

Abstract
The disruption of the intestinal epithelial barrier function occurs commonly in various pathologies, but the exact mechanisms responsible are unclear. The H19 long noncoding RNA (lncRNA) regulates the expression of different genes and has been implicated in human genetic disorders and cancer. Here, we report that H19 plays an important role in controlling the intestinal epithelial barrier function by serving as a precursor for microRNA 675 (miR-675). H19 overexpression increased the cellular abundance of miR-675, which in turn destabilized and repressed the translation of mRNAs encoding tight junction protein ZO-1 and adherens junction E-cadherin, resulting in the dysfunction of the epithelial barrier. Increasing the level of the RNA-binding protein HuR in cells overexpressing H19 prevented the stimulation of miR-675 processing from H19, promoted ZO-1 and E-cadherin expression, and restored the epithelial barrier function to a nearly normal level. In contrast, the targeted deletion of HuR in intestinal epithelial cells enhanced miR-675 production in the mucosa and delayed the recovery of the gut barrier function after exposure to mesenteric ischemia/reperfusion. These results indicate that H19 interacts with HuR and regulates the intestinal epithelial barrier function via the H19-encoded miR-675 by altering ZO-1 and E-cadherin expression posttranscriptionally.
INTRODUCTION
The majority of the mammalian genome is transcribed into a vast number of noncoding RNAs, whereas protein-coding transcripts account for only a minority of transcriptional output (1, 2). Long noncoding RNAs (lncRNAs) are defined as transcribed RNAs spanning >200 nucleotides in length that lack protein-coding capacity and are distinct from well-characterized structural RNAs (rRNAs, tRNAs, snRNAs, and snoRNAs) or small regulatory RNAs (3, 4). lncRNAs arise from intergenic, antisense, or promoter-proximal regions, and they share many features with mRNAs. Both classes of RNA can be transcribed from multiexonic genes and possess a 5′-methyl-guanosine cap and 3′-poly(A) tail (1, 3, 5). Some lncRNAs are ubiquitous, but others are dynamically expressed in tissue-, differentiation stage-, and cell type-specific patterns (4, 6). lncRNAs have been involved in a variety of cellular functions, physiologic processes, and disease states by modulating gene expression at different levels, including chromatin remodeling, transcriptional and posttranscriptional control, and protein metabolism (1, 3, 4). lncRNAs can serve as repressors or activators of gene transcription, as has been extensively reported (1, 4), but lncRNAs also can regulate mRNA decay and translation, working jointly with microRNAs (miRNAs) and RNA-binding proteins (RBPs) (7–9).
Intercellular junction complexes, comprising tight junctions (TJs) and adherens junctions (AJs), fence the paracellular space in simple epithelia, such as those lining the intestine, kidneys, and lung, and form an important barrier against a wide array of noxious substances present in the lumen (10, 11). In the intestine, the disruption of this barrier function occurs commonly in various critically pathological conditions, such as trauma, burns, hemorrhage, sepsis, inflammation, and massive surgical operations, leading to the translocation of luminal toxic substances and bacteria to the bloodstream (12). The TJ is the most apical of these junctional complexes, and it seals epithelial cells together in a way that prevents even small molecules from leaking between cells (10, 13). TJ complexes primarily consist of TJ-transmembrane proteins, such as the claudin family of proteins, that associate with a cytosolic plaque of TJ-membrane-associated proteins, such as ZO-1, that links tightly to the cortical cytoskeleton (14). Immediately below the TJs are the cadherin-rich AJs that mediate strong cell-to-cell adhesion and have functional roles in forming and regulating the epithelial barrier (15). The AJ E-cadherin integrates various intracellular and extracellular signals and plays an important role in the control of epithelial paracellular permeability (16, 17). TJs and AJs are highly dynamic, and their constituent proteins undergo continuous remodeling and turnover under tight regulation by numerous extracellular and intracellular factors. The maintenance of dynamic levels of TJ and AJ proteins is critical for the normal function of the epithelial barrier, whereas the disrupted expression of TJs and AJs results in gut epithelial barrier dysfunction (10, 18–20).
The H19 lncRNA is a 2.3-kb capped, spliced, and polyadenylated noncoding RNA that is transcribed from the conserved imprinted H19/igf2 gene cluster located on human chromosome 11p5.5 (21, 22). H19 is highly expressed during embryogenesis and is strongly downregulated after birth (23, 24). Emerging evidence also indicates that the increased expression of H19 is commonly detected in a broad spectrum of pathological conditions, such as various malignancies (25–27), and after estrogen treatment (28) or exposure to hypoxia (29). H19 represses embryonic placental growth and trans regulates a network of imprinted genes during fetal development (23, 30), but its role in cancer development can be tumor suppressive or oncogenic, depending on cellular content and tumor type (25, 29, 31). Mice with targeted H19 deletion (H19Δ3) exhibit an overgrowth phenotype, which can be rescued by the transgenic reexpression of the H19 gene (23, 32). To understand how H19 acts to modulate distinct cellular processes, several studies have suggested that H19 functions as a primary miRNA template for miRNA 675 (miR-675) (30, 33) and also acts as a molecular sponge for the miRNA let-7 (34, 35). Here, we report a novel function of H19 in the regulation of the intestinal epithelial barrier and present evidence that H19 overexpression specifically decreases the stability and translation of mRNAs encoding the TJ ZO-1 and AJ E-cadherin via miR-675 release, resulting in barrier dysfunction. Our results also reveal that the RBP HuR inhibits miR-675 processing from H19 and prevents H19-induced barrier dysfunction, whereas HuR deletion in intestinal epithelial cells (IECs) increases miR-675 abundance in the intestinal mucosa and delays the recovery of gut barrier function in mice exposed to pathological stress.
MATERIALS AND METHODS
Cell culture and animals.
The Caco-2 human colon carcinoma cell line was purchased from the American Type Culture Collection (Manassas, VA) and was maintained under standard culture conditions (18). Tissue culture medium and dialyzed fetal bovine serum (FBS) were obtained from Invitrogen (Carlsbad, CA), and biochemicals were obtained from Sigma (St. Louis, MO). Antibodies recognizing HuR, ZO-1, ZO-2, claudin-1, claudin-3, JAM-1, E-cadherin, β-catenin, and β-actin were obtained from Santa Cruz Biotechnology (Santa Cruz, CA) and BD Biosciences (Sparks, MD), and the secondary antibody conjugated to horseradish peroxidase was obtained from Sigma.
Intestinal epithelial tissue-specific HuR deletion (IE-HuR−/−) mice were generated by crossing the HuRflox/flox (HuRfl/fl) mouse (from the Jackson Laboratory) with the villin-Cre mouse as described previously (36). HuRfl/fl-Cre− mice served as a littermate control. Mice were housed and handled in a specific-pathogen-free breeding barrier and cared for by trained technicians and veterinarians. All animal experiments were conducted in accordance with NIH guidelines and were approved by the Institutional Animal Care and Use Committee of the University Maryland School of Medicine and Baltimore VA hospital.
Plasmid construction.
An expression vector containing a 1.5-kb fragment flanking the human H19 locus (including the entire exon 1) under the control of the pCMV promoter was purchased from OriGene (Rockville, MD), and the HuR expression vector was described previously (37). Transient transfections were performed using the Lipofectamine reagent by following the manufacturer's recommendations (Invitrogen). Forty-eight hours after transfection using Lipofectamine, cells were harvested for analysis.
Quantitative real-time PCR and immunoblot analyses.
Total RNA was isolated by using the RNeasy minikit (Qiagen, Valencia, CA) and used in reverse transcription (RT) and PCR amplification reactions as described previously (38). Quantitative real-time PCR (qPCR) was performed using StepOnePlus systems with specific primers, probes, and software (Applied Biosystems, Foster City, CA).
To examine protein levels, whole-cell lysates were prepared using 2% SDS, sonicated, and centrifuged at 4°C for 15 min. The supernatants were boiled for 5 min and size fractionated by SDS-PAGE. After transferring proteins onto nitrocellulose filters, the blots were incubated with primary antibodies recognizing TJ or AJ proteins. Following incubations with secondary antibodies, immunocomplexes were visualized by using chemiluminescence.
Analysis of newly translated protein and polysome analysis.
De novo synthesis of nascent proteins was detected by a Click-iT protein analysis detection kit (Life Technologies, Grand Island, NY) by following the manufacturer's instructions (39). Briefly, cells were incubated in methionine-free medium and then exposed to l-azidohomoalaine (AHA). After mixing cell lysates with the reaction buffer containing biotin-alkyne reagent and CuSO4 for 20 min, the biotin-alkyne-azide-modified protein complex was pulled down using paramagnetic streptavidin-conjugated Dynabeads. The pulldown material was resolved by 10% SDS-PAGE and analyzed by Western immunoblot analysis using antibodies that recognized ZO-1, E-cadherin, or glyceraldehyde-3-phosphate dehydrogenase (GAPDH) proteins.
Polysome analysis was performed as described previously (40). Briefly, cells at ∼70% confluence were incubated for 15 min in 0.1 mg/ml cycloheximide, lifted by scraping in 1 ml of polysome extraction buffer, and lysed on ice for 10 min. Nuclei were pelleted, and the resulting supernatant was fractionated through a 15 to 60% linear sucrose gradient to fractionate cytoplasmic components according to their molecular weights. The eluted fractions were prepared with a fraction collector (Brandel, Gaithersburg, MD), and their quality was monitored at 254 nm using a UV-6 detector (ISCO, Louisville, KY). After RNA was extracted from each fraction, the levels of each individual mRNA were quantified by RT followed by qPCR analysis of each of the fractions.
Biotin-labeled miR-675 pulldown and ribonucleoprotein immunoprecipitation (RNP-IP) assays.
The binding of miR-675 to target mRNAs was examined by biotin-labeled miR-675-3p as described previously (41). Briefly, biotin-labeled miR-675-3p was transfected into cells and 24 h later whole-cell lysates were collected, mixed with streptavidin-Dynal beads (Invitrogen, Carlsbad, CA), and incubated at 4°C on a rotator overnight. After the beads were washed thoroughly, the bead-bound RNA was isolated and subjected to RT followed by qPCR analysis. Input RNA was extracted and used as a control.
To assess the association of endogenous HuR with endogenous H19, the IP of RNP complexes was performed as described previously (19). Twenty million cells were collected per sample, and lysates were used for IP for 4 h at room temperature in the presence of excess (30 μg) IP antibody (IgG or anti-HuR). RNA in IP materials was used in RT reactions followed by PCR and qPCR analyses to detect the presence of lncRNAs.
Measurements of gut epithelial barrier function.
The epithelial barrier function in vitro was examined by paracellular tracer flux assays using the 12-mm Transwell as described previously (37, 42). Fluorescein isothiocyanate (FITC)-dextran, a membrane-impermeable molecule, served as the paracellular tracer and was added to the apical bathing wells. The basal bathing well had no added tracers and contained the same flux assay medium as that in the apical compartment. All flux assays were performed at 37°C, and the basal medium was collected 2 h after the addition of FITC-dextran. The concentration of FITC-dextran in the basal medium was determined using a fluorescence plate reader with an excitation wavelength of 490 nm and an emission wavelength of 530 nm. Transepithelial electrical resistance (TEER) was measured with an epithelial voltmeter under open circuit conditions (WPI, Sarasota, FL) as described previously (37), and the TEER of all monolayers was normalized to that of control monolayers in the same experiment.
Gut permeability in vivo was determined by examining the appearance in blood of FITC-dextran administered by gavage as described previously (43). Briefly, mice were gavaged with FITC-dextran at a dose of 60 mg/100 g of body weight 4 h before harvest. Blood samples were collected by cardiac puncture. The serum concentration of the FITC-dextran was determined using a fluorescence plate reader as described above.
Surgical procedures.
The ischemia/reperfusion (I/R) model was performed as described previously (44). Mice were anesthetized by pentobarbital sodium (Nembutal; 5.5 mg/100 g, intraperitoneally [i.p.]), and a midline abdominal incision was performed. Mice were exposed to 30 min of superior mesenteric artery ischemia followed by 2 h of reperfusion. Sham operation for controls involves laparotomy without mesenteric ischemia. The incision was closed using a 2-layer procedure: 5-0 silk suture on the muscle layer and the skin, respectively. Mice received 1 ml of saline i.p. for fluid resuscitation at the time of closure and 0.1 mg/100 g Buprenex subcutaneously (s.c.) 4 times at 12-h intervals to minimize distress.
Statistics.
Values are means ± standard errors of the means (SEM) from three to six samples. Immunoblot results were repeated three times. The significance of the difference between means was determined by analysis of variance (ANOVA). The level of significance was determined by using Duncan's multiple-range test (45).
RESULTS
H19 represses ZO-1 and E-cadherin expression and disrupts epithelial barrier function.
To define the role of H19 in the regulation of intestinal epithelial barrier function, we determined the effect of overexpressing H19 on the expression of TJs and AJs in Caco-2 cells, a widely used cell model for gut epithelial barrier studies in vitro (46, 47). As shown (Fig. 1A, left), the abundance of H19 increased dramatically when cells were transfected with the H19 expression vector compared to that of cells transfected with empty vector. Transfection with the H19 expression vector did not alter the levels of lncRNA SPRY4-IT1 (Fig. 1A, middle) or U6 RNA (Fig. 1A, right). Increasing H19 levels specifically inhibited the expression of the TJ protein ZO-1 and the AJ protein E-cadherin (Fig. 1B) but failed to alter the expression levels of TJ proteins ZO-2, claudin-1, claudin-3, and JAM-1 and AJ protein β-catenin. The levels of ZO-1 and E-cadherin proteins in H19-transfected cells decreased by ∼91% and ∼75% (n = 3; P < 0.05), respectively, compared to those for cells transfected with empty vector. Consistent with this, ZO-1 and E-cadherin proteins, as detected by immunostaining, also decreased remarkably after ectopic H19 overexpression (Fig. 1C, top and middle). In contrast, there were no differences in the extent of immunostaining of β-catenin (Fig. 1C, bottom) and ZO-2 (data not shown) in H19-transfected cells relative to that in cells transfected with empty vector. Importantly, the elevation of H19 abundance disrupted the epithelial barrier function, as evidenced by a decrease in TEER values (Fig. 1D, left) and an increase in the levels of paracellular flux of FITC-dextran (Fig. 1D, right). On the other hand, H19 overexpression did not affect cell viability, as measured by Trypan blue staining (data not shown), and it only slightly inhibited Caco-2 cell proliferation, as determined by MTT assay and the measurement of cell numbers (see Fig. S1 in the supplemental material). In addition, the expression of neither ZO-1 nor E-cadherin was affected by the ectopic overexpression of lncRNA SPRY4-IT1 or HULC (data not shown).
FIG 1.
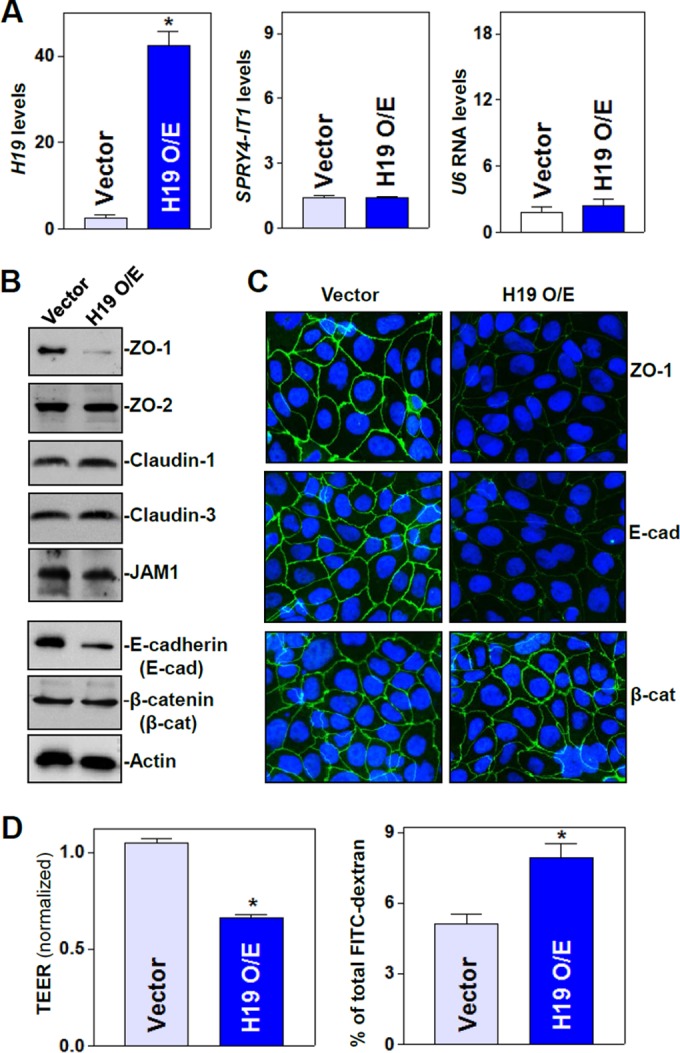
H19 lncRNA inhibits expression of ZO-1 and E-cadherin and disrupts epithelial barrier function. (A) The levels of H19 (left), SPRY4-IT1 (middle), and U6 RNA (right) transcripts after overexpression (O/E) of H19 were measured by qPCR analysis. Caco-2 cells were transfected with the H19 expression vector or control vector, and total RNA was harvested 48 h thereafter. Values are means ± SEM of data from triplicate experiments. *, P < 0.05 compared to results for the vector. (B) Representative immunoblots of tight junctions and adherens junctions as examined by Western blotting of cells described for panel A. Three experiments were performed that showed similar results. (C) Cellular distribution of ZO-1 and E-cadherin in cells described for panel A. Forty-eight hours after transfection, cells were fixed, permeabilized, incubated with the antibody against ZO-1 or E-cadherin, and then incubated with anti-IgG conjugated with FITC. Original magnification, ×500. (D) Changes in epithelial barrier function as indicated by changes in TEER (left) and FITC-dextran paracellular permeability (right) in cells described for panel A. TEER assays were performed on 12-mm Transwell filters. Paracellular permeability was assayed by using the membrane-impermeable trace molecule FITC-dextran, which was added to the insert medium. Values are means ± SEM of data from six samples. *, P < 0.05 compared to results for the vector.
To examine the mechanism underlying the repression of ZO-1 and E-cadherin by H19, we examined changes in the levels of ZO-1 and E-cadherin (E-cad) mRNAs in cells overexpressing H19. As shown in Fig. 2A and B, H19 overexpression decreased levels of ZO-1 and E-cad mRNAs by enhancing their degradation, since ectopic H19 overexpression selectively lowered the half-lives of ZO-1 and E-cad mRNAs. We also determined the translation rate of ZO-1 and E-cad mRNAs using the Click-iT technology (Materials and Methods), pulling down biotin-alkyne-azide-modified protein complexes by paramagnetic streptavidin-conjugated Dynabeads. We found that the levels of newly synthesized ZO-1 and E-cadherin proteins decreased significantly in cells overexpressing H19 compared to levels in cells transfected with an empty vector (Fig. 2C). The levels of newly synthesized ZO-1 and E-cadherin proteins in H19-transfected cells decreased by ∼84% and ∼88% (n = 3; P < 0.05), respectively, compared with those of cells transfected with empty control vector. The inhibition of ZO-1 and E-cadherin protein synthesis by H19 was specific, since there was no change in nascent GAPDH synthesis after transfection with H19. To further define the role of H19 in the regulation of translation of ZO-1 and E-cad mRNAs, we examined the relative distribution of ZO-1 and E-cad mRNAs in individual fractions from polyribosome gradients after the ectopic overexpression of H19 as reported previously (40, 48). Although increasing the levels of H19 did not affect global polysomal profiles (data not shown), the association of ZO-1 and E-cad mRNAs with actively translating fractions (fractions 8 and 9) decreased dramatically, shifting to low-translating fractions (fractions 6 and 7) (Fig. 2D, left and middle). In contrast, housekeeping Gapdh mRNA distributed similarly in both groups (Fig. 2D, right). These data indicate that H19 inhibits the expression of ZO-1 and E-cadherin by destabilizing the ZO-1 and E-cad mRNAs and repressing their translation, resulting in the disruption of the epithelial barrier.
FIG 2.

H19 enhances decay of the mRNAs encoding ZO-1 and E-cadherin and represses their translation. (A) Steady-state levels of ZO-1 mRNA (a) and ZO-1 mRNA half-life (b) 48 h after transfection with either an H19 expression vector or a control vector. ZO-1 mRNA levels were examined at different times after the administration of actinomycin D. Values are means ± SEM of data from triplicate experiments. *, P < 0.05 compared to results with the vector. (B) The level of E-cadherin (E-cad) mRNA and its stability in cells described for panel A. (C) Newly synthesized ZO-1 and E-cadherin proteins in cells overexpressing H19. After cells were exposed to l-azidohomoalaine (AHA), cell lysates were incubated with the reaction buffer containing biotin/alkyne reagent; the biotin-alkyne-azide-modified protein complex was pulled down by paramagnetic streptavidin-conjugated Dynabeads. (D) Distributions of ZO-1 and E-cad mRNAs in each gradient fraction prepared from polysomal profiles in cells after H19 overexpression. Nuclei were pelleted, and the resulting supernatants were fractionated through a 10 to 50% linear sucrose gradient. Total RNA was isolated from different fractions, and the levels of ZO-1, E-cad, and Gapdh mRNAs were measured and plotted as a percentage of each of the total levels of ZO-1, E-cad, and Gapdh mRNA in each sample.
H19 inhibits the epithelial barrier through the release of miR-675.
Since H19 exon 1 encodes two conserved miRNAs (miR-675-5p and miR-675-3p) (Fig. 3A) and can function as the primary miRNA (pri-miRNA) transcript for miR-675, we investigated the possibility that the function of H19 is linked to the release of miR-675. Increasing the levels of H19 by transfecting Caco-2 cells with the H19 expression vector specifically induced the expression levels of miR-675-5p and miR-675-3p (Fig. 3B and C) but did not alter the expression of miR-133a and miR-222, suggesting that the processing of miRNA-675 from H19 increases in cells overexpressing H19. To determine the role of miR-675 in H19-induced barrier dysfunction, we examined whether increasing the levels of miR-675-3p through transfection with its precursor (pre-miR-675-3p) repressed the expression of ZO-1 and E-cadherin. Transient transfection with pre-miR-675-3p dramatically increased the levels of miR-675-3p (Fig. 4A, left), although there were no changes in the levels of miR-222 (Fig. 4A, right). Similar to the effect of H19 induction, increasing the levels of miR-675-3p by pre-miR-675-3p transfection also specifically inhibited the expression of ZO-1 and E-cadherin (Fig. 4B), but it did not affect the expression levels of ZO-2, claudin-1, claudin-3, JAM-1, or β-catenin. The levels of ZO-1 and E-cadherin proteins in pre-miR675-3p-transfected cells decreased by ∼86% and ∼77% (n = 3; P < 0.05), respectively, compared to those of cells transfected with the scrambled control RNA. Moreover, miR-675-3p overexpression disrupted the epithelial barrier function, as revealed by a decrease in TEER values (Fig. 4C, top) and an increase in paracellular permeability (Fig. 4C, bottom). Consistent with the effect of miR-675-3p, the ectopic overexpression of miR-675-5p by transfection with the pre-miR-675-5p also reduced ZO-1 and E-cadherin expression levels and caused epithelial barrier dysfunction (data not shown). Since H19 also reduces the bioavailability of the let-7 miRNA by acting as a molecular sponge (34, 35), we examined if let-7 was involved in the repression of ZO-1 and E-cadherin repression by H19. However, the inhibition of let-7 by the transfection of specific antagomirs (antisense oligomers) directed at let-7a and let-7b (highly expressed members of the let-7 family in IECs) failed to alter the expression levels of ZO-1 and E-cadherin proteins (see Fig. S2 in the supplemental material), suggesting that the impact of H19 on let-7 does not contribute to the H19-induced ZO-1 and E-cadherin repression. These results support the idea that H19 represses ZO-1 and E-cadherin and disrupts the barrier function from a specific induction in miR-675 processing from H19.
FIG 3.
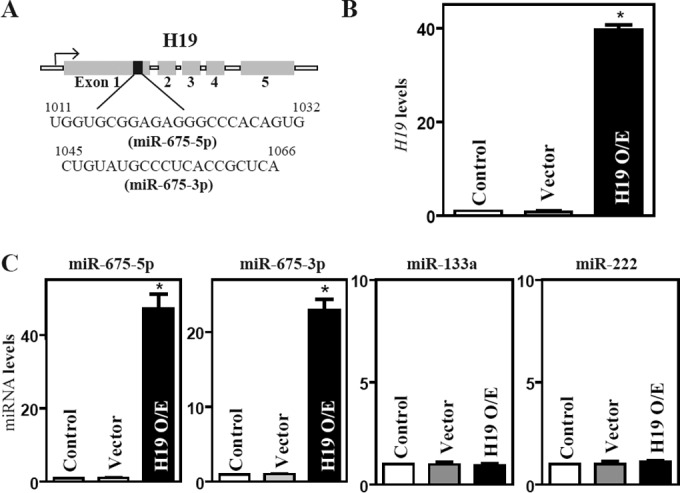
H19 overexpression increases the levels of miR-675. (A) Schematic representation of the H19 transcriptional unit with miR-675, shown in black, embedded within H19 exon 1. (B) Levels of H19 in Caco-2 cells 48 h after transfection of H19 expression vector or control vector. Values are means ± SEM of data from triplicate experiments. *, P < 0.05 compared to results with the vector. (C) Levels of miR-675-5p, miR-675-3p, miR-133a, and miR-222 as measured in cells described for panel B.
FIG 4.
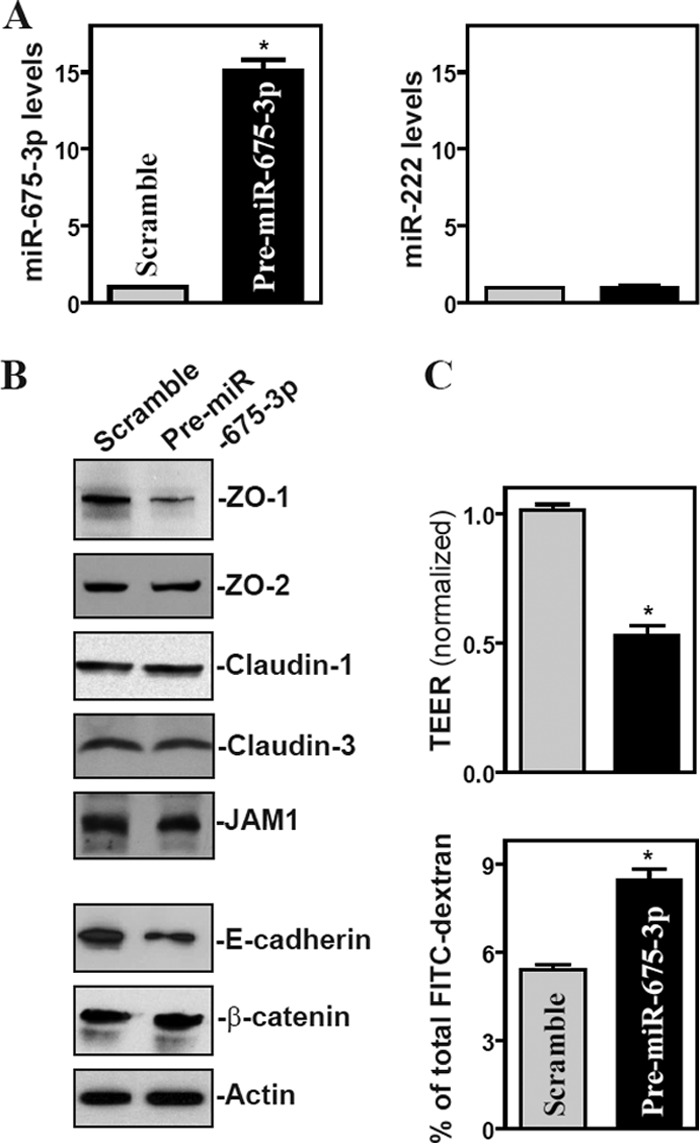
Ectopic overexpression of miR-675 inhibits expression of ZO-1 and E-cadherin and disrupts epithelial barrier function. (A) Levels of miR-675-3p (left) and miR-222 (right) 48 h after transfecting cells with the pre-miR-675-3p or scrambled control RNA. Values are means ± SEM of data from triplicate experiments. *, P < 0.05 compared to results with scrambled RNA. (B) Representative immunoblots of tight junctions and adherens junctions in cells described for panel A. (C) Changes in epithelial barrier function as indicated by changes in TEER (top) and FITC-dextran paracellular permeability (bottom) in cells described for panel A. TEER assays were performed on 12-mm Transwell filters. Paracellular permeability was assayed by using the membrane-impermeable trace molecule FITC-dextran, which was added to the insert medium. Values are means ± SEM of data from six samples. *, P < 0.05 compared to results with scrambled RNA.
ZO-1 and E-cad mRNAs are novel targets of miR-675.
ZO-1 and E-cad mRNAs are potential targets of miR-675-3p, as there are predicted binding sites for miR-675-3p within their 3′ untranslated regions (3′-UTRs) (Fig. 5A) using standard online software (RNA22). Thus, we examined the association of miR-675-3p with the ZO-1 and E-cad mRNAs by RNA pulldown assays using biotin-labeled miR-675-3p (Dharmacon) (shown in Fig. 5Ba). Twenty-four hours after transfection, miR-675-3p levels increased significantly, but the levels of the housekeeping noncoding RNA U6 did not (Fig. 5Bb and c). The ZO-1 and E-cad mRNAs were enriched in the materials pulled down by biotin-miR-675-3p but not in materials from cells transfected with a control scrambled RNA (Fig. 5C, left). The association of miR-675-3p with the ZO-1 or E-cad mRNA was specific, since biotin-miR-675-3p did not interact with the claudin-1 or claudin-3 mRNAs. These results indicate that miR-675-3p can target ZO-1 and E-cad mRNAs.
FIG 5.
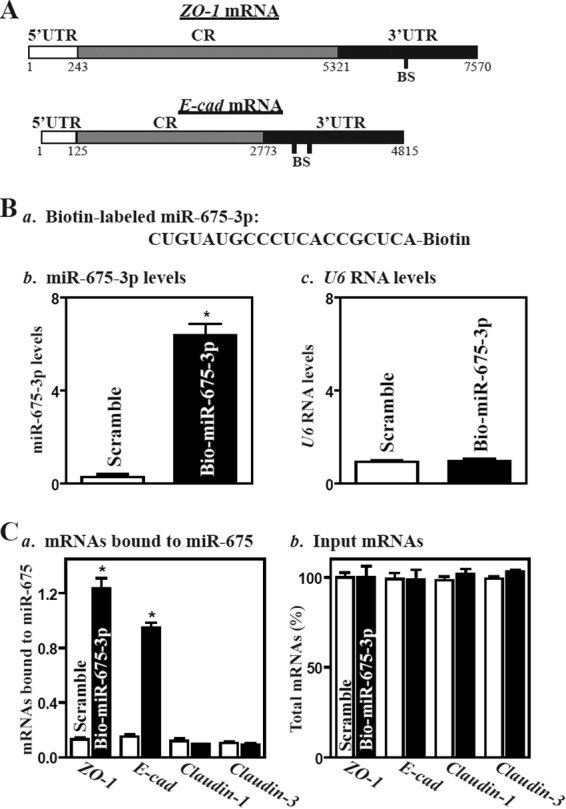
miR-675-3p associates with the ZO-1 and E-cadherin (E-cad) mRNAs. (A) Schematic representations of the ZO-1 and E-cad mRNAs depicting predicted target sites for miR-675-3p in their 3′ untranslated regions (3′-UTRs). BS, predicted miR-675-3p-binding site. (B) Levels of biotinylated miR-675-3p 24 h after transfection. (a) Schematic representation of biotinylated miR-675-3p; (b) miR-miR-675-3p levels as measured by qPCR analysis; (c) U6 RNA levels. Values are means ± SEM of data from triplicate experiments. *, P < 0.05 compared with cells transfected with control scrambled oligomer. (C) Binding of biotinylated miR-675-3p to mRNAs encoding ZO-1, E-cad, claudin-1, and claudin-3. (a) Levels of mRNAs in the materials pulled down by biotin-miR-675-3p; (b) levels of total input mRNAs. *, P < 0.05 compared to results with cells transfected with scrambled oligomer.
To examine the functional consequences of the miR-675-3p/ZO-1 or E-cad mRNA association, we designed the experiments to investigate whether increasing the levels of miR-675-3p affected the expression of ZO-1 or E-cadherin posttranscriptionally. As shown, increasing the levels of miR-675-3p by transfection with pre-miR-675-3p not only destabilized mRNAs encoding ZO-1 and E-cadherin but also repressed their translation. The half-lives of ZO-1 and E-cad mRNAs decreased in cells overexpressing miR-675-3p (Fig. 6A and B) and reduced the levels of newly synthesized proteins of ZO-1 (by ∼81%; n = 3; P < 0.05) and E-cadherin (by ∼76%; n = 3; P < 0.05) (Fig. 6C). Furthermore, miR-675-3p overexpression decreased the association of ZO-1 and E-cad mRNAs with actively translating fractions (fractions 8 and 9) from polyribosome gradients, with shifting to weakly translating fractions (fractions 6 and 7) (Fig. 6D), although it did not affect global polysomal profiles (data not shown). On the other hand, Gapdh mRNA showed a similar distribution in both groups. These findings indicate that miR-675-3p interacts directly with the ZO-1 and E-cad mRNAs and decreases their stability and translation.
FIG 6.

Elevating miR-675-3p enhances decay of the ZO-1 and E-cad mRNAs and represses their translation. (A) The level of ZO-1 mRNA (a) and its stability (b) 48 h after transfection with the pre-miR-675-3p or scrambled oligomer. ZO-1 mRNA levels were examined at different times after incubation with actinomycin D. Values are means ± SEM of data from triplicate experiments. *, P < 0.05 compared to results with scrambled oligomer. (B) The level of E-cad mRNA and its stability in cells described for panel A. (C) Newly synthesized ZO-1 and E-cadherin proteins in cells overexpressing miR-675-3p. Three experiments were performed that showed similar results. (D) Distributions of ZO-1 and E-cad mRNAs in each gradient fraction prepared from polysomal profiles in cells after miR-675-3p overexpression. The levels of ZO-1, E-cad, and Gapdh mRNAs were measured and plotted as a percentage of each of total ZO-1 or E-cad mRNAs and Gapdh mRNA levels in the samples.
HuR blocks miR-675 processing from H19 and prevents H19-induced barrier dysfunction.
The ubiquitous RBP HuR plays an important role in the control of gut barrier function (11, 19) and recently has been shown to interact with H19 (30). The ensuing studies further examined if HuR altered the H19-induced disruption of the epithelial barrier function. Similar to the findings observed in other cell types (30), HuR was found to bind directly to H19 in Caco-2 cells as measured by RNP-IP assays using a specific antibody against HuR, since H19 was highly enriched in HuR IP but not control IgG IP (see Fig. S3 in the supplemental material). In contrast, there were no significant changes in the levels of lncRNAs HULC and PTENP1 in HuR IP samples. To assess if HuR blocked the miR-675 release from H19, we cotransfected Caco-2 cells with the H19 and HuR expression vectors and found that the increase in miR-675-5p and miR-675-3p levels elicited by H19 overexpression was significantly abolished by HuR overexpression (Fig. 7A), suggesting that HuR suppressed miR-675 processing from H19. Consistent with this effect, HuR overexpression prevented the inhibition of expression of ZO-1 and E-cad mRNAs (see Fig. S4 in the supplemental material) and their proteins (Fig. 7B), and it also largely restored the epithelial barrier function (Fig. 7C) seen in cells overexpressing H19. The expression levels of ZO-1 and E-cadherin and epithelial barrier function, as indicated by the levels of TEER and FITC-dextran flux in cells cotransfected with H19 and HuR, were indistinguishable from those observed in cells transfected with control vector. In contrast, cotransfection with the H19 and HuR expression vectors did not alter the expression levels of ZO-2, claudin-1, claudin-3, JAM-1, and β-catenin proteins.
FIG 7.
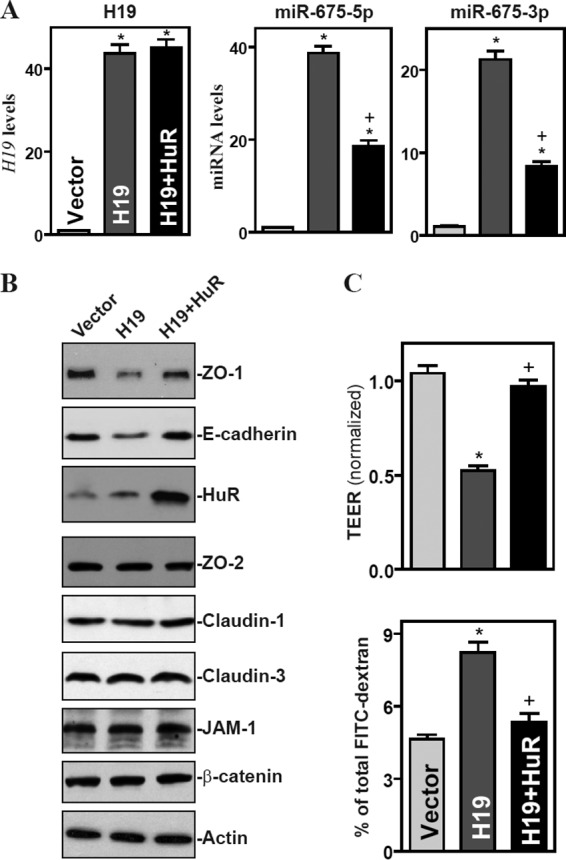
HuR prevents H19-induced epithelial barrier dysfunction by reducing the release of miR-675 from H19. (A) Levels of H19 (left), miR-675-5p (middle), and miR-675-3p (right) in cells overexpressing both H19 and HuR. Cells were cotransfected with the H19 expression vector and HuR expression vector, and RNA was harvested 48 h after the cotransfection. Values are means ± SEM of data from triplicate experiments. *, P < 0.05 compared to results with cells transfected with control vector (Vector). +, P < 0.05 compared to results with cells transfected with HuR expression. (B) Representative immunoblots of ZO-1, E-cadherin, and HuR proteins in cells described for panel A. Three experiments were performed that showed similar results. (C) Changes in the epithelial barrier function as indicated by changes in TEER (top) and FITC-dextran paracellular permeability (bottom) in cells described for panel A. Values are the means ± SEM of data from six samples. *, P < 0.05 compared to results with the vector.
HuR deletion enhances miR-675 processing and delays the recovery of gut barrier function in mice exposed to pathological stress.
To determine the in vivo function of H19-derived miR-675 in the regulation of the gut barrier function, we employed IE-HuR−/− mice that were generated recently in our groups. As reported previously (36), HuR mRNA and protein in the small intestinal and colonic mucosa were undetectable in IE-HuR−/− mice but were expressed at wild-type levels in stomach mucosa, lung, liver, and pancreas compared with those observed in control littermates (data not shown). HuR deletion in IECs significantly increased the basal levels of miR-675-5p and miR-675-3p in the small intestinal mucosa, although it only marginally reduced mucosal H19 abundance (Fig. 8A). Increased levels of miR-675 in the mucosa were associated with an inhibition of ZO-1 and E-cadherin expression, as shown by a decrease in their protein levels (Fig. 8B). The mucosal ZO-1 and E-cadherin proteins decreased by ∼51% and ∼82% in IE-HuR−/− mice (n = 4; P < 0.05), respectively, compared with levels in control littermates. To examine if the miR-675-induced inhibition of ZO-1 and E-cadherin altered the gut barrier function after exposure to stress, an acute mesenteric ischemia/reperfusion (I/R) model (see Materials and Methods) was used. As expected, both littermates and IE-HuR−/− mice subjected to mesenteric I/R displayed a dramatic increase in gut permeability to FITC-dextran (Fig. 8C), but there were no significant differences in the degree of increased gut permeability between IE-HuR−/− mice and littermates when measured immediately and 2 h after I/R. However, HuR deletion repressed the recovery of gut barrier function after I/R-induced stress. The barrier function recovered quickly in littermates, as evidenced by a rapid decrease in the gut permeability after I/R, whereas this recovery was slow in the absence of HuR. The levels of gut permeability in IE-HuR−/− mice were higher than those observed in littermates when examined at 6 and 12 h after I/R. These results indicate that HuR deletion in IECs enhances miR-675 processing from H19 and inhibits the recovery of gut barrier function after exposure to pathological stress. Taken together, our findings suggest a model whereby H19 regulates intestinal epithelial barrier function by releasing miR-675, which destabilizes ZO-1 and E-cad mRNAs and represses their translation (Fig. 8D). The ensuing reduction in ZO-1 and E-cadherin causes the dysfunction of the epithelial barrier. By preventing the release of miR-675 from H19, the RBP HuR increases ZO-1 and E-cadherin protein production and maintains barrier function.
FIG 8.
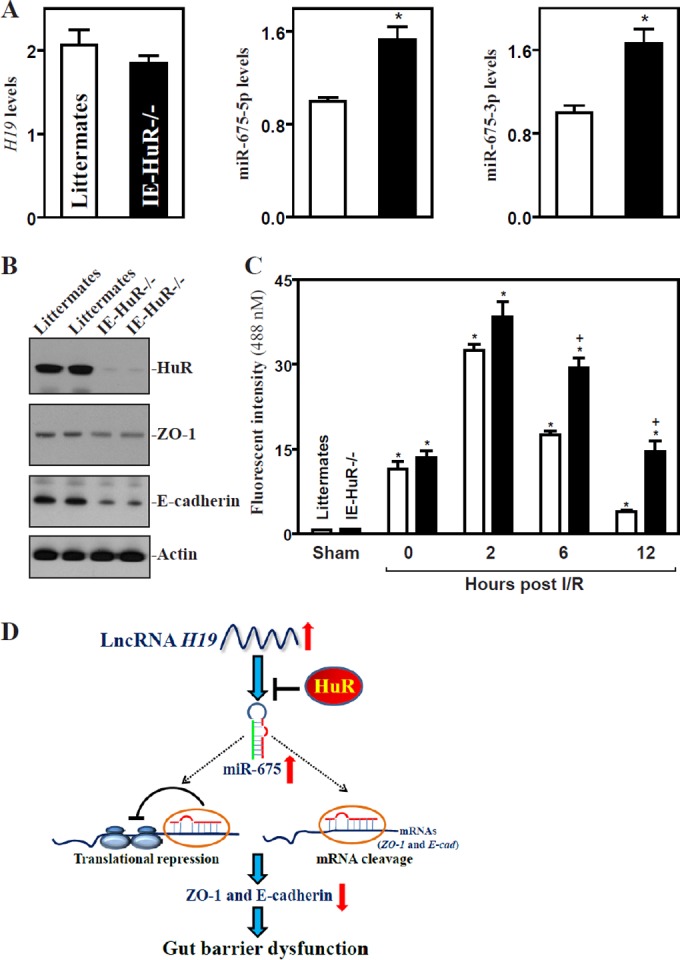
Intestinal epithelial tissue-specific HuR deletion increases the basal level of miR-675 and causes a delay in the recovery of gut barrier dysfunction after exposure to intestinal ischemia/reperfusion (I/R). (A) Levels of H19 (left), miR-675-5p (middle), and miR-675-3p (right) in the small intestinal mucosa in IE-HuR−/− mice. Values are means ± SEM of data from 5 animals. *, P < 0.05 compared to results with littermates. (B) Immunoblots of HuR, ZO-1, and E-cadherin proteins in the small intestinal mucosa described for panel A. (C) Changes in gut permeability in mice exposed to 30 min of superior mesenteric artery ischemia followed by 2 h of reperfusion. FITC-dextran was given orally, and blood samples were collected 4 h thereafter for measurement. Values are means ± SEM of data from 5 animals. *, P < 0.05 compared to results with littermates exposed to I/R. (D) Model proposed to explain the influence of H19 upon epithelial barrier function. H19 inhibits ZO-1 and E-cadherin expression and disrupts the barrier function through the release of miR-675; HuR blocks miR-675 processing from H19 and promotes epithelial barrier function.
DISCUSSION
The effectiveness and stability of the gut epithelial barrier depend on specialized structures, TJs and AJs, but the exact mechanisms that regulate the expression of their constituent proteins are not fully known. The therapies to preserve the integrity of the epithelial barrier are limited, especially in surgical intensive care patients supported with total parenteral nutrition (12, 49). Efforts to understand the control of TJ and AJ expression and to define the potential targets for developing effective therapeutics to protect the barrier function are extremely important. Here, we have identified a novel mechanism by which H19 regulates the intestinal epithelial barrier function by repressing the production of TJ protein ZO-1 and the AJ protein E-cadherin at the posttranscriptional level via the H19-encoded miR-675. HuR overexpression prevented the stimulation of miR-675 processing from H19, promoted ZO-1 and E-cadherin production, and abolished H19-induced barrier dysfunction. In contrast, the targeted deletion of HuR increased miR-675 production in the intestinal mucosa and delayed the recovery of the gut barrier function after exposure to mesenteric I/R. These findings make a significant conceptual advance by linking H19 with gut permeability and underscore the impact of the H19/miR-675 axis on the pathogenesis of acute gut barrier dysfunction.
H19 has a conserved secondary RNA structure and is predominantly cytoplasmic, but it lacks a conserved open reading frame (1, 21, 50). H19 can have multiple functions by acting on specific genes through distinct molecular mechanisms, depending on the biological and spatiotemporal contexts during development and disease states. The expression of the Igf2 gene is affected by the deletion of the H19 gene; eight other genes belonging to an imprinted gene network (IGN) also show increased expression levels in the absence of H19 (32, 51). H19 controls the gene expression of this IGN by recruiting repressive histone markers to their differentially methylated regions (52). H19 also acts through the processing of miR-675 to inhibit placental growth (30) and to promote skeletal muscle differentiation and regeneration (33), but it regulates muscle glucose metabolism by acting as an upstream regulator of the miRNA let-7, which inhibits the expression of key metabolic genes, such as Insr and Lpl (34). Full-length H19 interacts with EZH2 (a key component of polycomb repressive complex 2) and inhibits the transcription of a select group of genes, thereby promoting bladder cancer metastasis (26) and glioma cell invasion (53). Our present results provide new evidence that H19 represses ZO-1 and E-cadherin expression in IECs by acting as a regulatable reservoir of miR-675 and compromises the function of the gut epithelial barrier.
Our results indicate that miR-675 interacts with the ZO-1 and E-cad mRNAs, and this association destabilizes ZO-1 and E-cad mRNAs and also inhibits their translation. Although the specific molecular mechanism underlying the posttranscriptional inhibition of ZO-1 and E-cadherin by miR-675 is unknown, our previous studies demonstrated that miR-222-induced repression of CDK4 translation (54), miR-503-induced CUGBP1 inhibition (41), and miR-195-induced Stim1 mRNA destabilization (55) in IECs are directly linked to increased translocation of these target mRNAs to processing bodies (PBs), where mRNAs are sorted for translation repression and/or degradation (56). Silencing PB-resident proteins such as AGO2 or RCK by transient transfection with their specific short interfering RNAs prevents this repressive influence in cells overexpressing miR-222, miR-503, or miR-195, whereas AGO2 overexpression enhances these inhibitory effects on target transcripts. It is unclear, however, whether induced miR-675 from H19 alters the subcellular localization of the ZO-1 and E-cad mRNAs to PBs and whether an increase in the recruitment of ZO-1 and E-cad mRNAs to PBs by miR-675 affects their stability and translation rates. We are actively investigating these possibilities at present.
Our findings also show that increasing the levels of HuR inhibits miR-675 processing from H19 and prevents H19-induced barrier dysfunction. HuR has three RNA recognition motifs through which it directly interacts with numerous mRNAs to modulate their translation and/or stability (7, 36, 57). Recent studies revealed that HuR also interacts with several lncRNAs to jointly regulate target transcripts antagonistically or synergistically (7, 8, 57–60). HuR binds lncRNA-p21 and induces the recruitment of let-7/AGO to lncRNA-p21, leading to the destabilization of lncRNA-p21 (7). HuR also competes with ncRNA 7SL to modulate p53 translation (8). In this study, HuR overexpression in H19-transfected cells not only blocked the production of miR-675 but also restored ZO-1 and E-cadherin expression and epithelial barrier function to nearly normal levels. Consistent with our current findings, HuR was shown to suppress the processing of human miR-7 from the highly expressed HNRNPK transcripts (61). On the other hand, HuR also can regulate posttranscriptional events of TJs and AJs either through direct interaction with 3′-UTRs of target transcripts (19, 62) or through association with the lncRNA SPRY4-IT1 (63). Taken together, these findings suggest a wider repertoire of targets and miRNAs through which HuR modulates barrier dysfunction in cells overexpressing H19.
The H19-induced repression of ZO-1 and E-cadherin expression has pathophysiological significance, since IE-HuR−/− mice displayed increased miR-675 production in the intestinal mucosa and delayed recovery of the gut barrier function after exposure to mesenteric I/R. Although the complete sets of mRNAs and processes controlled by H19 in the intestinal epithelium remain to be fully elucidated, the fact that the prevention of miR-675 processing from H19 by HuR protected the epithelial barrier function highlights the impact of an individual lncRNA in controlling gut permeability in specific clinical settings. These findings are particularly significant in patients with traumatic damage, thermal injury, or shock and in patients recovering from major surgical operations, as acute gut barrier dysfunction occurs commonly in these clinical situations (12, 64). In summary, our results indicate that the H19/miR-675 axis downregulates the intestinal barrier function by inhibiting ZO-1 and E-cadherin expression posttranscriptionally, whereas HuR prevents miR-675 processing from H19 and is crucial for maintaining a normal barrier function under biological and pathological conditions.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by a Merit Review Award (to Jian-Ying Wang) from the U.S. Department of Veterans Affairs, grants from National Institutes of Health (DK57819, DK61972, and DK68491 to Jian-Ying Wang), and funding from the National Institute on Aging-Intramural Research Program, NIH (to Myriam Gorospe). Jian-Ying Wang is a Senior Research Career Scientist, Biomedical Laboratory Research & Development Service, U.S. Department of Veterans Affairs.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.01030-15.
REFERENCES
- 1.Ponting CP, Oliver PL, Reik W. 2009. Evolution and functions of long noncoding RNAs. Cell 136:629–641. doi: 10.1016/j.cell.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 2.Wang KC, Chang HY. 2011. Molecular mechanisms of long noncoding RNAs. Mol Cell 43:904–914. doi: 10.1016/j.molcel.2011.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ulitsky I, Bartel DP. 2013. LincRNAs: genomics, evolution, and mechanisms. Cell 154:26–46. doi: 10.1016/j.cell.2013.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Batista PJ, Chang HY. 2013. Long noncoding RNAs: cellular address codes in development and disease. Cell 152:1298–1307. doi: 10.1016/j.cell.2013.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Esteller M. 2011. Non-coding RNAs in human disease. Nat Rev Genet 12:861–874. doi: 10.1038/nrg3074. [DOI] [PubMed] [Google Scholar]
- 6.Ørom UA, Derrien Beringer T, Gumireddy M, Gardini K, Bussotti A, Lai G, Zytnicki F, Notredame M, Huang CQ, Guigo R, Shiekhattar R. 2010. Long noncoding RNAs with enhancer-like function in human cells. Cell 143:46–58. doi: 10.1016/j.cell.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yoon JH, Abdelmohsen K, Srikantan S, Yang X, Martindale JL, De S, Huarte M, Zhan M, Becker KG, Gorospe M. 2012. LncRNA-p21 suppresses target mRNA translation. Mol Cell 47:648–655. doi: 10.1016/j.molcel.2012.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Abdelmohsen K, Panda AC, Kang MJ, Guo R, Kim J, Grammatikakis I, Yoon JH, Dudekula DB, Noh JH, Yang X, Martindale JL, Gorospe M. 2014. 7SL RNA represses p53 translation by competing with HuR. Nucleic Acids Res 42:10099–10111. doi: 10.1093/nar/gku686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu X, Li D, Zhang W, Guo M, Zhan Q. 2012. Long noncoding RNA gadd7 interacts with TDP-43 and regulates Cdk6 mRNA decay. EMBO J 31:4415–4427. doi: 10.1038/emboj.2012.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Turner JR. 2009. Intestinal mucosal barrier function in health and disease. Nat Rev Immunol 9:799–809. doi: 10.1038/nri2653. [DOI] [PubMed] [Google Scholar]
- 11.Yang H, Rao JN, Wang JY. 2014. Posttranscriptional regulation of intestinal epithelial tight junction barrier by RNA-binding proteins and microRNAs. Tissue Barriers 2:e28320. doi: 10.4161/tisb.28320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carter SR, Zahs A, Palmer JL, Wang L, Ramirez L, Gamelli RL, Kovacs EJ. 2013. Intestinal barrier disruption as a cause of mortality in combined radiation and burn injury. Shock 40:281–289. doi: 10.1097/SHK.0b013e3182a2c5b5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schneeberger EE, Lynch RD. 2004. The tight junction: a multifunctional complex. Am J Physiol Cell Physiol 286:C1213–C1228. doi: 10.1152/ajpcell.00558.2003. [DOI] [PubMed] [Google Scholar]
- 14.Furuse M, Izumi Y, Oda Y, Higashi T, Iwamoto N. 2014. Molecular organization of tricellular tight junctions. Tissue Barriers 2:e28960. doi: 10.4161/tisb.28960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tian X, Liu Z, Niu B, Zhang J, Tan TK, Lee SR, Zhao Y, Harris DC, Zheng G. 2011. E-cadherin/β-catenin complex and the epithelial barrier. J Biomed Biotechnol 2011:567305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bhatt T, Rizvi A, Batta SP, Kataria S, Jamora C. 2013. Signaling and mechanical roles of E-cadherin. Cell Commun Adhes 20:189–199. doi: 10.3109/15419061.2013.854778. [DOI] [PubMed] [Google Scholar]
- 17.Oldenburger A, Poppinga WJ, Kos F, de Bruin HG, Rijks WF, Heijink IH, Timens W, Meurs H, Maarsingh H, Schmidt M. 2014. A-kinase anchoring proteins contribute to loss of E-cadherin and bronchial epithelial barrier by cigarette smoke. Am J Physiol Cell Physiol 306:C585–C597. doi: 10.1152/ajpcell.00183.2013. [DOI] [PubMed] [Google Scholar]
- 18.Chen J, Xiao L, Rao JN, Zou T, Liu L, Bellavance E, Gorospe M, Wang JY. 2008. JunD represses transcription and translation of the tight junction protein zona occludens-1 modulating intestinal epithelial barrier function. Mol Biol Cell 19:3701–3712. doi: 10.1091/mbc.E08-02-0175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu TX, Wang PY, Rao JN, Zou T, Liu L, Xiao L, Gorospe M, Wang JY. 2011. Chk2-dependent HuR phosphorylation regulates occludin mRNA translation and epithelial barrier function. Nucleic Acids Res 39:8472–8487. doi: 10.1093/nar/gkr567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ye D, Guo S, Al-Sadi R, Ma TY. 2011. MicroRNA regulation of intestinal epithelial tight junction permeability. Gastroenterology 141:1323–1333. doi: 10.1053/j.gastro.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leighton PA, Ingram RS, Eggenschwiler J, Efstratiadis A, Tilghman SM. 1995. Disruption of imprinting caused by deletion of the H19 gene region in mice. Nature 375:34–39. doi: 10.1038/375034a0. [DOI] [PubMed] [Google Scholar]
- 22.Brannan CI, Dees EC, Ingram RS, Tilghman SM. 1990. The product of the H19 gene may function as an RNA. Mol Cell Biol 10:28–36. doi: 10.1128/MCB.10.1.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gabory A, Jammes H, Dandolo L. 2010. The H19 locus: role of an imprinted non-coding RNA in growth and development. Bioessays 32:473–480. doi: 10.1002/bies.200900170. [DOI] [PubMed] [Google Scholar]
- 24.Onyango P, Feinberg AP. 2011. A nucleolar protein, H19 opposite tumor suppressor (HOTS), is a tumor growth inhibitor encoded by a human imprinted H19 antisense transcript. Proc Natl Acad Sci U S A 108:16759–16764. doi: 10.1073/pnas.1110904108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yan L, Zhou J, Gao Y, Ghazal S, Lu L, Bellone S, Yang Y, Liu N, Zhao X, Santin AD, Taylor H, Huang Y. 2015. Regulation of tumor cell migration and invasion by the H19/let-7 axis is antagonized by metformin-induced DNA methylation. Oncogene 34:3076–3084. doi: 10.1038/onc.2014.236. [DOI] [PubMed] [Google Scholar]
- 26.Luo M, Li Z, Wang W, Zeng Y, Liu Z, Qiu J. 2013. Long non-coding RNA H19 increases bladder cancer metastasis by associating with EZH2 and inhibiting E-cadherin expression. Cancer Lett 333:213–221. doi: 10.1016/j.canlet.2013.01.033. [DOI] [PubMed] [Google Scholar]
- 27.Zhu M, Chen Q, Liu X, Sun Q, Zhao X, Deng R, Wang Y, Huang J, Xu M, Yan J, Yu J. 2014. lncRNA H19/miR-675 axis represses prostate cancer metastasis by targeting TGFBI. FEBS J 281:3766–3775. doi: 10.1111/febs.12902. [DOI] [PubMed] [Google Scholar]
- 28.Ghazal S, McKinnon B, Zhou J, Mueller M, Men Y, Yang L, Mueller M, Flannery C, Huang Y, Taylor HS. 2015. H19 lncRNA alters stromal cell growth via IGF signaling in the endometrium of women with endometriosis. EMBO Mol Med 7:996–1003. doi: 10.15252/emmm.201505245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matouk IJ, DeGroot N, Mezan S, Ayesh S, Abu-lail R, Hochberg A, Galun E. 2007. The H19 non-coding RNA is essential for human tumor growth. PLoS One 2:e845. doi: 10.1371/journal.pone.0000845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Keniry A, Oxley D, Monnier P, Kyba M, Dandolo L, Smits G, Reik W. 2012. The H19 lincRNA is a developmental reservoir of miR-675 that suppresses growth and Igf1r. Nat Cell Biol 14:659–665. doi: 10.1038/ncb2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yoshimizu T, Miroglio A, Ripoche MA, Gabory A, Vernucci M, Riccio A, Colnot S, Godard C, Terris B, Jammes H, Dandolo L. 2008. The H19 locus acts in vivo as a tumor suppressor. Proc Natl Acad Sci U S A 105:12417–12422. doi: 10.1073/pnas.0801540105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gabory A, Ripoche MA, Le Digarcher A, Watrin F, Ziyyat A, Forné T, Jammes H, Ainscough JF, Surani MA, Journot L, Dandolo L. 2009. H19 acts as a trans-regulator of the imprinted gene network controlling growth in mice. Development 136:3413–3421. doi: 10.1242/dev.036061. [DOI] [PubMed] [Google Scholar]
- 33.Dey BK, Pfeifer K, Dutta A. 2014. The H19 long noncoding RNA gives rise to microRNAs miR-675-3p and miR-675-5p to promote skeletal muscle differentiation and regeneration. Genes Dev 28:491–501. doi: 10.1101/gad.234419.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gao Y, Wu F, Zhou J, Yan L, Jurczak MJ, Lee HY, Yang L, Mueller M, Zhou XB, Dandolo L, Szendroedi J, Roden M, Flannery C, Taylor H, Carmichael GG, Shulman GI, Huang Y. 2014. The H19/let-7 double-negative feedback loop contributes to glucose metabolism in muscle cells. Nucleic Acids Res 42:13799–13811. doi: 10.1093/nar/gku1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kallen AN, Zhou XB, Xu J, Qiao C, Ma J, Yan L, Lu L, Liu C, Yi JS, Zhang H, Min W, Bennett AM, Gregory RI, Ding Y, Huang Y. 2013. The imprinted H19 lncRNA antagonizes let-7 microRNAs. Mol Cell 52:101–112. doi: 10.1016/j.molcel.2013.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu L, Christodoulou-Vafeiadou E, Rao JN, Zou T, Xiao L, Chung HK, Yang H, Gorospe M, Kontoyiannis D, Wang JY. 2014. RNA-binding protein HuR promotes growth of small intestinal mucosa by activating the Wnt signaling pathway. Mol Biol Cell 25:3308–3318. doi: 10.1091/mbc.E14-03-0853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu TX, Rao JN, Zou T, Liu L, Xiao L, Ouyang M, Cao S, Gorospe M, Wang JY. 2013. Competitive binding of CUGBP1 and HuR to occludin mRNA controls its translation and modulates epithelial barrier function. Mol Biol Cell 24:85–99. doi: 10.1091/mbc.E12-07-0531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cao S, Xiao L, Rao JN, Zou T, Liu L, Zhang D, Turner DJ, Gorospe M, Wang JY. 2014. Inhibition of Smurf2 translation by miR-322/503 modulates TGF-β/Smad2 signaling and intestinal epithelial homeostasis. Mol Biol Cell 25:1234–1243. doi: 10.1091/mbc.E13-09-0560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xiao L, Rao JN, Zou T, Liu L, Cao S, Martindale JL, Su W, Chung HK, Gorospe M, Wang JY. 2013. miR-29b represses intestinal mucosal growth by inhibiting translation of cyclin-dependent kinase 2. Mol Biol Cell 24:3038–3046. doi: 10.1091/mbc.E13-05-0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu L, Ouyang M, Rao JN, Zou T, Xiao L, Chung HK, Wu J, Donahue JM, Gorospe M, Wang JY. 2015. Competition between RNA-binding proteins CELF1 and HuR modulates MYC translation and intestinal epithelium renewal. Mol Biol Cell 26:1797–1810. doi: 10.1091/mbc.E14-11-1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cui YH, Xiao L, Rao JN, Zou T, Liu L, Chen Y, Turner DJ, Gorospe M, Wang JY. 2012. miR-503 represses CUG-binding protein 1 translation by recruiting CUGBP1 mRNA to processing bodies. Mol Biol Cell 23:151–162. doi: 10.1091/mbc.E11-05-0456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guo X, Rao JN, Liu L, Zou TT, Turner DJ, Bass BL, Wang JY. 2003. Regulation of adherens junctions and epithelial paracellular permeability: a novel function for polyamines. Am J Physiol Cell Physiol 285:C1174–C1187. doi: 10.1152/ajpcell.00015.2003. [DOI] [PubMed] [Google Scholar]
- 43.Chung HK, Chen Y, Rao JN, Liu L, Xiao L, Turner DJ, Yang P, Gorospe M, Wang JY. 2015. Transgenic expression of miR-222 disrupts intestinal epithelial regeneration by targeting multiple genes including Frizzled-7. Mol Med 21:676–687. doi: 10.2119/molmed.2015.00147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hart ML, Grenz A, Gorzolla IC, Schittenhelm J, Dalton JH, Eltzschig HK. 2011. Hypoxia-inducible factor-1α-dependent protection from intestinal ischemia/reperfusion injury involves ecto-5′-nucleotidase (CD73) and the A2B adenosine receptor. J Immunol 186:4367–4374. doi: 10.4049/jimmunol.0903617. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 45.Harter JL. 1960. Critical values for Duncan's new multiple range tests. Biometric 16:671–685. doi: 10.2307/2527770. [DOI] [Google Scholar]
- 46.Pochard C, Coquenlorge S, Jaulin J, Cenac N, Vergnolle N, Meurette G, Freyssinet M, Neunlist M, Rolli-Derkinderen M. 2015. Defects in 15-HETE production and control of epithelial permeability by human enteric glial cells from patients with Crohn's disease. Gastroenterology 150:168–180. [DOI] [PubMed] [Google Scholar]
- 47.Jain S, Suzuki T, Seth A, Samak G, Rao R. 2011. Protein kinase Cζ phosphorylates occludin and promotes assembly of epithelial tight junctions. Biochem J 437:289–299. doi: 10.1042/BJ20110587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zou T, Rao JN, Liu L, Xiao L, Chung HK, Li Y, Chen G, Gorospe M, Wang JY. 2015. JunD enhances miR-29b levels transcriptionally and posttranscriptionally to inhibit proliferation of intestinal epithelial cells. Am J Physiol Cell Physiol 308:C813–C824. doi: 10.1152/ajpcell.00027.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mosenthal AC, Xu D, Deitch EA. 2002. Elemental and intravenous total parenteral nutrition diet-induced gut barrier failure is intestinal site specific and can be prevented by feeding nonfermentable fiber. Crit Care Med 30:396–402. doi: 10.1097/00003246-200202000-00022. [DOI] [PubMed] [Google Scholar]
- 50.Cai X, Cullen BR. 2007. The imprinted H19 noncoding RNA is a primary microRNA precursor. RNA 13:313–316. doi: 10.1261/rna.351707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Varrault A, Gueydan C, Delalbre A, Bellmann A, Houssami S, Aknin C, Severac D, Chotard L, Kahli M, Le Digarcher A, Pavlidis P, Journot L. 2006. Zac1 regulates an imprinted gene network critically involved in the control of embryonic growth. Dev Cell 11:711–722. doi: 10.1016/j.devcel.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 52.Monnier P, Martinet C, Pontis J, Stancheva I, Ait-Si-Ali S, Dandolo L. 2013. H19 lncRNA controls gene expression of the Imprinted Gene Network by recruiting MBD1. Proc Natl Acad Sci U S A 110:20693–20698. doi: 10.1073/pnas.1310201110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shi Y, Wang Y, Luan W, Wang P, Tao T, Zhang J, Qian J, Liu N, You Y. 2014. Long non-coding RNA H19 promotes glioma cell invasion by deriving miR-675. PLoS One 9:e86295. doi: 10.1371/journal.pone.0086295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xiao L, Cui YH, Rao JN, Zou T, Liu L, Smith A, Turner DJ, Gorospe M, Wang JY. 2011. Regulation of cyclin-dependent kinase 4 translation through CUG-binding protein 1 and microRNA-222 by polyamines. Mol Biol Cell 22:3055–3069. doi: 10.1091/mbc.E11-01-0069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhuang R, Rao JN, Zou T, Liu L, Xiao L, Cao S, Hansraj NZ, Gorospe M, Wang JY. 2013. miR-195 competes with HuR to modulate stim1 mRNA stability and regulate cell migration. Nucleic Acids Res 41:7905–7919. doi: 10.1093/nar/gkt565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kulkarni M, Ozgur S, Stoecklin G. 2010. On track with P-bodies. Biochem Soc Trans 38:242–251. doi: 10.1042/BST0380242. [DOI] [PubMed] [Google Scholar]
- 57.Mukherjee N, Corcoran DL, Nusbaum JD, Reid DW, Georgiev S, Hafner M, Ascano M Jr, Tuschl T, Ohler U, Keene JD. 2011. Integrative regulatory mapping indicates that the RNA-binding protein HuR couples pre-mRNA processing and mRNA stability. Mol Cell 43:327–339. doi: 10.1016/j.molcel.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Srikantan S, Gorospe M. 2012. HuR function in disease. Front Biosci 17:189–205. doi: 10.2741/3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kim HH, Kuwano Y, Srikantan S, Lee EK, Martindale JL, Gorospe M. 2009. HuR recruits let-7/RISC to repress c-Myc expression. Genes Dev 23:1743–1748. doi: 10.1101/gad.1812509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu L, Rao JN, Zou T, Xiao L, Wang PY, Turner DJ, Gorospe M, Wang JY. 2009. Polyamines regulate c-Myc translation through Chk2-dependent HuR phosphorylation. Mol Biol Cell 20:4885–4898. doi: 10.1091/mbc.E09-07-0550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Choudhury NR, de Lima Alves F, de Andrés-Aguayo L, Graf T, Cáceres JF, Rappsilber J, Michlewski G. 2013. Tissue-specific control of brain-enriched miR-7 biogenesis. Genes Dev 27:24–38. doi: 10.1101/gad.199190.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yu TX, Gu BL, Yan JK, Zhu J, Yan WH, Chen J, Qian LX, Cai W. 2016. CUGBP1 and HuR regulate E-cadherin translation by altering recruitment of E-cadherin mRNA to processing bodies and modulate epithelial barrier function. Am J Physiol Cell Physiol 310:C54–C65. [DOI] [PubMed] [Google Scholar]
- 63.Xiao L, Rao JN, Cao S, Liu L, Chung HK, Zhang Y, Zhang J, Liu Y, Gorospe M, Wang JY. 2016. Long noncoding RNA SPRY4-IT1 regulates intestinal epithelial barrier function by modulating the expression levels of tight junction proteins. Mol Biol Cell 27:617–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang J, Yuan C, Hua G, Tong R, Luo X, Ying Z. 2010. Early gut barrier dysfunction in patients with severe acute pancreatitis: attenuated by continuous blood purification treatment. Int J Artif Organs 33:706–715. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.