

Abstract
Background:
Overexpression of G-protein coupled receptor 34 (GPR34) affects the progression and prognosis of human gastric adenocarcinoma, however, the role of GPR34 in gastric cancer development and progression has not been well-determined. The current study aimed to investigate the effect of GPR34 knockdown on the proliferation, migration, and apoptosis of HGC-27 gastric cancer cells and the underlying mechanisms.
Methods:
The expression of GPR34 in gastric cancer cell line HGC-27 was detected by quantitative real-time reverse transcription-polymerase chain reaction (RT-PCR) and Western blotting. HGC-27 cells were employed to construct the stable GPR34 knockdown cell model in this study. Real-time RT-PCR and Western blotting were applied to validate the effect of short hairpin RNA (ShRNA) on the expression of GPR34 in HGC-27 gastric cells. The proliferation, migration of these cells were examined by Cell Counting Kit-8 and transwell. We also measured expression profile of PI3K/PDK1/AKT and ERK using Western blotting.
Results:
The ShRNA directed against GPR34 effectively inhibited both endogenous mRNA and protein expression levels of GPR34, and significantly down-regulated the expression of PIK3CB (P < 0.01), PIK3CD (P < 0.01), PDK1 (P < 0.01), phosphorylation of PDK1 (P < 0.01), Akt (P < 0.01), and ERK (P < 0.01). Furthermore, GPR34 knockdown resulted in an obvious reduction in HGC-27 cancer cell proliferation and migration activity (P < 0.01).
Conclusions:
GPR34 knockdown impairs the proliferation and migration of HGC-27 gastric cancer cells in vitro and provides a potential implication for therapy of gastric cancer.
Keywords: Gastric Cancer, GPR34, Knockdown, Migration, Proliferation
INTRODUCTION
Gastric cancer is one of the most common epithelial malignancies and the secondary leading cause of cancer-related death in the world.[1] There are about 952,000 new gastric cancer cases each year world-wide, 47% of which are in China. Gastric cancer is ranked second in the incidence of malignant tumors in China, and some 352,300 people in China die of gastric cancer each year, ranking third in cancer mortality in the country.[2]
Despite the advances in treatment and research efforts over the past few decades, the poor overall 5-year survival rate of gastric cancer is largely because many gastric cancers are fail to diagnosed at an early stage, tumor recurrence and metastasis[3]. Thus, it is necessary to further explore and investigate the tumorigenesis of gastric cancer and its novel therapy targets.[4]
G protein-coupled receptors (GPCRs) represent a large family of proteins and regulate key biological functions including cellular growth, motility, differentiation, and gene transcription, and also appear to be involved in cancer progression.[5] GPCRs are one of the most important drug targets for the pharmaceutical industry and >30% of all marketed therapeutics act on them.[6] Additional, short hairpin RNA (ShRNA)-mediated GPCR gene silencing has promising therapeutic potential for gastric cancer therapy.[7,8]
G protein - coupled receptor 34 (GPR34) is a 7-transmembrane receptor. Only a few studies have addressed elevated expression levels of GPR34 in malignancies, including metastatic melanoma[9] and MALT lymphoma[10,11] and its essential roles in tumor development and progression. Our previous studies have demonstrated that increased expression of GPR34 in the membrane of gastric cancer tissues was likely associated with metastatic ability, as well as invasion of NCI-N87 gastric cancer cells.[12] However, the relationship between down-regulation of GPR34 and proliferation and the underlying mechanism remains largely unknown.
In this study, we investigated the effects of down-regulation of GPR34 by ShRNA on proliferation, apoptosis, and migration of HGC-27 gastric cancer cells, as well as PI3K/AKT and ERK expression, with the aim of evaluating whether the expression of GPR34 may be linked to gastric cancer progression, and exploring the underlying mechanisms.
METHODS
Cell culture
The human gastric adenocarcinoma cell line HGC-27 was purchased from the American Type Culture Collection and HGC-27 in DMEM (Gibco, USA) containing 10% fetal bovine serum (FBS) (Hyclone, German) and penicillin/streptomycin (10 μl/ml) (Gibco, USA) at 37°C in a humidified atmosphere with 5% CO2.
Stable knockdown of GPR34
One MSCV-based retroviral vector containing-shRNA that specifically target GPR34 transcript was purchased from Open Biosystems (Huntsville, AL, USA). Sense sequence of GPR34 shRNA is as follows: CAGTTTGGATCGCTATATA. The expression of shRNAs was driven by the U6 promoter. Amplification and purification of plasmid DNAs were performed as specified by the manufacturer's instructions. Lipofectin 2000 was used to transfect MSCV-empty vector (Clontech, USA)-or MSCV-ShRNA plasmids-into HGC-27 gastric cancer cells according to manufacturer's instruction (Invitrogen, USA). The stable transformed clones were selected by puromycin (2 mg/mL) (Invitrogen, USA) and used for subsequent experiments.
Quantitative real-time reverse transcription-polymerase chain reaction and Western blotting analyses
Quantitative real-time reverse transcription-polymerase chain reaction (RT-PCR) was performed using an OPTICON 2 real-time PCR detection system (Bio-Rad, USA) using human GPR34 primers[12] combined with SYBR Green Master Mix (Applied Biosystems, USA). In brief, total RNA was extracted from cells with the RNeasy mini kit (Qiagen, USA) and cDNA was subsequently generated with Super-Script III (Invitrogen, USA). The PCR reactions began with 94°C for 2 min, followed by 40 cycles of 94°C for 20 s, 59°C for 20 s, 72°C for 30 s, 74°C for 1 s.
(GADPH primers: Forward, 5’-CTGGTAAAGT GGATATTGTTGCCAT-3’, reverse, 5’-TGGAATCATA TTGGAACATGTAAACC-3’; human GPR34 primers (forward: 5’-CTCCCACAGAATGCGCTTTATA-3’, reverse: 5’-CAACCAGTCCCACGATGAAAA-3’).
Western blotting analyses were performed by conventional protocols as described by Cui et al.[13] In brief, Cells were lysed in ice-cold lysis buffer. Whole cell lysates were then clarified by centrifugation at 16,000 ×g for 30 min at 4°C and mixed with loading buffer and boiled for 5 min. Samples were resolved by 8% sodium dodecyl sulfate polyacrylamide gel electrophoresis and proteins were transferred onto Hybond™-N Nylon membranes (Pharmacia, UK).
Nylon membranes were then blocked with 5% skimmed milk in tris buffered saline containing 0.1% Tween 20 (TBST), followed by an overnight incubation with primary antibodies (1:1000) at 4°C. Membranes were then washed with TBST and incubated with secondary antibodies conjugated to HRP (Jackson, USA). Signals were detected using the SuperSignal West Pico Trial Kit (Thermo Scientific, USA). Images were acquired using an Image Qluant 350 (GE HEALTHCARE, USA) and analyzed by ImageJ software (NIH, USA). β-actin, (1:2000 dilution) (Proteintech, USA) PI3 Kinase Ab Sampler Kit (#9655), P-AKT pathway Sampler Kit (#9916), and ERK/P-ERK (#9102/#4370) (1:1000 dilution; CST, USA).
Cell proliferation assay
Cell proliferation assays were performed with Cell Counting Kit-8 (CCK-8, Dojindo Rockville, USA) according to the manufacturer's instructions. The HGC-27 parental cells (HGC-27), the empty-vector transfected HGC-27 cells (HGC-27-V), and GPR34-ShRNA transfected HGC-27 cells (HGC-27-K) were seeded at 5 × 103 into each well of a 96-well plate and cultured in 100 μl of DMEM supplemented with 10% FBS. At the indicated time-points, culture medium was exchanged for 110 μl of DMEM supplemented with CCK-8 reagent (10 μl CCK-8 plus 100 μl DMEM), and the cells were incubated for an additional 2 h. Absorbances were measured for each well at a wavelength of 450 nm, with a reference wavelength set at 600 nm. An increase or decrease in absorbance values at 450 nm in the experimental wells relative to the initial value indicated cell growth or death respectively. Cell growth was monitored at 24 h, 48 h, 72 h, and 96 h (and was repeated in replicates of 3 wells per time-point.
Apoptosis assay
Apoptosis was monitored using the Annexin V-FITC Apoptosis Detection Kit (KeyGen Biotech, China). For FACS analysis, 1 × 105 cells were co-stained with Annexin V-and with propidium iodide (PI). Cells that stained negative for both PI and annexin V were considered viable cells; PI-negative and annexin V-positive stained cells were considered early apoptotic cells; PI-positive and annexin V-positive stained cells were considered primarily in the later stages of apoptosis.
Migration assay
The activity of migration of cells was performed using 24-well transwell chamber (Corning, USA) with 8.0-μm pore polycarbonate filter inserts. Cells (5 × 104 cells/well) suspended in serum-free DMEM containing 0.2% BSA were overlaid in the upper chamber of each transwell. In each lower chamber, 600 μl of DMEM supplemented with 10% FBS was added. Then, the inserts were incubated at 37°C in a humidified atmosphere containing 5% CO2 for overnight. The cells that had not penetrated the filters were removed using cotton swabs. The migrated cells attached to the bottom side were fixed in 4% paraformaldehyde for 10 min and stained in 0.1% crystal violet for 30 min, rinsed in PBS and examined under a bright-field microscope with × 100 magnification. The value of migratory activity was expressed as the average number of migrated cells per microscopic field over the 5 fields in each assay from three independent experiments.
Statistical analysis
Data from all quantitative assays are expressed as the mean ± standard deviation (SD) and were analyzed statistically using one-way analysis of variance (ANOVA) and the independent-samples t test. Statistical calculations were performed using SPSS 16.0 (SPSS Inc., USA). P < 0.05 were considered as statistically significant.
RESULTS
Construction of HGC-27 GPR34 knock-down cell models
HGC-27, a low-phosphatase and tensin homolog gastric cancer cell line, was selected and used as a cell model in vitro to determine the correlation between GPR34 expression and proliferation, apoptosis and migration of gastric cancer cells. Both western blotting and real-time RT-PCR results indicated that the GPR34 knockdown HGC-27 cell model (one clone) was successfully constructed [Figure 1a–c].
Figure 1.
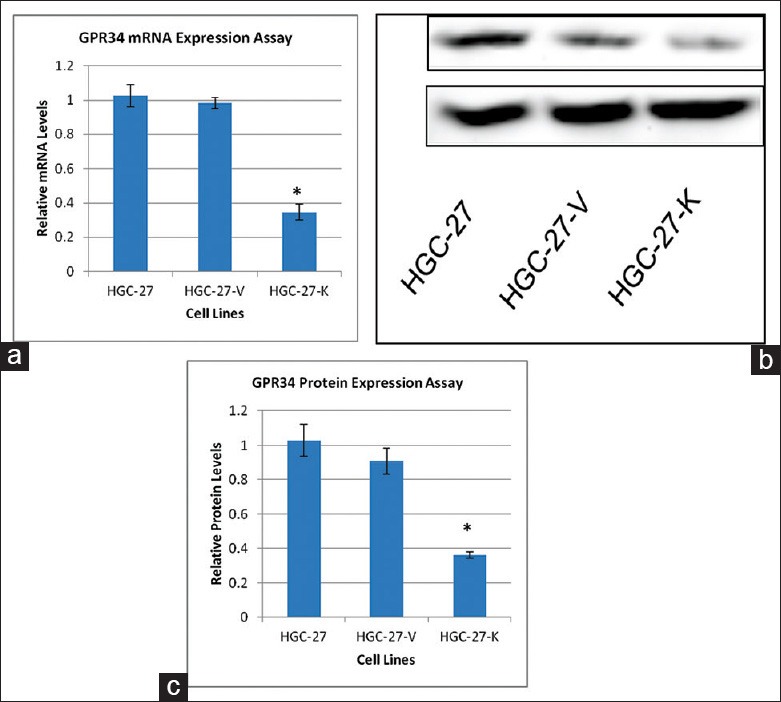
Validation of GPR34 knockdown in HGC-27 cells. (a) Knockdown of GPR34 expression in HGC-27 cells. The vertical axis shows the GPR34 mRNA levels relative to that of GAPDH. The data represent the mean ± SD of triplicate experiments; (b) Western blot validation of GPR34 knock-down p185YR cell model construction; (c) Densitometry analysis of Western blotting of GPR34 in HGC-27 cells (ImageJ) (*P< 0.01).
GPR34 knockdown impairs the proliferation of HGC-27 gastric cancer cells
Significant up-regulation of GPR34 protein in gastric cancer tissues suggested a potential oncogenic role of this gene. To investigate the possible pro-proliferative effects of GPR34 in vitro, a CCK-8 assay was performed and a cell growth curve was generated. As shown in Figure 2, the proliferation of HGC-27 cells transfected with GPR34 specific ShRNA was significantly suppressed [Figure 2].
Figure 2.
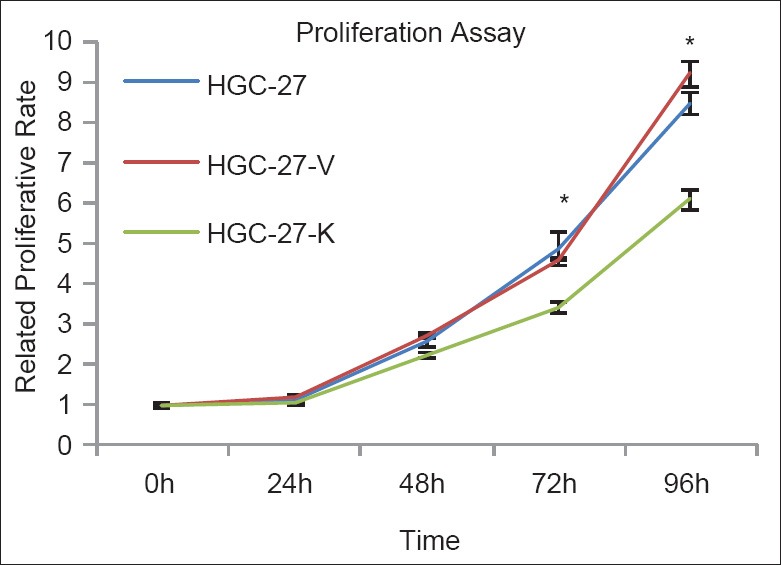
GPR34 knockdown impairs the proliferation of HGC-27 cells in vitro. Proliferative assay by Cell Counting Kit-8. GPR34 knock-down impairs the proliferative ability of HGC-27 cells. The proliferative value is shown in vertical axis as the mean ± SD of triplicate wells (*P < 0.01 vs. controls, n = 3).
GPR34 knockdown dose not induce apoptosis of HGC-27 gastric cancer cells
To further evaluate the effects of GPR34 on HGC-27 cells apoptosis, the combination of annexin V-FITC and PI was applied. We found that GPR34 knockdown did not induce the apoptosis of HGC-27 gastric cancer cells [Figure 3].
Figure 3.
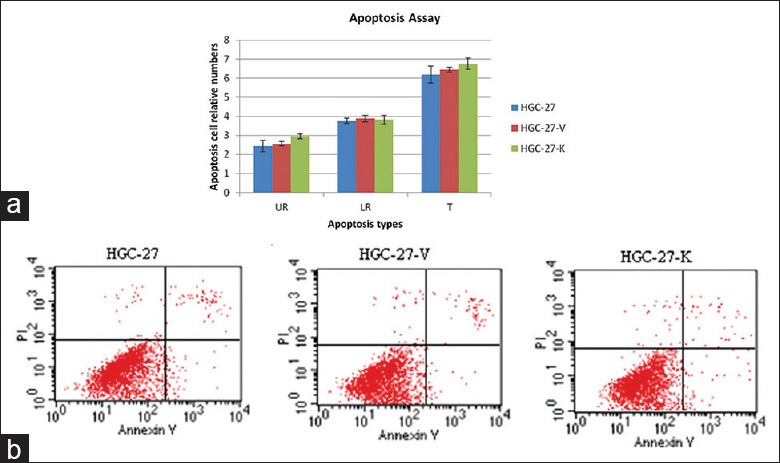
GPR34 knockdown does not induce the apoptosis of HGC-27 cell in vitro. Cell apoptosis was assessed by Annextin V assay. (a) The apoptotic cells in UR, LR, and total (T) were counted and expressed as the percentage of total cells. There is significant difference between HGC-27, HGC-27-V, and HGC-27-K (P > 0.05 vs. controls, n = 3); (b) GPR34 knockdown did not induce the apoptosis in HGC-27 cells.
GPR34 knockdown inhibits the migration of HGC-27 gastric cancer cells
To evaluate the function of GPR34 on HGC-27 cells migration, transwell chambers were utilized. We found that GPR34 knockdown led to a significantly decreased migratory ability compared with the control group (P < 0.01, Figure 4).
Figure 4.
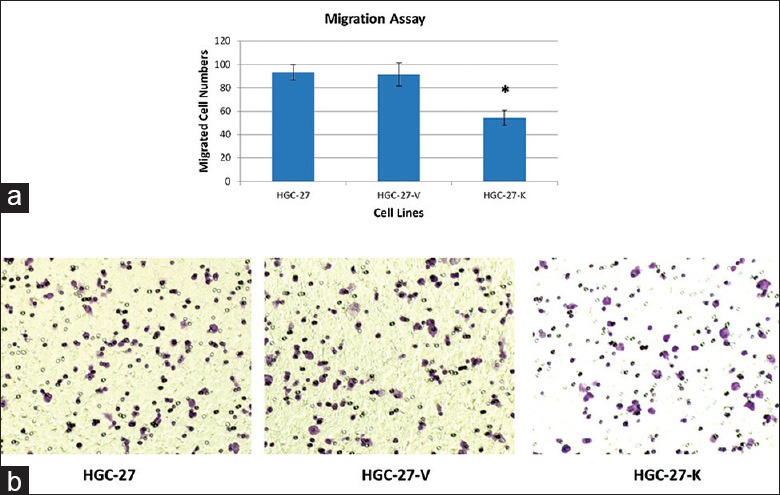
The effect of GPR34 on migration of HGC-27 cells In vitro. Migration assay was conducted using transwell chamber. (a) The number of migrated cells that penetrated transwell chamber was expressed as the mean number of cells in the 5 random fields identified within (*P < 0.01 vs. controls). The results shown here were for one representative experiment of three with similar results; (b) Cells on the lower surface of the filter were photographed.
GPR34 knockdown suppresses the ERK and PI3K/PDK1/AKT pathways
Combined with our previous study,[14] to understand decreased expression of GPR34 in HGC-27 represents a potential molecular mechanism by which HGC-27-K results in the decreased abilities of proliferation and migration any differently from that seen with HGC-27 parental cells in vitro. We studied alterations in ERK, the class I PI3K subunits (p110α, p110β, and p110δ), and PDK1/AKT. When compared with HGC-27 and HGC-27-V cells, both p110β and p110δ were found to be down-regulated in HGC-27-K cells. We did not observe any alteration in the expression of p110α and detect the expression of p110γ in either cell-line [Figure 5]. Furthermore, low expression levels of p-PDK1, p-AKT, and p-ERK were observed in HGC-27-K cells [Figure 5].
Figure 5.
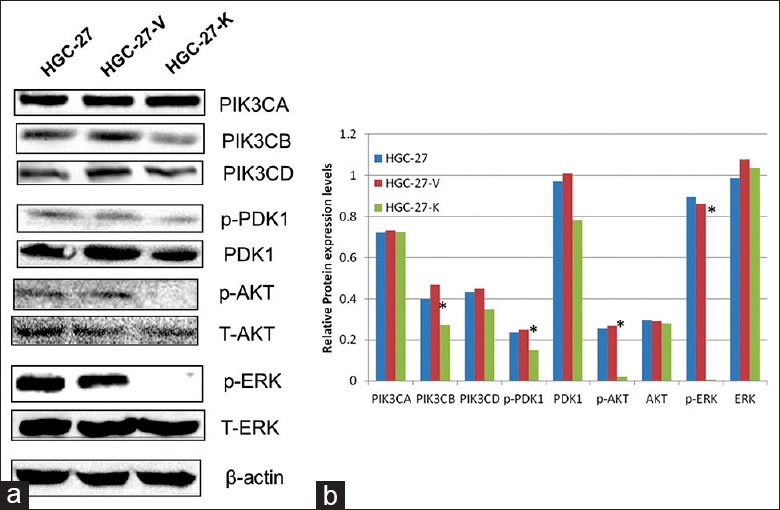
GPR34 expression involved in gastric cell proliferation and migration (HGC-27 cell) in vitro by ERK and PIK3/AKT pathways. (a) Basal expression of p110 (α, β, and δ) as compared to HGC-27 and HGC-27-V. Only p110β was found to be significantly downregulated in HGC-27-K (*P< 0.01). The phosphorylated PDK1, AKT, and ERK were constitutively suppressed; (b) Densitometry analysis of all tested proteins in Western blotting was performed using ImageJ software (*P< 0.01).
DISCUSSION
The activation of oncogenes is currently thought as one of the most important factors in development and progression of gastric cancer.[13] However, the critical underlying molecular mechanism of its progression is largely unclear.
GPCRs act as cell-surface mediators of a diverse spectrum of biological signals and are able to activate or inhibit PI3K pathways. By regulating PI3K p110β, G protein-coupled receptors are able to “examine” resulting biochemical and physiological changes, such as cellular proliferation, apoptosis, and migration.[15,16] Given the importance of GPR34-mediated signaling in regulating the proliferation and migration of HGC-27 cells, it is conceivable that GPR34 serves a novel, and potentially pathway in HGC-27 induced increased proliferation and migration.
The PI3Ks have been linked to an extraordinarily diverse group of cellular functions, including proliferation, apoptosis and tumor cell migration.[14] Many of these functions relate to the activation of the PI3Ks upstream molecules like GPCRs, the phosphoralation activation of ERK and the ability of PI3K to activate its key downstream effector AKT.[17,18] Many studies have shown that PI3Ks/AKT and or ERK activity are detectable in gastric cancer.[19,20] Our previous study[12] showed that up-regulation of GPR34 in primary gastric cancer tissues/cell lines might play a critical role in tumor progression and in determining patient prognosis. Furthermore, alteration of GPR34 expression also involved in the NCI-N87 invasion in vitro by PI3K/PDK1/AKT pathway.
In this study, our results revealed that both GPR34 mRNA and protein were markedly inhibited in HGC-27 cell line transfected with GPR34-ShRNA, which is consistent with our studies showing in NCI-N87.[12] Furthermore, ShRNA-directed targeting of GPR34 in these cells could reduce cellular proliferation and migration. More importantly, a low level of p-ERK, p110β and p110δ, p-PDK1, p-AKT in the HGC-27-K group was detected compared with HGC-27 and HGC-27-V control groups. The above data indicate that knockdown of GPR34 may result in a reduction in ERK-phosphorylation, decreased catalytic activity of PI3K, subsequent low-phosphorylation of the downstream effector AKT, and thus low activity or aberrant inactivation of ERK and PI3K/AKT pathways in these cells.
Taken together, ShRNA targeting GPR34 effectively impairs the proliferation and migration of HGC-27 cells and suppresses the ERK and PI3K/Akt pathways, and would be expected to become a potential strategy for the therapy of gastric cancer regardless of GPR34. However, further studies will be required to understand the detail molecular mechanism and develop efficient approaches for the delivery of ShRNA into target cells.
Footnotes
Edited by: Jian Gao
Source of Support: This study was supported by grants from the National Natural Science Foundation of China (No. 30872923), the Peking University People's Hospital Research and Development Foundations (No. RDB2007-47, No. RDK2008-01 and No. RDB2011-26).
Conflict of Interest: None declared.
REFERENCES
- 1.Anderson WF, Camargo MC, Fraumeni JF, Jr, Correa P, Rosenberg PS, Rabkin CS. Age-specific trends in incidence of noncardia gastric cancer in US adults. JAMA. 2010;303:1723–8. doi: 10.1001/jama.2010.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Available from: http://www.news.xinhuanet.com/english/health/2014-06/03/c_133379921.htm .
- 3.D’Ugo D, Persiani R, Zoccali M, Cananzi F, Vigorita V, Mazzeo P, et al. Surgical issues after neoadjuvant treatment for gastric cancer. Eur Rev Med Pharmacol Sci. 2010;14:315–9. [PubMed] [Google Scholar]
- 4.Zhou XK, Tang SS, Yi G, Hou M, Chen JH, Yang B, et al. RNAi knockdown of PIK3CA preferentially inhibits invasion of mutant PIK3CA cells. World J Gastroenterol. 2011;17:3700–8. doi: 10.3748/wjg.v17.i32.3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gao Y, Kitagawa K, Hiramatsu Y, Kikuchi H, Isobe T, Shimada M, et al. Up-regulation of GPR48 induced by down-regulation of p27Kip1 enhances carcinoma cell invasiveness and metastasis. Cancer Res. 2006;66:11623–31. doi: 10.1158/0008-5472.CAN-06-2629. [DOI] [PubMed] [Google Scholar]
- 6.Hopkins AL, Groom CR. The druggable genome. Nat Rev Drug Discov. 2002;1:727–30. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- 7.Xi HQ, Cai AZ, Wu XS, Cui JX, Shen WS, Bian SB, et al. Leucine-rich repeat-containing G-protein-coupled receptor 5 is associated with invasion, metastasis, and could be a potential therapeutic target in human gastric cancer. Br J Cancer. 2014;110:2011–20. doi: 10.1038/bjc.2014.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu CY, Liang GB, Du P, Liu YH. Lgr4 promotes glioma cell proliferation through activation of Wnt signaling. Asian Pac J Cancer Prev. 2013;14:4907–11. doi: 10.7314/apjcp.2013.14.8.4907. [DOI] [PubMed] [Google Scholar]
- 9.Qin Y, Verdegaal EM, Siderius M, Bebelman JP, Smit MJ, Leurs R, et al. Quantitative expression profiling of G-protein-coupled receptors (GPCRs) in metastatic melanoma: The constitutively active orphan GPCR GPR18 as novel drug target. Pigment Cell Melanoma Res. 2011;24:207–18. doi: 10.1111/j.1755-148X.2010.00781.x. [DOI] [PubMed] [Google Scholar]
- 10.Ansell SM, Akasaka T, McPhail E, Manske M, Braggio E, Price-Troska T, et al. t (X; 14) (p11;q32) in MALT lymphoma involving GPR34 reveals a role for GPR34 in tumor cell growth. Blood. 2012;120:3949–57. doi: 10.1182/blood-2011-11-389908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baens M, Finalet Ferreiro J, Tousseyn T, Urbankova H, Michaux L, de Leval L, et al. t (X; 14)(p11.4;q32.33) is recurrent in marginal zone lymphoma and up-regulates GPR34. Haematologica. 2012;97:184–8. doi: 10.3324/haematol.2011.052639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu W, Ma S, Wang L, Zuo B, Li M, Qiao Z, et al. Upregulation of GPR34 expression affects the progression and prognosis of human gastric adenocarcinoma by PI3K/PDK1/AKT pathway. Histol Histopathol. 2013;28:1629–38. doi: 10.14670/HH-28.1629. [DOI] [PubMed] [Google Scholar]
- 13.Cui M, Yu W, Dong J, Chen J, Zhang X, Liu Y. Downregulation of ABI1 expression affects the progression and prognosis of human gastric carcinoma. Med Oncol. 2010;27:632–9. doi: 10.1007/s12032-009-9260-6. [DOI] [PubMed] [Google Scholar]
- 14.Ji D, Zhang Z, Cheng L, Chang J, Wang S, Zheng B, et al. The combination of RAD001 and MK-2206 exerts synergistic cytotoxic effects against PTEN mutant gastric cancer cells: Involvement of MAPK-dependent autophagic, but not apoptotic cell death pathway. PLoS One. 2014;9:e85116. doi: 10.1371/journal.pone.0085116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.New DC, Wu K, Kwok AW, Wong YH. G protein-coupled receptor-induced Akt activity in cellular proliferation and apoptosis. FEBS J. 2007;274:6025–36. doi: 10.1111/j.1742-4658.2007.06116.x. [DOI] [PubMed] [Google Scholar]
- 16.Tang SY, Xie H, Yuan LQ, Luo XH, Huang J, Cui RR, et al. Apelin stimulates proliferation and suppresses apoptosis of mouse osteoblastic cell line MC3T3-E1 via JNK and PI3-K/Akt signaling pathways. Peptides. 2007;28:708–18. doi: 10.1016/j.peptides.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 17.Arrighi N, Bodei S, Zani D, Michel MC, Simeone C, Cosciani Cunico S, et al. Different muscarinic receptor subtypes modulate proliferation of primary human detrusor smooth muscle cells via Akt/PI3K and map kinases. Pharmacol Res. 2013;74:1–6. doi: 10.1016/j.phrs.2013.04.007. [DOI] [PubMed] [Google Scholar]
- 18.Tveteraas IH, Müller KM, Aasrum M, Ødegård J, Dajani O, Guren T, et al. Mechanisms involved in PGE2-induced transactivation of the epidermal growth factor receptor in MH1C1 hepatocarcinoma cells. J Exp Clin Cancer Res. 2012;31:72. doi: 10.1186/1756-9966-31-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fu X, Feng J, Zeng D, Ding Y, Yu C, Yang B. PAK4 confers cisplatin resistance in gastric cancer cells via PI3K/Akt- and MEK/Erk-dependent pathways. Biosci Rep. 2014 doi: 10.1042/BSR20130102. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takahashi N, Yamada Y, Taniguchi H, Fukahori M, Sasaki Y, Shoji H, et al. Clinicopathological features and prognostic roles of KRAS, BRAF, PIK3CA and NRAS mutations in advanced gastric cancer. BMC Res Notes. 2014;7:271. doi: 10.1186/1756-0500-7-271. [DOI] [PMC free article] [PubMed] [Google Scholar]