

Abstract
Overexpression of the nuclear receptor 4A1 (NR4A1) in breast cancer patients is a prognostic factor for decreased survival and increased metastasis, and this has been linked to NR4A1-dependent regulation of transforming growth factor β (TGF-β) signaling. Results of RNA interference studies demonstrate that basal migration of aggressive SKBR3 and MDA-MB-231 breast cancer cells is TGF-β independent and dependent on regulation of β1-integrin gene expression by NR4A1 which can be inhibited by the NR4A1 antagonists 1,1-bis(3′-indolyl)-1-(p-hydroxyphenyl)methane (DIM-C-pPhOH) and a related p-carboxymethylphenyl [1,1-bis(3′-indolyl)-1-(p-carboxymethylphenyl)methane (DIM-C-pPhCO2Me)] analog. The NR4A1 antagonists also inhibited TGF-β-induced migration of MDA-MB-231 cells by blocking nuclear export of NR4A1, which is an essential step in TGF-β-induced cell migration. We also observed that NR4A1 regulates expression of both β1- and β3-integrins, and unlike other β1-integrin inhibitors which induce prometastatic β3-integrin, NR4A1 antagonists inhibit expression of both β1- and β3-integrin, demonstrating a novel mechanism-based approach for targeting integrins and integrin-dependent breast cancer metastasis.
INTRODUCTION
Cell adhesion and attachment are essential for tissue integrity and cellular homeostasis, and the heterodimeric integrin cell surface receptors play a critical role in these processes (1–3). There are 18 different α subunits and 8 different β subunits that form 24 αβ-integrin receptor heterodimers, and the large 12-member β1-integrin subgroup bind multiple extracellular matrix (ECM) molecules to activate multiple intracellular pathways and also induce cross talk with other signaling systems (1–3). The functions of integrin heterodimers are highly tissue specific, and many human pathologies also involve integrin signaling (reviewed in references 4 and 5). β1-Integrin is highly expressed in most tumors and is associated with a negative prognostic significance such as overall and disease-free survival, recurrence, and metastasis for head and neck and squamous cell carcinoma, melanoma, lung, breast, prostate, laryngeal, and pancreatic cancers (6–17). A recent immunostaining study of 225 breast invasive ductal carcinomas (IDCs) showed that β1-integrin was overexpressed in 32.8% of patients with IDCs (13). Numerous studies show that focal adhesion kinase (FAK) which is downstream from β1-integrin is also a negative prognostic factor for breast cancer patients (18–20). The important functional role of β1-integrin has been demonstrated in mouse models expressing erbB2 under the control of the mouse mammary tumor virus and crossed with mammary tissue-specific β1-integrin-deficient mice. These mice exhibit a decrease in tumor volume, increased apoptosis, and decreased lung metastasis compared to animals expressing wild-type β1-integrin (21–23). Although small molecules, peptides, and antibodies that inhibit β1-integrin signaling have been developed, clinical agents that target β1-integrin for cancer chemotherapy are not currently available.
The orphan nuclear receptor 4A1 (NR4A1) (also called TR3 or Nur77) is overexpressed in breast cancer and other tumors, and functional studies show that NR4A1 exhibits prooncogenic activity (reviewed in reference 24). Studies in this laboratory have characterized a series of 1,1-bis(3′-indolyl)-1-(p-substituted phenyl)methane (C-DIM) analogs that bind NR4A1 and act as receptor antagonists to inhibit growth and induce apoptosis in several cancer cell lines and in tumors from mouse xenografts (25–30). A recent study demonstrated functional interactions between NR4A1 and transforming growth factor β (TGF-β) and in estrogen receptor (ER)-negative MDA-MB-231 cells, knockdown of NR4A1 decreased migration and also inhibited TGF-β-induced migration of this cell line (31). Results of gene array studies in pancreatic cancer cells identified β1-integrin as a potential NR4A1-regulated gene (27). In this study, we demonstrate that NR4A1 regulates β1-integrin expression and β1-integrin-dependent migration of breast cancer cells, and this is accompanied by decreased expression of β3-integrin. In MDA-MB-231 cells, results of our studies show that both constitutive and TGF-β-induced migration are dependent on nuclear and extranuclear NR4A1-regulated pathways, respectively. C-DIM/NR4A1 antagonists inhibit NR4A1-dependent expression of β1- and β3-integrins and other prooncogenic NR4A1-regulated genes and pathways and represent a novel class of mechanism-based anticancer agents.
MATERIALS AND METHODS
Cell lines and antibodies.
SKBR3, MDA-MB-231, and MCF-7 breast cancer cells were purchased from American Type Culture Collection (Manassas, VA). The cells were maintained at 37°C in the presence of 5% CO2 in Dulbecco's modified Eagle's medium (DMEM)–Ham's F-12 medium with 10% fetal bovine serum with antibiotic. NR4A1 antibody was purchased from Novus Biologicals (Littleton, CO). TGF-β was purchased from BD Biosystems (Bedford, MA). β-Actin antibody, Dulbecco's modified Eagle's medium, RPMI 1640 medium, and 36% formaldehyde were purchased from Sigma-Aldrich (St. Louis, MO). Hematoxylin was purchased from Vector Laboratories (Burlingame, CA). β3-Integrin, phosphorylated focal adhesion kinase (p-FAK), FAK, axin 2, leptomycin B, and NR4A1 immunofluorescent antibody were purchased from Cell Signaling Technologies (Manassas, VA). β1-Integrin antibody was purchased from Santa Cruz Biotech (Santa Cruz, CA), p84 antibody was purchased from GeneTex (Irvine, CA), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody was purchased from Biotium (Hayward, CA).
Cell adhesion assay.
SKBR3, MDA-MB-231, and MCF-7 cancer cells (3.0 × 105 per well) were seeded in Dulbecco's modified Eagle's medium–Ham's F-12 medium supplemented with 2.5% charcoal-stripped fetal bovine serum and were allowed to attach for 24 h. The cells were seeded and subsequently treated with various concentrations of 1,1-bis(3′-indolyl)-1-(p-hydroxyphenyl)methane (DIM-C-pPhOH) or p-carboxymethylphenyl (1,1-bis(3′-indolyl)-1-(p-carboxymethylphenyl)methane [DIM-C-pPhCO2Me]) for 24 h or 1 h prior to treatment with TGF-β (5 ng/ml) (4-h cotreatment) or without TGF-β or with 100 nM siβ1-integrin (small interfering RNA against β1-integrin) or siNR4A1 for 48 h. The cells were trypsinized, counted, and then placed for 90 min on BD BioCoat human fibronectin cellware 24-well plates (BD Biosciences, Bedford, MA). The medium was then aspirated, and the wells were gently washed with phosphate-buffered saline (PBS) and stained with 0.5% crystal violet stain. The cells were then counted for adhesion to fibronectin. Wells coated with bovine serum albumin (BSA) and poly-l-lysine were used as negative controls.
Boyden chamber assay.
SKBR3, MDA-MB-231, and MCF-7 cancer cells (3.0 × 105 per well) were seeded in Dulbecco's modified Eagle's medium–Ham's F-12 medium supplemented with 2.5% charcoal-stripped fetal bovine serum and were allowed to attach for 24 h. The cells were seeded and subsequently treated with various concentrations of DIM-C-pPhOH or DIM-C-pPhCO2Me for 24 h or 1 h prior to treatment with TGF-β (5 ng/ml) (4 h cotreatment) or without TGF-β or with 100 nM siβ1-integrin, siNR4A1, siSp1 (Sp1 stands for specificity protein 1), or sip300 for 48 h. The cells were trypsinized, counted, placed in 24-well 8.0-μm-pore ThinCerts from BD Biosciences (Bedford, MA), allowed to migrate for 24 h, fixed with formaldehyde, and then stained with hematoxylin. Cells that migrated through the pores were then counted.
Real-time PCR.
RNA was isolated using Zymo Research Quick-RNA MiniPrep kit (Irvine, CA). Quantification of mRNA (β1-integrin, β3-integrin) was performed using Bio-Rad iTaq universal SYBR green one-step kit (Richmond, CA) using the manufacturer's protocol with real-time PCR. TATA binding protein (TBP) mRNA was used as a control to determine relative mRNA expression.
Immunoprecipitation.
MDA-MB-231 cancer cells (3.0 × 105 per well) were seeded in Dulbecco's modified Eagle's medium–Ham's F-12 medium supplemented with 2.5% charcoal-stripped fetal bovine serum and allowed to attach for 24 h. The medium was then changed to DMEM–Ham's F-12 medium containing 2.5% charcoal-stripped fetal bovine serum, and either dimethyl sulfoxide (DMSO) or TGF-β (5 ng/ml) was added for 4 h (after pretreatment with leptomycin B [20 nM] for 24 h or pretreatment with 20 μM DIM-C-pPhOH or DIM-C-pPhCO2Me or no pretreatment). Protein A Dynabeads were prepared, and binding of antibody with protein and protein-protein interactions were isolated by Life Technologies immunoprecipitation kit using Dynabeads coated with protein A (Grand Island, NY) following the manufacturer's protocol. Protein-protein interactions of interest were determined by Western blot analysis.
Chromatin immunoprecipitation.
The chromatin immunoprecipitation (ChIP) assay was performed using the ChIP-IT Express magnetic chromatin immunoprecipitation kit (Active Motif, Carlsbad, CA) according to the manufacturer's protocol. SKBR3 and MDA-MB-231 cells were treated with DMSO, DIM-C-pPhOH, or DIM-C-pPhCO2Me (15 or 20 μM) for 24 h. The cells were then fixed with 1% formaldehyde, and the cross-linking reaction was stopped by the addition of 0.125 M glycine. After the cells were washed twice with phosphate-buffered saline, the cells were scraped and pelleted. Collected cells were hypotonically lysed, and nuclei were collected. Nuclei were then sonicated to the desired chromatin length (∼200 to 1,500 bp). The sonicated chromatin was immunoprecipitated with normal IgG, p300 (Santa Cruz), siSp1 (Abcam), NR4A1 (Novus Biologicals), or RNA polymerase II (Pol II) (Active Motif) antibodies and protein A-conjugated magnetic beads at 4°C overnight. After the magnetic beads were extensively washed, protein-DNA cross-links were reversed and eluted. DNA was prepared by proteinase K digestion followed by PCR amplification. The primers for detection of the β1-integrin promoter region were 5′-TCACCACCCTTCGTGACAC-3′ (sense) and 5′-GAGATCCTGCATCTCGGAAG-3′ (antisense), and the primers for detection of the β3-integrin promoter region were 5′-TCTCAGGCGCAGGGTCTAGAGAA-3′ (sense) and 5′-TCGCGGCGCCCACCGCCTGCTCTACGCT-3′ (antisense). PCR products were resolved on a 2% agarose gel in the presence of RGB-4103 GelRed nucleic acid stain.
Nuclear/cytosolic extraction.
MDA-MB-231 cancer cells (3.0 × 105 per well) were seeded in Dulbecco's modified Eagle's medium–Ham's F-12 medium supplemented with 2.5% charcoal-stripped fetal bovine serum and were allowed to attach for 24 h. The medium was then changed to DMEM–Ham's F-12 medium containing 2.5% charcoal-stripped fetal bovine serum, and either DMSO or TGF-β (5 ng/ml) was added for 4 h (after pretreatment with 20 nM leptomycin B for 24 h or pretreatment with 20 μM DIM-C-pPhOH or DIM-C-pPhCO2Me or no pretreatment). Nuclear and cytosolic fractions were then isolated using Thermo Scientific NE-PER nuclear and cytoplasmic extraction kit (Rockford, IL) according to the manufacturer's protocol. Fractions were then analyzed by Western blotting. GAPDH and p84 were used as cytoplasmic and nuclear positive controls, respectively.
Immunofluorescence.
MDA-MB-231 (1.0 × 105 per well) were seeded in two-well Nunc Lab-Tek chambered borosilicate cover glass slides (no. 1 borosilicate cover glass) from Thermo Scientific and were allowed to attach for 24 h. The medium was then changed to DMEM–Ham's F-12 medium containing 2.5% charcoal-stripped fetal bovine serum, and either DMSO or TGF-β (5 ng/ml) was added for 4 h (after pretreatment with leptomycin B [20 nM] for 24 h or pretreatment with 20 μM DIM-C-pPhOH or DIM-C-pPhCO2Me or no pretreatment). The cells were then treated with fluorescent NR4A1 primary antibody [Nur77 (D63C5) XP], and immunofluorescence was observed according to Cell Signaling Technology's immunofluorescence protocol. 4′,6′-Diamidino-2-phenylindole (DAPI) staining was observed using Hoechst staining according to Biotium's apoptotic and necrotic assay kit by following the manufacturer's protocol. The cells were visualized by microscopy (Advanced Microscopy), and NR4A1 localization was determined by green fluorescence. DAPI was used to stain the nucleus, and images were taken sequentially of NR4A1, DAPI, and then merged (28–30).
Western blot analysis.
SKBR3, MDA-MB-231, and MCF-7 cancer cells (3.0 × 105 per well) were seeded in Dulbecco's modified Eagle's medium–Ham's F-12 medium supplemented with 2.5% charcoal-stripped fetal bovine serum and were allowed to attach for 24 h. The cells were transfected with 100 nM siβ1-integrin, siNR4A1, siSp1, or sip300 for 72 h or treated with various C-DIM compounds. Cell lysates were analyzed by Western blotting as described previously (28–30).
Small interfering RNA interference assay.
Small interfering RNA (siRNA) experiments were conducted as described previously (28–30). The siRNA complexes used in the study are as follows: siGL2-5′, CGU ACG CGG AAU ACU UCG A; siNR4A1, SASI_Hs02_00333289[1], SASI_Hs02_00333290[2]; siβ1-integrin, SASI_Hs02_00333437[1], SASI_Hs01_00159474; siSp1, SASI_Hs02_003; sip300, SASI_Hs01_00052818.
Triple-negative breast cancer (TNBC) orthotopic xenograft studies.
Female BALB/c nude mice (6 to 8 weeks old) were obtained (Charles River Laboratory, Wilmington, MA) and maintained and treated as previously described (30). Tumor volumes and tumor weights were determined as previously described (30). Tumor lysates were obtained and analyzed by Western blotting.
Statistical analysis.
Statistical significance of differences between the treatment groups was determined by Student's t test. The results are expressed as means with error bars representing 95% confidence intervals (95% CIs) for at least three experiments for each group unless otherwise indicated. A P value of <0.05 was considered statistically significant. All statistical tests were two sided.
RESULTS
NR4A1 regulates β1-integrin expression.
β1-Integrin is expressed in ER-positive MCF-7, ER-negative MDA-MB-231, and erbB2-overexpressing SKBR3 breast cancer cells, and knockdown of NR4A1 (siNR4A1) by RNA interference (RNAi) decreased expression of β1-integrin protein and mRNA (Fig. 1A). Previous studies identified 1,1-bis(3′-indolyl)-1-(p-hydroxyphenyl)methane (DIM-C-pPhOH; C-DIM8) and 1,1-bis(3′-indolyl)-1-(p-carboxymethylphenyl)methane (DIM-C-pPhCO2Me; C-DIM14) as NR4A1 ligands that act as antagonists in breast and other cancer cell lines (25–30), and both compounds also decreased expression of β1-integrin protein (Fig. 1B) and mRNA (Fig. 1C) in MCF-7, MDA-MB-231, and SKBR3 cells. Moreover, Western blot analysis of tumor lysates from mice bearing MDA-MB-231 cells (orthotopic) (30) showed that DIM-C-pPhCO2Me significantly decreases β1-integrin protein expression (Fig. 1D). β1-Integrin regulates phosphorylation of FAK (p-FAK), and transfection of MCF-7, MDA-MB-231, and SKBR3 cells with siNR4A1 (Fig. 2A) or treatment with DIM-CpPhOH (Fig. 2B) or DIM-C-pPhCO2Me (Fig. 2C) decreased phosphorylation of FAK. In addition, results from the in vivo orthotopic study (30) showed that p-FAK is decreased in tumors from mice bearing MDA-MB-231 cells and treated with DIM-C-pPhCO2Me (Fig. 2D). Fibronectin-induced cell adhesion is also a prototypical β1-integrin-regulated response, and cell adhesion was significantly decreased in MCF-7, MDA-MB-231, and SKBR3 cells after transfection with siNR4A1 (Fig. 2E) or after treatment with DIM-C-pPhOH or DIM-C-pPhCO2Me (Fig. 2F). For a positive control, we showed that knockdown of β1-integrin (siβ1-integrin) by RNA also decreased cell adhesion (Fig. 2E) (see Fig. S1 in the supplemental material).
FIG 1.

NR4A1 regulates β1-integrin expression in breast cancer cells and tumors. (A) Breast cancer cells were transfected with siNR4A1, and cell extracts were analyzed for protein and mRNA expression by Western blotting or real-time PCR, respectively, as outlined in Materials and Methods. (B and C) Breast cancer cells were treated with DMSO, DIM-C-pPhOH, or DIM-C-pPhCO2Me for 24 h, and extracts were analyzed for protein (B) or mRNA (C) levels by Western blotting and real-time PCR, respectively, as outlined in Materials and Methods. (D) Cell lysates from tumors (MDA-MB-231 orthotopic) (30) derived from animals treated with corn oil (control) or DIM-C-pPhCO2Me (C-DIM-14; 40 mg/kg of body weight/day) were analyzed by Western blotting, and decreased protein expression was determined and normalized against the β-actin protein loading control. Quantified data are presented as means plus standard errors (SE) (error bars) (at least three replicates), and significant (P < 0.05) decreases are indicated by an asterisk.
FIG 2.

NR4A1 regulates β1-integrin-dependent responses. Breast cancer cells were transfected with siNR4A1 (A), treated with DMSO and DIM-C-pPhOH (B) or DIM-C-pPhCO2Me (C) for 24 h, and whole-cell lysates were analyzed by Western blotting as outlined in Materials and Methods. (D) Tumor lysates from mice (MDA-MB-231 orthotopic-derived [30]) treated with corn oil or DIM-C-pPhCO2Me (40 mg/kg/day) were analyzed by Western blotting and quantitated as outlined in the legend to Fig. 1D. (E and F) The effects of siNR4A1 and siβ1-integrin (E) or DIM-C-pPhOH and DIM-C-pPhCO2Me (F) on fibronectin-induced adhesion of breast cancer cells was determined as outlined in Materials and Methods. Results in panels D to F are means plus SE (error bars) (at least three replicates), and a significant (P < 0.05) decrease is indicated by an asterisk. Western blots in Fig. 1 and 2 were derived from the same experiment showing effects on β1-integrin (Fig. 1) and β1-integrin-regulated responses (Fig. 2).
Mechanisms of NR4A1 regulation of β1-integrin and β3-integrin.
NR4A1 regulates gene expression through direct interactions with genomic nerve growth factor Bα (NGFBα) response elements (NBRE) and Nur response elements (NuRE) or by interactions with specificity protein 1 (Sp1) bound to GC-rich promoter elements (32, 33). NBRE and NuRE were not identified in the β1-integrin promoter, whereas two GC-rich sequences were located at −760 and −676 in the proximal region of the β1-integrin promoter (Fig. 3A). Previous studies show that NR4A1, Sp1, and the nuclear coregulatory gene p300 interact with the GC-rich region of the survivin promoter to regulate survivin gene expression (25). Using the more aggressive SKBR3 and MDA-MB-231 cells as models, cells were treated with dimethyl sulfoxide (DMSO), DIM-C-pPhOH, or DIM-C-pPhCO2Me and analyzed in a chromatin immunoprecipitation (ChIP) assay using primers targeted to the GC-rich region of the β1-integrin promoter. The results show that Pol II, NR4A1, Sp1, and p300 interact with the GC-rich promoter regions, and after treatment with DIM-C-pPhOH or DIM-C-pPhCO2Me for 24 h, the band for Pol II was decreased in both cell lines (Fig. 3A), and this was consistent with decreased β1-integrin expression. Ligand-induced inactivation of NR4A1 also decreased NR4A1 binding to the promoter; however, changes in the Sp1 and p300 bands were somewhat variable and dependent on cell context and ligand. For example, the loss of p300 was observed in SKBR3 cells but not MDA-MB-231 cells, and it is possible that p300 was interacting with the trans-acting factors in the proximal region of the β1-integrin promoter. We further investigated the roles of Sp1 and p300 in regulating β1-integrin expression in SKBR3 and MDA-MB-231 cells by RNAi, and knockdown of Sp1 (siSp1) and p300 (sip300) also decreased β1-integrin expression (Fig. 3B), suggesting that like survivin (25), NR4A1 regulates β1-integrin expression through a NR4A1/p300/Sp1 complex. p300 knockdown also decreases Sp1 expression, suggesting that p300 plays a role in regulating expression of this gene. These results do not exclude a role for other factors in NR4A1 regulation of β1-integrin, and this is currently being investigated.
FIG 3.

Role of NR4A1/p300/Sp1 in regulation of β1- and β3-integrin. (A) Analysis of Pol II, NR4A1, Sp1, and p300 binding to the β1-integrin promoter was determined in a ChIP assay using the indicated primers (ITGB1 F [ITGB1 stands for β1-integrin and F stands for forward] and ITGB1 R [R stands for reverse]). TSS, transcription start site. (B) Cells were treated with oligonucleotides that knock down Sp1 (siSp1) and p300 (sip300), and whole-cell lysates were analyzed by Western blotting as outlined in Materials and Methods. (C) Cells were transfected with siNR4A1 or treated with DIM-C-pPhOH (C-DIM8) or DIM-C-pPhCO2Me (C-DIM14), and whole-cell lysates were analyzed by Western blotting as outlined in Materials and Methods. (D) Cells were transfected with siNR4A1 or treated with C-DIM8 or C-DIM14, and their effects on β3-integrin (ITGB3) mRNA levels were determined. The treatments significantly (P < 0.05) decrease mRNA levels. (E) Analysis of Pol II, NR4A1, Sp1, and p300 binding to the proximal GC-rich region of the β3-integrin promoter was determined in a ChIP assay as outlined in Materials and Methods. (F) Cells were transfected with siSp1 and sip300 and analyzed by real-time PCR for β3-integrin mRNA levels. Both oligonucleotides significantly (P < 0.05) decreased β3-integrin mRNA levels.
Previous reports show that inhibition of β1-integrin by RNAi or other β1-integrin inhibitors increases expression of β3-integrin resulting in enhanced metastasis (34–36). The β3-integrin promoter is also GC rich (37), and therefore, we investigated the possible regulation of β3-integrin by NR4A1. Western blot analysis showed that constitutive β3-integrin protein levels were barely detectable and remained low after treatment with C-DIM/NR4A1 antagonists or siNR4A1 (Fig. 3C), whereas knockdown of β1-integrin by RNAi increased β3-integrin protein as previously reported (36). There was more robust expression of β3-integrin mRNA in MDA-MB-231 and SKBR3 cells, and transfection of siNR4A1 or treatment with C-DIM/NR4A1 antagonists significantly decreased β3-integrin mRNA levels (Fig. 3D). ChIP assays showed that NR4A1, Sp1, and p300 bound the proximal GC-rich region of the β3-integrin gene, and treatment with DIM-C-pPhOH or DIM-C-pPhCO2Me decreased binding of Pol II, NR4A1, and Sp1 but differentially affected p300 binding to the promoter. In addition, we also observed that knockdown of Sp1 (siSp1) or p300 (sip300) in MDA-MB-231 and SKBR3 cells decreased β3-integrin mRNA levels (Fig. 3E). These results demonstrate that NR4A1 regulates both β1- and β3-integrin expression, and in contrast to β1-integrin-specific inhibitors, NR4A1 antagonists downregulate expression of both β1- and β3-integrin.
Migration of MDA-MB-231 and SKBR3 cells: roles of NR4A1 and β1-integrin.
Both MDA-MB-231 and SKBR3 cells undergo migration (constitutive) in a Boyden chamber assay in the absence of a stimulus. Transfection of these cells with siNR4A1 (Fig. 4A) or siβ1-integrin (Fig. 4B) decreased migration of both cell lines, and similar results were observed with two oligonucleotides targeting NR4A1 and β1-integrin. Treatment of SKBR3 and MDA-MB-231 cells with DIM-C-pPhOH (CDIM8) or DIM-C-pPhCO2Me (CDIM14) also decreased migration (Fig. 4C), and the effects of DIM-C-pPhOH as an inhibitor of cell migration was not affected by cotreatment with leptomycin B (LMB), confirming that the inhibitory effects of this NR4A1 antagonist did not require nuclear export (25). We also investigated the role of NR4A1 in mediating DIM-C-pPhCO2Me-dependent inhibition of migration of MDA-MB-231 and SKBR3 cells by knocking down NR4A1 and then treating with the NR4A1 antagonist DIM-C-pPhcO2Me (Fig. 4E). Treatment of the NR4A1-depleted cells with DIM-C-pPhCO2Me resulted in minimal inhibition of cell migration. Similar results were observed after treatment of β1-integrin-depleted cells with DIM-C-pPhCO2Me, and we also observed that DIM-C-pPhOH did not inhibit invasion in cells depleted of NR4A1 or β1-integrin (see Fig. S2 in the supplemental material). This would suggest that induction of β3-integrin after knockdown of β1-integrin (Fig. 3B) does not play a very significant role in cell migration using the Boyden chamber assay. Thus, inhibition of breast cancer cell migration by C-DIMs/NR4A1 antagonists is dependent on both NR4A1 and β1-integrin and consistent with regulation of β1-integrin by NR4A1. Overexpression of β1-integrin in SKBR3 and MDA-MB-231 cells slightly increases cell migration, and in NR4A1-depleted cells which exhibit decreased migration, overexpression of β1-integrin significantly reverses this response (Fig. 4F). In addition, NR4A1 ligand-mediated inhibition of breast cancer cell migration was also rescued by β1-integrin overexpression (Fig. 4G), further confirming that β1-integrin-mediated migration is NR4A1 dependent. Thus, the constitutive or basal migration of SKBR3 and MDA-MB-231 cells in the absence of endogenous stimuli is linked to nuclear NR4A1 regulation of β1-integrin.
FIG 4.

NR4A1 regulates β1-integrin-dependent breast cancer cell migration. (A to D) Cells were transfected with siNR4A1 (A) or siβ1-integrin (B) or treated with DIM-C-pPhOH and DIM-C-pPhCO2Me (C) and DIM-C-pPhOH with or without leptomycin B (LMB) (D), and breast cancer cell migration was determined in a Boyden chamber assay as outlined in Materials and Methods. (E) Cells were transfected with a nonspecific oligonucleotide (siCtl), siNR4A1, or siβ1-integrin and treated with DIM-C-pPhCO2Me, and cell migration was determined in a Boyden chamber assay as outlined in Materials and Methods. (F and G) Cells were transfected with siNR4A1 alone (F) or treated with DIM-C-pPhOH/DIM-C-pPhCO2Me (G) in combination with a β1-integrin (ITGB1) expression plasmid, and effects on cell migration were determined in a Boyden chamber assay as outlined in Materials and Methods. Results are expressed as means plus SE (error bars) for at least three replicates for each treatment group, and significantly (P < 0.05) decreased migration (*) or rescue by β1-integrin overexpression (**) is indicated.
TGF-β-induced migration of MDA-MB-231 cells: role of extranuclear NR4A1.
A recent study reported that TGF-β-induced migration of MDA-MB-231 cells was also NR4A1 dependent and involved a pathway associated with SMAD7 degradation resulting in activation of TGF-βR receptor 1 (TGF-βR1) (31). Treatment of MDA-MB-231 cells with 5 ng/ml TGF-β significantly induced cell migration (Fig. 5A) as previously described (31), and knockdown of NR4A1 or treatment with DIM-C-pPhOH or DIM-C-pPhCO2Me blocked TGF-β-induced migration and significantly decreased overall migration, similar to that observed after knockdown of NR4A1 or treatment with the NR4A1 antagonists alone (Fig. 4A to C). TGF-β-induced migration was inhibited after cotreatment with the TGF-βR1 inhibitor ALK5i and also the nuclear export inhibitor LMB, and ALK5i had no effect on endogenous cell migration (data not shown). Analysis of cytosolic and nuclear extracts show that TGF-β induced expression and nuclear export of NR4A1 which was blocked by LMB (Fig. 5B) indicating that TGF-β-induced migration requires cytosolic NR4A1, whereas constitutive migration which is not inhibited by ALK5i is due to nuclear NR4A1-dependent regulation of β1-integrin. We also examined SMAD7 expression and observed minimal endogenous expression in MDA-MB-231 and SKBR3 cells, and TGF-β increased SMAD7 only in SKBR3 cells (Fig. 5C). In contrast, cotreatment with TGF-β plus LMB, CDIM8, or CDIM14 dramatically increased SMAD7 protein expression of both cell lines, suggesting that nuclear localization of NR4A1 inhibits degradation of SMAD7, which is a cytosolic protein. These results are consistent with previous studies, suggesting that NR4A1 (cytosolic) plays a role in proteasome-dependent degradation of SMAD7 (31). Immunostaining of NR4A1 in MDA-MB-231 cells confirms that NR4A1 is nuclear, and treatment with TGF-β induces nuclear export of this receptor, and this is blocked by LMB (Fig. 5C).
FIG 5.
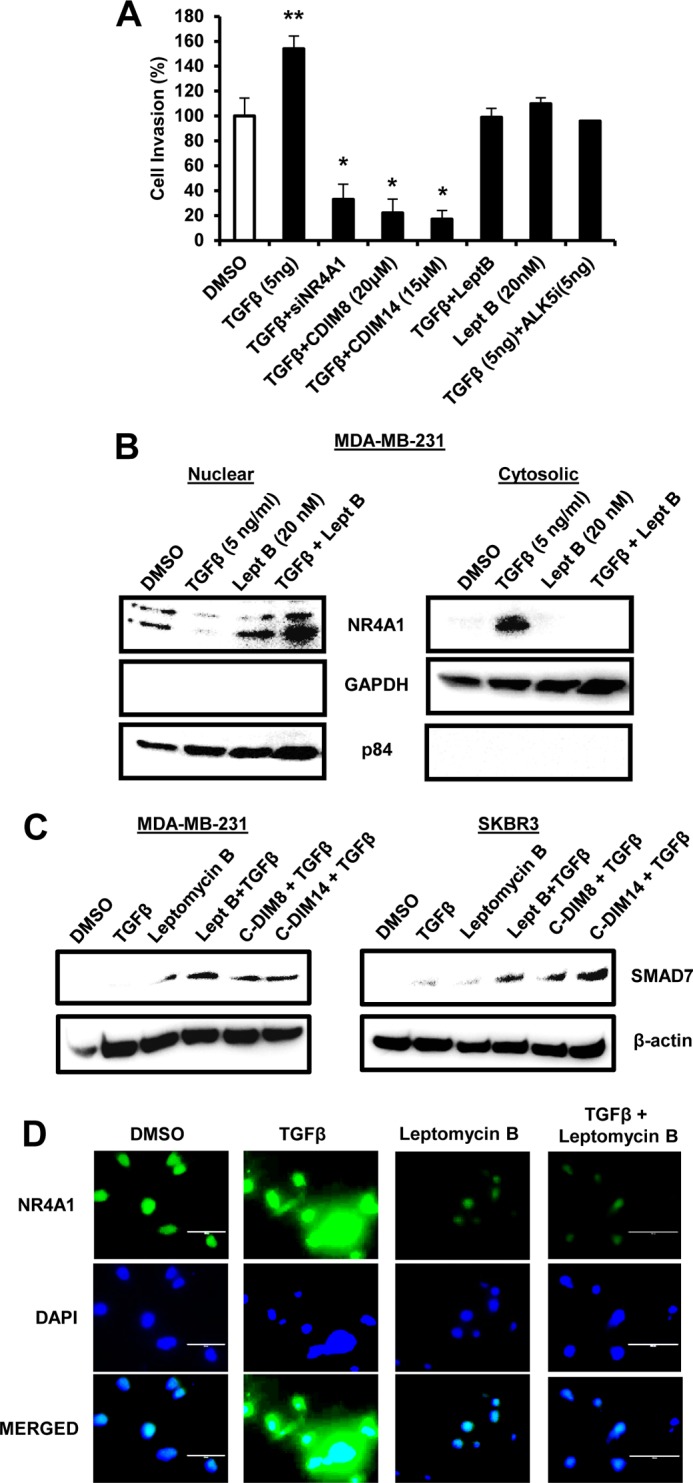
Role of NR4A1 on TGF-β-induced migration of MDA-MB-231 cells. (A) MDA-MB-231 cells were treated with TGF-β alone for 4 h or in combination with siNR4A1, DIM-C-pPhOH, and DIM-C-pPhCO2Me (24-h treatment), LMB and ALK5i, and LMB (alone). Cell migration was determined in a Boyden chamber assay. (B) MDA-MB-231 cells were treated with DMSO, TGF-β, and LMB (alone) and in combination for 4 h. Nuclear and cytosolic extracts were analyzed by Western blotting using nuclear (p84) and cytosolic (GADPH) loading controls. (C) Cells were treated with DMSO, TGF-β, LMB alone, and TGF-β in combination with LMB, DIM-C-pPhOH (CDIM8), or DIM-C-pPhCO2Me (CDIM14) for 4 h, and whole-cell lysates were analyzed for SMAD7 expression by Western blot analysis. (D) Cells were treated with DMSO, 5 ng/ml TGF-β, LMB, and LMB plus TGF-β for 4 h and immunostained with both NR4A1 antibodies and DAPI as outlined in Materials and Methods.
Since C-DIM/NR4A1 antagonists act through binding nuclear NR4A1, we examined the effects of short-term (4-h) treatment of MDA-MB-231 with DIM-C-pPhOH or DIM-C-pPhCO2Me on TGF-β-induced migration. Like LMB, both compounds blocked TGF-β-induced migration (Fig. 6A), and this was accompanied by inhibition of TGF-β-induced nuclear export of NR4A1 (Fig. 6B) and paralleled results observed for LMB (Fig. 5A and B). The inhibitory effects observed after treatment with the C-DIM/NR4A1 ligands for 4 h were not due to decreased β1-integrin expression (Fig. 6C), suggesting that bound NR4A1 was resistant to TGF-β-induced nuclear export, and the factors that regulate nuclear export are currently being investigated. A previous report showed that TGF-β-induced NR4A1 interacts with axin 2 and other factors (e.g., E3 ligases Arkadia and RNF12) to form a polyubiquitination complex (31), and after treatment of MDA-MB-231 cells with TGF-β, LMB, C-DIMs, and their combinations, Western blot analysis of the cytosolic fraction immunoprecipitated with axin 2 antibodies gave a strong band for NR4A1 only in cells treated with TGF-β alone (Fig. 6D). In contrast, treatment with DIM-C-pPhOH, DIM-C-pPhCO2Me, or LMB, which inhibit TGF-β-induced nuclear export of NR4A1, resulted in decreased intensities of cytosolic NR4A1 bands associated with the axin 2 antibody immunoprecipitates. The results demonstrate that NR4A1 plays an important role in breast cancer cell migration by regulation of β1-integrin (endogenous activity) and TGF-β-induced migration which is dependent on NR4A1 nuclear export (Fig. 6E).
FIG 6.

(A) MDA-MB-231 cells were treated with TGF-β, DIM-C-pPhOH, and DIM-C-pPhCO2Me alone and TGF-β plus C-DIMs for 4 h, and cell migration was determined in a Boyden chamber assay and immunostaining (NR4A1) and DAPI staining was determined as outlined in the legend to Fig. 5C. (B) Cells were treated as described in the legend to Fig. 5B, and the cytosolic and nuclear extracts were further examined by Western blot analyses. (C) MDA-MB-231 cells were treated with DIM-C-pPhCO2Me or DIM-C-pPhOH for different times, and whole-cell lysates were analyzed by Western blotting for β1-integrin expression. (D) Cells were treated as outlined above for panel B, and whole-cell lysates were immunoprecipitated with axin 2 antibodies (IP:Axin2) and analyzed by Western blotting (immunoblotting [IB]). (E) Schematic outline of the role of NR4A1 in constitutive and TGF-β-induced migration in breast cancer cells.
DISCUSSION
The NR4A family of orphan nuclear receptors NR4A1, NR4A2, and NR4A3 were initially identified as stress-induced immediate early genes with a characteristic domain structure observed for nuclear receptors. NR4A receptors have both unique and overlapping functions, and there is increasing evidence that they play an important role in cellular homeostasis and diseases associated with metabolism, cardiovascular and neurological functions, inflammation, and the immune system (38–40). Endogenous ligands for NR4A1 have not been identified; however, synthetic ligands that are structurally related to cytosporone B have been developed (41–43) and have potential clinical applications. For example, ethyl[2,3,4-trimethoxy-6-(i-octanoyl)phenyl]acetate is an NR4A1 ligand that acts as a receptor antagonist to decrease NR4A1-dependent hepatic gluconeogenesis and lower blood glucose levels in a rodent model for type 2 diabetes (43). NR4A1 is also overexpressed in solid tumors including both ER-positive and ER-negative breast tumors and is a negative prognostic factor for lung, colon, and breast cancer patients (26, 31, 44).
Initial studies targeting NR4A1 for cancer chemotherapy showed that cell death observed in some cancer cell lines treated with several apoptosis agents was due to nuclear export of NR4A1 and the subsequent interactions of NR4A1 with bcl-2 to form a proapoptotic complex that disrupted mitochondria (45, 46). The proapoptotic effects were also observed using peptides and paclitaxel that mimic NR4A1 interactions with bcl-2 (42, 47). Studies in this laboratory have identified C-DIMs as NR4A1 ligands that act as antagonists in cancer cell lines, and previous studies have demonstrated that C-DIM/NR4A1 antagonists inhibit growth and induce cell death through inactivation of nuclear NR4A1-dependent prooncogenic pathways in pancreatic, lung, colon, kidney, and breast cancer cell lines (24–30).
A recent report showed that high expression of NR4A1 in breast tumors correlated with decreased relapse-free survival, and this was linked to the role of NR4A1 in TGF-β and TGF-β/cytokine-induced migration/invasion and metastasis (31). Results of ongoing genomic and functional studies in several cancer cell lines identified β-integrin as a possible NR4A1-regulated promigration/invasion gene, and this correlated with previous in vivo studies showing that β1-integrin was important for metastasis of mammary tumors overexpressing the erbB2 oncogene (9, 21–23). Results in Fig. 1, 2, and 4 demonstrate that knockdown of NR4A1 or treatment with the NR4A1 antagonists DIM-C-pPhOH and DIM-C-pPhCO2Me decreased expression of β1-integrin protein and mRNA and β1-integrin-dependent responses in MCF7, MDA-MB-231, and SKBR3 cells and also inhibited migration of the latter two cell lines.
The mechanism of NR4A1 regulation of β-integrin in SKBR3 and MDA-MB-231 cells did not involve direct binding to cis-acting genomic sequences but through an indirect mechanism in which NR4A1/p300 act as a coregulatory complex to activate Sp1-regulated genes. The ChIP assays show that NR4A1, Sp1, and p300 interacted at the GC-rich region of the β1-integrin gene promoter (Fig. 3), and knockdown of any one of these factors or treatment with C-DIM/NR4A1 antagonists resulted in decreased β1-integrin expression. These results are similar to those previously observed for NR4A1/p300/Sp1-mediated regulation of survivin in pancreatic cancer cells (25) and are consistent with other reports showing that other nuclear receptors also regulate expression of other Sp-dependent genes through NR4A1/Sp1 complexes (48–50). Previous studies show that knockdown or inhibition of β1-integrin in breast cancer cells results in the expression of β3-integrin, and this “integrin-switching” enhances TGF-β-induced metastasis (34–36), which presents a problem for applications of β1-integrin inhibitors in treatment of breast cancer. Like β1-integrin, the 5′-promoter region of the β3-integrin gene contains GC-rich sequences (37), and our results demonstrate that NR4A1 also regulates β3-integrin expression, and NR4A1 antagonists or NR4A1 knockdown decreases expression of both genes (Fig. 3). Thus, coregulation of β1- and β3-integrin by NR4A1 negates the “integrin-switching” phenomena (34–36) and further demonstrates that the C-DIM/NR4A1 antagonists represent a novel therapeutic approach for inhibiting β1/β3-integrin-induced signaling and metastasis in breast cancer cells.
MDA-MB-231 and SKBR3 cells readily migrate in the absence of TGF-β or cytokine stimulus, and results of RNAi studies show that inhibition of cell migration by C-DIM/NR4A1 antagonists was observed only in cells expressing NR4A1 or β1-integrin (Fig. 4). Moreover, since the inhibitory effects of C-DIMs were similar in the presence or absence of the nuclear export inhibitor LMB (Fig. 4F), our results indicate that constitutive migration of these cells was due to nuclear NR4A1-dependent regulation of β1-integrin. This is also supported by the observation that the TGF-β receptor inhibitor ALK5i inhibits TGF-β-induced migration but does not affect the high rate of constitutive migration of MDA-MB-231 cells (Fig. 5A). A recent study showed that NR4A1 was also required for TGF-β-induced migration of MDA-MB-231 and other cell lines, and this was due to interactions of NR4A1, axin 2, and E3 ligases which enhanced SMAD7 degradation, resulting in activation of the TGF-βR1 pathway (31). We also observed that TGF-β induced NR4A1 expression and migration of MDA-MB-231 cells; however, the key essential element in this pathway was that TGF-β induced nuclear export of NR4A1 (Fig. 5B and D and 6C). Moreover, inhibition of nuclear export by the NR4A1 ligands (DIM-C-pPhOH or DIM-C-pPhcO2Me) or LMB also blocked TGF-β-induced migration and enhanced SMAD7 expression (Fig. 5C). Previous studies on SMAD7 degradation in MDA-MB-231 cells used transfected FLAG-SMAD7 (31), whereas in this study, we observed low to nondetectable SMAD7 expression in MDA-MBA-231 and SKBR3 cells. However, LMB, DIM-C-pPhOH, and DIM-C-pPhCO2Me which prevent NR4A1 export also increased SMAD7 expression in cells cotreated with these compounds plus TGF-β (Fig. 5C), and this is consistent with a role for cytosolic NR4A1 in SMAD7 degradation as previously reported (31). Thus, TGF-β-induced migration of MDA-MB-231 cells is due to nuclear export of NR4A1, and the C-DIM/NR4A1 antagonist blocks this pathway presumably by inhibiting factors/pathways required for nuclear export, and these factors/pathways are currently being investigated.
In summary, results of this study show that nuclear NR4A1 regulates β1-integrin expression in breast cancer cells, and C-DIM/NR4A1 antagonists inhibit expression of β1-integrin and β1-integrin-mediated responses, including cell migration, and the antagonists also inhibit NR4A1-regulated expression of β3-integrin. In contrast, TGF-β-induced migration of MDA-MB-231 cells requires nuclear export of NR4A1 which is inhibited not only by LMB but also by C-DIM/NR4A1 antagonists. Thus, constitutive migration and TGF-β-induced migration are dependent on nuclear and extranuclear NR4A1, respectively, and the C-DIM/NR4A1 antagonists inhibit both pathways by decreasing NR4A1-dependent expression of β1-integrin and by inhibition of TGF-β-induced nuclear export of NR4A1 (Fig. 6E). This study expands on the prooncogenic functions of NR4A1 and indicates that C-DIM compounds and other NR4A1 antagonists represent an important new class of mechanism-based anticancer drugs for treating patients with tumors overexpressing this receptor.
Supplementary Material
ACKNOWLEDGMENTS
The financial assistance of the National Institutes of Health (P30-ES023512, S. Safe), the DOD-CDRMP (BC103116, M. Singh), Texas AgriLife Research and Sid Kyle endowment, is gratefully acknowledged.
E.H. carried out the in vitro studies and assisted in writing the manuscript. S.-O.L. carried out some of the in vitro studies and initially identified β1-integrin as an NR4A1-regulated gene. R.D. carried out the in vivo studies. M.S. supervised the in vivo studies and carried out data analysis. S.S. developed the C-DIMs as NR4A1 antagonists, supervised the studies, and wrote the manuscript.
We declare that we have no conflicts of interest that would prejudice the impartiality of this research.
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00912-15.
REFERENCES
- 1.Shattil SJ, Kim C, Ginsberg MH. 2010. The final steps of integrin activation: the end game. Nat Rev Mol Cell Biol 11:288–300. doi: 10.1038/nrm2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arnaout MA, Goodman SL, Xiong JP. 2007. Structure and mechanics of integrin-based cell adhesion. Curr Opin Cell Biol 19:495–507. doi: 10.1016/j.ceb.2007.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hynes RO. 2002. Integrins: bidirectional, allosteric signaling machines. Cell 110:673–687. doi: 10.1016/S0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 4.Goodman SL, Picard M. 2012. Integrins as therapeutic targets. Trends Pharmacol Sci 33:405–412. doi: 10.1016/j.tips.2012.04.002. [DOI] [PubMed] [Google Scholar]
- 5.Desgrosellier JS, Cheresh DA. 2010. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer 10:9–22. doi: 10.1038/nrc2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brakebusch C, Fassler R. 2005. β1-Integrin function in vivo: adhesion, migration and more. Cancer Metastasis Rev 24:403–411. doi: 10.1007/s10555-005-5132-5. [DOI] [PubMed] [Google Scholar]
- 7.Barkan D, Chambers AF. 2011. β1-Integrin: a potential therapeutic target in the battle against cancer recurrence. Clin Cancer Res 17:7219–7223. doi: 10.1158/1078-0432.CCR-11-0642. [DOI] [PubMed] [Google Scholar]
- 8.Howe GA, Addison CL. 2012. β1 Integrin: an emerging player in the modulation of tumorigenesis and response to therapy. Cell Adh Migr 6:71–77. doi: 10.4161/cam.20077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lahlou H, Muller WJ. 2011. Beta1-integrins signaling and mammary tumor progression in transgenic mouse models: implications for human breast cancer. Breast Cancer Res 13:229. doi: 10.1186/bcr2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang D, Muller S, Amin AR, Huang D, Su L, Hu Z, Rahman MA, Nannapaneni S, Koenig L, Chen Z, Tighiouart M, Shin DM, Chen ZG. 2012. The pivotal role of integrin beta1 in metastasis of head and neck squamous cell carcinoma. Clin Cancer Res 18:4589–4599. doi: 10.1158/1078-0432.CCR-11-3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oshita F, Kameda Y, Hamanaka N, Saito H, Yamada K, Noda K, Mitsuda A. 2004. High expression of integrin beta1 and p53 is a greater poor prognostic factor than clinical stage in small-cell lung cancer. Am J Clin Oncol 27:215–219. doi: 10.1097/01.COC.0000054894.64867.80. [DOI] [PubMed] [Google Scholar]
- 12.Yao ES, Zhang H, Chen YY, Lee B, Chew K, Moore D, Park C. 2007. Increased beta1 integrin is associated with decreased survival in invasive breast cancer. Cancer Res 67:659–664. doi: 10.1158/0008-5472.CAN-06-2768. [DOI] [PubMed] [Google Scholar]
- 13.dos Santos PB, Zanetti JS, Ribeiro-Silva A, Beltrao EI. 2012. Beta 1 integrin predicts survival in breast cancer: a clinicopathological and immunohistochemical study. Diagn Pathol 7:104. doi: 10.1186/1746-1596-7-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wei G, Du Y, Yang C, Zhang X. 2008. The expression and significance of integrin beta1 and focal adhesion kinase and its clinical value in laryngeal carcinoma. Lin Chung Er Bi Yan Hou Tou Jing Wai Ke Za Zhi 22:1112–1114. (In Chinese.) [PubMed] [Google Scholar]
- 15.Nikkola J, Vihinen P, Vlaykova T, Hahka-Kemppinen M, Heino J, Pyrhonen S. 2004. Integrin chains beta1 and alphav as prognostic factors in human metastatic melanoma. Melanoma Res 14:29–37. [DOI] [PubMed] [Google Scholar]
- 16.Pontes-Junior J, Reis ST, de Oliveira LC, Sant'anna AC, Dall'oglio MF, Antunes AA, Ribeiro-Filho LA, Carvalho PA, Cury J, Srougi M, Leite KR. 2010. Association between integrin expression and prognosis in localized prostate cancer. Prostate 70:1189–1195. doi: 10.1002/pros.21153. [DOI] [PubMed] [Google Scholar]
- 17.Bottger TC, Maschek H, Lobo M, Gottwohl RG, Brenner W, Junginger T. 1999. Prognostic value of immunohistochemical expression of beta-1 integrin in pancreatic carcinoma. Oncology 56:308–313. doi: 10.1159/000011984. [DOI] [PubMed] [Google Scholar]
- 18.Luo M, Guan JL. 2010. Focal adhesion kinase: a prominent determinant in breast cancer initiation, progression and metastasis. Cancer Lett 289:127–139. doi: 10.1016/j.canlet.2009.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Theocharis SE, Klijanienko JT, Padoy E, Athanassiou S, Sastre-Garau XX. 2009. Focal adhesion kinase (FAK) immunocytochemical expression in breast ductal invasive carcinoma (DIC): correlation with clinicopathological parameters and tumor proliferative capacity. Med Sci Monit 15:BR221–BR226. [PubMed] [Google Scholar]
- 20.Yom CK, Noh DY, Kim WH, Kim HS. 2011. Clinical significance of high focal adhesion kinase gene copy number and overexpression in invasive breast cancer. Breast Cancer Res Treat 128:647–655. doi: 10.1007/s10549-010-1150-2. [DOI] [PubMed] [Google Scholar]
- 21.Lahlou H, Sanguin-Gendreau V, Zuo D, Cardiff RD, McLean GW, Frame MC, Muller WJ. 2007. Mammary epithelial-specific disruption of the focal adhesion kinase blocks mammary tumor progression. Proc Natl Acad Sci U S A 104:20302–20307. doi: 10.1073/pnas.0710091104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.White DE, Kurpios NA, Zuo D, Hassell JA, Blaess S, Mueller U, Muller WJ. 2004. Targeted disruption of beta1-integrin in a transgenic mouse model of human breast cancer reveals an essential role in mammary tumor induction. Cancer Cell 6:159–170. doi: 10.1016/j.ccr.2004.06.025. [DOI] [PubMed] [Google Scholar]
- 23.Huck L, Pontier SM, Zuo DM, Muller WJ. 2010. β1-Integrin is dispensable for the induction of ErbB2 mammary tumors but plays a critical role in the metastatic phase of tumor progression. Proc Natl Acad Sci U S A 107:15559–15564. doi: 10.1073/pnas.1003034107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Safe S, Jin UH, Hedrick E, Reeder A, Lee SO. 2014. Role of orphan nuclear receptors in cancer and potential as drug targets. Mol Endocrinol 28:157–172. doi: 10.1210/me.2013-1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee SO, Abdelrahim M, Yoon K, Chintharlapalli S, Papineni S, Kim K, Wang H, Safe S. 2010. Inactivation of the orphan nuclear receptor TR3/Nur77 inhibits pancreatic cancer cell and tumor growth. Cancer Res 70:6824–6836. doi: 10.1158/0008-5472.CAN-10-1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee SO, Andey T, Jin UH, Kim K, Singh M, Safe S. 2012. The nuclear receptor TR3 regulates mTORC1 signaling in lung cancer cells expressing wild-type p53. Oncogene 31:3265–3276. doi: 10.1038/onc.2011.504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee SO, Jin UH, Kang JH, Kim SB, Guthrie AS, Sreevalsan S, Lee JS, Safe S. 2014. The orphan nuclear receptor NR4A1 (Nur77) regulates oxidative and endoplasmic reticulum stress in pancreatic cancer cells. Mol Cancer Res 12:527–538. doi: 10.1158/1541-7786.MCR-13-0567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee SO, Li X, Hedrick E, Jin UH, Tjalkens RB, Backos DS, Li L, Zhang Y, Wu Q, Safe S. 2014. Diindolylmethane analogs bind NR4A1 and are NR4A1 antagonists in colon cancer cells. Mol Endocrinol 28:1729–1739. doi: 10.1210/me.2014-1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hedrick E, Lee SO, Kim G, Abdelrahim M, Jin UH, Safe S, Abudayyeh A. 2015. Nuclear receptor 4A1 (NR4A1) as a drug target for renal cell adenocarcinoma. PLoS One 10:e0128308. doi: 10.1371/journal.pone.0128308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hedrick E, Lee SO, Doddapaneni R, Singh M, Safe S. 2015. Nuclear receptor 4A1 as a drug target for breast cancer chemotherapy. Endocr Relat Cancer 22:831–840. doi: 10.1530/ERC-15-0063. [DOI] [PubMed] [Google Scholar]
- 31.Zhou F, Drabsch Y, Dekker TJ, de Vinuesa AG, Li Y, Hawinkels LJ, Sheppard KA, Goumans MJ, Luwor RB, de Vries CJ, Mesker WE, Tollenaar RA, Devilee P, Lu CX, Zhu H, Zhang L, Dijke PT. 2014. Nuclear receptor NR4A1 promotes breast cancer invasion and metastasis by activating TGF-beta signalling. Nat Commun 5:3388. doi: 10.1038/ncomms4388. [DOI] [PubMed] [Google Scholar]
- 32.Wilson TE, Padgett KA, Johnston M, Milbrandt J. 1993. A genetic method for defining DNA-binding domains: application to the nuclear receptor NGFI-B. Proc Natl Acad Sci U S A 90:9186–9190. doi: 10.1073/pnas.90.19.9186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Philips A, Lesage S, Gingras R, Maira MH, Gauthier Y, Hugo P, Drouin J. 1997. Novel dimeric Nur77 signaling mechanism in endocrine and lymphoid cells. Mol Cell Biol 17:5946–5951. doi: 10.1128/MCB.17.10.5946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parvani JG, Galliher-Beckley AJ, Schiemann BJ, Schiemann WP. 2013. Targeted inactivation of β1 integrin induces β3 integrin switching, which drives breast cancer metastasis by TGF-β. Mol Biol Cell 24:3449–3459. doi: 10.1091/mbc.E12-10-0776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Truong HH, Xiong J, Ghotra VP, Nirmala E, Haazen L, Le Devedec SE, Balcioglu HE, He S, Snaar-Jagalska BE, Vreugdenhil E, Meerman JH, van de Water B, Danen EH. 2014. β1 Integrin inhibition elicits a prometastatic switch through the TGFβ-miR-200-ZEB network in E-cadherin-positive triple-negative breast cancer. Sci Signal 7:ra15. doi: 10.1126/scisignal.2004751. [DOI] [PubMed] [Google Scholar]
- 36.Madamanchi A, Zijlstra A, Zutter MM. 2014. Flipping the switch: integrin switching provides metastatic competence. Sci Signal 7:pe9. doi: 10.1126/scisignal.2005236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Villa-Garcia M, Li L, Riely G, Bray PF. 1994. Isolation and characterization of a TATA-less promoter for the human β3 integrin gene. Blood 83:668–676. [PubMed] [Google Scholar]
- 38.Kurakula K, Koenis DS, van Tiel CM, de Vries CJ. 2014. NR4A nuclear receptors are orphans but not lonesome. Biochim Biophys Acta 1843:2543–2555. doi: 10.1016/j.bbamcr.2014.06.010. [DOI] [PubMed] [Google Scholar]
- 39.Pearen MA, Muscat GE. 2010. Nuclear hormone receptor 4A signaling: implications for metabolic disease. Mol Endocrinol 24:1891–1903. doi: 10.1210/me.2010-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Safe S, Jin UH, Morpurgo B, Abudayyeh A, Singh M, Tjalkens RB. 23 April 2015. Nuclear receptor 4A (NR4A) family - orphans no more. J Steroid Biochem Mol Biol doi: 10.1016/j.jsbmb.2015.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu JJ, Zeng HN, Zhang LR, Zhan YY, Chen Y, Wang Y, Wang J, Xiang SH, Liu WJ, Wang WJ, Chen HZ, Shen YM, Su WJ, Huang PQ, Zhang HK, Wu Q. 2010. A unique pharmacophore for activation of the nuclear orphan receptor Nur77 in vivo and in vitro. Cancer Res 70:3628–3637. doi: 10.1158/0008-5472.CAN-09-3160. [DOI] [PubMed] [Google Scholar]
- 42.Kolluri SK, Zhu X, Zhou X, Lin B, Chen Y, Sun K, Tian X, Town J, Cao X, Lin F, Zhai D, Kitada S, Luciano F, O'Donnell E, Cao Y, He F, Lin J, Reed JC, Satterthwait AC, Zhang XK. 2008. A short Nur77-derived peptide converts Bcl-2 from a protector to a killer. Cancer Cell 14:285–298. doi: 10.1016/j.ccr.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhan YY, Chen Y, Zhang Q, Zhuang JJ, Tian M, Chen HZ, Zhang LR, Zhang HK, He JP, Wang WJ, Wu R, Wang Y, Shi C, Yang K, Li AZ, Xin YZ, Li TY, Yang JY, Zheng ZH, Yu CD, Lin SC, Chang C, Huang PQ, Lin T, Wu Q. 2012. The orphan nuclear receptor Nur77 regulates LKB1 localization and activates AMPK. Nat Chem Biol 8:897–904. doi: 10.1038/nchembio.1069. [DOI] [PubMed] [Google Scholar]
- 44.Wu H, Lin Y, Li W, Sun Z, Gao W, Zhang H, Xie L, Jiang F, Qin B, Yan T, Chen L, Zhao Y, Cao X, Wu Y, Lin B, Zhou H, Wong AS, Zhang XK, Zeng JZ. 2011. Regulation of Nur77 expression by beta-catenin and its mitogenic effect in colon cancer cells. FASEB J 25:192–205. doi: 10.1096/fj.10-166462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lin B, Kolluri SK, Lin F, Liu W, Han YH, Cao X, Dawson MI, Reed JC, Zhang XK. 2004. Conversion of Bcl-2 from protector to killer by interaction with nuclear orphan receptor Nur77/TR3. Cell 116:527–540. doi: 10.1016/S0092-8674(04)00162-X. [DOI] [PubMed] [Google Scholar]
- 46.Zhang XK. 2007. Targeting Nur77 translocation. Expert Opin Ther Targets 11:69–79. doi: 10.1517/14728222.11.1.69. [DOI] [PubMed] [Google Scholar]
- 47.Ferlini C, Cicchillitti L, Raspaglio G, Bartollino S, Cimitan S, Bertucci C, Mozzetti S, Gallo D, Persico M, Fattorusso C, Campiani G, Scambia G. 2009. Paclitaxel directly binds to Bcl-2 and functionally mimics activity of Nur77. Cancer Res 69:6906–6914. doi: 10.1158/0008-5472.CAN-09-0540. [DOI] [PubMed] [Google Scholar]
- 48.Pipaon C, Tsai SY, Tsai MJ. 1999. COUP-TF upregulates NGFI-A gene expression through an Sp1 binding site. Mol Cell Biol 19:2734–2745. doi: 10.1128/MCB.19.4.2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shimada J, Suzuki Y, Kim SJ, Wang PC, Matsumura M, Kojima S. 2001. Transactivation via RAR/RXR-Sp1 interaction: characterization of binding between Sp1 and GC box motif. Mol Endocrinol 15:1677–1692. doi: 10.1210/mend.15.10.0707. [DOI] [PubMed] [Google Scholar]
- 50.Sugawara A, Uruno A, Kudo M, Ikeda Y, Sato K, Taniyama Y, Ito S, Takeuchi K. 2002. Transcription suppression of thromboxane receptor gene by peroxisome proliferator-activated receptor-gamma via an interaction with Sp1 in vascular smooth muscle cells. J Biol Chem 277:9676–9683. doi: 10.1074/jbc.M104560200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.