and 5 mM EDTA plus protease inhibitor mixture; Roche Diagnostics, Indianapolis, IN) were precleared with protein A-agarose beads for 30 min and incubated overnight with anti-GFP antibody (Santa Cruz Biotechnology), anti-NF-κB p65 antibody (Abcam), or mouse IgG (Santa Cruz Biotechnology) at 4°C. The bead-bound immunoprecipitates were captured by centrifugation at 2,500 g, washed twice with IP buffer, and then dissociated from the beads, after which the recovered supernatant (using 2× Laemmli's sample buffer at 98°C) was used for Western blot analysis after fractionation on 10% SDS-PAGE. For the immunoblot, GFP and NF-κB p65 antibodies were used to detect the protein.
Statistics.
Data are expressed as mean ± SE and analyzed by the two-tailed paired Student' t-test using Prism software. P < 0.05 was considered statistically significant. All experiments using cultured cells or mouse livers were repeated at least three times.
RESULTS
Functional NF-κB binding sites are present in the promoters of genes encoding liver transport proteins.
On in silico analysis (Table 2) we identified previously unknown NF-κB binding sites in the human (mouse) BSEP (Bsep), MRP2 (Mrp2), FXR (Fxr), MDR3 (Mdr2), SHP (Shp), ABCG5/G8 (AbcG5/G8), and OSTa (Ostα)/OSTβ (Ostβ) promoters that suggest a novel regulatory mechanism for effects of NF-κB on FXR targets. As a representative example, experiments were first done in Huh-7 cells to show that the NF-κB binding site in the BSEP promoter was functional.
Table 2.
NF-κB binding sites in the promoters of human and mouse genes encoding liver transport proteins
| Gene | Position | Sequence | Position |
|---|---|---|---|
| hOSTα | −404 | GTGGGGTTTCCCAG | −390 |
| mOstβ | −416 | GTAGCTAAGTCCCTT | −412 |
| hOSTα | −604 | CAGGGGGTGCCCTCT | −590 |
| mOstβ | −270 | CTGTGGATGCCCTGA | −258 |
| hABCG5/G8 | INTERGENIC REGION | GGCCAAGTCCCA | |
| mAbcg5/g8 | INTERGENIC REGION | CAGGGCACTCCCA | |
| hFXR | −887 | TGGGGATTTTCGATG | −878 |
| mFxr | −99 | GCTGGCAATTCCAAG | −85 |
| hSHP | −99 | TTGGGCCATTCCC | −85 |
| mShp | −62 | TTTGGCCAGTCCCCT | −56 |
| hMRP2 | −190 | CCTAGGGCTTTTTAG | −176 |
| mMrp2 | −135 | TCTGGTGATTCCCAG | −121 |
| hBSEP | −371 | GGGATTTTCC | −362 |
| mBsep- | −165 | TCAGAAGGTCCCCA | −150 |
| hMDR3 | −115 | GTAGGCGTTTCCGG | −100 |
| mMdr2 | −469 | GAAGAAAAGCCCCTG | −455 |
An EMSA demonstrated the specific interaction between the NF-κB p65 protein and a 32P-labeled BSEP NF-κBE. The addition of nuclear extract from NF-κB p65-transfected Huh-7 cells led to the appearance of a shifted DNA-protein complex (Fig. 1A, lane 2). This shifted band was not detected using extracts prepared from untransfected cells (Fig. 1A, lane 1). NF-κB p65 binding was specifically competed off by the addition of a 25-fold molar excess of wild-type oligo (Fig. 1A, lane 3), but not by an NF-κB p65 mutant oligonucleotide (Fig. 1A, lane 4), demonstrating the specificity of the complex.
Fig. 1.
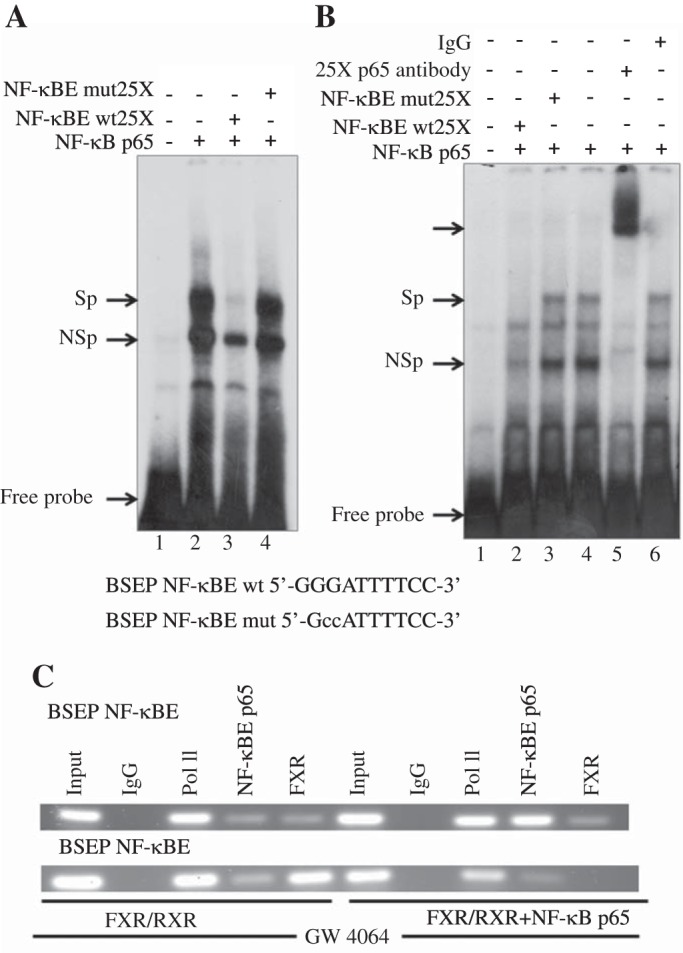
NF-κB p65 binds to an NF-κB binding element (NF-κBE) in human BSEP promoter. A: Huh-7 cells were transfected with an expression plasmid for human NF-κB p65. Nuclear extracts were prepared using the NE-PERkit (Thermo Fisher Scientific) according to the manufacturer's directions. EMSAs were carried out as described in materials and methods using a 32P-labeled human BSEP NF-κBE. A, top: cDNA used for transfection of Huh-7 cells from which the nuclear extracts were prepared and the probes and competitors used (at 25-fold excess) in the assay. Sp, specific DNA-protein complex; NSp, nonspecific complex. DNA sequence of the wild-type (wt) and mutant (mut) NF-κBE shown with the mutated residues indicated in lowercase. B: in vitro translated NF-κB p65 was used in EMSA with BSEP NF-κBE as a probe as described in materials and methods.“Unprogrammed” reticulocyte lysate (lane 1) and reticulocyte lysates programmed with NF-κB p65 (lanes 2–6) cDNA were incubated with BSEP NF-κBE probe. Competition analysis was performed with unlabeled 25-fold molar excess of wild-type (lane 2) or mutant NF-κBE (lane 3). Lane 5 was incubated with NF-κB p65 antibody. C: chromatin immunoprecipitation (ChIP) analysis using Huh-7 cells transfected with NF-κB p65 confirmed binding of NF-κB p65 to the BSEP NF-κBE but not the farnesoid X receptor (FXR) element. Gel shows RT-PCR reaction products generated using primers designed to amplify BSEP NF-κB p65 binding site. In these cells transfected with FXR/RXR recruitment or binding of FXR/RXR to the FXRE in BSEP promoter was blocked by expression of NF-κB p65 There was no recruitment of NF-κB p65 to the FXRE. IgG and Pol II are negative and positive controls, respectively.
The EMSA results were verified in a cell-free system using in vitro translated NF-κB p65 (Fig. 1B). Binding of the NF-κB p65 protein to the 32P-labeled BSEP NF-κBE was again demonstrated (Fig. 1B, lanes 4 and 6). NF-κB p65 binding was specifically competed off by the addition of a 25-fold molar excess of wild-type oligo (Fig. 1B, lane 2) but not an NF-κB p65 mutant (Fig. 1B, lane 3). A super-shifted band was demonstrated with the addition of an NF-κB p65 antibody (Fig. 1B, lane 5).
ChIP was next used to explore interactions between NF-κB p65 and DNA within the natural chromatin context of the cell. ChIP analysis (Fig. 1C) using Huh-7 cells transfected with NF-κB p65 and FXR/RXR showed recruitment of NF-κB p65 to the NF-κB binding site in BSEP promoter compared with cells without NF-κB overexpression (Fig. 1C, top row). In these cells NF-κB p65 was not recruited to the FXRE, and FXR binding or recruitment to the FXRE in BSEP promoter was blocked by overexpression of NF-κB p65 (Fig. 1C, bottom row).
Since NF-κB is found at low levels in nuclei of unstimulated cells (7, 25), it may act to repress and/or activate transcription in the absence of an activation signal. Indeed, siRNA knockdown of NF-κB p65 expression (Fig. 2, A and B) was confirmed by a marked decrease in NF-κB p65 protein and mRNA in Huh-7 cells. We found that siRNA knockdown of NF-κB p65 led to a two- to threefold increase in BSEP and FXR mRNA levels in Huh-7 cells (Fig. 2, C and D), indicating that NF-κB may have some regulatory role under basal conditions.
Fig. 2.

Effect of siRNA knockdown of NF-κB p65 on endogenous expression of BSEP and FXR mRNA in Huh-7 cells. Immunoblot analysis (A) of Huh-7 cell extracts confirms siRNA depletion of NF-κB p65 protein compared with cells treated with a control siRNA. Knockdown of NF-κB p65 (B) is associated with a significant increase in both FXR and BSEP mRNA levels (C and D) in Huh-7 cells compared with cells transfected with a nonsilencing siRNA as assessed by real-time quantitative PCR. *P < 0.05.
Effect of NF-κB expression on transactivation of the BSEP (bsep), MRP2 (Mrp2), and ABCG5-ABCG8 promoters.
Next, Huh-7 cells were transiently transfected with FXR/RXR together with plasmids containing the BSEP, ABCG5-ABCG8, MRP2, or FXR or promoter sequences that included the NF-κBE linked to luciferase as reporter. As expected, transfection with FXR/RXR in the presence of the FXR ligand GW4064 led to a marked transactivation of these promoters (Fig. 3, A–D). In contrast, NF-κB p65 overexpression markedly repressed ligand-induced FXR/RXR transactivation of the BSEP, MRP2, FXR, and ABCG5/G8 promoters, which was totally reversed by expression of the IκBα super repressor promoters (Fig. 3, A–D). However, when the NF-κB binding site in the BSEP promoter was mutated, NF-κB p65 dose dependently inhibited ligand-induced transactivation of the BSEP promoter, and this effect was reversed by expression of the IκBα super-repressor (Fig. 4, A and B). However, it was clear that at the lowest dose of NF-κB p65 overexpression (25 ng) the majority of inhibition could be attributed to the NF-κB binding site (∼57% inhibition with the wild-type promoter vs. ∼17% with the promoter after mutation of the NF-κB binding site).
Fig. 3.

NF-κB p65 expression suppresses the transactivation of the BSEP, MRP2, FXR, and ABCG5-ABCG8 promoters. Huh-7 cells were transiently transfected with FXR/RXR and plasmid vectors containing the BSEP (A), MRP2 (C), FXR (D), or ABCG5-ABCG8 (B) promoter sequences that included the NF-κBE linked to luciferase as reporter. FXR transactivation of the promoter constructs was monitored by luciferase activity. As expected, transfection with FXR/RXR in the presence of the FXR ligand GW4064 led to a marked transactivation of these promoters (A–D). In contrast, NF-κB p65 overexpression (50 ng) markedly repressed ligand-induced FXR/RXR transactivation of the BSEP and ABCG5/G8 promoters, which was totally reversed by expression of the IκBα super-repressor. RLU, relative light units. #P < 0.05.
Fig. 4.

Effect of mutation of the BSEP NF-κB binding site on the transactivation of BSEP promoter. Huh-7 cells were transiently transfected with FXR/RXR and plasmid vectors containing the BSEP promoter sequences that either included the NF-κBE or with a construct of a mutated NF-κB binding site linked to luciferase as reporter. A: suppression of the BSEP promoter by NF-κB p65 was dose dependent and reversed by expression of the IκBα super-repressor. When The NF-κB binding site in the BSEP promoter was mutated (B), NF-κB p65 still dose dependently inhibited ligand-induced transactivation of the mutant BSEP promoter, and this effect was reversed by expression of the IκBα super-repressor. However, at the lowest dose of NF-κB p65 overexpression (25 ng), the majority of inhibition could be attributed to the NF-κB binding site (∼57% inhibition with the wild-type promoter vs. ∼17% with the promoter after mutation of the NF-κB binding site). Each histogram is ±SE from representative experiments done at least 3 times. #P < 0.05.
Since the significance of the NF-κB expression was assessed in mouse models of cholestasis, the effects of NF-κB p65 expression on the transactivation of the mouse Bsep and Mrp2 promoters were also examined in the mouse hepatoma cell line Hepa-1. Similar to the experiments with human promoters, NF-κB p65 overexpression markedly repressed ligand-induced FXR/RXR transactivation of the Bsep and Mrp2 promoters, which was totally reversed by expression of the IκBα super repressor (Fig. 5, A and B).
Fig. 5.

Expression of the mouse Bsep and Mrp2 promoters is suppressed by NF-κB p65. The mouse liver cell Hepa-1 was transiently transfected with FXR/RXR and plasmid vectors containing the Bsep (A) and Mrp2 (B) promoter sequences that included the NF-κBE linked to luciferase as reporter. Transfection with FXR/RXR in the presence of the FXR ligand GW4064 led to a marked transactivation of these promoters (A and B). In contrast, NF-κB p65 overexpression (50 ng) markedly repressed ligand-induced FXR/RXR transactivation of these promoters, which was totally reversed by expression of the IκBα super-repressor. #P < 0.05.
NF-κB p65 interacts directly with FXR.
Previous studies have suggested that NF-κB p65 interacts physically with FXR based on glutathione S-transfersase (GST)-pull down assays using NF-κB p65 and FXR transcribed and translated in vitro (12). Using a different approach, we next asked whether FXR and NF-κB p65 directly interact with each other by coimmunoprecipitation. NF-κB p65 was first immunoprecipitated using an NF-κB p65 antibody from a whole cell lysate of Huh-7 cells overexpressing FXR-GFP and NF-κB p65. FXR association with NF-κB p65 was demonstrated on Western blot analysis with anti-GFP antibody (Fig. 6A). FXR-GFP also coimmunoprecipitated with NF-κB p65 from a Huh-7 cell lysate when FXR-GFP was first immunoprecipitated with anti-GFP antibody, run on a Western blot, and probed with a anti- NF-κB p65 antibody (Fig. 6B). These data further support the association between NF-κB p65 and FXR, but the importance of this mechanism will likely depend on the relative affinity of NF-κB for FXR compared with its binding site as well as the relative amounts of FXR and NF-κB found in vivo in normal and cholestatic livers.
Fig. 6.
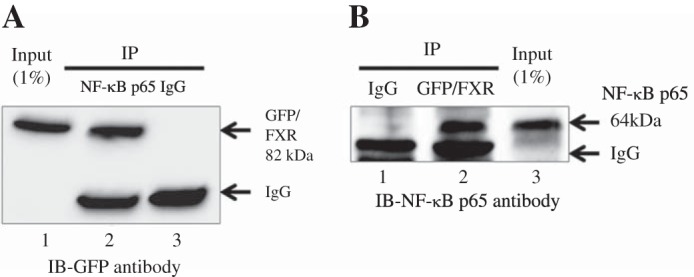
NF-κB p65 associates with FXR in vivo Huh-7 cells were transiently cotransfected with the FXR-GFP and NF-κB p65 constructs 1 day before collecting the cells. A: when NF-κB p65 was immunoprecipitated (IP) from a whole cell lysate of Huh-7 cells, association was demonstrated with FXR on Western blotting with a anti-GFP antibody (lane 2). B: FXR-GFP was also coimmunoprecipitated with NF-κB p65 from a Huh-7 cell lysate when FXR-GFP was first immunoprecipitated, run on a Western blot, and probed with a NF-κB p65 antibody (lane 2). The input lysate was at 1% of the lysate used for immunoblot (IB).
Identification of corepressors involved in NF-κB-mediated gene repression.
To determine if corepressors contribute to the inhibitory effects of NF-κB, we tested the effects of the corepressors SMRT and NCoR1 on ligand-induced transactivation of the BSEP promoter. Figure 7 shows that NF-κB p65 overexpression in Huh-7 cells markedly repressed ligand-induced transactivation of a BSEP-luciferase promoter construct that included the NF-κB binding site. Expression of the corepressor SMRT but not NCoR further enhanced NF-κB repression of the BSEP promoter. SMRT had some effect when expressed alone, possibly acting with endogenously expressed NF-κB or through an unidentified repressive factor.
Fig. 7.

The corepressor silencing mediator of retinoic acid and thyroid hormone receptor (SMRT) acts synergistically with NF-κB p65 to suppress ligand-induced transactivation of the BSEP promoter. Huh-7 cells were transiently transfected with FXR/RXR and plasmid vectors containing the BSEP- promoter sequence that included the NF-κBE linked to luciferase as reporter. Transfection with FXR/RXR in the presence of the FXR ligand GW4064 led to a marked transactivation of these promoters. NF-κB p65 markedly suppressed ligand-induced transactivation of the FXR promoter. SMRT but not nuclear receptor corepressor (NCoR) expression further enhanced the inhibitory effect of NF-κB p65. #P < 0.05.
The role of NF-κB and associated corepressors in experimental cholestasis.
The role of NF-κB binding sites in regulating FXR-target gene expression was further investigated using in vivo mouse models of experimental cholestasis. Figure 8 depicts in vivo ChIP assays done with liver nuclei obtained from mice after 3 days of common BDL or 6 h post-LPS injection. As shown in liver cell lines, NF-κB p65 can be detected at the NF-κB binding site of the Bsep promoter in sham-operated and vehicle-treated mice. On in vivo ChIP assays there was a markedly increased recruitment of NF-κB p65 to the Bsep promoter in both common BDL and LPS-treated mice compared with controls (Fig. 8, A and B). There was also increased recruitment of the corepressor SMRT but not NCoR to the NF-κB site in both models. We also found increased recruitment of HDAC 2 and 3 but not HDAC1 to this locus. HDAC3 is often found as part of the SMRT corepressor complex (22, 31). HDAC2 is known to interact directly with NF-κB (4, 17, 22), but how it participates in the process is unknown.
Fig. 8.

Chromatin recruitment of NF-κB p65, SMRT, and histone deacetylases (HDACs) to the Bsep promoter after common bile duct ligation or treatment with LPS in mice. In vivo ChIP analysis of the Bsep promoter using mouse liver after 3 days of common bile duct ligation (BDL) or after sham surgery (A) and 6 h after treatment with lipopolysaccharide (LPS; 2.0 mg/kg; B) or saline vehicle by intraperitoneal injection in male C57BL6 mice using antibodies to NF-κB p65, SMRT, NCoR, HDAC1, HDAC2, HDAC3, and RNA polymerase II. On in vivo ChIP assays using quantitative RT-PCR there was a markedly increased recruitment of NF-κB p65 to the Bsep promoter in both BDL and LPS treated mice compared with controls. There was also increased recruitment of the corepressor SMRT but not NCoR to the NF-κB site. Additionally increased recruitment of HDAC 2 and 3 but not HDAC1 to this locus was observed. *P < 0.05.
We also found increased recruitment of NF-κB p65 to the FXR promoter in BDL and mice, providing evidence that NF-κB can interfere with FXR signaling at several levels (not shown).
These findings indicate that NF-κB activation in the context of cholestasis establishes a repressive chromatin environment at the promoters of several FXR-target genes through recruitment of HDACs and corepressors. This may be the predominant mechanism for the inhibitory effects of NF-κB in vivo, particularly for many promoters where FXR is already bound.
Activation of OSTα/OSTβ by NF-κB.
The mechanisms underlying the upregulation OSTα/OSTβ in cholestasis have been enigmatic (26, 34). We first examined the effect of siRNA knockdown of NF-κB p65 on the expression of endogenous expression of OSTα/OSTβ in Huh-7 cells. Depletion of NF-κB p65 led to a significant decrease in endogenous OSTα and OSTβ mRNA in Huh-7 cells (Fig. 9).
Fig. 9.

Effect of siRNA knockdown of NF-κB p65 on expression of OSTα siRNA depletion of NF-κB p65 led to a marked decrease in OSTα (A) and OSTβ (B) mRNA in Huh-7 cells as measured by RT-PCR compared with untreated cells or cells treated a nontargeting siRNA. *P < 0.05.
In vitro and In vivo studies then were used to further examine the effect of NF-κB on expression of OSTα and OSTβ. Both the hOSTα and mOSTβ promoters have NF-κB binding sites (Table 2). In contrast to the inhibitory effect of p65 on expression of the BSEP, ABCG5/G8, MRP2, and FXR promoters, we found that both NF-κB p65 and p50 expression (Fig. 10, A and B) individually or together dose dependently activated the OSTα and OSTβ promoters in Huh-7 cells.
Fig. 10.

Activation of OSTα and OSTβ by NF-κB. Huh-7 cells were transiently transfected with NF-κB p65, p50, and plasmid vectors containing OSTα or OSTβ promoter sequences that included the NF-κBE linked to luciferase as reporter. Transactivation of the OSTα (A) and OSTβ (B) promoter constructs by NF-κB subunits was monitored by luciferase activity. NF-κB p65 and p50 expression (A and B) individually or together dos dependently activated the OSTα and OSTβ promoters in Huh-7 cells. *P < 0.05.
Next we confirmed on ChIP analysis that there was increased recruitment of NF-κB p65 to the Osta and Ostβ promoters in BDL mice compared with sham-operated controls (Fig. 11, A and B). Previous studies have shown that NF-κB, acting as a transcriptional activator, recruits a coactivator complex, which usually includes the cAMP response element binding protein (CREB)-binding protein (CBP), p300, members of the p160 family of coactivators, and the CBP-associated factor (p/CAF) (11, 16). CBP/p300 is particularly important in providing a platform for recruitment of a variety of these proteins required for NF-κB-dependent gene expression. Therefore, we tested whether there was increased recruitment of CBP/p300 to the NF-κB locus of Ostα and Ostβ promoters in the BDL mice. On ChIP analysis using an antibody that recognizes the homologous CBP and p300 proteins, there was increased recruitment of CBP/p300 to the NF-κB site in the Ostα and Ostβ promoters in BDL mice compared with controls (Fig. 11, A and B). In contrast, there was significant depletion of CBP/p300 at the FXRE of Ostα and Ostβ in these mice compared with controls (Fig. 11, C and 11D).
Fig. 11.

Effect BDL on recruitment of NF-κB p65 and cAMP response element binding protein (CREB)-binding protein (CBP)/p300 to NF-κB binding site in Ostα and Ostβ promoters. In vivo ChIP assays of mouse liver after 3 days of common BDL or after sham surgery in male C57BL6 mice using antibodies to NF-κB p65, CBP/p300, and RNA polymerase II. On quantitative RT-PCR there was a markedly increased recruitment of NF-κB p65 and CBP/p300 to the NF-κB binding site in Ostα (A) and Ostβ (B) promoters in BDL mice compared with controls. In contrast there was significant depletion of CBP/p300 at the FXRE of Ostα (C) and Ostβ (D) in these mice compared with control. *P < 0.05.
DISCUSSION
NF-κB is an inducible transcription factor that regulates the expression of a variety of genes involved in inflammatory responses and cell survival (5). NF-κB is known to downregulate FXR-target genes in cholestasis (15, 34), but surprisingly the mechanisms underlying this effect have not been well defined in liver. After discovery of NF-κB binding sites in the promoters of genes encoding liver transport proteins, we confirmed that these sites were functional in binding NF-κB and in affecting the transcription of FXR target genes in vitro and in vivo and contributed to the suppression of transporter gene expression in experimental cholestasis. The significance of the direct physical interaction between NF-κB p65 and FXR demonstrated by us by coimmunoprecipitation remains uncertain. A straightforward explanation may be that the interaction contributes to transcriptional inhibition by preventing the binding of the FXR to DNA. Indeed, on ChIP analysis NF-κB overexpression is associated with blocking of FXR binding to the FXR, which may be operative for FXR not bound to the promoter.
Overexpression studies in cell culture provide a proof of principle that NF-κB is important in repressing the expression of some FXR-target genes while activating others. However, it is difficult to replicate the complex biology of NF-κB subunits that in the basal state are largely inactive in a complex with IκBs inhibitors. The super-repressor form of IκBα used by us is resistant to both phosphorylation and proteolytic degradation and therefore permanently prevents the nuclear translocation of NF-κB. The majority of inhibition could be attributed to the NF-κB binding site at the lowest dose of NF-κB p65 overexpression in our experiments. Even at this dose the normal interaction of IκBs with NF-κB subunits is likely to be overwhelmed allowing more direct binding of NF-κB p65 with FXR in the cytoplasm and after translocation into the nucleus unimpeded by members of the IκB family. FXR can also interfere with the inhibitory effects of NF-κB, indicating that there can be significant cross talk between these pathways depending on the gene, the cellular context, and the relative amount of each molecule within the hepatocyte (29, 32). In a previous study by Wang et al. (29), NF-κB activation suppressed FXR-mediated gene expression, and conversely FXR activation reduced binding of NF-κB to DNA without regard to the identification of specific NF-κB binding site. There is evidence for cross talk between NF-κB and other nuclear receptors such the glucorticoid receptor, the estrogen receptor, and the peroxisome proliferator-activated receptor (10).
NF-κB has typically been thought of as residing in the cytoplasm in an inactive form bound by its inhibitory proteins, members of the IκB family. However, ∼17% of NF-κB p65 is not complexed with IκBα in a resting cell and may be found in the nuclei of unstimulated cells (7). Evidence indicates that NF-κB may shuttle between the nucleus and cytoplasm in unstimulated cells and, as we have also shown here and others have shown, can be present at promoters in the basal state where it may have some poorly defined regulatory role in repressing and/or activating basal gene expression (7, 24).
Probing the mouse genome using ChIP and DNA sequencing (ChIP-seq) has shown that FXR is prebound to several thousand response elements awaiting the presence of ligands to initiate gene expression (9, 27). In cholestasis we found that NF-κB does not bind to FXR at the FXRE but rather binds specifically to NF-κB sites in the promoters of FXR-target genes with subsequent recruitment of coregulators important for either repression or activation in a gene and cell context specific manner. It is unknown how NF-κB is specifically recruited to a subset of these sites and selectively recruits corepressors or coactivators in cholestasis.
Numerous studies emphasize the crucial involvement of transcriptional corepressor complexes linked to histone deacetylation in inflammatory and metabolic gene regulation (17, 22, 30). NF-κB activation suppresses target gene transcription by recruitment of a corepressor complex consisting of the NF-κB subunits, corepressors NCoR/SMRT, and various combinations of HDACs (4, 17). HDACs catalyze the removal of acetyl groups from lysine residues in histones and nonhistone proteins, most often resulting in transcriptional repression. The HDAC3 protein itself has little enzyme activity but acquires its HDAC function from association with deacetylase-activating domain of SMRT or NCoR (31).
The organic solute transporter Ostα/Ostβ, a unique heteromeric transporter localized to the hepatocyte basolateral membrane, is upregulated in cholestasis where it appears to play a protective role (15, 26, 33). Although FXR can transactivate the promoters of Ostα/Ostβ (21), FXR signaling is compromised at several levels in our cholestatic models including coactivator recruitment to the FXRE. We found that NF-κB is recruited to well conserved sites in the promoters of OSTα and OSTβ and unexpectedly activated rather repressed gene expression. Moreover, NF-κB activation of Ostα/Ostβ in BDL mice was associated with markedly increased recruitment of the potent coactivator CBP/p300 to NF-κB binding site and depletion of CBP/p300 at the FXRE. CBP/p300 is also known to bind to NF-κB p65 (11, 16) and contributes to gene expression by catalyzing acetylation of lysine 9 and 14 of histone H3, which are activating histone modifications. This ubiquitous activator is also critical for ligand-activated induction of FXR target genes such as SHP (11).
In conclusion, we have discovered a new paradigm for adaptation to cholestasis in which the transcription factor NF-κB has a central role acting through specific binding sites in the promoters of FXR-target genes and possibly through direct interaction with FXR. It is well known that a number of critically important liver transport proteins are downregulated at the transcriptional level contributing to the cholestatic process. The resulting retention of bile acids and other cholephiles likely contributes to ongoing hepatocyte injury. However, downregulation of canalicular transporters could also have a protective role when bile flow in compromised by bile duct obstruction. In contrast, Ostα/Ostβ on the basolateral membrane is upregulated through the action of NF-κB and associated coactivators to provide an alternate route for removal of biliary constituents.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grant DK-084434 (to F. J. Suchy).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
N.B., M.A., and F.J.S. conception and design of research; N.B. performed experiments; N.B., M.A., and F.J.S. analyzed data; N.B. interpreted results of experiments; N.B. prepared figures; N.B. and F.J.S. drafted manuscript; N.B. and M.A. approved final version of manuscript; M.A. and F.J.S. edited and revised manuscript.
ACKNOWLEDGMENTS
The technical support of Shuhua Xu and An-Qiang Sun is gratefully acknowledged.
REFERENCES
- 1.Ananthanarayanan M, Balasubramanian N, Makishima M, Mangelsdorf DJ, Suchy FJ. Human bile salt export pump promoter is transactivated by the farnesoid X receptor/bile acid receptor. J Biol Chem 276: 28857–28865, 2001. [DOI] [PubMed] [Google Scholar]
- 2.Ananthanarayanan M, Li S, Balasubramaniyan N, Suchy FJ, Walsh MJ. Ligand-dependent activation of the farnesoid X-receptor directs arginine methylation of histone H3 by CARM1. J Biol Chem 279: 54348–54357, 2004. [DOI] [PubMed] [Google Scholar]
- 3.Ananthanarayanan M, Li Y, Surapureddi S, Balasubramaniyan N, Ahn J, Goldstein JA, Suchy FJ. Histone H3K4 trimethylation by MLL3 as part of ASCOM complex is critical for NR activation of bile acid transporter genes and is downregulated in cholestasis. Am J Physiol Gastrointest Liver Physiol 300: G771–G781, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ashburner BP, Westerheide SD, Baldwin AS Jr. The p65 (RelA) subunit of NF-kappaB interacts with the histone deacetylase (HDAC) corepressors HDAC1 and HDAC2 to negatively regulate gene expression. Mol Cell Biol 21: 7065–7077, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baker RG, Hayden MS, Ghosh S. NF-kappaB, inflammation, and metabolic disease. Cell Metab 13: 11–22, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Balasubramaniyan N, Luo Y, Sun AQ, Suchy FJ. SUMOylation of the farnesoid X receptor (FXR) regulates the expression of FXR target genes. J Biol Chem 288: 13850–13862, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carlotti F, Dower SK, Qwarnstrom EE. Dynamic shuttling of nuclear factor kappa B between the nucleus and cytoplasm as a consequence of inhibitor dissociation. J Biol Chem 275: 41028–41034, 2000. [DOI] [PubMed] [Google Scholar]
- 8.Chaya D, Zaret KS. Sequential chromatin immunoprecipitation from animal tissues. Methods Enzymol 376: 361–372, 2004. [DOI] [PubMed] [Google Scholar]
- 9.Chong HK, Infante AM, Seo YK, Jeon TI, Zhang Y, Edwards PA, Xie X, Osborn TF. Genome-wide interrogation of hepatic FXR reveals an asymmetric IR-1 motif and synergy with LRH-1. Nucleic Acids Res 38: 6007–6017, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Bosscher K, Vanden Berghe W, Haegeman G. Cross-talk between nuclear receptors and nuclear factor kappaB. Oncogene 25: 6868–6886, 2006. [DOI] [PubMed] [Google Scholar]
- 11.Fang S, Tsang S, Jones R, Ponugoti B, Yoon H, Wu SY, Chiang CM, Willson TM, Kemper JK. The p300 acetylase is critical for ligand-activated farnesoid X receptor (FXR) induction of SHP. J Biol Chem 283: 35086–35095, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gadaleta RM, Oldenburg B, Willemsen EC, Spit M, Murzilli S, Salvatore L, Klomp LW, Siersema PD, van Erpecum KJ, van Mil SW. Activation of bile salt nuclear receptor FXR is repressed by pro-inflammatory cytokines activating NF-kappaB signaling in the intestine. Biochim Biophys Acta 1812: 851–858, 2011. [DOI] [PubMed] [Google Scholar]
- 13.Gartung C, Ananthanarayanan M, Rahman MA, Schuele S, Nundy S, Soroka CJ, Stolz A, Suchy FJ, Boyer JL. Downregulation of expression and function of the rat liver Na+/bile acid cotransporter in extrahepatic cholestasis. Gastroenterology 110: 199–209, 1996. [DOI] [PubMed] [Google Scholar]
- 14.Geier A, Dietrich CG, Voigt S, Ananthanarayanan M, Lammert F, Schmitz A, Trauner M, Wasmuth HE, Boraschi D, Balasubramaniyan N, Suchy FJ, Matern S, Gartung C. Cytokine-dependent regulation of hepatic organic anion transporter gene transactivators in mouse liver. Am J Physiol Gastrointest Liver Physiol 289: G831–G841, 2005. [DOI] [PubMed] [Google Scholar]
- 15.Geier A, Wagner M, Dietrich CG, Trauner M. Principles of hepatic organic anion transporter regulation during cholestasis, inflammation and liver regeneration. Biochim Biophys Acta 1773: 283–308, 2007. [DOI] [PubMed] [Google Scholar]
- 16.Gerritsen ME, Williams AJ, Neish AS, Moore S, Shi Y, Collins T. CREB-binding protein/p300 are transcriptional coactivators of p65. Proc Natl Acad Sci USA 194: 2927–2932, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ghisletti S, Huang W, Jepsen K, Benner C, Hardiman G, Rosenfeld MG, Glass CK. Cooperative NCoR/SMRT interactions establish a corepressor-based strategy for integration of inflammatory and anti-inflammatory signaling pathways. Genes Dev 23: 681–693, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hellman LM, Fried MG. Electrophoretic mobility shift assay (EMSA) for detecting protein-nucleic acid interactions. Nat Protoc 2: 1849–1861, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kemper JK. Regulation of FXR transcriptional activity in health and disease: emerging roles of FXR cofactors and post-translational modifications. Biochim Biophys Acta 1812: 842–850, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim MS, Shigenaga J, Moser A, Feingold K, Grunfeld C. Repression of farnesoid X receptor during the acute phase response. J Biol Chem 278: 8988–8995, 2003. [DOI] [PubMed] [Google Scholar]
- 21.Landrier JF, Eloranta JJ, Vavricka SR, Kullak-Ublick GA. The nuclear receptor for bile acids, FXR, transactivates human organic solute transporter-α and -β genes. Am J Physiol Gastrointest Liver Physiol 290: G476–G485, 2006. [DOI] [PubMed] [Google Scholar]
- 22.Mottis A, Mouchiroud L, Auwerx J. Emerging roles of the corepressors NCoR1 and SMRT in homeostasis. Genes Dev 27: 819–835, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sheppard KA, Rose DW, Haque ZK, Kurokawa R, McInerney E, Westin S, Thanos D, Rosenfeld MG, Glass CK, Collins T. Transcriptional activation by NF-kappaB requires multiple coactivators. Mol Cell Biol 19: 6367–6378, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Siggers T, Chang AB, Teixeira A, Wong D, Williams KJ, Ahmed B, Ragoussis J, Udalova IA, Smale ST, Bulyk ML. Principles of dimer-specific gene regulation revealed by a comprehensive characterization of NF-kappaB family DNA binding. Nat Immunol 13: 95–102, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smale ST. Hierarchies of NF-kappaB target-gene regulation. Nat Immunol 12: 689–694, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Soroka CJ, Ballatori N, Boyer JL. Organic solute transporter, OSTalpha-OSTbeta: its role in bile acid transport and cholestasis. Semin Liver Dis 30: 178–185, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thomas AM, Hart SN, Kong B, Fang J, Zhong XB, Guo GL. Genome-wide tissue-specific farnesoid X receptor binding in mouse liver and intestine. Hepatology 51: 1410–1419, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trauner M, Arrese M, Soroka CJ, Ananthanarayanan M, Koeppel TA, Schlosser SF, Suchy FJ, Keppler D, Boyer JL. The rat canalicular conjugate export pump (Mrp2) is downregulated in intrahepatic and obstructive cholestasis. Gastroenterology 113: 255–264, 1997. [DOI] [PubMed] [Google Scholar]
- 29.Wang YD, Chen WD, Wang M, Yu D, Forman BM, Huang W. Farnesoid X receptor antagonizes nuclear factor kappaB in hepatic inflammatory response. Hepatology 48: 1632–1643, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Watson PJ, Fairall L, Schwabe JW. Nuclear hormone receptor co-repressors: structure and function. Mol Cell Endocrinol 348: 440–449, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.You SH, Lim HW, Sun Z, Broache M, Won KJ, Lazar MA. Nuclear receptor co-repressors are required for the histone-deacetylase activity of HDAC3 in vivo. Nat Struct Mol Biol 20: 182–187, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vavassori P, Mencarelli A, Renga B, Distrutti E, Fiorucci S. The bile acid receptor FXR is a modulator of intestinal innate immunity. J Immunol 183: 6251–6261, 2009. [DOI] [PubMed] [Google Scholar]
- 33.Wagner M, Zollner G, Trauner M. Nuclear receptor regulation of the adaptive response of bile acid transporters in cholestasis. Semin Liver Dis 30: 160–177, 2010. [DOI] [PubMed] [Google Scholar]
- 34.Zollner G, Marschall HU, Wagner M, Trauner M. Role of nuclear receptors in the adaptive response to bile acids and cholestasis: pathogenetic and therapeutic considerations. Mol Pharm 3: 231–251, 2006. [DOI] [PubMed] [Google Scholar]