

ABSTRACT
SpoIIQ is an essential component of a channel connecting the developing forespore to the adjacent mother cell during Bacillus subtilis sporulation. This channel is generally required for late gene expression in the forespore, including that directed by the late-acting sigma factor σG. Here, we present evidence that SpoIIQ also participates in a previously unknown gene regulatory circuit that specifically represses expression of the gene encoding the anti-sigma factor CsfB, a potent inhibitor of σG. The csfB gene is ordinarily transcribed in the forespore only by the early-acting sigma factor σF. However, in a mutant lacking the highly conserved SpoIIQ transmembrane amino acid Tyr-28, csfB was also aberrantly transcribed later by σG, the very target of CsfB inhibition. This regulation of csfB by SpoIIQ Tyr-28 is specific, given that the expression of other σF-dependent genes was unaffected. Moreover, we identified a conserved element within the csfB promoter region that is both necessary and sufficient for SpoIIQ Tyr-28-mediated inhibition. These results indicate that SpoIIQ is a bifunctional protein that not only generally promotes σG activity in the forespore as a channel component but also specifically maximizes σG activity as part of a gene regulatory circuit that represses σG-dependent expression of its own inhibitor, CsfB. Finally, we demonstrate that SpoIIQ Tyr-28 is required for the proper localization and stability of the SpoIIE phosphatase, raising the possibility that these two multifunctional proteins cooperate to fine-tune developmental gene expression in the forespore at late times.
IMPORTANCE Cellular development is orchestrated by gene regulatory networks that activate or repress developmental genes at the right time and place. Late gene expression in the developing Bacillus subtilis spore is directed by the alternative sigma factor σG. The activity of σG requires a channel apparatus through which the adjacent mother cell provides substrates that generally support gene expression. Here we report that the channel protein SpoIIQ also specifically maximizes σG activity as part of a previously unknown regulatory circuit that prevents σG from activating transcription of the gene encoding its own inhibitor, the anti-sigma factor CsfB. The discovery of this regulatory circuit significantly expands our understanding of the gene regulatory network controlling late gene expression in the developing B. subtilis spore.
INTRODUCTION
Cellular development requires that complex molecular and morphological events occur in a precisely controlled spatiotemporal manner. Gene regulatory networks underlie and orchestrate these events, ensuring that the appropriate suites of developmental genes are activated or repressed at the right time and place (1). Endospore formation (sporulation) by the bacterium Bacillus subtilis is an ancient differentiation process and premier model system for studies of how gene regulatory networks drive prokaryotic development. Under favorable conditions, B. subtilis demonstrates vegetative growth by binary fission; however, when nutrients are depleted, B. subtilis cells embark upon an alternate cellular differentiation pathway toward quiescence, sporulation (2). Early in sporulation, an asymmetric cell division creates two compartments: a smaller forespore, which becomes the spore, and a larger mother cell, which aids in the development of the forespore but ultimately dies. The mother cell then engulfs the forespore in a phagocytosis-like process that results in a cell-within-a-cell configuration. As a consequence of engulfment, the forespore is separated from the mother cell by two membranes, the inner forespore membrane and the outer engulfing mother cell membrane. A protective peptidoglycan cortex and a protein coat are then deposited around the engulfed forespore, which is finally released as a mature spore into the environment upon lysis of the mother cell.
The aforementioned molecular and morphological events of sporulation, like those of developmental pathways in higher organisms, are orchestrated by a complex gene regulatory network (3, 4). At the core of the sporulation gene regulatory network are four RNA polymerase sigma (σ) subunits that coordinate distinct programs of gene expression in the two developing cells. σF and σE direct gene expression at early times in the forespore and mother cell, respectively; at later times, σG replaces σF in the forespore, while σK replaces σE in the mother cell. This study is focused upon the gene regulatory circuitry that orchestrates the transition from early, σF-directed gene expression to late, σG-directed gene expression in the developing forespore. This σF-to-σG switch is tightly regulated such that overlap between the activities of the two sigma factors has not been detected (5); however, the molecular mechanisms that control the switch are not fully understood.
σF is made early in sporulation (prior to cell division) but is held inactive by the anti-sigma factor SpoIIAB (6, 7). Upon asymmetric cell division, σF is released from SpoIIAB inhibition in the forespore via a complex circuit involving the anti-anti-sigma factor SpoIIAA (8) and the membrane-embedded phosphatase SpoIIE, which dephosphorylates and activates SpoIIAA (9). SpoIIE also plays an earlier role in asymmetric division prior to σF activation (10, 11) and has further been observed to directly associate at later times with the forespore membrane protein SpoIIQ (Q), which is produced under the control of σF, though the function of this interaction is unknown (12). In addition to spoIIQ (Q), σF directs the transcription of other genes required for early forespore development, as well as sigG, the gene that encodes σG (sigG is also transcribed at later times in an autoregulatory loop by σG itself) (13). Another member of the σF regulon, csfB, encodes the anti-sigma factor CsfB (also called Gin, for σG inhibitor), which helps to delay σG activity until the early phase of σF-directed gene expression is complete (14–16). Still, deletion of csfB does not generally lead to premature/elevated activation of σG in the majority of sporulating cells (17, 18), indicating that other redundant regulatory mechanisms are also in place to keep σG activity in check at early times.
Upon the completion of engulfment, the early, σF-directed program of developmental gene expression is replaced by the late, σG-directed program. The inhibition of σF prior to this switch, among other unidentified mechanisms, is mediated by a small protein called Fin (previously called YabK) (19). To complete the switch, σG must escape direct inhibition by CsfB, though how this occurs is unknown. Interestingly, previous work has predicted that this involves, at least in part, a mechanism to prevent σG from activating further transcription of csfB during sporulation (20).
σG-directed gene activation also requires the assembly of a channel apparatus that connects the two cells and is comprised of the eight mother cell proteins SpoIIIAA-AH (AA-AH) and the forespore protein Q (21–26). In contrast to CsfB, this AA-AH·Q channel does not specifically regulate σG but, rather, is required more generally for any gene expression (i.e., even that directed by the heterologous phage T7 RNA polymerase) in the forespore (21). These findings, as well as the shrunken and collapsed forespores observed in mutants lacking channel genes (22), have led to a model in which the AA-AH·Q channel functions as a feeding tube through which the mother cell provides the forespore with essential nutrients and osmolytes at later stages of development.
As mentioned above, the forespore membrane protein Q is an essential component of the AA-AH·Q channel required for late, σG-directed gene expression in the forespore. The assembly of Q into this channel relies upon its extracellular C-terminal domain, which directly interacts with the AH channel protein anchored in the opposing mother cell membrane (24, 27). Additional interactions that have been experimentally detected between Q and other mother cell proteins await further characterization (28, 29). Q is anchored in the forespore membrane by an N-terminal transmembrane domain (TMD) that harbors several conserved amino acids (Fig. 1). One of these amino acids, Tyr-28, is conserved with 100% identity among Bacillaceae species with annotated Q orthologs. We previously found that phage T7 RNA polymerase (T7 RNAP), expressed in the forespore, was significantly more active during times coinciding with σG activity when Q Tyr-28 was replaced by Ala (QY28A) (21). Remarkably, this QY28A mutant phenotype was complemented with just an N-terminal fragment of Q that spans the TMD but lacks the C-terminal, AH-interacting domain (QN) (21). This finding suggested that the Q TMD and Tyr-28 in particular perform a secondary function that is distinct from the primary role of Q in channel formation and to which T7 RNAP is especially sensitive. However, the normal, physiological role for Q Tyr-28 in sporulation remains unclear.
FIG 1.

A highly conserved amino acid (Tyr-28) in SpoIIQ. (Top) Cartoon of the B. subtilis SpoIIQ (Q) protein, indicating its transmembrane domain (TMD) and the extracellular, SpoIIIAH (AH)-interacting domain. Q N-terminal residues 1 to 53 correspond to the QN truncation (middle) used in this study. (Bottom) A multiple-sequence alignment of the Q N terminus is shown for Q orthologs from various Bacillus and related species. Accession numbers for protein sequences are listed in Materials and Methods. Similar or identical residues are shaded light gray (60% identity), dark gray (80% identity), or black (100% identity). The conserved tyrosine (Tyr-28) residue is indicated.
Here we report that Q Tyr-28 participates in a previously unknown regulatory circuit required to prevent σG-dependent transcription of the gene encoding the σG inhibitor, CsfB. Consistent with the overproduction of CsfB, a QY28A mutant fails to activate σG to wild-type (WT) levels. Importantly, other σF-dependent promoters do not display aberrant σG-dependent activation in the QY28A mutant, indicating that Q Tyr-28 specifically regulates csfB expression. Bioinformatic and mutational analyses of the csfB promoter reveal a conserved promoter element that is both necessary and sufficient for Q Tyr-28-mediated inhibition. Together, these results support a model in which Q is a bifunctional protein that promotes the switch to σG activity at late times in the forespore in two ways: generally, as a component of the AA-AH·Q intercellular channel apparatus, and specifically, as a component of a regulatory circuit that represses csfB expression. Our data further suggest that Q may execute the latter function in collaboration with the multifunctional SpoIIE phosphatase, thereby coupling anti-sigma factor gene regulation not only to the AA-AH·Q channel but also to the earlier, SpoIIE-dependent processes of asymmetric cell division and σF activation.
MATERIALS AND METHODS
General methods.
B. subtilis strains were maintained with Luria-Bertani (LB) medium in liquid cultures or on solid plates with 1.6% agar. Escherichia coli DH5α strains, each with a specific plasmid, were similarly maintained with LB medium including ampicillin (100 μg/ml). For all experiments in which sporulating cells were analyzed for lacZ reporter expression (i.e., assays for β-galactosidase activity) or for SpoIIE-green fluorescent protein (GFP) content (i.e., Western blotting), sporulation was induced by the resuspension method (30, 31). Cells were collected by centrifugation at hourly intervals and stored at −80°C for further processing. β-Galactosidase activity was measured as previously described (21). The results of all β-galactosidase reporter assays are graphed as the averages from three experiments, unless stated otherwise in the corresponding figure legend.
Strain and plasmid construction.
All B. subtilis strains were derived from the laboratory strain PY79 (32). Details of the strain and plasmid sources and construction are given in the supplemental material. The full genotypes of the experimental strains used in this study are listed in Table S1 in the supplemental material, the plasmids used in this study are listed in Table S2 in the supplemental material, and the primers and gene fragments used in this study are listed in Tables S3 and S4 in the supplemental material, respectively.
Sequence analysis.
Plasmid, genomic, and protein sequences were visualized, edited, and aligned with Geneious software. The following SpoIIQ reference sequences were selected as representative orthologs (GenBank accession numbers are given in parentheses): B. subtilis (NP_391536.1), Bacillus licheniformis (WP_003185941.1), Bacillus anthracis (NP_847683.1), Geobacter kaustophilus (WP_011232806.1), Bacillus halodurans (WP_010899871.1), Bacillus clausii (WP_011248679.1), and Oceanobacillus iheyensis (WP_011067358.1). The multiple-sequence alignment of SpoIIQ was generated with the ClustalW program in Geneious software. Full-length P1P2csfB sequences (where P1 is the σF/σG-dependent promoter and P2 is the σK-dependent promoter) were identified by NCBI BLAST analysis using the Geneious Linnaeus search function. P1P2csfB sequences were recovered from the following species: B. subtilis (query sequence), Bacillus amyloliquefaciens, Bacillus atrophaeus, B. licheniformis, and Bacillus methylotrophicus. A nonredundant consensus P1P2csfB sequence from each of these species was extracted and used as input in a multiple-sequence Geneious alignment in order to produce the sequence logo for the 37-nucleotide (nt) repressor sequence.
Microscopy.
General microscopy methods have been previously described (19). Briefly, cells expressing the spoIIE-gfp fusion gene were collected at hours 2.5 and 3.5 of sporulation to acquire images of each forespore engulfment stage. Harvested samples were resuspended in phosphate-buffered saline containing 1 μg/ml of the membrane stain FM 4-64 (Life Technologies) and mounted on 3% agarose pads. Fluorescence microscopy was performed with an Olympus BX61 microscope fitted with filter sets U-M41001 and U-MWG2 for GFP and FM 4-64 detection, respectively. Images were captured with an Orca-R2 digital charge-coupled-device camera using Simple PCI software, v6.0 (Hamamatsu Corp.). Images were falsely colored, overlaid, and identically adjusted for brightness and contrast with Fiji software (33).
Western blot analysis.
Cell pellets, each from 1 ml sporulating culture, were resuspended in B-PER lysis buffer (with DNase and lysozyme; Pierce) and 1× HALT protease inhibitor (Pierce). Lysis buffer volumes were normalized to cell densities using measurements of the optical density at 600 nm acquired during cell collection. Samples were incubated for 15 min at 37°C, followed by addition of 4× reducing sodium dodecyl sulfate (SDS) sample buffer (Amresco). Samples in 1× sample buffer were heated for 10 min at 80°C, and equal volumes were separated by SDS-PAGE on precast TGX gels (Bio-Rad). Resolved proteins were transferred to a polyvinylidene difluoride membrane using Turbo RDF transfer kits (Bio-Rad). The membranes were blocked with 5% bovine serum albumin and incubated first with rabbit polyclonal anti-GFP antibody (diluted 1:15,000; Abcam) and then with an anti-rabbit immunoglobulin peroxidase-conjugated secondary antibody (diluted 1:2,000; Immunostar) in between washes in Tris-buffered saline with 0.1% Tween 20 (TBST). Chemiluminescence was provided by the West Pico chemiluminescent substrate (Pierce) and visualized with an LAS-3000 image reader (Fujifilm). Densitometry was completed with Fiji software with quantified averages (n = 3) after background (hour 1) subtraction (33).
RESULTS
The highly conserved Q Tyr-28 is required for maximal σG activity and sporulation.
We previously demonstrated that alteration of a highly conserved tyrosine (Tyr-28) in the Q TMD (Fig. 1) significantly stimulates the activity of T7 RNAP engineered to be expressed in the B. subtilis forespore (21). However, the relevance of this phenotype to the normal progression of sporulation remained unclear. To begin, we asked whether Q Tyr-28 is required for the normal activity of the late-acting forespore sigma factor σG. Interestingly, we observed a modest but significant reduction in the expression of lacZ reporters fused to the σG-dependent sspB promoter (Fig. 2A) and spoVT promoter (see Fig. S1 in the supplemental material) (34, 35). More specifically, QY28A strains produced only 70 to 75% of the β-galactosidase levels seen in isogenic Q+ strains, as measured at hour 5 of sporulation. This reduction in σG activity was reversed in QY28A strains into which a second, wild-type copy of Q was introduced (data not shown). Interestingly, full complementation was also observed with an N-terminal fragment encoding only amino acids 1 to 53 of the product of Q (QN) (Fig. 1), which includes the TMD but lacks the C-terminal AH interaction domain of Q (21) (Fig. 2B). Introduction of the Y28A mutation in either context (in the full-length or N-terminal [QN,Y28A] fragment) prevented complementation (data not shown and Fig. 2B). Together, these results indicate that Q Tyr-28 is required for maximal activation of σG at late times in the forespore and that a fragment of Q containing merely its TMD can restore this function.
FIG 2.
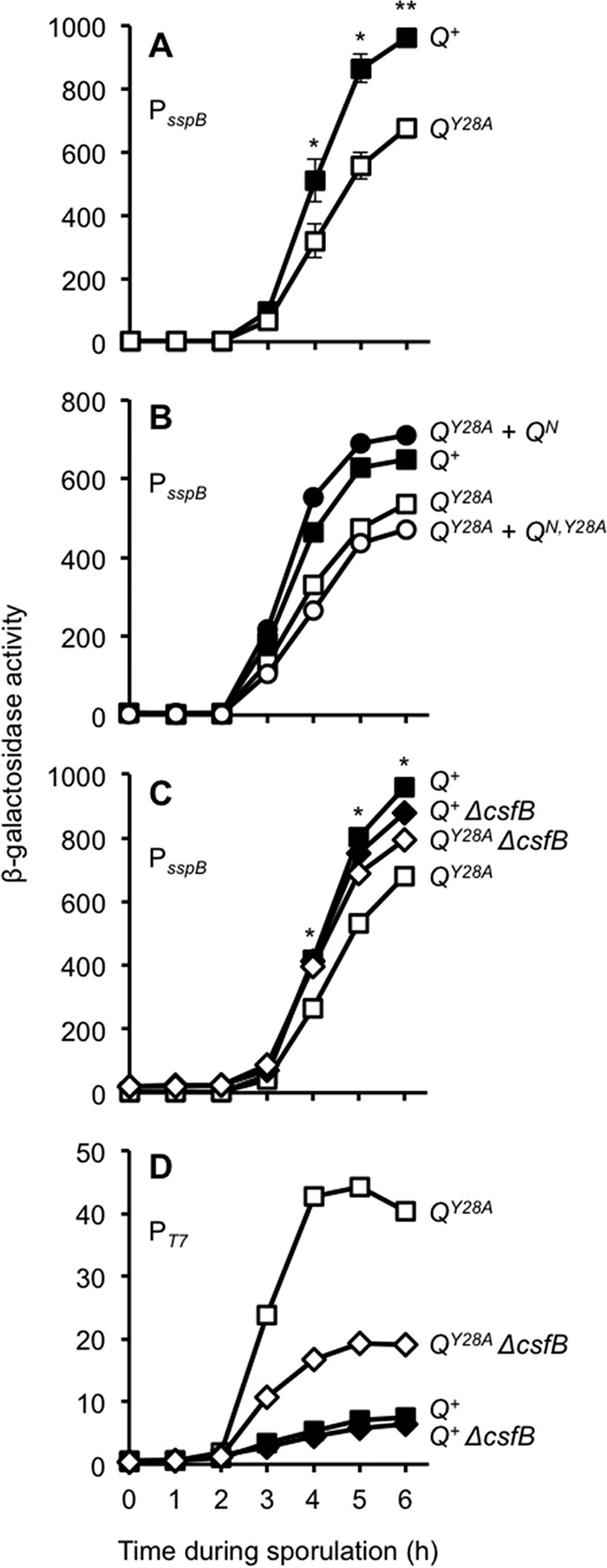
Q Tyr-28 regulates σG activity in a csfB-dependent manner. (A) Q Tyr-28 is required for maximal levels of σG activity. The σG-dependent activation of a PsspB-lacZ reporter at the ywrK locus (also used in the assay for which the results are shown in panel C) was monitored during sporulation of Q+ cells and QY28A cells (strains AHB1586 and AHB1589, respectively). In each of these strains, the native Q gene was deleted and either wild-type Q (Q+) or the Q Tyr-28 mutant (QY28A) was expressed from the sacA locus. Significance was determined by a post hoc Student's t test. *, P < 0.05; **, P < 0.001. Error bars indicate SEMs (n = 7). (B) The QY28A defect in σG activity can be complemented by the Q N terminus. The σG-dependent activation of a PsspB-lacZ reporter at the amyE locus was monitored during sporulation of Q+ cells, QY28A cells, or QY28A cells harboring complementation constructs encoding QN (QY28A + QN) or QN,Y28A (QY28A + QN,Y28A) (strains KF26, KF27, KF32, and KF33, respectively). In each of these strains, the native Q gene was deleted and either Q+ or QY28A was expressed from the lacA locus. The QN or QN,Y28A complementation constructs were inserted at the sacA locus, encode the 53-residue N terminus of Q, and have either tyrosine or alanine at position 28, respectively. (C) The reduction of σG activity exhibited by the QY28A strain is partially reversed by removing the anti-σG factor CsfB. β-Galactosidase production from the PsspB-lacZ reporter was monitored during sporulation of Q+ cells, QY28A cells, Q+ cells from which csfB was deleted (Q+ ΔcsfB), or QY28A cells from which csfB was deleted (QY28A ΔcsfB) (strains AHB1586, AHB1589, AHB1801, and AHB1802, respectively). In these strains, the native Q gene was deleted and either Q+ or QY28A was encoded at the sacA locus. *, P < 0.05, post hoc Student's t test. (D) The hyperactivity of T7 RNAP in the QY28A mutant forespore is also partially reversed by deletion of csfB. T7 RNAP-directed PT7-lacZ expression was monitored during sporulation of Q+ cells, QY28A cells, Q+ cells from which csfB was deleted (Q+ ΔcsfB), or QY28A cells from which csfB was deleted (QY28A ΔcsfB) (strains AHB1542, AHB1543, AHB1799, and AHB1800, respectively). In these strains, the PspoIIQ-T7 RNAP was inserted at the ylnF locus, the T7 RNAP reporter PT7-lacZ was inserted at the ywrK locus, the native Q gene was deleted, and Q+ or QY28A was encoded at the sacA locus.
Given the reduction of σG activity in the QY28A mutant, we predicted that this strain should also be less capable of forming heat-resistant spores. We previously reported that QY28A cells exhibit apparently wild-type levels of spore formation after single-round sporulation assays (21). However, a subtle defect may be undetectable in a single round of sporulation. We therefore performed competition assays in which the QY28A mutant was competed directly with control Q+ cells through multiple rounds of growth and sporulation. At the end of each round, the ratio of surviving heat-resistant spores of each genotype (assessed by use of a lacZ reporter in one of the strains) was determined, and surviving spores were back-diluted into fresh medium for another round of growth and sporulation. As predicted, the QY28A mutant was unable to compete efficiently with the Q+ strain, evidenced by an ∼7% decrease in the mutant population per round (see Fig. S2A in the supplemental material). Importantly, strains with identical Q genotypes competed equally well (see Fig. S2B in the supplemental material), and the QY28A defect was observed regardless of which strain (the QY28A or Q+ strain) was marked with lacZ (see Fig. S2A in the supplemental material). Finally, the defect was specific to sporulation, given that the Q+ and QY28A strains grew equally well in a growth medium that does not induce sporulation (see Fig. S2C in the supplemental material). We therefore conclude that Q Tyr-28 is ordinarily required for maximal σG activation and sporulation efficiency.
Decreased σG activity in the absence of Q Tyr-28 is partially dependent on CsfB.
We hypothesized that the reduction in σG activity in QY28A cells might be attributed to the inappropriate activity of a σG inhibitor. We first considered whether the reduction in σG activity might be due to inhibition by SpoIIAB. Due to the structural similarity of σF and σG (36, 37), the anti-σF factor SpoIIAB is also capable of binding and inhibiting σG (38, 39), although previous experiments have argued against a role for SpoIIAB-mediated inhibition of σG in the forespore during sporulation (37). Nevertheless, to test for the involvement of SpoIIAB, we introduced a mutation (E156K) into the gene (sigG) encoding σG that renders it insensitive to SpoIIAB-mediated inhibition (37). This strategy allowed us to dissect the role of SpoIIAB specifically in σG inhibition without confounding effects on the earlier, primary role of SpoIIAB in σF inhibition. We found that σG activation of the PsspB-lacZ reporter was unaltered by the sigGE156K genotype in either Q background (see Fig. S3A in the supplemental material). The hyperactivity of the T7 RNAP in QY28A cells (as monitored by use of a T7 RNAP-dependent lacZ reporter) was similarly unaffected by introduction of the sigGE156K mutation (see Fig. S3B in the supplemental material). As such, we conclude that σG activity in the QY28A mutant is reduced (and T7 RNAP activity is increased) by a mechanism that does not involve the anti-sigma factor SpoIIAB.
We next tested whether the reduction of σG activity in the QY28A strain was caused by inappropriate activity of the σG inhibitor, CsfB. Remarkably, we found that the QY28A-associated reduction in σG activity was significantly (albeit not completely) rescued by the deletion of csfB (Fig. 2C). Deletion of csfB in a Q+ background did not increase σG activity, suggesting that removal of this anti-sigma factor does not simply cause a generalized disinhibition of σG. We also found that csfB deletion significantly reduced the hyperactivity of T7 RNAP in QY28A cells (Fig. 2D). These data support a model in which σG is subject to excessive inhibition by CsfB in QY28A cells. Moreover, we speculate here that T7 RNAP hyperactivity, the very phenotype that originally drew our attention to the QY28A mutant, may be an indirect consequence of reduced σG activity. In this engineered strain, T7 RNAP (expressed under σF control) and σG coexist in the forespore and compete for finite resources (21). Although we cannot exclude other models, it seems plausible that a modest reduction in σG activity, when multiplied across the ∼100 endogenous σG target genes (4, 40, 41), would free up limiting resources and therefore highly stimulate T7 RNAP activation of its one target gene, the PT7-lacZ reporter.
SpoIIQ Tyr-28 prevents σG-dependent activation of the csfB promoter.
The genetic interaction between QY28A and csfB suggests that Q Tyr-28 may promote maximal σG activity in the developing forespore by a mechanism that downregulates CsfB expression and/or activity. We first tested whether Q Tyr-28 influences csfB expression. The csfB gene is expressed under the control of σF at early times in the forespore and later by σK in the mother cell (15, 42). The transcription of csfB is also activated by σG during vegetative growth (20). This complex regulation appears to be mediated by two promoters in the regulatory region upstream of the csfB gene (Fig. 3A): one matching the consensus sequence for both σF and σG and a second, upstream promoter that matches the consensus sequence for σK (42).
FIG 3.

Q Tyr-28 is required to prevent σG-dependent activation of the csfB promoter during sporulation. (A) Cartoon of the B. subtilis csfB upstream regulatory region. The −35 and −10 promoter elements recognized by σK (P2csfB) or σF and σG (P1csfB) are depicted as black boxes. The P1P2csfB-lacZ and P2csfB-lacZ reporter constructs are drawn to scale below. (B) P1csfB and P2csfB are activated by σF and σK, respectively, during sporulation. The accumulation of β-galactosidase from P1P2csfB-lacZ (P1P2; closed circles) and P1csfB-lacZ (P1; triangles) was measured during sporulation of otherwise wild-type cells. The activity of the P1P2csfB-lacZ reporter was also monitored in a strain lacking σK (P1P2 ΔsigK; open circles). Reporters were inserted at the amyE locus. P1P2, P1P2 ΔsigK, and P1 were carried by strains AHB1702, JDC5, and JDC138, respectively. (C) σG-dependent activation of PcsfB is unmasked in the QY28A mutant. Activation of the P1csfB-lacZ reporter was monitored during sporulation of strains harboring Q+ or QY28A (strains JDC142 and JDC143, respectively). P1csfB-lacZ activity was also measured in a QY28A strain lacking σG (QY28A ΔsigG; strain JDC150). In each of these strains, the native Q gene was deleted and either Q+ or QY28A was inserted at the sacA locus. Note that the y axis in panel C is the same as that in panel B. (D) The Q Tyr-28 substitution does not unmask σG activation of other σF-activated promoters. Four representative σF-dependent promoters (those of csfC, yyaC, spoIIQ, and lonB) were fused to lacZ and assayed for expression in both wild-type Q+ and QY28A mutant strains. Q+ and QY28A strains are strains EBM49 and EBM50, respectively, for PcsfC; strains EBM44 and EBM47, respectively, for PyyaC; strains EBM42 and EBM45, respectively, for PspoIIQ; and strains EBM43 and EMB46, respectively, for PlonB; in all of these strains, the lacZ reporter genes were inserted at amyE. Data are from representative, single experiments. (E) The 53-residue wild-type Q N terminus (QN) can restore proper P1csfB expression in the QY28A mutant. P1csfB-lacZ activity was monitored in strains harboring either Q+ or QY28A encoded at the lacA locus and was also monitored in QY28A strains that were complemented with either wild-type QN (QY28A + QN) or mutant QN,Y28A (QY28A + QN,Y28A) at the sacA locus (strains KF5, KF6, KF11, and KF12, respectively).
Given the complexity of csfB expression, we built and characterized two different PcsfB-lacZ reporter constructs. The first included the σF/σG-dependent promoter (P1) and the σK-dependent promoter (P2) (see the P1P2 construct in Fig. 3A), while the second was shortened such that it harbored only the σF/σG-dependent promoter (see the P1 construct in Fig. 3A). As shown in Fig. 3B, both reporter constructs displayed nearly identical activity at early times of sporulation (at about hour 2), consistent with the σF-dependent activation of the P1 σF/σG-dependent promoter. We also observed a weaker, second phase of β-galactosidase production at about hour 5 of sporulation in strains harboring the P1P2csfB-lacZ reporter but not the P1csfB-lacZ, reporter, consistent with the σK-dependent activation of P2csfB. This later activity was eliminated by deletion of spoIVCB, which is part of the composite gene, sigK, that encodes σK (Fig. 3B). Importantly, our data revealed no evidence for σG-mediated activation of the P1 σF/σG-dependent promoter during sporulation, in accordance with the fact that csfB activation by σG has been reported only during vegetative growth (20). Given our specific interest in csfB function in the forespore (i.e., the compartment in which σG is ordinarily active), as well as to exclude the potentially confounding σK-driven activation, we used the P1csfB-lacZ reporter construct or variations thereof for all subsequent experiments, unless otherwise noted.
To determine whether Q Tyr-28 was necessary for proper csfB expression in the forespore, we monitored P1csfB-lacZ expression in Q+ and QY28A strains. As expected, β-galactosidase produced from P1csfB-lacZ in Q+ cells peaked at hour 2 of sporulation (coinciding with the timing of σF activation), after which time there was no additional β-galactosidase production (Fig. 3C). The QY28A mutant demonstrated levels of σF-dependent activation of P1csfB-lacZ similar to those of the Q+ control strain but also displayed a robust second wave of expression such that by hour 5 of sporulation, the QY28A mutant had 3-fold higher levels of β-galactosidase than the Q+ control strain (Fig. 3C). We confirmed by immunoblot analysis that a functional GFP-CsfB fusion protein under the control of the P1csfB promoter also accumulated to approximately 40% higher levels in the QY28A strain than the Q+ strain at later times during sporulation (data not shown). Together, these results indicate that aberrant late activation of the P1csfB promoter occurs in the absence of Q Tyr-28, leading to an increase in the steady-state levels of the anti-σG factor CsfB.
We next wondered whether the late activation of P1csfB during sporulation in QY28A cells was due to σG, the very target of CsfB inhibition. Supporting this idea, the timing of the late activity coincided with the known timing of σG activity in the forespore (Fig. 2A). Indeed, we found that the aberrant activity of P1csfB in QY28A cells (after hour 3) disappeared when sigG was deleted (Fig. 3C). We therefore conclude that the σF-dependent P1csfB promoter can be recognized and activated by σG during sporulation but that σG is ordinarily prevented from doing so by a mechanism that requires Q Tyr-28.
The ability of σG to activate csfB expression in the QY28A mutant could be the result of the general misregulation of σF target promoters or a specific misregulation of P1csfB. To distinguish between these possibilities, each of the σF-controlled promoters of spoIIQ, lonB, yyaC, and csfC (15, 40, 41, 43) was fused to the lacZ reporter gene and assayed in sporulating cells of the Q+ and QY28A strains. As shown in Fig. 3D, none of these reporters were aberrantly expressed in the QY28A strain. As such, the activation of P1csfB by σG in the absence of Q Tyr-28 is not due to a general misregulation of σF target genes but is instead specific to csfB.
Finally, given that the N-terminal fragment of Q (QN; Fig. 1) restored wild-type levels of σG activity to the QY28A mutant (Fig. 2B), we hypothesized that QN might also restore proper regulation of P1csfB in this mutant. As shown in Fig. 3E, csfB misexpression by σG was significantly reduced when QN (but not QN,Y28A) was introduced into QY28A cells. Altogether, these results support a model in which Q Tyr-28 specifically prevents σG from inappropriately activating the transcription of a gene (csfB) that encodes its own inhibitor.
A conserved sequence within the csfB promoter is required to prevent σG-dependent activation.
The ability of the QY28A mutation to unmask the σG-dependent activation of csfB suggested to us that the csfB promoter itself may harbor features that make it a target for Q Tyr-28-dependent regulation. We reasoned that such features would likely be conserved among other spore-forming bacteria related to B. subtilis. At least four species have a csfB promoter arrangement similar to that found in B. subtilis (i.e., a σK-dependent promoter followed by a σF/σG-dependent promoter): Bacillus amyloliquefaciens, Bacillus atrophaeus, Bacillus licheniformis, and Bacillus methylotrophicus. Using sequences from all these species, we created a consensus logo sequence for the csfB regulatory region corresponding to P1csfB in these species (Fig. 4A). As expected, the −35 and −10 binding sites for σF/σG, as well as the ribosome binding site, displayed significant conservation. Sequences between these functional elements were less conserved, with the notable exception of an ∼37-nt stretch mostly upstream of but also including 3 nt downstream of the −35 element of the σF/σG-dependent promoter.
FIG 4.

A regulatory element near P1csfB is necessary to prevent its activation by σG during sporulation. (A) A 37-nt sequence adjacent to the B. subtilis P1csfB −35 element is well conserved. The csfB regulatory sequences corresponding to the P1csfB-lacZ construct from B. subtilis, B. amyloliquefaciens, B. atrophaeus, B. licheniformis, and B. methylotrophicus were analyzed by the use of Geneious software to generate a sequence logo depiction of nucleotide conservation. Shown below the sequence logo are the 5′ boundaries of the trunc1P1csfB-lacZ and trunc2P1csfB-lacZ constructs. Nucleotides mutated in the mut1P1csfB-lacZ, mut2P1csfB-lacZ, and mut3P1csfB-lacZ constructs are indicated by mut1, mut2, and mut3, respectively. Note that the data obtained for the mut2P1csfB-lacZ and mut3P1csfB-lacZ constructs are presented in Fig. S4 in the supplemental material. RBS, ribosome binding site. (B) Activity of P1csfB variants during sporulation. β-Galactosidase production was monitored during the sporulation of strains harboring P1csfB-lacZ (WTP), trunc1P1csfB-lacZ, trunc2P1csfB-lacZ, and mut1P1csfB-lacZ reporter constructs integrated at amyE (strains KF76, KF159, AHB2088, and KF13, respectively). (C) The late activity of mut1P1csfB-lacZ is due to inappropriate activation by σG. β-Galactosidase production from the mut1P1csfB-lacZ reporter was assessed in a strain with native sigG (mut1P) or a strain from which sigG was deleted (mut1P ΔsigG). As a control, the wild-type P1csfB-lacZ reporter was also assessed in a strain with a native sigG (WTP) or a strain from which sigG was deleted (WTP ΔsigG). Reporter genes were inserted at the amyE locus in these strains (strains KF13, KF15, JDC138, and KF109, respectively). (D) Mutation of the csfB promoter element and substitution of Q Tyr-28 misregulate csfB expression to a similar extent and in a nonadditive manner. β-Galactosidase activity from the P1csfB-lacZ or mut1P1csfB-lacZ reporter constructs was measured in strains harboring Q+ or QY28A. The strains were P1csfB-lacZ Q+ (WTP Q+), P1csfB-lacZ QY28A (WTP QY28A), mut1P1csfB-lacZ Q+ (mut1P Q+), and mut1P1csfB-lacZ QY28A (mut1P QY28A) (strains KF1, KF2, KF3, and KF4, respectively). In each of these strains, the native Q gene was deleted and either Q+ or QY28A was inserted at the sacA locus; lacZ reporters were inserted at the amyE locus. Note that the y axes for panels C and D are the same as the y axis for panel B.
To ascertain if this conserved region contributes to the regulation of csfB expression, we first made progressive deletions from the 5′ end of our P1csfB-lacZ reporter construct to produce two P1csfB truncations (trunc1P1csfB and trunc2P1csfB; Fig. 4A). As shown in Fig. 4B, the levels of β-galactosidase produced from the trunc1P1csfB-lacZ and trunc2P1csfB-lacZ fusions were similar to those produced from P1csfB-lacZ at hour 2 of sporulation, indicating normal σF-dependent activation. At later times, however, we observed a substantial 2- to 3-fold increase in expression in the trunc1P1csfB-lacZ and trunc2P1csfB-lacZ reporter fusions (Fig. 4B). The more extensive deletion of the 5′ end (trunc2P1csfB) promoted the highest level of β-galactosidase expression. We next generated a new P1csfB-lacZ variant in which seven of the most highly conserved nucleotides that were removed in trunc2P1csfB-lacZ were mutated (mut1P1csfB-lacZ; Fig. 4A). The mut1P1csfB-lacZ reporter gene was also aberrantly expressed during late sporulation in a manner that was indistinguishable from that of the trunc2P1csfB-lacZ reporter (Fig. 4B). As expected, the late activity of the mut1P1csfB-lacZ and trunc2P1csfB-lacZ reporters was significantly reduced when sigG was deleted (Fig. 4C and data not shown), confirming dependence upon σG, as in the QY28A mutant strain. Finally, we wondered whether conserved nucleotides closer to the P1csfB −35 element might also be necessary to prevent σG-dependent activation. Indeed, we found that substitution of nucleotides upstream or downstream of the −35 element (mut2P1csfB or mut3P1csfB, respectively; Fig. 4A) caused aberrant late activation to the same extent that trunc2P1csfB and mut1P1csfB did (see Fig. S4 in the supplemental material). Altogether, these results suggest that conserved nucleotides mostly upstream of but also downstream of the −35 element of P1csfB are required to prevent its activation by σG during late sporulation.
Q Tyr-28 and the csfB promoter element operate in the same genetic pathway.
Our data thus far indicate that σG can activate csfB expression in the forespore under two circumstances: (i) when we substitute Tyr-28 of Q (as in QY28A) and (ii) when we mutate a conserved sequence near the σF/σG −35 binding site (as in mut1P1csfB). These findings may reflect two separate mechanisms of csfB regulation or could indicate that Q Tyr-28 and the conserved P1csfB regulatory element operate together in a single pathway to block σG activation of PcsfB during sporulation. To distinguish these possibilities, we generated a double mutant strain harboring the mut1P1csfB-lacZ reporter in a QY28A mutant background. As shown in Fig. 4D, the mut1P1csfB-lacZ QY28A double mutant produced the same levels of β-galactosidase produced by both single mutants alone. The nonadditivity of these phenotypes argues against separate mechanisms and instead supports a model in which Q Tyr-28 and the conserved promoter element operate in the same pathway to block σG activation of P1csfB.
The conserved csfB promoter element is sufficient to repress promoter activation by σG in late forespore development.
Our data indicate that a conserved promoter element near the −35 element bound by σF in P1csfB is necessary to block subsequent σG-dependent expression of csfB during sporulation. We were curious if this conserved sequence might also be sufficient to repress σG activity in a promoter ordinarily activated by both σF and σG. We chose as our test promoter that of the putative σF inhibitor gene fin (previously yabK) (19). Not only has Pfin been demonstrated to be activated by both σF and σG (19), but Pfin also harbors a −35 element identical to that found in P1csfB (GTATA). This identity facilitated the construction of chimeric promoters (see below) in which surrounding nucleotides were swapped without altering core promoter nucleotides.
To begin, we constructed a Pfin-lacZ promoter fusion that harbored 29 nt upstream of the σF/σG-dependent −35 element, the same number of nucleotides found upstream of the matching −35 element in our P1csfB-lacZ promoter fusion (Fig. 5A). As expected, this Pfin-lacZ promoter fusion was activated first during the height of σF activity (approximately hour 2) and then later under the control of σG (approximately hours 3 to 5) (Fig. 5B). We then replaced the 5′ 35 nt of Pfin with the corresponding 35 nt of P1csfB, yielding a chimeric promoter, PcsfB-fin (Fig. 5A). (The final 2 nt of the 37 nt conserved sequence were already identical in Pfin.) As shown in Fig. 5B, the chimeric PcsfB-fin-lacZ reporter displayed significantly reduced σG-dependent activation compared to that of the original Pfin-lacZ reporter. Importantly, as shown in Fig. 5B, σG-dependent activation of PcsfB-fin was restored when we mutated the same seven highly conserved nucleotides altered in mut1PcsfB (Fig. 4A). Altogether, these data indicate that these 37 nt, which include the P1csfB −35 element, are sufficient to interfere with late σG activity when placed in the context of another σF/σG-dependent promoter.
FIG 5.
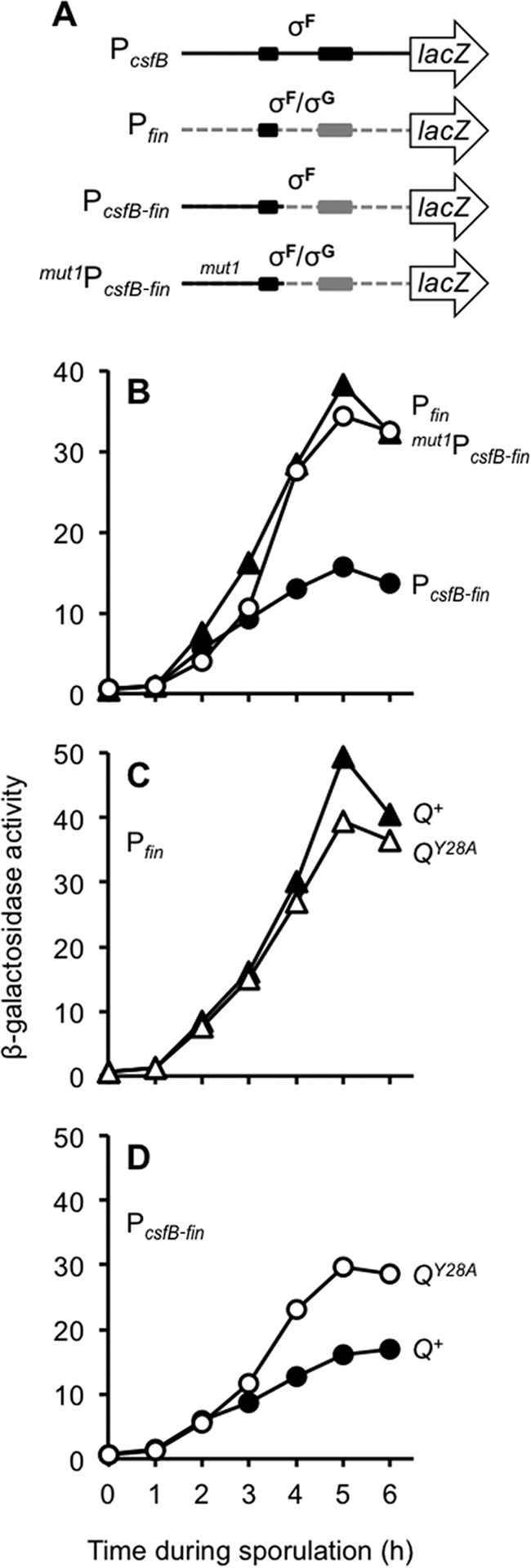
The conserved 37-nt sequence from P1csfB is sufficient to reduce σG-dependent activation of a promoter ordinarily recognized by σF and σG. (A) Cartoon of P1csfB, Pfin, and a chimeric promoter (PcsfB-fin) in which the 37-nt conserved sequence from P1csfB was substituted for the corresponding 37 nt in Pfin. Also shown is the mut1PcsfB-fin promoter construct in which the 7-nt sequence mutated in mut1P1csfB is introduced into PcsfB-fin (marked by mut1). The −35 and −10 elements for each promoter are indicated as shaded boxes; note that PcsfB and Pfin have identical −35 elements. (B) The PcsfB-fin promoter but not mut1PcsfB-fin displays significantly reduced activation by σG. β-Galactosidase production from Pfin (triangles), PcsfB-fin (closed circles), and mut1PcsfB-fin (open circles) fusions to lacZ (inserted at the amyE locus) was measured during sporulation of otherwise wild-type strains (strains KF76, KF129, KF130, and KF166, respectively). (C) Q Tyr-28 is required for maximal levels of σG activation of Pfin. Pfin-lacZ reporter gene expression was monitored in strains harboring Q+ or QY28A (strains KF146 and KF152, respectively). (D) σG-dependent activation of PcsfB-fin is unmasked in the QY28A mutant. PcsfB-fin-lacZ reporter gene expression was monitored in strains harboring Q+ or QY28A (strains KF147 and KF153, respectively). In all strains for which the results are shown in panels C and D, the native Q gene was deleted and either Q+ or QY28A was encoded at the sacA locus. Reporters were inserted at amyE.
Finally, we tested whether the 37-nt repressor sequence derived from P1csfB was sufficient to confer sensitivity to Q Tyr-28. In other words, is the σG-dependent activation of PcsfB-fin unmasked in a QY28A mutant like that of PcsfB was? As shown in Fig. 5C, Pfin-lacZ activity is ordinarily slightly diminished in the presence of the QY28A mutation (i.e., relative to that in the control Q+ strain), consistent with the ∼25 to 30% reduction in σG activity that we have reported here (Fig. 2A). In contrast, the PcsfB-fin reporter, which was engineered to harbor the 37-nt repressor sequence, demonstrated an increase in expression in the QY28A strain relative to that in the Q+ strain (Fig. 5D). We therefore conclude that a conserved 37-nt sequence found near P1csfB is necessary and sufficient to prevent late, σG-dependent promoter activation by a mechanism that requires Q Tyr-28.
Misregulation of csfB expression is insufficient to explain the QY28A-mediated reduction in σG activity.
We have demonstrated that QY28A mutant forespores exhibit diminished σG activity and that this is partially reversed by csfB deletion. Moreover, we found that the QY28A mutation causes csfB to be aberrantly expressed by σG, which likely contributes to higher steady-state levels of the csfB-encoded σG inhibitor, CsfB. We therefore hypothesized that the diminished σG activity in the QY28A mutant is due at least in part to misexpression of csfB by σG. A key prediction of this hypothesis is that expression of csfB from a mutant promoter activated by both σF and σG should cause a reduction in σG activity similar to that caused by the QY28A mutation. To test this hypothesis, we directly engineered the misexpression of csfB by mutating seven highly conserved nucleotides upstream of the P1csfB (σF/σG) −35 element (the same mutations found in the mut1P1csfB-lacZ construct) at the native csfB locus (i.e., without disrupting any other aspect of the csfB promoter or coding sequence). Surprisingly, however, altering csfB expression in this manner failed to detectably reduce the activity of σG (see Fig. S5A in the supplemental material). Moreover, expression of csfB from wild-type or mutated promoters at both the native locus and an additional, ectopic locus had no observable effect on σG activity (see Fig. S5B in the supplemental material), nor did any of these alterations to csfB expression cause late hyperactivity of T7 RNAP in the forespore, which we speculate may be a sensitive indicator of subtle changes in σG activity (data not shown). We therefore conclude that the misexpression of the anti-σG gene csfB cannot fully explain the reduction of σG activity in the QY28A mutant and, as such, that csfB expression is unlikely the only target of Q Tyr-28 regulation.
Q Tyr-28 is required for the proper localization and stability of the phosphatase SpoIIE.
Our data support a model in which an amino acid within the Q TMD, Tyr-28, somehow blocks σG-dependent expression of csfB. One possibility is that Q Tyr-28 exerts this effect by modulating the activity of the AA-AH·Q channel, which globally promotes late forespore gene expression, including that directed by σG (21). We find this scenario unlikely, however, given the specificity of csfB misregulation, i.e., given that no other tested promoters were similarly misregulated in the QY28A mutant. Instead, we posit here that Tyr-28 executes a separate function of Q, by which it modulates the expression of specific genes (including csfB) to maximize the activity of σG. We further predict that Q does so in collaboration with one or more intermediary proteins that topologically link the membrane-embedded Tyr-28 to regulatory events occurring in the cytosol. An intriguing candidate for such an intermediary protein is the membrane phosphatase SpoIIE. It has been reported previously that SpoIIE and Q interact in the forespore membrane and that the relocalization of SpoIIE to the forespore septal membrane during engulfment requires Q (12). These events occur after the well-established functions of SpoIIE in polar septum formation and σF activation, suggesting that SpoIIE and Q together carry out an undetermined function in the forespore at later times.
Given the earlier requirement for SpoIIE in σF activation, we were unable to delete spoIIE to directly test for a later role in proper PcsfB regulation. Nevertheless, to explore a possible link between Q Tyr-28-mediated gene regulation and SpoIIE, we tested whether Tyr-28 is required for the dynamic localization of SpoIIE in the forespore membrane. As previously reported, a functional SpoIIE-GFP fusion protein localized almost exclusively to the septal membrane during engulfment in wild-type cells but mislocalized around the entire forespore membrane in the absence of Q (Fig. 6A). When ΔQ cells were complemented with Q+ at an ectopic locus, proper SpoIIE-GFP localization to the forespore septal membrane was restored (Fig. 6A). In contrast, when ΔQ cells were complemented with QY28A, SpoIIE-GFP localization failed to be restored, with the majority of the protein remaining distributed throughout the forespore membrane throughout engulfment (Fig. 6A). We therefore conclude that the SpoIIE·Q complex at the forespore septal membrane during engulfment requires Q Tyr-28.
FIG 6.

Q Tyr-28 is required for the proper localization and stability of SpoIIE-GFP. (A) SpoIIE-GFP is mislocalized in QY28A mutant forespores during engulfment. The localization of a functional SpoIIE-GFP fusion protein (encoded at the native spoIIE locus) was monitored by fluorescence microscopy during sporulation (hour 2.5) of a strain harboring wild-type Q at its native locus (WT), a strain in which Q was deleted (ΔQ), or strains in which Q was deleted and complemented with either Q+ or QY28A at the sacA locus (strains AHB1648, AHB1649, AHB1650, and AHB1651, respectively). A representative set of cells at an early stage of engulfment is shown for each strain, with both SpoIIE-GFP (IIE-GFP) fluorescence (green) and FM 4-64 membrane (memb) fluorescence (red) being shown in pseudocolor. (Bottom) Two cartoons illustrate SpoIIE-GFP localization to the septal forespore membrane in strains with wild-type Q (left) and SpoIIE-GFP mislocalization around the forespore membrane in strains lacking either Q or Q Tyr-28 (right). (B) QY28A forespores demonstrate diminished SpoIIE-GFP signal intensity (arrowheads) compared to Q+ cells. Representative images of Q+ and QY28A cells (strains AHB1650 and AHB1651, respectively) at hour 3.5 of sporulation, after engulfment was complete, are shown. SpoIIE-GFP fluorescence (green) and FM 4-64 membrane fluorescence (red) are shown merged. Membranes surrounding engulfed forespores are not stained because the FM 4-64 dye is not able to permeate the membrane. GFP images for each strain were captured and processed identically. (C) SpoIIE-GFP steady-state protein levels are quantifiably reduced in the QY28A mutant. Immunoblot analysis of whole-cell extracts from sporulating Q+ or QY28A cells (strains AHB1650 and AHB1651, respectively) was performed using anti-GFP antibodies. The time during sporulation is indicated. Equal protein loading was confirmed by Coomassie staining (not shown). Densitometry was used to quantify the average levels of SpoIIE-GFP in the QY28A mutant as a fraction of those in the Q+ strain at each time point after the background was subtracted. Error bars are SEMs (n = 3).
In addition to the mislocalization of SpoIIE-GFP during engulfment, we also observed a notable decrease in the intensity of SpoIIE-GFP fluorescence in QY28A forespores relative to that in Q+ forespores after engulfment was complete (Fig. 6B). Consistent with the microscopy findings, the steady-state levels of SpoIIE-GFP measured by immunoblot analysis were consistently lower in the QY28A cells than Q+ cells, with the difference becoming more pronounced at later times such that by hour 4 of sporulation, the QY28A cells harbored only ∼40% of the SpoIIE-GFP levels present in Q+ cells (Fig. 6C). Altogether, these results indicate that Q Tyr-28 is required for the localization and stability of SpoIIE-GFP in the forespore membrane, raising the intriguing possibility that Q Tyr-28 modulates the expression of csfB by a mechanism that involves the multifunctional SpoIIE phosphatase.
DISCUSSION
In the developing B. subtilis spore, the transition from early, σF-directed gene expression to late, σG-directed gene expression is precisely orchestrated but still relatively poorly understood. One key player in the σF-to-σG switch is CsfB, an anti-sigma factor capable of inhibiting σG (14, 16). The csfB gene is transcribed early in the forespore under σF control (15). Interestingly, σG can also direct csfB transcription during vegetative growth (i.e., in nonsporulating cells) from the same promoter utilized by σF during sporulation (P1csfB) (Fig. 3A) (20, 42). The ability of σG to activate csfB expression in vegetative cells is proposed to serve a quality control purpose, to shut down inappropriate σG activity under nonsporulation conditions (20). However, these findings raise an important question: if σG is able to recognize P1csfB (i.e., like it does during vegetative growth), why does it not also do so in the forespore during sporulation, at which point a negative-feedback loop would be detrimental? In this study, we report the discovery of a regulatory circuit involving amino acid Tyr-28 of the forespore membrane protein Q and a conserved element in the csfB promoter that blocks the σG-dependent activation of P1csfB in the forespore. Removing either Q Tyr-28 or the csfB promoter element leads to inappropriate σG-dependent csfB activation during sporulation. The nonadditivity of these two mutant phenotypes is consistent with Q Tyr-28 and the csfB promoter element operating in the same pathway to ensure that csfB is transcribed exclusively under σF control at early times in the forespore.
The Q Tyr-28–csfB regulatory circuit.
By what mechanism might Q Tyr-28 and the csfB promoter element prevent σG-dependent activation of csfB? Figure 7 provides our working model for this regulatory pathway. First, we propose that the conserved csfB promoter element is a binding site for an unidentified DNA-binding repressor protein (Rep in Fig. 7). Given the proximity of the conserved nucleotides to the −35 element of P1csfB (Fig. 4A), as well as our findings that alterations to either side of the −35 element cause promoter misregulation, we speculate that a protein docked at this site could simply prevent RNA polymerase holoenzyme binding due to obstruction of the −35 element. But then how does binding of a repressor specifically block σG-mediated activation of P1csfB without also interfering with its earlier activation by σF? To account for this, we predict that the putative repressor protein is present and active specifically in the forespore only after the earlier phase of σF activity is complete. One possibility is that the gene encoding the repressor protein is expressed under the control of σF or σG. Alternatively, the repressor protein may be differentially regulated by a posttranslational modification or some other protein-protein interaction in the forespore at later times. Efforts are ongoing in our laboratory to identify this putative csfB transcriptional repressor through candidate and unbiased approaches. To date we have excluded the possibility of the involvement of several candidates (our unpublished results), including two DNA-binding proteins known to regulate gene expression in the forespore, RsfA and SpoVT (35, 44).
FIG 7.
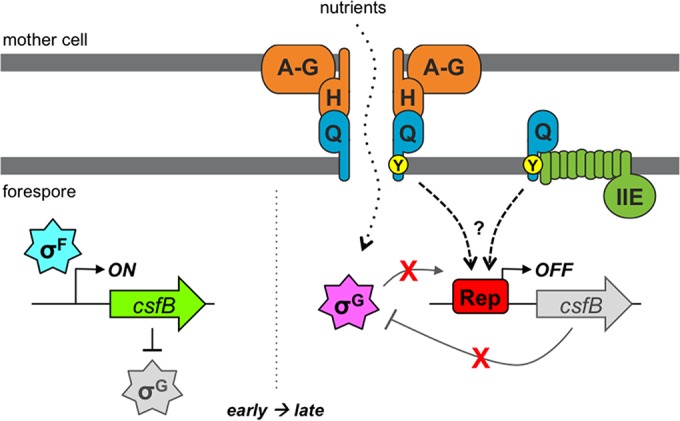
A model for the Q Tyr-28-mediated csfB regulatory pathway. After asymmetric division and during engulfment (early, left), transcription of the csfB gene is activated by σF in the forespore. In turn, the produced CsfB protein acts as an inhibitor of σG, helping to prevent its premature activation. After the completion of forespore engulfment (late, right), the transcription of csfB by σG is prevented by a conserved csfB promoter element, which we propose is a binding site for a repressor protein (Rep). The cessation of csfB transcription limits CsfB production to promote maximal σG activation. A highly conserved tyrosine (Tyr-28; Y) in the TMD of Q (whose gene is also activated by σF [not shown]) is also required for the inhibition of σG-dependent csfB transcription. Q is known to assemble into a channel apparatus with the mother cell proteins AA-AH (A-H) and to form a direct interaction with AH (H). This channel is generally required for gene expression at late times in the forespore, including that directed by σG; as such, it has been proposed to serve as a feeding tube through which the mother cell provides nutrients required for macromolecular synthesis (as shown). A separate subpopulation of Q is also known to interact with the SpoIIE phosphatase (IIE), an interaction that we have shown in the current study to be dependent specifically on Q Tyr-28. We propose that this subpopulation of Q, in collaboration with SpoIIE, positively regulates the csfB repressor protein by an unknown mechanism. However, we cannot rule out other scenarios, including the possibility that SpoIIE is not involved and/or that Q interacts with yet another partner via Tyr-28 to effect csfB regulation. Regardless, our data support the conclusion that Q is a bifunctional protein that (i) generally activates σG, through its assembly into the AA-AH·Q channel, and (ii) specifically maximizes the activity of σG, through participation in a gene regulatory circuit that represses expression of the gene encoding the anti-σG factor CsfB.
The mechanism by which Q Tyr-28 blocks σG-dependent csfB expression is less clear. The best-understood function of Q is its assembly with the mother cell proteins AA-AH into a channel apparatus that connects the forespore and mother cell (23). This AA-AH·Q channel is generally required for gene expression at late times in the forespore, including that directed by σF, σG, or the heterologous T7 RNAP (21). The current model suggests that the channel serves as a portal (feeding tube) through which the mother cell delivers small-molecule nutrients required for transcription and/or translation (21, 22). One possibility is that the misexpression of csfB by σG in QY28A mutant cells is the result of altered channel activity. For example, the AA-AH·QY28A mutant channel may have enhanced activity that generally stimulates gene expression in the forespore, including that directed by σG. We find this explanation unlikely for several reasons. First, and in contrast to the activity of P1csfB, the well-characterized σG-dependent promoters PsspB and PspoVT are not more active in the QY28A mutant but, rather, display an ∼25 to 30% reduction in expression. Second, no other tested σF-target promoter displayed evidence of misexpression by σG in the absence of Q Tyr-28. Together, these findings indicate that csfB expression is specifically misregulated in QY28A cells, in contrast to the general effect on forespore gene expression expected from altered channel activity. Finally, the misexpression of csfB in QY28A cells is complemented by an N-terminal fragment of Q entirely lacking the C-terminal domain required to interact with mother cell channel components. As such, we propose here that the best explanation for our data is that the QY28A variant is defective for a second function of Q that specifically regulates expression of csfB in the forespore and for which the N-terminal TMD region of Q is sufficient.
If Q Tyr-28 does not affect csfB expression via AA-AH·Q channel activity (as we have argued against above), then by what mechanism does it do so? We speculate that Q Tyr-28 is part of a mechanism required to activate the csfB repressor protein (Fig. 7, dashed arrows). We expect that the repressor protein is regulated such that it is present and active only at late times in the forespore. Given that Q itself is expressed under σF control, its involvement in this pathway could provide a delay function, creating a window of time during early sporulation in which the repressor protein is not yet active and σF-mediated activation of csfB can occur. It is tempting to speculate further that the involvement of Q in this pathway could also serve a checkpoint function, coupling the status of the AA-AH·Q channel function to σG activation. For example, perhaps the forespore stops the synthesis of the σG inhibitor CsfB only once the channel, which provides general resources required for gene expression, is assembled and active.
A role for SpoIIE?
One question raised by our model for csfB regulation is a topological one: how can Q Tyr-28, an amino acid buried within the forespore membrane, regulate gene expression in the forespore cytosol? Only the N-terminal ∼20 amino acids of Q are predicted to be exposed to the forespore cytosol, and these are followed by the Q TMD within which Tyr-28 is embedded. Given this topology, as well as the relative lack of conservation among its cytosolic N-terminal amino acids (Fig. 1), it seems unlikely that any part of the full-length, membrane-anchored Q protein interacts directly with any cytosolic components of the csfB regulatory circuit. As such, we envision two possible explanations for Q Tyr-28-mediated csfB regulation in the cytosol. The first is that Q may undergo a change in topology such that Tyr-28 gains access to the cytosol. Intriguingly, Q is subject to proteolysis by the secreted protease SpoIVB at a site not far downstream of the TMD (after Val-72), resulting in an N-terminal fragment that becomes cytosolic (45, 46). However, we have found that csfB expression is not misregulated in a strain in which spoIVB is deleted (our unpublished results), arguing against a model in which a SpoIVB-generated fragment of Q is responsible for csfB regulation. Nevertheless, it remains a formal possibility that Q (or a subpopulation of Q) undergoes another change in topology that has yet to be characterized.
The second possible explanation for Q Tyr-28-mediated csfB regulation in the cytosol, which we currently favor, is that Q laterally interacts (via Tyr-28) with an intermediary protein in the membrane and that this intermediary protein has a cytosolic domain that either regulates the putative csfB repressor or acts directly as the repressor. The multifunctional, membrane-anchored sporulation protein SpoIIE, which plays critical roles in earlier sporulation events, including asymmetric division and σF activation (9–11), is an intriguing candidate for this hypothetical intermediary protein linking Q Tyr-28 to csfB promoter regulation. The SpoIIE protein has the expected topology, with both an N-terminal membrane-spanning region (with 10 TMDs) (47) and two cytosolic domains: a central regulatory domain (48) and a C-terminal protein phosphatase 2C domain (9, 47). SpoIIE persists and dynamically localizes to the forespore membrane throughout and after engulfment (12) and is therefore appropriately positioned to participate in the regulatory circuit controlling csfB expression. Moreover, this late forespore population of SpoIIE interacts with a subpopulation of Q (apparently distinct from the subpopulation forming channel interactions), as evidenced by biochemical copurification, microscopic colocalization, and the loss of SpoIIE dynamic localization in a ΔQ mutant (12). These data indicate that SpoIIE, in cooperation with Q, performs a third function (in addition to its roles in asymmetric division and σF activation) at late times in the forespore.
Could it be that csfB gene regulation in collaboration with Q Tyr-28 is the mysterious third function of SpoIIE? Consistent with this idea, we found here that SpoIIE was mislocalized and unstable in the QY28A mutant, thus correlating proper SpoIIE localization and stability with proper regulation of the csfB promoter. On the other hand, the mislocalization and instability of SpoIIE in the QY28A strain were not detectably complemented by the wild type N-terminal domain of Q (QN) (our unpublished results), in contrast to the robust restoration of σG activity and csfB repression found using the same complementation strategy. This lack of correlation may argue against a role for SpoIIE in csfB regulation or could indicate that, for example, only a small subpopulation of properly localized and/or stable SpoIIE is sufficient to fully restore csfB regulation and σG activity. Unfortunately, a direct test for a causal link between SpoIIE and proper csfB regulation is complicated by the earlier functions of SpoIIE in sporulation: a ΔspoIIE mutant is unable to progress to the stage of sporulation at which this regulatory circuit operates (10, 49). Efforts to find SpoIIE variants that are specifically defective in csfB regulation (for example, by identifying and substituting highly conserved amino acids in the SpoIIE TMDs) have so far been unsuccessful. Current work in our laboratory is focused on other approaches, such as targeted protein degradation, to determine the role, if any, of SpoIIE in csfB regulation. In the event that SpoIIE is found to not participate in csfB regulation, our findings here will instead serve as evidence of another function of Q Tyr-28 (in partnership with SpoIIE) at later times in the developing forespore.
Other targets for Q Tyr-28?
Our identification of the regulatory circuit controlling csfB expression was serendipitous, given that this study began as an investigation of a puzzling variant of Q lacking the amino acid Tyr-28. We were first able to link QY28 to csfB through our finding that the 25 to 30% reduction in σG activity in the QY28A mutant was significantly alleviated in the absence of csfB. Based on this genetic interaction, we hypothesized that σG was subject to excessive inhibition by CsfB in the absence of Q Tyr-28. This in turn led us to discover that Q Tyr-28 participates in a regulatory circuit that ordinarily prevents σG-dependent activation of P1csfB in the forespore. The simplest explanation for the QY28A mutant phenotype, then, is that σG activity is reduced due to excessive synthesis of csfB by σG itself; in other words, it is reduced due to an inappropriately timed negative-feedback loop in which σG directs the synthesis of its own inhibitor. The ability of ΔcsfB to partially restore σG activity to QY28A cells is consistent with this model. A second prediction of this model is that expression of csfB itself from a mutant promoter activated by both σF and σG (such as the mut1PcsfB promoter) should also cause a reduction in σG activity, similar to that caused by the QY28A mutation. However, we observed no detectable defect in σG activity in cells expressing mut1PcsfB-csfB. One explanation could be that the alterations to the csfB promoter in the strain with mut1PcsfB do not cause misexpression to the same extent that they do in the QY28A mutant, although we find this unlikely, given that misexpression appears to be equivalent in the two strains, as measured from lacZ reporters. Instead, we interpret these data to indicate that the misexpression of csfB is not sufficient to explain the reduction of σG activity in QY28A cells.
By what other mechanisms might Q Tyr-28 promote maximal σG activity? One possibility is that Q Tyr-28 also regulates CsfB at the posttranscriptional level (CsfB activity or protein stability, for example). In addition, we predict that there must be other targets of Q Tyr-28 regulation aside from csfB, given that the deletion of csfB does not fully restore σG activity to QY28A cells. It is tempting to speculate that these targets are other genes whose expression is similarly regulated by Q Tyr-28 in the forespore; in this scenario, the reduction of σG activity in the QY28A mutant would be due to the collective misregulation of several genes and, as such, would not be phenocopied by misexpression of any one individual gene. We anticipate that transcriptional profiling of wild-type Q versus QY28A strains will help to identify any additional targets of Q Tyr-28 transcriptional regulation that, like csfB, modulate σG activity in the forespore.
Q is a multifunctional protein.
One final conclusion from this study is that Q is a bifunctional protein. In addition to its role as a component of the AA-AH·Q feeding tube channel, which generally promotes late gene expression in the forespore, we have found here that Q also functions in a gene regulatory circuit to specifically limit forespore expression of csfB (Fig. 7). This finding adds Q to a growing list of bi-/multifunctional proteins that serve to coordinate two or more events or processes during bacterial cell growth and development (50). For example, during the switch from flagellum-mediated motility to sessile growth in biofilms, the B. subtilis protein EpsE functions as both a flagellar clutch that halts cellular motility and an extracellular polysaccharide (EPS) biosynthetic enzyme, helping to form the matrix that binds the biofilm community together (51, 52). In another example, Caulobacter crescentus CtpS functions as both a CTP biosynthetic enzyme and a regulator of cell shape via interaction with the cytoskeletal protein crescentin (53). Our discovery that Q is also a bifunctional protein helps to further highlight this important class of proteins and raises the intriguing idea that other critical regulatory mechanisms and pathways driving B. subtilis sporulation (or any number of other biological processes for that matter) are hidden in plain sight within some of the most well-studied proteins.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge Richard Losick, David Rudner, Patrick Stragier, and Bjorn Traag for kindly sharing plasmids and strains. We also thank Kumaran Ramamurthi, Niels Bradshaw, Shawn Massoni, and other members of the A. H. Camp lab for valuable conversations and comments on the manuscript. Finally, we are especially grateful to Richard Losick for his insight and generous support.
This work was funded by National Institutes of Health grants DP2 GM105439 and R15 GM101559 to A.H.C. J.D.C. was supported by funding from Lehigh University, and A.F.W.E. was supported by National Institutes of Health grant GM18568.
The funders had no role in study design, data collection and interpretation, the decision to publish, or preparation of the manuscript.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00958-15.
REFERENCES
- 1.Davidson EH, Levine MS. 2008. Properties of developmental gene regulatory networks. Proc Natl Acad Sci U S A 105:20063–20066. doi: 10.1073/pnas.0806007105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tan IS, Ramamurthi KS. 2014. Spore formation in Bacillus subtilis. Environ Microbiol Rep 6:212–225. doi: 10.1111/1758-2229.12130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de Hoon M, Eichenberger P, Vitkup D. 2010. Hierarchical evolution of the bacterial sporulation network. Curr Biol 20:R735–R745. doi: 10.1016/j.cub.2010.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arrieta-Ortiz ML, Hafemeister C, Bate AR, Chu T, Greenfield A, Shuster B, Barry SN, Gallitto M, Liu B, Kacmarczyk T, Santoriello F, Chen J, Rodrigues CD, Sato T, Rudner DZ, Driks A, Bonneau R, Eichenberger P. 2015. An experimentally supported model of the Bacillus subtilis global transcriptional regulatory network. Mol Syst Biol 11:839. doi: 10.15252/msb.20156236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li Z, Piggot PJ. 2001. Development of a two-part transcription probe to determine the completeness of temporal and spatial compartmentalization of gene expression during bacterial development. Proc Natl Acad Sci U S A 98:12538–12543. doi: 10.1073/pnas.221454798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Duncan L, Losick R. 1993. SpoIIAB is an anti-σ factor that binds to and inhibits transcription by regulatory protein σF from Bacillus subtilis. Proc Natl Acad Sci U S A 90:2325–2329. doi: 10.1073/pnas.90.6.2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Min K, Hilditch CM, Diederich B, Errington J, Yudkin MD. 1993. σF, the first compartment-specific transcription factor of B. subtilis, is regulated by an anti-σ factor that is also a protein kinase. Cell 74:735–742. doi: 10.1016/0092-8674(93)90520-Z. [DOI] [PubMed] [Google Scholar]
- 8.Diederich B, Wilkinson JF, Magnin T, Najafi M, Erringston J, Yudkin MD. 1994. Role of interactions between SpoIIAA and SpoIIAB in regulating cell-specific transcription factor σF of Bacillus subtilis. Genes Dev 8:2653–2663. doi: 10.1101/gad.8.21.2653. [DOI] [PubMed] [Google Scholar]
- 9.Duncan L, Alper S, Arigoni F, Losick R, Stragier P. 1995. Activation of cell-specific transcription by a serine phosphatase at the site of asymmetric division. Science 270:641–644. doi: 10.1126/science.270.5236.641. [DOI] [PubMed] [Google Scholar]
- 10.Feucht A, Magnin T, Yudkin MD, Errington J. 1996. Bifunctional protein required for asymmetric cell division and cell-specific transcription in Bacillus subtilis. Genes Dev 10:794–803. doi: 10.1101/gad.10.7.794. [DOI] [PubMed] [Google Scholar]
- 11.Carniol K, Ben-Yehuda S, King N, Losick R. 2005. Genetic dissection of the sporulation protein SpoIIE and its role in asymmetric division in Bacillus subtilis. J Bacteriol 187:3511–3520. doi: 10.1128/JB.187.10.3511-3520.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Campo N, Marquis KA, Rudner DZ. 2008. SpoIIQ anchors membrane proteins on both sides of the sporulation septum in Bacillus subtilis. J Biol Chem 283:4975–4982. doi: 10.1074/jbc.M708024200. [DOI] [PubMed] [Google Scholar]
- 13.Sun DX, Cabrera-Martinez RM, Setlow P. 1991. Control of transcription of the Bacillus subtilis spoIIIG gene, which codes for the forespore-specific transcription factor σG. J Bacteriol 173:2977–2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rhayat L, Duperrier S, Carballido-Lopez R, Pellegrini O, Stragier P. 2009. Genetic dissection of an inhibitor of the sporulation sigma factor σG. J Mol Biol 390:835–844. doi: 10.1016/j.jmb.2009.05.073. [DOI] [PubMed] [Google Scholar]
- 15.Decatur A, Losick R. 1996. Identification of additional genes under the control of the transcription factor σF of Bacillus subtilis. J Bacteriol 178:5039–5041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karmazyn-Campelli C, Rhayat L, Carballido-Lopez R, Duperrier S, Frandsen N, Stragier P. 2008. How the early sporulation sigma factor σF delays the switch to late development in Bacillus subtilis. Mol Microbiol 67:1169–1180. doi: 10.1111/j.1365-2958.2008.06121.x. [DOI] [PubMed] [Google Scholar]
- 17.Chary VK, Xenopoulos P, Piggot PJ. 2007. Expression of the σF-directed csfB locus prevents premature appearance of σG activity during sporulation of Bacillus subtilis. J Bacteriol 189:8754–8757. doi: 10.1128/JB.01265-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Camp AH, Losick R. 2008. A novel pathway of intercellular signalling in Bacillus subtilis involves a protein with similarity to a component of type III secretion channels. Mol Microbiol 69:402–417. doi: 10.1111/j.1365-2958.2008.06289.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Camp AH, Wang AF, Losick R. 2011. A small protein required for the switch from σF to σG during sporulation in Bacillus subtilis. J Bacteriol 193:116–124. doi: 10.1128/JB.00949-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Serrano M, Real G, Santos J, Carneiro J, Moran CP Jr, Henriques AO. 2011. A negative feedback loop that limits the ectopic activation of a cell type-specific sporulation sigma factor of Bacillus subtilis. PLoS Genet 7:e1002220. doi: 10.1371/journal.pgen.1002220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Camp AH, Losick R. 2009. A feeding tube model for activation of a cell-specific transcription factor during sporulation in Bacillus subtilis. Genes Dev 23:1014–1024. doi: 10.1101/gad.1781709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Doan T, Morlot C, Meisner J, Serrano M, Henriques AO, Moran CP Jr, Rudner DZ. 2009. Novel secretion apparatus maintains spore integrity and developmental gene expression in Bacillus subtilis. PLoS Genet 5:e1000566. doi: 10.1371/journal.pgen.1000566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crawshaw AD, Serrano M, Stanley WA, Henriques AO, Salgado PS. 2014. A mother cell-to-forespore channel: current understanding and future challenges. FEMS Microbiol Lett 358:129–136. doi: 10.1111/1574-6968.12554. [DOI] [PubMed] [Google Scholar]
- 24.Levdikov VM, Blagova EV, McFeat A, Fogg MJ, Wilson KS, Wilkinson AJ. 2012. Structure of components of an intercellular channel complex in sporulating Bacillus subtilis. Proc Natl Acad Sci U S A 109:5441–5445. doi: 10.1073/pnas.1120087109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meisner J, Wang X, Serrano M, Henriques AO, Moran CP Jr. 2008. A channel connecting the mother cell and forespore during bacterial endospore formation. Proc Natl Acad Sci U S A 105:15100–15105. doi: 10.1073/pnas.0806301105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meisner J, Maehigashi T, André I, Dunham CM, Moran CP Jr. 2012. Structure of the basal components of a bacterial transporter. Proc Natl Acad Sci U S A 109:5446–5451. doi: 10.1073/pnas.1120113109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meisner J, Moran CP Jr. 2011. A LytM domain dictates the localization of proteins to the mother cell-forespore interface during bacterial endospore formation. J Bacteriol 193:591–598. doi: 10.1128/JB.01270-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fredlund J, Broder D, Fleming T, Claussin C, Pogliano K. 2013. The SpoIIQ landmark protein has different requirements for septal localization and immobilization. Mol Microbiol 89:1053–1068. doi: 10.1111/mmi.12333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rodrigues CDA, Marquis KA, Meisner J, Rudner DZ. 2013. Peptidoglycan hydrolysis is required for assembly and activity of the transenvelope secretion complex during sporulation in Bacillus subtilis. Mol Microbiol 89:1039–1052. doi: 10.1111/mmi.12322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sterlini JM, Mandelstam J. 1969. Commitment to sporulation in Bacillus subtilis and its relationship to development of actinomycin resistance. Biochem J 113:29–37. doi: 10.1042/bj1130029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nicholson WL, Setlow P. 1990. Sporulation, germination, and outgrowth, p 391–450. In Harwood CR, Cutting SM (ed), Molecular biological methods for Bacillus. John Wiley & Sons, Inc, New York, NY. [Google Scholar]
- 32.Youngman P, Perkins JB, Losick R. 1984. Construction of a cloning site near one end of Tn917 into which foreign DNA may be inserted without affecting transposition in Bacillus subtilis or expression of the transposon-borne erm gene. Plasmid 12:1–9. doi: 10.1016/0147-619X(84)90061-1. [DOI] [PubMed] [Google Scholar]
- 33.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. 2012. Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun D, Fajardo-Cavazos P, Sussman MD, Tovar-Rojo F, Cabrera-Martinez RM, Setlow P. 1991. Effect of chromosome location of Bacillus subtilis forespore genes on their spo gene dependence and transcription by EσF: identification of features of good EσF-dependent promoters. J Bacteriol 173:7867–7874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bagyan I, Hobot J, Cutting S. 1996. A compartmentalized regulator of developmental gene expression in Bacillus subtilis. J Bacteriol 178:4500–4507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Campbell EA, Masuda S, Sun JL, Muzzin O, Olson CA, Wang S, Darst SA. 2002. Crystal structure of the Bacillus stearothermophilus anti-σ factor SpoIIAB with the sporulation σ factor σF. Cell 108:795–807. doi: 10.1016/S0092-8674(02)00662-1. [DOI] [PubMed] [Google Scholar]
- 37.Serrano M, Neves A, Soares CM, Moran CP Jr, Henriques AO. 2004. Role of the anti-sigma factor SpoIIAB in regulation of σG during Bacillus subtilis sporulation. J Bacteriol 186:4000–4013. doi: 10.1128/JB.186.12.4000-4013.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kellner EM, Decatur A, Moran CP Jr. 1996. Two-stage regulation of an anti-sigma factor determines developmental fate during bacterial endospore formation. Mol Microbiol 21:913–924. doi: 10.1046/j.1365-2958.1996.461408.x. [DOI] [PubMed] [Google Scholar]
- 39.Evans L, Clarkson J, Yudkin MD, Errington J, Feucht A. 2003. Analysis of the interaction between the transcription factor σG and the anti-sigma factor SpoIIAB of Bacillus subtilis. J Bacteriol 185:4615–4619. doi: 10.1128/JB.185.15.4615-4619.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Steil L, Serrano M, Henriques AO, Völker U. 2005. Genome-wide analysis of temporally regulated and compartment-specific gene expression in sporulating cells of Bacillus subtilis. Microbiology 151:399–420. doi: 10.1099/mic.0.27493-0. [DOI] [PubMed] [Google Scholar]
- 41.Wang ST, Setlow B, Conlon EM, Lyon JL, Imamura D, Sato T, Setlow P, Losick R, Eichenberger P. 2006. The forespore line of gene expression in Bacillus subtilis. J Mol Biol 358:16–37. doi: 10.1016/j.jmb.2006.01.059. [DOI] [PubMed] [Google Scholar]
- 42.Serrano M, Gao J, Bota J, Bate AR, Meisner J, Eichenberger P, Moran CP Jr, Henriques AO. 2015. Dual-specificity anti-sigma factor reinforces control of cell-type specific gene expression in Bacillus subtilis. PLoS Genet 11:e1005104. doi: 10.1371/journal.pgen.1005104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Serrano M, Hovel S, Moran CP Jr, Henriques AO, Völker U. 2001. Forespore-specific transcription of the lonB gene during sporulation in Bacillus subtilis. J Bacteriol 183:2995–3003. doi: 10.1128/JB.183.10.2995-3003.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu LJ, Errington J. 2000. Identification and characterization of a new prespore-specific regulatory gene, rsfA, of Bacillus subtilis. J Bacteriol 182:418–424. doi: 10.1128/JB.182.2.418-424.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jiang X, Rubio A, Chiba S, Pogliano K. 2005. Engulfment-regulated proteolysis of SpoIIQ: evidence that dual checkpoints control sigma activity. Mol Microbiol 58:102–115. doi: 10.1111/j.1365-2958.2005.04811.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chiba S, Coleman K, Pogliano K. 2007. Impact of membrane fusion and proteolysis on SpoIIQ dynamics and interaction with SpoIIIAH. J Biol Chem 282:2576–2586. doi: 10.1074/jbc.M606056200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Arigoni F, Guerout-Fleury AM, Barak I, Stragier P. 1999. The SpoIIE phosphatase, the sporulation septum and the establishment of forespore-specific transcription in Bacillus subtilis: a reassessment. Mol Microbiol 31:1407–1415. doi: 10.1046/j.1365-2958.1999.01282.x. [DOI] [PubMed] [Google Scholar]
- 48.Feucht A, Abbotts L, Errington J. 2002. The cell differentiation protein SpoIIE contains a regulatory site that controls its phosphatase activity in response to asymmetric septation. Mol Microbiol 45:1119–1130. doi: 10.1046/j.1365-2958.2002.03082.x. [DOI] [PubMed] [Google Scholar]
- 49.Margolis P, Driks A, Losick R. 1991. Establishment of cell type by compartmentalized activation of a transcription factor. Science 254:562–565. doi: 10.1126/science.1948031. [DOI] [PubMed] [Google Scholar]
- 50.Radhakrishnan SK, Viollier P. 2012. Two-in-one: bifunctional regulators synchronizing developmental events in bacteria. Trends Cell Biol 22:14–21. doi: 10.1016/j.tcb.2011.09.004. [DOI] [PubMed] [Google Scholar]
- 51.Blair KM, Turner L, Winkelman JT, Berg HC, Kearns DB. 2008. A molecular clutch disables flagella in the Bacillus subtilis biofilm. Science 320:1636–1638. doi: 10.1126/science.1157877. [DOI] [PubMed] [Google Scholar]
- 52.Guttenplan SB, Blair KM, Kearns DB. 2010. The EpsE flagellar clutch is bifunctional and synergizes with EPS biosynthesis to promote Bacillus subtilis biofilm formation. PLoS Genet 6:e1001243. doi: 10.1371/journal.pgen.1001243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ingerson-Mahar M, Briegel A, Werner JN, Jensen GJ, Gitai Z. 2010. The metabolic enzyme CTP synthase forms cytoskeletal filaments. Nat Cell Biol 12:739–746. doi: 10.1038/ncb2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.