

ABSTRACT
Accumulating data suggest that tripartite-motif-containing (TRIM) proteins participate in host responses to viral infections, either by acting as direct antiviral restriction factors or through regulating innate immune signaling of the host. Of >70 TRIMs, TRIM56 is a restriction factor of several positive-strand RNA viruses, including three members of the family Flaviviridae (yellow fever virus, dengue virus, and bovine viral diarrhea virus) and a human coronavirus (OC43), and this ability invariably depends upon the E3 ligase activity of TRIM56. However, the impact of TRIM56 on negative-strand RNA viruses remains unclear. Here, we show that TRIM56 puts a check on replication of influenza A and B viruses in cell culture but does not inhibit Sendai virus or human metapneumovirus, two paramyxoviruses. Interestingly, the anti-influenza virus activity was independent of the E3 ligase activity, B-box, or coiled-coil domain. Rather, deletion of a 63-residue-long C-terminal-tail portion of TRIM56 abrogated the antiviral function. Moreover, expression of this short C-terminal segment curtailed the replication of influenza viruses as effectively as that of full-length TRIM56. Mechanistically, TRIM56 was found to specifically impede intracellular influenza virus RNA synthesis. Together, these data reveal a novel antiviral activity of TRIM56 against influenza A and B viruses and provide insights into the mechanism by which TRIM56 restricts these medically important orthomyxoviruses.
IMPORTANCE Options to treat influenza are limited, and drug-resistant influenza virus strains can emerge through minor genetic changes. Understanding novel virus-host interactions that alter influenza virus fitness may reveal new targets/approaches for therapeutic interventions. We show here that TRIM56, a tripartite-motif protein, is an intrinsic host restriction factor of influenza A and B viruses. Unlike its antiviral actions against positive-strand RNA viruses, the anti-influenza virus activity of TRIM56 was independent of the E3 ligase activity. Rather, expression of a short segment within the very C-terminal tail of TRIM56 inhibited the replication of influenza viruses as effectively as that of full-length TRIM56 by specifically targeting viral RNA synthesis. These data reveal the remarkable multifaceted activity of TRIM56, which has developed multiple domains to inhibit multiple viral families. They also raise the possibility of developing a broad-spectrum, TRIM56-based antiviral approach for addition to influenza prophylaxis and/or control strategies.
INTRODUCTION
Classified within the family Orthomyxoviridae, influenza A (IAV) and B (IBV) viruses are important respiratory pathogens responsible for significant morbidity and mortality worldwide (1–4). These viruses possess segmented negative-strand RNA genomes that are encapsidated with nucleoprotein (NP) and bound with polymerase proteins (PB2, PB1, and PA), collectively known as viral ribonucleoprotein (vRNP) complexes (3, 4). One striking feature of the influenza virus life cycle is that virus RNA (vRNA) synthesis takes place in the nucleus, rare for RNA viruses (4–6). After virion entry and uncoating, trafficking of incoming vRNPs into the nucleus is an active process dependent on the nuclear import machinery, followed by initiation of virus transcription and replication (4–6). As is the case with most RNA viruses, replication of influenza viruses is error prone, generating mutant viruses that can escape host immune responses and/or antiviral drugs. Despite intense research and antiviral drug development efforts, options to combat influenza are for the most part limited to those targeting the viral neuraminidase and M2 ion channel, and emergence of drug-resistant viral mutants is a pressing issue (1, 3, 4). Thus, studies on novel virus-host interactions that alter influenza virus fitness are warranted and may reveal new targets for therapeutic interventions.
In response to virus assaults, including infection by influenza viruses, host cells employ exquisite intrinsic mechanisms to fend off the invader. The induction of type I and type III inteferons (IFNs) represents a major immediate antiviral response in mammalian cells and is initiated by several classes of pattern recognition receptors (PRRs), including the Toll-like receptors (TLRs) and retinoic-inducible gene I (RIG-I)-like receptors (7–10). These PRRs engage viral pathogen-associated molecular patterns, most notably viral RNAs, triggering signaling cascades that culminate in activation of two critical transcription factors—interferon regulatory factor 3 (IRF3) and NF-κB—which play pivotal roles in the transcription of interferons and proinflammatory cytokines/chemokines, respectively (7–10). To circumvent this host defense mechanism, IAV blocks IRF3 activation by its multifunctional nonstructural protein (NS-1) to stop IFN production, whereas it exploits activated NF-κB to regulate its viral RNA synthesis and promote inflammation (11, 12). In addition to the IFN system, host cells express, often constitutively, a number of antiviral proteins dubbed restriction factors, which act to curb different viral infections via distinct mechanisms (13). Among these, members of the tripartite-motif-containing protein (TRIM) family have garnered increasing attention in recent years, with several reported to participate in host defense against influenza virus (14–17).
TRIMs are characterized by a highly conserved N-terminal RBCC (RING B-box coiled-coil) motif and are believed to possess E3 ligase activity conferred by the RING domain. In contrast, TRIM's C-terminal portions exhibit high variability in domain structures, based on which they are divided into up to 11 subfamilies (16, 17). Of these, subfamily C-IV members, exemplified by TRIM25 and TRIM22, harbor SPRY and/or PRY domains facilitating protein-protein interactions; TRIM-NHL proteins in subfamily C-VII, such as TRIM32 and TRIM71, possess NHL (NCL-1, HT2A, and LIN-41) repeats mediating binding to RNAs/proteins (18). Functionally, TRIMs have been implicated in a broad array of cellular processes that range from cell growth, differentiation, and oncogenesis to, more recently, immunity (15, 17–20). Of the immunity-regulating TRIMs reported to date, several play important roles in orchestrating PRR-mediated signaling cascades in response to viral invasions. A prime example is TRIM25, which promotes ubiquitination and activation of RIG-I to sense IAV infection and induce IFN expression (21). Acting in a different functional category, several TRIMs exhibit direct antiviral effects. For example, TRIM22 inhibits IAV by polyubiquitinating viral NP and targets it for proteasome-dependent degradation (22). Notably, both TRIM25 and TRIM22 depend on the RING-type E3 ligase activity to operate (15).
Recent evidence suggests that TRIM56 (also referred to as T56) is a novel player in innate antiviral immunity (23–25). This TRIM belongs to subfamily C-V because it lacks a well-characterized domain in the C-terminal half (14, 15). TRIM56 promotes TLR3- and STING-mediated antiviral signaling pathways toward IFN induction via E3 ligase-independent and -dependent mechanisms, respectively (23, 26). Additionally, TRIM56 exerts direct antiviral effects, which are dependent on its E3 ligase activity, on four positive-strand RNA viruses, including three members of the family Flaviviridae, i.e., bovine viral diarrhea virus (BVDV), yellow fever virus (YFV), and dengue virus serotype 2 (DENV2), and a coronavirus, human coronavirus OC43 (HCoV-OC43) (24, 25). However, little is known regarding TRIM56's antiviral spectrum against negative-strand RNA viruses, especially respiratory viruses, such as influenza viruses, given the fact that TRIM56 is most abundantly expressed in the lung (24).
In this study, we demonstrate that TRIM56 is an intrinsic host restriction factor of influenza viruses. Intriguingly, this attribute was found to be uncoupled from TRIM56's E3 ligase activity, and the entire N-terminal TRIM motif was dispensable. Instead, the C-terminal 63 residues were both necessary and sufficient to inhibit replication of both IAV and IBV. Moreover, we show that TRIM56 impedes influenza virus infection by hindering viral RNA synthesis via its C-terminal-tail portion.
MATERIALS AND METHODS
Plasmids.
The coding sequences of the C-terminal-tail 135 and 63 residues (CTT135 and CTT63) of TRIM56 were amplified via PCR from a plasmid encoding full-length human TRIM56 and subsequently ligated into the pcDNA5/FRT/TO (Invitrogen) vector to generate the pcDNA5/FRT/TO-HA-TRIM56-CTT135 or -CTT63 construct, respectively. In these constructs, TRIM56-CTT135 and -CTT63 were expressed as peptides fused in frame to an N-terminal 2-hemagglutinin (2×HA) epitope tag. The identities of all the plasmids were confirmed by DNA sequencing. Plasmids for the luciferase (Luc) reporter-based IAV RNA synthesis assay were gifts from Richard Webby (St. Jude Children's Research Hospital); they included polymerase II (Pol II)-driven pHW plasmids expressing PB2, PB1, PA, and NP of influenza A/PR/8/34 virus and a polymerase I (Pol I)-driven Luc reporter plasmid (pHY-Luci), which expresses in the negative sense the firefly luciferase flanked by noncoding sequences from the IAV matrix (M) segment (27).
Cell lines.
Human embryo kidney (HEK293), HeLa, and Madin-Darby canine kidney (MDCK) epithelial cells were maintained in Dulbecco's modified Eagle medium supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin, and 100 μg/ml streptomycin. Madin-Darby bovine kidney (MDBK) cells and their derivative cells were grown in medium containing horse serum in lieu of FBS. MDBK cell pools stably expressing C-terminally Flag-tagged wild-type (WT) or various mutant (Mut) human TRIM56 proteins (MDBK-T56 WT/Mut) or an empty vector (MDBK-Bsr) have been described previously (24). Human osteosarcoma U2OS cell pools stably expressing TRIM56-Flag (referred to as U2OS-T56) and control U2OS-Bsr cells expressing an empty retroviral vector (pCX4-Bsr) were developed by retroviral gene transfer, followed by stable selection with blasticidin.
HEK293 cell lines with tetracycline (Tet)-regulated expression of HA-tagged CTT135 or CTT63 of TRIM56 were developed using the Flp-In T-REx (FIT) expression system (Invitrogen) following the manufacturer's recommended protocol (25). In brief, HEK293-FIT cells were cotransfected with pOG44 encoding the Flp recombinase and pcDNA5/FRT/TO-HA-TRIM56-CTT135 or -CTT63 at a ratio of 9:1, followed by stable selection of cells in medium containing 200 μg/ml hygromycin. The resultant cell lines were designated 293-FIT-T56-CTT135 and -CTT63, respectively. To induce the expression of HA-tagged TRIM56-CTT135 or -CTT63, these cell lines were cultured in Tet-containing medium for 24 to 48 h. Other Tet-inducible 293-FIT-derived cells conditionally expressing HA-tagged WT TRIM56 and various TRIM56 mutants (HEK293-FIT-T56-WT or -Mut), as well as HeLa-FIT-hACE2-derived cells with Tet-regulated expression of HA-tagged TRIM56 (designated HeLa-FitA2-T56), have been described previously (23, 25).
Viruses, viral infections, and replication assays.
Viral stocks of influenza A/PR/8/34 (H1N1) virus (ATCC VR-1469) and B/Florida/4/06 (Yamagata lineage) virus (provided by Richard Webby) were propagated, and viral titers were determined in MDCK cells. A recombinant vesicular stomatitis virus (VSV) expressing firefly luciferase, VSV-Luc, was a gift from Sean Whelan (Harvard Medical School). VSV-Luc stocks were prepared in Vero cells. Stocks of Sendai virus (SeV) (strain Cantell) were purchased from Charles River Laboratories. Human metapneumovirus (hMPV) stocks were propagated and viral titers were determined in LLC-MK2 cells (28). Viral infections were conducted as described previously (29, 30). Endpoint dilution-based 50% tissue culture infective dose (TCID50) assays to determine progeny infectious-virus titers of IAV/IBV in cell-free culture supernatants were performed in 96-well plates on monolayers of MDCK cells (24, 30, 31). The cytopathic effect was recorded and used for calculation of infectivity at 24 h postinfection (hpi) for IAV and 48 hpi for IBV. All viral titers were expressed as TCID50/ml.
RNA interference.
MDBK cells with stable knockdown of endogenous bovine TRIM56 (boT56) have been described previously (24). We created HeLa cell pools with stable knockdown of human TRIM56 (HeLa-shT56) by lentiviral delivery of a short hairpin RNA (shRNA) targeting the coding region of human TRIM56 (designated shT56-#094) (25). To knock down exogenously expressed human TRIM56 in MDBK-T56 cells, shT56-#094 or a nontargeting scrambled shRNA (as a negative control) was packaged into replication-incompetent lentiviral particles in HEK293FT cells and subsequently used to transduce MDBK-T56 cells. Following selection in medium containing 2 to 5 μg/ml puromycin for 2 to 3 weeks, the surviving cell colonies were pooled for further analysis.
RNA analyses.
Extraction of total RNA by TRIzol, cDNA synthesis by reverse transcription (RT), and quantitative PCR (qPCR) were implemented as described previously (12, 24, 32, 33). To analyze the abundances of three different influenza virus RNA species, RT was performed using strand- and sense-specific oligonucleotides for vRNA, anti-genomic cRNA, and mRNA as previously described (12). A 28S-specific primer (5′ CTTAACGGTTTCACGCCCTC 3′) was also included in the RT reaction mixture for vRNA or cRNA analysis. Subsequently, the following primers were used for qPCR: 28S (23), M segment of influenza A/PR/8/34 virus, 5′ GACCRATCCTGTCACCTCTGAC 3′ (forward) and 5′ AGGGCATTYTGGACAAAKCGTCTA 3′ (reverse), and HA segment of influenza A/PR/8/34 virus, 5′ GGCCCAACCACAACACAAAC 3′ (forward) and 5′ AGCCCTCCTTCTCCGTCAGC 3′ (reverse). The qPCR primers for measuring the mRNA abundance of the SeV P gene were 5′-CTCTGGGAGAACAAGCAAGC-3′ (forward) and 5′-TCGCCCAGATCCTGAGATAC-3′ (reverse). The relative abundance of each target was normalized to that of 28S rRNA.
Luciferase reporter-based minigenome replication assay of IAV RNA synthesis.
A luciferase reporter-based minigenome replication assay for monitoring IAV transcription and replication was performed as previously described (27, 33, 34). In brief, HEK293-FIT-T56-WT or -Mut cells cultured in the presence or absence of Tet (to induce or repress HA-T56 WT/Mut expression, respectively) were cotransfected using Lipofectamine 3000 (Invitrogen) with polymerase II-driven pHW plasmids expressing RNP complex genes (PB2, PB1, PA, and NP) of influenza A/PR/8/34 virus and a polymerase I-driven luciferase reporter plasmid, pHY-Luci (27). Also cotransfected was a cytomegalovirus (CMV) promoter-driven plasmid expressing Renilla luciferase (pRL-CMV; Promega), which served as an internal control for normalization of transfection efficiency. At the indicated time points posttransfection, cells were lysed and processed for dual-luciferase assay.
Immunoblotting, immunofluorescence staining, and confocal microscopy.
Cell lysates were prepared in RIPA buffer and subjected to SDS-PAGE and immunoblot analysis as previously described (24, 35). Immunofluorescence staining and confocal microscopy were performed as previously described (24). The following monoclonal (MAb) and polyclonal (PAb) antibodies were utilized: mouse anti-influenza A/WSN/33 (H1N1) virus NP 5/1 MAb (confirmed to react with the NP of A/PR/8/34 virus) and goat anti-M protein antiserum (gifts from Richard Webby); goat anti-influenza B/Hong Kong/8/73 virus HA PAb (BEI Resources; NR-3165), which also reacts with the HA of B/Florida/4/06 virus; rabbit anti-SeV PAb (a gift from Ilkka Julkumen, National Institute for Health and Welfare, Helsinki, Finland); rabbit anti-hMPV PAb (28); rabbit anti-TRIM56 PAb (24, 25); mouse anti-HA tag MAb (Invivogen), which we have confirmed not to react with the HA of either A/PR/8/34 or B/Florida/4/06 virus; mouse anti-HA tag MAb (clone 12CA5; Roche); mouse anti-actin MAb (Sigma); rabbit anti-β-tubulin PAb (Santa Cruz); peroxidase-conjugated secondary goat anti-rabbit, goat anti-mouse, and rabbit anti-goat PAbs; fluorescein isothiocyanate (FITC)-conjugated secondary goat anti-mouse PAb (Southern Biotech); and Alexa Fluor 594-conjugated secondary donkey anti-mouse and chicken anti-goat PAbs (Invitrogen).
Statistical analysis.
SPSS 11.5 software was employed to perform Student's t test for analysis of statistical differences where appropriate. All P values were two tailed, and a P value of <0.05 was considered to be statistically significant.
RESULTS
Ectopic expression of TRIM56 inhibits propagation of IAV and IBV, but not that of SeV or hMPV.
We had previously shown that TRIM56 is a restriction factor of four positive-strand RNA viruses, including three members of the family Flaviviridae (BVDV, YFV, and DENV2) and a member of the family Coronaviridae (HCoV-OC43) (24, 25). However, whether TRIM56 participates in host defense against negative-strand RNA viruses is largely unknown. VSV (a rhabdovirus) is the only negative-strand RNA virus that has been examined to date; the propagation of the virus was not affected by ectopic expression of TRIM56 (24). In this study, we set out to determine if manipulation of TRIM56 abundance alters the propagation of influenza viruses, which are medically important viruses classified within the family Orthomyxoviridae.
We first utilized HEK293-FIT-derived cells with Tet-inducible expression of N-terminal HA-tagged TRIM56 (25) to examine the impact of TRIM56 overexpression on IAV. As shown in Fig. 1A (left), negligible expression of HA-TRIM56 protein was observed in these cells when cultured in the absence of Tet (−Tet), but considerable HA-TRIM56 protein was detected following Tet addition to the culture medium (+Tet) (Fig. 1A, compare lanes 1 and 2). When infected with influenza A/PR/8/34 virus at a multiplicity of infection (MOI) of 0.1, there was abundant viral NP antigen in −Tet cells at 9 hpi (lane 3). However, NP expression was diminished in cells induced for HA-TRIM56 expression (+Tet, lane 4). Consistent with this, the progeny virus titers in supernatants of 293-FIT-T56 +Tet cells were significantly decreased compared with −Tet cells (P < 0.001) (Fig. 1A, right). This antiviral effect of TRIM56 persisted at later time points (i.e., 16, 24, and 48 hpi) (Fig. 1B). Additionally, MDBK-T56 cells stably expressing Flag-tagged human TRIM56 were substantially less susceptible to influenza A/PR/8/34 virus than MDBK-Bsr cells that express a control retroviral vector, as evidenced by substantially reduced intracellular NP antigen expression (Fig. 1C, bottom right, immunoblots), ∼1 log unit fewer (P < 0.01) progeny virions yielded in culture supernatants (Fig. 1C, upper right panel), and a significantly lower percentage of NP-positive cells (Fig. 1C, left). Furthermore, TRIM56's antiviral effect against IAV was also observed in U2OS cells stably expressing TRIM56-Flag (U2OS-T56) compared with control cells (U2OS-Bsr) expressing a control vector (data not shown). To further corroborate this finding, we knocked down the ectopically expressed human TRIM56 in MDBK-T56 cells by lentiviral delivery of a TRIM56 shRNA (shT56). As shown in Fig. 1D (bottom immunoblot), TRIM56-Flag expression was reduced by 80% in the knockdown cells (MDBK-T56 shT56) compared with the control cells (MDBK-T56 ctrl) transduced with a scrambled, negative-control shRNA, concomitant with a 6-fold increase (P < 0.05) in progeny virus production (MOI = 0.5; 24 hpi) (Fig. 1D, top). Altogether, we conclude that ectopic expression of TRIM56 impedes IAV propagation. Importantly, this is a reproducible phenomenon, regardless of the expression system or cell type.
FIG 1.

Ectopic expression of TRIM56 hinders propagation of IAV and IBV, but not infection by hMPV or Sendai virus. (A) (Left) HEK293-Flp-In T-REx-TRIM56 (293-FIT-T56) cells were cultured without (lanes 1 and 3, −tet) or with (lanes 2 and 4, +tet) tetracycline (2 μg/ml) for 48 h, followed by mock infection (lanes 1 and 2) or infection with influenza A/PR/8/34 (H1N1) virus for 9 h (MOI = 0.1; lanes 3 and 4). +Tet cells (lanes 2 and 4) were kept in Tet-containing media until they were lysed for Western blotting. Expression of N-terminally HA-tagged TRIM56 was detected using an anti-HA tag antibody (Invivogen) that does not cross-react with viral HA of H1N1 influenza A viruses, and expression of IAV NP was detected with an anti-influenza A/WSN/33 (H1N1) virus NP MAb. Actin served as a loading control to demonstrate equal sample loading. (Right) Progeny virus production in culture supernatants of 293-FIT-T56 cells with or without Tet treatment at 9 hpi with influenza A/PR/8/34 virus (MOI = 0.1). (B) Progeny virus production in culture supernatants of 293-FIT-T56 cells cultured with or without Tet at 0, 8, 16, 24, and 48 hpi with influenza A/PR/8/34 virus (MOI = 0.1). (C) (Left) Immunofluorescence staining of NP (using anti-influenza A/WSN/33 [H1N1] virus NP MAb) in MDBK cells stably expressing TRIM56-Flag (T56) or a control retroviral vector (Bsr) at 24 hpi with A/PR/8/34 virus (MOI = 5). DAPI, 4′,6-diamidino-2-phenylindole. (Right) Progeny virus titers in culture supernatants (top) and NP and actin expression (bottom) in lysates of infected cells under the same conditions used for immunofluorescence staining. (D) (Bottom) Immunoblot analysis of ectopically expressed human TRIM56 (using anti-TRIM56 antibody) in MDBK-T56 cells transduced with lentiviruses encoding either an shRNA specifically targeting human TRIM56 (MDBK-T56 shT56) or a nontargeting, scrambled control shRNA (MDBK-T56 ctrl). (Top) Progeny virus titers in culture supernatants of the indicated cells infected with influenza A/PR/8/34 virus for 24 h (MOI = 0.5). (E) (Top) Progeny virus production in culture supernatants of 293-FIT-T56 cells with or without Tet treatment at 24 hpi with influenza B/Florida/4/06 (Yamagata) virus (MOI = 0.1). (Bottom) Immunoblot analysis of HA-tagged TRIM56 (using an anti-HA tag MAb [Invivogen] not cross-reacting with IBV HA) and viral HA (using an anti-influenza B/Hong Kong/8/73 virus HA PAb). Tubulin served as a loading control to demonstrate equal sample loading. (F) HeLa-FitA2-T56 cells with Tet-regulated expression of HA-tagged TRIM56 were cultured with (lanes 3 and 4) or without (lanes 1 and 2) Tet, followed by mock infection (lanes 1 and 3) or infection with hMPV (MOI = 1; lanes 2 and 4) for 15 h. The cell lysates were subjected to immunoblot analysis of HA-tagged TRIM56 (using anti-HA tag MAb [clone 12CA5; Roche]) and viral proteins (using anti-hMPV PAb). The asterisk denotes a nonspecific band. (G) 293-FIT-T56 cells cultured in the absence (−tet) or presence (+tet) of tetracycline were infected with SeV at the indicated doses (1.6, 16, and 160 HA units (HAU)/ml) for 8 h, followed by immunoblot analysis of HA-tagged TRIM56 (using anti-HA MAb [Invivogen]) and viral proteins (using anti-SeV PAb, which mainly reacted with SeV hemagglutinin-neuraminidase and nucleocapsid). Actin served as a loading control. The asterisks indicate that statistical differences exist between the indicated cells; *, P < 0.05; **, P < 0.01; ***, P < 0.001. The error bars represent standard deviations.
We next investigated whether TRIM56's antiviral effect could be extended to IBV. It was found that induction of TRIM56 expression in 293-FIT-T56 cells led to a sharp decline in viral HA antigen abundance compared with −Tet (uninduced) cells at 24 hpi with influenza B/Florida/4/06 virus (MOI = 0.1) (Fig. 1E, bottom). Concordant with this, progeny virus production in +Tet cells was 7-fold (P < 0.05) lower than in −Tet cells (Fig. 1E, top). A similar anti-IBV effect of TRIM56 overexpression was also observed at 8 and 12 hpi (MOI = 0.01) in HeLa-FitA2-T56 cells with Tet-inducible expression of HA-TRIM56 (data not shown), again confirming that the phenomenon was not cell type specific. Collectively, it was established that TRIM56, when ectopically expressed, curbs IBV infection. Of note, influenza viruses (IAV and IBV) represent the first negative-strand RNA virus family TRIM56 was found to inhibit.
To determine whether TRIM56 also restricts other negative-strand RNA viruses, we studied the effects of TRIM56 overexpression on hMPV (Fig. 1F) and SeV (Fig. 1G), paramyxoviruses that cause respiratory infections in humans and rodents, respectively. There was no inhibitory effect on viral protein expression following infection by either virus when cells induced for HA-TRIM56 expression and uninduced cells were compared. The lack of an antiviral effect on these paramyxoviruses suggests that TRIM56 does not target negative-strand RNA viruses indiscriminately. Consistent with this concept, we previously demonstrated that TRIM56 overexpression does not inhibit VSV propagation (24).
Knockdown of TRIM56 augments multiplication of influenza viruses.
To determine whether TRIM56 expressed at physiological levels poses a barrier to infection by influenza viruses, we dissected the effect of TRIM56 knockdown on influenza virus propagation. Compared to control MDBK cells, MDBK-T56i cells with stable knockdown of endogenous TRIM56 (24) (Fig. 2A, left) supported substantially higher levels of viral NP expression and produced >1 log unit more (P < 0.01) progeny influenza A/PR/8/34 virions (Fig. 2A, middle). In contrast, MDBK-T56i cells were not more permissive than control MDBK cells to infection by VSV-Luc, a recombinant VSV expressing luciferase as a readout of viral replication (Fig. 2A, right). These data lend further support to the notion that endogenous TRIM56 specifically hinders influenza virus infection. The same can be said regarding the impact of TRIM56 depletion on IAV infection in HeLa cells (Fig. 2B). We found that HeLa cells with stable knockdown of TRIM56 (shT56) yielded ∼6-fold (P < 0.01) more progeny IAV than control cells. HeLa-shT56 cells also supported higher levels of viral HA protein expression (Fig. 2C, immunoblots) and ∼1 log unit more (P < 0.01) progeny virus production (Fig. 2C, top) than did control cells following IBV infection. Altogether, these data demonstrate that TRIM56 expressed at physiological levels puts a check on infection by influenza A and B viruses.
FIG 2.

TRIM56 knockdown augments influenza virus multiplication. (A) (Left) qPCR analysis of boT56 mRNA expression in control MDBK cells (Ctrl) and their derived cells (T56i) stably transfected with a boT56 shRNA. (Middle, top) Progeny virus production in culture supernatants of MDBK-Ctrl and MDBK-T56i cells at 24 hpi with influenza A/PR/8/34 virus (MOI = 5). (Middle, bottom) Immunoblot analysis of NP and actin expression in cell lysates under the same conditions. (Right) MDBK-Ctrl cells and MDBK-T56i cells were infected with VSV-Luc at an MOI of 0.2. At the indicated times, cells were lysed for firefly luciferase assay. (B) (Bottom) Immunoblot analysis of human TRIM56 expression (using anti-TRIM56 antibody) in HeLa-plkO.1 (Ctrl) cells stably transduced with a nontargeting, scrambled control shRNA or HeLa-shT56 (shT56) cells transduced with an shRNA specifically targeting TRIM56. (Top) Progeny virus production in culture supernatants of the two cell lines at 8 hpi with influenza A/PR/8/34 virus (MOI = 0.01). (C) (Bottom) Immunoblot analysis of TRIM56 expression (using anti-TRIM56 antibody) and viral HA levels (using anti-influenza B/Hong Kong/8/73 virus HA PAb) in HeLa-plkO.1 (Ctrl) or HeLa-shT56 (shT56) cells at 8 hpi with influenza B/Florida/4/06 virus (MOI = 0.01). (Top) Progeny virus production in culture supernatants of the cells under the same conditions. The asterisks indicate that statistical differences exist between the indicated cells; **, P < 0.01. The error bars represent standard deviations.
TRIM56's C-terminal tail, but not its E3 ligase activity or other portions of the protein, is required for the ability to impede influenza virus infection.
TRIM56 exerts antiviral actions against positive-strand RNA viruses of distinct families via overlapping and distinct molecular determinants (24, 25). Specifically, while the RING and the E3 ligase activity conferred by this domain are required to inhibit flaviviruses and HCoV-OC43, the C-terminal portion (residues 621 to 755) is critical for restriction of flaviviruses but dispensable for HCoV-OC43 suppression (24, 25). We mapped the TRIM56 molecular determinants essential for the restriction of influenza viruses, using HEK293-FIT-derived cell lines with Tet-inducible expression of a series of HA-TRIM56 mutants. The mutants included for analysis were the E3 ligase-deficient CC21/24AA mutant; mutants lacking the RING (ΔRING), B-box (ΔB-box), or coiled-coil (Δcoiled-coil) domain; and mutants possessing different deletions in the C-terminal portion, i.e., Δ355-433, Δ621-695, and Δ693-750 (Fig. 3A, left). The expression levels of these mutants have been confirmed to be comparable to that of wild-type TRIM56 (25). When challenged with the A/PR/8/34 virus (MOI = 0.1), an inhibitory effect on NP accumulation following Tet induction (+Tet) was observed at 9 or 27 hpi in 293-FIT-T56 cells and in all of the mutant TRIM56-inducible cells except the line that expresses the Δ693-750 mutant (Fig. 3B). In agreement with the NP immunoblot data, progeny virus production was curtailed by ∼1 log unit by induction of WT TRIM56 (P < 0.001) or any of the TRIM56 mutants (P < 0.01), with the exception of the Δ693-750 mutant (Fig. 3A, right). Thus, the very C-terminal tail portion of TRIM56 contains the sole determinant crucial for the anti-IAV activity. In stark contrast with what is required for flavivirus and coronavirus restriction, the E3 ligase activity of TRIM56 is dispensable for containing IAV.
FIG 3.

The restriction of IAV by TRIM56 relies on its C-terminal-tail portion, but not on its E3 ligase activity or other parts of the protein. (A) (Left) Schematic representation of TRIM56 protein domains and the individual TRIM56 mutants investigated in this study. (Right) Progeny virus production in culture supernatants of 293-FIT-T56-WT or individual TRIM56 mutant cells cultured in the absence (−tet) or presence (+tet) of Tet at 9 hpi with influenza A/PR/8/34 virus (MOI = 0.1). (B) Immunoblot analysis of HA-tagged WT or Mut TRIM56 (using an anti-HA tag antibody [Ab] [Invivogen] that does not react with viral HA of H1N1 IAV) and NP protein abundance (using anti-influenza A/WSN/33 [H1N1] virus NP MAb) in 293-FIT-T56 WT/Mut cells in the absence (odd-numbered lanes) or presence (even-numbered lanes) of Tet and mock infected or infected with influenza A/PR/8/34 virus (MOI = 0.1) for 9 or 27 h. Data from each cell line are presented in an immunoblot block (8 blocks in total, representing 8 different WT/Mut TRIM56-inducible cell lines, as shown in panel A). (C) (Bottom) Immunoblot analysis of TRIM56 (using anti-human TRIM56 antibody) and NP protein abundance (using anti-influenza A/WSN/33 [H1N1] virus NP MAb) in parental MDBK cells, as well as MDBK-derived cells stably expressing a control vector (Bsr), C-terminal Flag-tagged human TRIM56 (WT), or its mutant versions infected with influenza A/PR/8/34 virus for 13 h (MOI = 3). (Top) Progeny viral titers in culture supernatants of the indicated MDBK-derived cell lines infected with influenza A/PR/8/34 virus (MOI = 3) for 13 h. The asterisks indicate that statistical differences exist between MDBK and WT/Mut T56-overexpressing cells or between −Tet and +Tet cells; **, P < 0.01; ***, P < 0.001. The error bars represent standard deviations.
To confirm these findings, we gauged NP expression and progeny virus production in MDBK cells stably expressing Flag-tagged WT or mutant human TRIM56 following infection by A/PR/8/34 virus. As shown in Fig. 3C (immuoblots), at 13 hpi (MOI = 3), abundant NP antigen was detected in MDBK-Bsr cells expressing a control vector (lane 1). However, there was a profound reduction in NP expression in cells expressing WT TRIM56 (lane 3), the E3 ligase-null CC21/24AA mutant (lane 4), and the ΔRING (lane 5) and Δcoiled-coil (lane 6) mutants. In contrast, NP expression was not reduced but increased (lane 7) compared with parental MDBK cells (lane 2). Progeny virus titer data (Fig. 3C, bar graph) were in accordance with the NP immunoblot results. Collectively, the data gleaned from MDBK-derived cells mirrored those obtained from HEK293-FIT-derived cells.
Consistently similar results were obtained when we determined progeny IBV production in HEK293-FIT-derived cells conditionally expressing WT TRIM56 or individual TRIM56 mutants (Fig. 4). The ∼1-log-unit reduction in progeny virus yields (P values ranging from <0.05 to <0.001) in +Tet cells compared with −Tet cells at 24 hpi with the B/Florida/4/06 virus (MOI = 0.1) was abrogated only in cells harboring the Δ693-750 mutant. Altogether, the domain-mapping experiments in HEK293-FIT- and MDBK-derived cells reached the same conclusion that the very C-terminal tail region contains the sole prerequisite for TRIM56's antiviral activity against IAV and IBV. The shared TRIM56 determinant in restricting both genera of influenza viruses is consistent with the similar replication strategies of these orthomyxoviruses and suggests that TRIM56 adopts the same antiviral mechanism against IAV and IBV.
FIG 4.
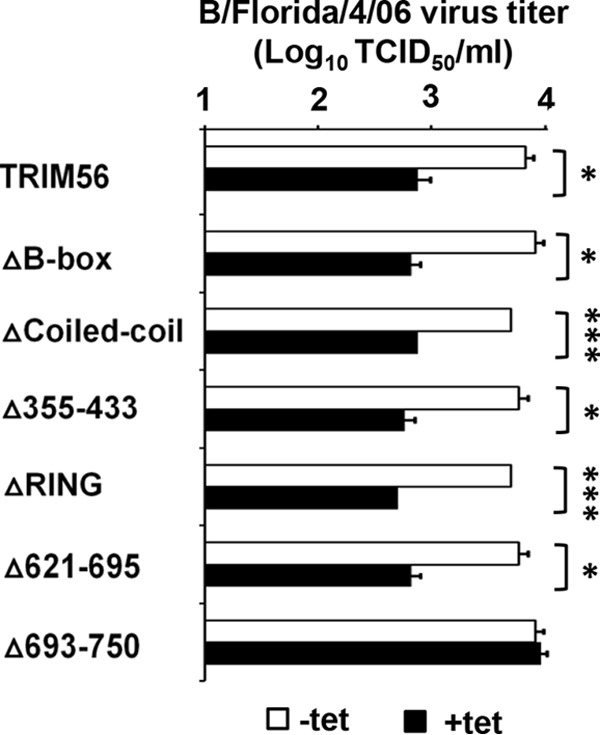
The TRIM56 C-terminal tail is the only prerequisite for its anti-IBV function. Shown is progeny virus production in culture supernatants of 293-FIT-derived T56 WT or individual mutant cells in the absence (−tet) or presence (+tet) of Tet at 24 hpi with influenza B/Florida/4/06 virus (MOI = 0.1). The asterisks indicate that statistical differences exist between −Tet and +Tet cells; *, P < 0.05; ***, P < 0.001. The error bars represent standard deviations.
Expression of the C-terminal tail of TRIM56 alone is sufficient to restrict influenza virus infection.
Since our data showed that residues 693 to 750 in the C-terminal-tail portion of TRIM56 are essential for the anti-influenza virus function, we asked if peptides derived from the TRIM56 C-terminal region are sufficient to confer influenza virus restriction. To this end, we established HEK293-FIT-derived cell lines with Tet-inducible expression of an HA-tagged TRIM56 fragment comprising the C-terminal 135 (i.e., amino acids [aa] 621 to 755, referred to as CTT135) and 63 (i.e., aa 693 to 755, referred to as CTT63) residues (Fig. 5A and B). When infected with A/PR/8/34 virus (MOI = 0.1), induction of CTT135 expression led to a substantial decline in NP expression and progeny virus production, which mirrored the effect of induction of WT TRIM56 expression (Fig. 5C). Induction of CTT63 produced a similar effect on NP expression (Fig. 5D), suggesting the 63 residues at the very C-terminal end of TRIM56 contain the minimal determinant for the antiviral function of the full-length protein. The same could be said when we infected cells with B/Florida/4/06 virus (Fig. 5E). The shared TRIM56 determinant governing the inhibitory effect on IAV and IBV implies that TRIM56 likely restricts both genera of influenza viruses via similar mechanisms. Thus, in subsequent experiments, we utilized IAV to probe how TRIM56 executes its anti-influenza virus actions.
FIG 5.

Expression of the TRIM56 C-terminal tail alone is sufficient to curb infection by IAV or IBV. (A) Schematic representation of putative NHL-like repeats in TRIM56's C-terminal portion and two peptides derived from the TRIM56 C-terminal tail, i.e., CTT135 and CTT63. (B) Immunoblot of HA-tagged WT TRIM56, CTT135, and CTT63 (using anti-HA tag MAb [Invivogen]) and actin in HEK293-Flp-In T-REx-derived cells with Tet-inducible expression of HA-tagged WT TRIM56 (293-FIT-T56-WT), CTT135 (293-FIT-T56-CTT135), or CTT63 (293-FIT-T56-CTT63) in the absence (−tet) or presence (+tet) of Tet. (C) (Top) Progeny virus titers in supernatants of 293-FIT-T56-WT or -CTT135 cells cultured with (+tet) or without (−tet) Tet at 8 hpi with influenza A/PR/8/34 virus (MOI = 0.1). (Bottom) Immunoblot analysis of NP (using anti-influenza A/WSN/33 [H1N1] virus NP MAb) and actin expression in the corresponding cell lysates. (D) Immunoblot analysis of NP (using anti-influenza A/WSN/33 [H1N1] virus NP MAb) and actin expression in 293-FIT-T56-WT (left) or -CTT63 cells (right) cultured with (+tet) or without (−tet) Tet at 8 hpi with influenza A/PR/8/34 virus (MOI = 0.1). (E) (Top) Progeny virus titers in supernatants of 293-FIT-T56-WT or -CTT63 cells cultured with (+tet) or without (−tet) Tet at 24 hpi with influenza B/Florida/4/06 virus (MOI = 0.1). (Bottom) Immunoblot analysis of viral HA (using anti-influenza B/Hong Kong/8/73 virus HA PAb) and actin expression in the corresponding cell lysates. The asterisks indicate that statistical differences exist between −Tet and +Tet cells; *, P < 0.05. A representative of three independent experiments is shown. The error bars represent standard deviations.
TRIM56 curtails influenza virus propagation by targeting virus RNA synthesis, but not the release step.
Following virus entry and uncoating steps, incoming influenza vRNPs are first transported into the nucleus, where viral RNA replication occurs (4–6). Directed by a trimeric viral RNA polymerase complex in association with NP, vRNAs serve as the templates for synthesis of mRNAs and cRNAs, with the latter becoming the templates for synthesis of progeny vRNAs. The viral mRNAs are then transported to the cytoplasm for translation into nascent viral proteins that are required for cRNA and vRNA synthesis (e.g., NP) and that perform structural and regulatory functions. At a late stage of the infection, vRNAs are exported out of the nucleus for virion packaging and budding (4, 12). To determine at which step(s) of the influenza virus life cycle TRIM56 acts, we performed single-cycle growth analysis of IAV infection. MDBK-T56 cells with stable expression of human TRIM56 and MDBK-Bsr cells (expressing the empty vector) were infected with influenza A/PR/8/34 virus at an MOI of 3, followed by a stringent wash in 0.9% NaCl (pH 2.2) to remove extracellular virions that had not entered the cells. At 1, 4, 8, and 12 hpi, the cells were washed extensively with phosphate-buffered saline (PBS) to eliminate extracellular viruses and viral RNAs before they were lysed for total RNA extraction and qPCR analysis of intracellular levels of vRNA, cRNA, and mRNA of the M segment. At the same time, yields of progeny viruses in cell-free culture supernatants at 8 and 12 hpi were determined by TCID50 assay.
At 1 hpi, intracellular vRNA levels of A/PR/8/34 virus in MDBK-T56 and MDBK-Bsr cells were indistinguishable (Fig. 6A). In line with previous reports (12, 33, 36, 37), we found that at this early time point IAV had already initiated mRNA transcription and cRNA synthesis in MDBK-Bsr cells. However, the abundances of cRNA (Fig. 6B) and mRNA (Fig. 6C) were 3-fold and 7-fold lower, respectively, in MDBK-T56 cells, suggesting that viral mRNA transcription and cRNA synthesis were hindered in TRIM56-overexpressing cells. At 4 hpi, when viral RNA synthesis occurs at a high rate (12, 36, 37), the abundance of vRNA rose by 5-fold compared to that at 1 hpi (Fig. 6A), while cRNA (Fig. 6B) and mRNA (Fig. 6C) both climbed by >3 log units in MDBK-Bsr cells. In contrast, vRNA levels did not increase in MDBK-T56 cells at 4 hpi, suggesting the onset of vRNA synthesis was delayed (Fig. 6A). Although cRNA and mRNA levels increased compared to 1 hpi, they were 61-fold and 88-fold lower, respectively, than those in MDBK-Bsr cells (Fig. 6B and C). At 8 hpi, when virus RNA synthesis had already finished (12, 37), we found that levels of all three RNA species reached plateaus in MDBK-Bsr cells, as well in MDBK-T56 cells (compared to 12 hpi). Nonetheless, vRNA, cRNA, and mRNA levels remained 4- to 7-fold lower in MDBK-T56 cells at both 8 and 12 hpi (Fig. 6A to C). These data show that there is a sustained influence on influenza virus vRNA, cRNA, and mRNA levels throughout the life cycle in TRIM56-overexpressing cells. However, it is unknown whether the influence on synthesis of viral RNAs occurs throughout this time frame or only in the early stages of infection.
FIG 6.
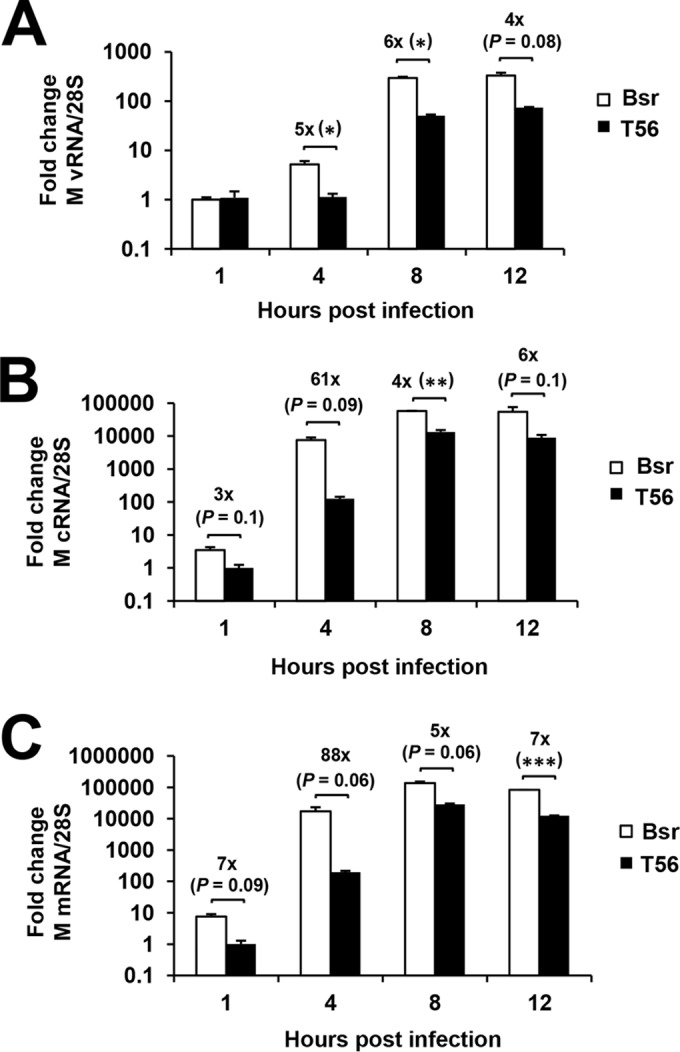
TRIM56 hinders IAV propagation by inhibiting intracellular virus RNA synthesis. Shown is qPCR analysis of vRNA (A), cRNA (B), and mRNA (C) levels of the M gene segment of influenza A/PR/8/34 virus in MDBK cells stably expressing a control vector (Bsr) or TRIM56 (T56) at 1, 4, 8, and 12 hpi (MOI = 3). The asterisks indicate that statistical differences exist between Bsr and T56 cells; *, P < 0.05; **, P < 0.01; ***, P < 0.001. A representative of three independent experiments is shown. The error bars represent standard deviations.
In the same experiments (at 8 and 12 hpi), we found that progeny virus production in MDBK-T56 cells was significantly reduced compared with MDBK-Bsr cells, and the extent of reduction was proportional to the decrease in vRNA synthesis (data not shown), indicating that the effect on viral RNA synthesis is primarily responsible for TRIM56's anti-influenza virus action. Confirming that this observation is not cell type specific, we obtained similar results when comparing the time courses of IAV RNA synthesis and progeny virus production in U2OS-T56 and U2OS-Bsr cells (data not shown).
The C-terminal tail of TRIM56 is both required and sufficient for the suppression of influenza virus RNA synthesis.
To corroborate that TRIM56 impedes influenza virus RNA synthesis, we performed a luciferase reporter-based minigenome replication assay to measure IAV transcription and replication in 293-FIT-T56 cells with (+Tet) and without (−Tet) induction of HA-tagged TRIM56. This system involves cotransfection of a total of six plasmids. Influenza virus RNA polymerase subunits (PB2, PB1, and PA) and NP derived from A/PR/8/34 virus are expressed from individual plasmids under the control of a Pol II promoter. In a fifth plasmid, the Pol I promoter controls transcription of the antisense strand of firefly luciferase RNA flanked by the untranslated regions of M segment vRNA (27). A sixth plasmid, pCMV-RL, expresses Renilla luciferase from the CMV promoter, serving as an internal control for normalization of transfection efficiency. Since this assay was designed to mimic IAV transcription and replication steps, the firefly/Renilla Luc ratio (i.e., relative luciferase activity) is proportional to the level of influenza virus RNA synthesis (27, 33, 34). We found that at both 24 and 48 h posttransfection (hpt), relative luciferase activity was consistently 2-fold higher (P < 0.01) in −Tet cells than in +Tet cells (Fig. 7A), corroborating the qPCR data on vRNA levels (Fig. 6) and lending further support for the notion that TRIM56 inhibits influenza virus RNA synthesis. Additional minigenome replication assays in Δ693-750 mutant TRIM56-inducible cells and its parental HEK293-FIT (Ctrl) cells (Fig. 7B, 48 hpt) showed that this C-terminal-tail deletion mutant loses the ability to suppress influenza virus RNA synthesis and ruled out any effect due to tetracycline treatment. Further, expression of CTT63 alone was sufficient to inhibit influenza virus RNA synthesis (Fig. 7C), demonstrating that the C-terminal tail of TRIM56 dictates the antiviral restriction of influenza viruses by hindering the viral RNA synthesis step.
FIG 7.
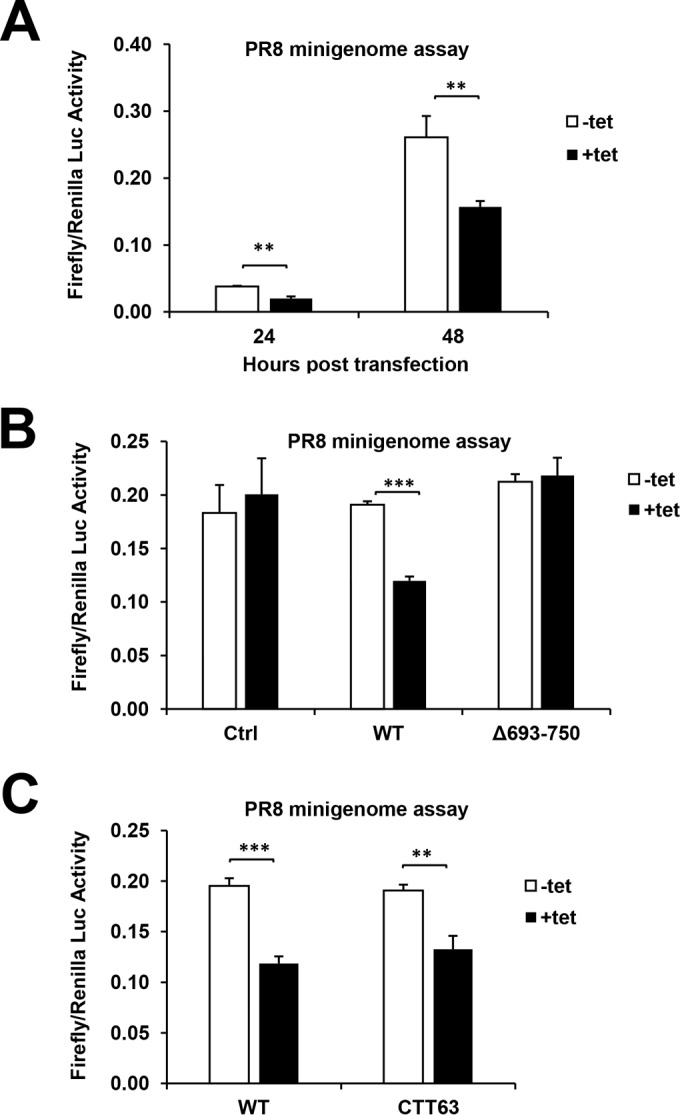
The C-terminal-tail portion is required for TRIM56 suppression of influenza virus RNA synthesis. (A) Luc reporter-based minigenome replication assay of IAV RNA synthesis was performed in 293-FIT-T56 cells cultured in the presence (+tet) or absence (−tet) of Tet at 24 and 48 hpt with polymerase II-driven pHW plasmids expressing PB2, PB1, PA, and NP; a polymerase I-driven Luc reporter plasmid, pHY-Luci; and a CMV promoter-driven plasmid expressing Renilla Luc, pRL-CMV (serving as an internal control for normalization of transfection efficiency). (B) Luciferase reporter-based minigenome replication assay of IAV RNA synthesis in parental 293-FIT (Ctrl), 293-FIT-T56 (WT), or 293-FIT-T56-Δ693-750 (Δ693-750) cells cultured with (+tet) or without (−tet) Tet at 48 hpt. (C) Luciferase reporter-based minigenome replication assay of IAV RNA synthesis in 293-FIT-T56 (WT) or 293-FIT-T56-CTT63 (CTT63) cells cultured with (+tet) or without (−tet) Tet at 48 hpt. The asterisks indicate that statistical differences exist between −Tet and +Tet cells; **, P < 0.01; ***, P < 0.001. A representative of three independent experiments is shown. The error bars represent standard deviations.
TRIM56 moves into the nucleus during influenza virus infection.
Because TRIM56 is a cytosolic protein in resting cells (24), we wondered how this protein impacts influenza virus RNA synthesis, which takes place in the nucleus. To address this question, we determined the subcellular localization of green fluorescent protein (GFP)-TRIM56 in transiently transfected HeLa cells prior to and after A/PR/8/34 virus infection. As shown by the representative confocal fluorescence microscopy images (Fig. 8A), while GFP-TRIM56 resided exclusively in the cytoplasm of mock-infected cells, GFP-TRIM56 fluorescence was detected in both the cytoplasm and nucleus in IAV-infected cells at different time points postinfection. Interestingly, the subcellular distribution pattern of HA-tagged TRIM56-CTT63 mirrored that of full-length HA-tagged TRIM56 (Fig. 8B). While there was no appreciable nuclear immunostaining of CTT63 in uninfected cells, a fraction of CTT63 was reproducibly found in the nucleus after IAV infection. The presence of TRIM56 and TRIM56-CTT63 in the nuclei of IAV-infected cells is consistent with their ability to impede influenza virus RNA synthesis.
FIG 8.

TRIM56 and TRIM56-CTT63 move into the nucleus after IAV infection. (A) HeLa cells transiently transfected with an N-terminally GFP-tagged TRIM56 (GFP-T56) plasmid were mock infected or infected with influenza A/PR/8/34 virus for 4, 8, and 16 h (MOI = 3), followed by immunostaining of NP (using anti-influenza A/WSN/33 [H1N1] virus NP MAb). The subcellular localizations of GFP-T56 (green fluorescence) and NP (red fluorescence) were examined by confocal microscopy. Nuclei were counterstained blue with DAPI. The scale bar represents 10 μm. (B) 293-FIT-T56 or 293-FIT-T56-CTT63 cells were cultured in the presence of Tet to induce expression of HA-tagged, TRIM56 (WT) or TRIM56-CTT63, followed by mock infection or infection with influenza A/PR/8/34 virus for 16 h (MOI = 3). HA-tagged TRIM56 and TRIM56-CTT63 were immunostained with an anti-HA tag MAb (Invivogen) and IAV M proteins with a goat anti-matrix antiserum. The subcellular localization of HA-tagged WT TRIM56 and TRIM56-CTT63 (green fluorescence) and M (red fluorescence) proteins was examined by confocal microscopy. Nuclei were counterstained blue with DAPI. Scale bars, 5 μm.
DISCUSSION
In this study, we have shown that TRIM56 is an intrinsic host restriction factor of influenza A and B viruses, expanding the antiviral spectrum of this TRIM to include the Orthomyxoviridae family of segmented, negative-strand RNA viruses. Ectopic expression of TRIM56 impeded the propagation of these medically important respiratory pathogens, while silencing of endogenous TRIM56 expression had the opposite effect. Importantly, these findings were reproduced in multiple cell types, irrespective of the expression system. Moreover, the antiviral effect was specific for influenza viruses, as TRIM56 did not inhibit SeV or hMPV, paramyxoviruses that cause respiratory infections in mice and humans, respectively. To our knowledge, this is the first study that demonstrates that TRIM56 targets negative-strand RNA viruses for inhibition.
TRIM56 positively regulates innate antiviral signaling through TLR3- and STING-dependent pathways. Specifically, TRIM56 forms a complex with TRIF, the TLR3 adaptor, via its C-terminal region, promoting IRF3 activation and subsequent IFN response in ways independent of its E3 ligase activity (23). In contrast, the mechanism underlying the augmentation of the STING-dependent IFN induction pathway is distinct. In this case, TRIM56 associates with STING and acts as an E3 ligase that promotes Lys63-linked ubiquitination of STING, thereby enhancing cytosolic DNA-induced IFN production (26). In previous studies (24, 25), we have shown that a heightened IFN response is not responsible for TRIM56-mediated inhibition of four positive-strand RNA viruses, BVDV, YFV, DENV2, and HCoV-OC43. This may also be the case for the novel anti-influenza virus activity of TRIM56 described in this study. First, we found no evidence of IFN induction or upregulation of interferon-stimulated genes (ISGs) in all cell types examined in this study following IAV (A/PR/8/34) infection, regardless of the TRIM56 expression status (data not shown). This was not surprising, given that IAV encodes multiple IFN antagonists, with NS1 being the most potent and best characterized (11, 38). Second, in cell culture, innate antiviral response to IAV is primarily mediated by RIG-I, while TLR3 plays a more proinflammatory role (39). However, TRIM56 does not contribute to or regulate RIG-I/MDA5 signaling (23). Third, although the STING-dependent pathway significantly contributes to host defense against DNA viruses and bacterial infections, direct evidence supporting the notion that the pathway plays a part in fending off RNA virus infections is lacking (26, 40). Also, it is worth noting that we demonstrated the anti-influenza virus effect of TRIM56 in cell lines derived from HEK293 cells, which have undetectable STING protein expression (25).
Of >70 TRIMs, 2 (TRIM22 and TRIM32) have been shown to inhibit influenza virus. Both critically hinge on the E3 ligase activity to operate by promoting viral protein degradation, with TRIM22 targeting NP (22) while TRIM32 acts on PB1 (41). However, the anti-influenza virus mechanism of TRIM56 is distinct and appears to be completely uncoupled from the E3 ligase activity and also does not require the RING, B-box, or coiled-coil domain. This E3 ligase-independent mechanism is also in stark contrast to what is known about the antiviral activities of TRIM56 against flaviviruses and HCoV-OC43 (25). Remarkably, a TRIM56 fragment with as few as 63 residues at the C terminus of the protein recapitulated the anti-influenza virus effect imposed by full-length TRIM56, indicating that a fully functional motif mediating influenza virus restriction is represented by this small portion of the protein. Given its small footprint, this anti-influenza virus determinant may be amenable to therapeutic manipulations. Additional refined mapping and codon optimization may lead to a TRIM56-derived peptide/small molecule that has broad activity against different influenza viruses, since both IAV and IBV are susceptible to the action of TRIM56.
Our time course viral RNA quantification data and influenza minigenome replication assay show that TRIM56 specifically inhibits influenza virus RNA synthesis with its C-terminal-tail portion. Consistent with the fact that influenza virus RNA synthesis occurs in the nucleus, we found that a fraction of TRIM56 localized in the nucleus in IAV-infected cells, in contrast with the exclusively cytosolic distribution of TRIM56 in uninfected cells. However, it is unknown whether the nuclear translocation of TRIM56 following influenza virus infection is an active, coordinated cellular process to combat the virus or whether TRIM56 is passively imported into or retained in the nucleus while it is associated with viral RNPs or host factors recruited to the RNPs. Further studies will be required to clarify this. Interestingly, TRIM32, which is another IAV-inhibitory TRIM, translocates to the nucleus following IAV infection as a result of its association with and retention by viral PB1 protein (41).
Precisely how the C-terminal-tail portion of TRIM56 impedes influenza virus RNA synthesis is unclear. The C-terminal region of TRIM56 has been shown to interact with viral and cellular proteins performing various biological functions, as exemplified by the TRIM56 association with the N-terminal protease of BVDV (24), TRIF (23), and STING (26). In light of this, TRIM56 may associate with a component(s) of viral RNP or a proviral host factor(s) recruited to the RNP complex, interfering with the partner's role in viral RNA synthesis. However, it is also possible that TRIM56 interacts with influenza virus RNAs, altering their incorporation into the viral transcription/replication complex and/or their stability. The possibility of TRIM56 associating with mRNAs of influenza virus or proviral cellular factors and regulating their translation also cannot be ruled out. Intriguingly, the C-terminal region of TRIM56 (residues 521 to 748) (Fig. 5A and 9) exhibits sequence homology with NHL repeats, based on a search of the NCBI conserved domain database. Sequence alignment of TRIM56 with several well-characterized TRIM-NHL proteins (TRIM2, TRIM3, TRIM32, and TRIM71) also showed conservation of numerous consensus residues shaping the NHL repeats (Fig. 9) (42, 43). The NHL repeats fold into a barrel-like β-propeller structure whose partial surface is positively charged and could directly interact with the phosphate backbone of RNA, which is negatively charged. As an example, TRIM71 can associate with microRNAs (miRNAs) and mRNAs and regulate translation and mRNA metabolism (44). Obviously, further investigation is required to elucidate how TRIM56 hinders influenza virus RNA synthesis, knowledge of which will provide new insights into factors and molecular interactions controlling this vital step in the influenza virus life cycle.
FIG 9.

Sequence alignment of the C-terminal portion of TRIM56 with the NHL repeats of well-characterized TRIM-NHL proteins. The C-terminal amino acid sequence of TRIM56 (GenBank identifier [ID], NP_112223) was aligned and compared with the NHL repeat sequences of the following TRIM-NHL proteins: NCL-1 (Swiss-Prot, P34611), LIN-41 (Swiss-Prot, Q9U489), KIAA0517 (GenBank ID, BAA25443), Brat (GenBank ID, AAF53771), TRIM3 (GenBank ID, NP_001234936), TRIM2 (GenBank ID, NP_056086), and TRIM71 (GenBank ID, NP_001034200). The numbering of the repeats in each protein are indicated as blades I to VI. The numbers show the beginning (left) and ending (right) positions of each repeat of the proteins. Residues that are >50% conserved are highlighted in color. Consensus residues at each conserved amino acid position across different blades are shown at the bottom in the consensus sequence, in which “h” represents the hydrophobic residues isoleucine, leucine, and valine. The asterisks denote the stop codon.
In summary, this study identifies TRIM56 as an intrinsic cellular restriction factor of influenza A and B viruses and describes a novel E3 ligase-independent antiviral mechanism that targets influenza virus RNA synthesis. Our data delineate novel antiviral properties of TRIM56 against orthomyxoviruses and may open new avenues to developing influenza antivirals.
ACKNOWLEDGMENTS
This work was supported in part by NIH grants AI101526 and AI069285 (to K.L.) and American Heart Association Predoctoral Fellowship 15PRE25080234 (to B.L.). B.L. was also supported by a Sigma Xi Grant-in-Aid of Research award (G20141015598932).
We are grateful to Mark Zanin at St. Jude Children's Research Hospital and Michelle Sims at the University of Tennessee Health Science Center for assistance with confocal microscopy.
REFERENCES
- 1.World Health Organization. 2014. Factsheet 211: influenza. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 2.Paddock CD, Liu L, Denison AM, Bartlett JH, Holman RC, Deleon-Carnes M, Emery SL, Drew CP, Shieh WJ, Uyeki TM, Zaki SR. 2012. Myocardial injury and bacterial pneumonia contribute to the pathogenesis of fatal influenza B virus infection. J Infect Dis 205:895–905. doi: 10.1093/infdis/jir861. [DOI] [PubMed] [Google Scholar]
- 3.Nelson MI, Holmes EC. 2007. The evolution of epidemic influenza. Nat Rev Genet 8:196–205. doi: 10.1038/nrg2053. [DOI] [PubMed] [Google Scholar]
- 4.Palese P, Shaw ML. 2007. Orthomyxoviridae: the viruses and their replication, p 1647–1689. In Knipe DM, Howley PM (ed), Fields virology. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 5.Boulo S, Akarsu H, Ruigrok RW, Baudin F. 2007. Nuclear traffic of influenza virus proteins and ribonucleoprotein complexes. Virus Res 124:12–21. doi: 10.1016/j.virusres.2006.09.013. [DOI] [PubMed] [Google Scholar]
- 6.Cros JF, García-Sastre A, Palese P. 2005. An unconventional NLS is critical for the nuclear import of the influenza A virus nucleoprotein and ribonucleoprotein. Traffic 6:205–213. doi: 10.1111/j.1600-0854.2005.00263.x. [DOI] [PubMed] [Google Scholar]
- 7.Kawai T, Akira S. 2006. Innate immune recognition of viral infection. Nat Immunol 7:131–137. doi: 10.1038/ni1303. [DOI] [PubMed] [Google Scholar]
- 8.Kawai T, Akira S. 2010. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol 11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 9.Yoneyama M, Fujita T. 2010. Recognition of viral nucleic acids in innate immunity. Rev Med Virol 20:4–22. doi: 10.1002/rmv.633. [DOI] [PubMed] [Google Scholar]
- 10.Lester SN, Li K. 2014. Toll-like receptors in antiviral innate immunity. J Mol Biol 426:1246–1264. doi: 10.1016/j.jmb.2013.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Talon J, Horvath CM, Polley R, Basler CF, Muster T, Palese P, García-Sastre A. 2000. Activation of interferon regulatory factor 3 is inhibited by the influenza A virus NS1 protein. J Virol 74:7989–7996. doi: 10.1128/JVI.74.17.7989-7996.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kumar N, Xin ZT, Liang Y, Ly H, Liang Y. 2008. NF-kappaB signaling differentially regulates influenza virus RNA synthesis. J Virol 82:9880–9889. doi: 10.1128/JVI.00909-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baumann JG. 2006. Intracellular restriction factors in mammalian cells—an ancient defense system finds a modern foe. Curr HIV Res 4:141–168. doi: 10.2174/157016206776055093. [DOI] [PubMed] [Google Scholar]
- 14.Munir M. 2010. TRIM proteins: another class of viral victims. Sci Signal 3:jc2. doi: 10.1126/scisignal.3118jc2. [DOI] [PubMed] [Google Scholar]
- 15.Rajsbaum R, García-Sastre A, Versteeg GA. 2014. TRIMmunity: the roles of the TRIM E3-ubiquitin ligase family in innate antiviral immunity. J Mol Biol 426:1265–1284. doi: 10.1016/j.jmb.2013.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kawai T, Akira S. 2011. Regulation of innate immune signalling pathways by the tripartite motif (TRIM) family proteins. EMBO Mol Med 3:513–527. doi: 10.1002/emmm.201100160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ozato K, Shin DM, Chang TH, Morse HC III. 2008. TRIM family proteins and their emerging roles in innate immunity. Nat Rev Immunol 8:849–860. doi: 10.1038/nri2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hatakeyama S. 2011. TRIM proteins and cancer. Nat Rev Cancer 11:792–804. doi: 10.1038/nrc3139. [DOI] [PubMed] [Google Scholar]
- 19.Loedige I, Filipowicz W. 2009. TRIM-NHL proteins take on miRNA regulation. Cell 136:818–820. doi: 10.1016/j.cell.2009.02.030. [DOI] [PubMed] [Google Scholar]
- 20.Kano S, Miyajima N, Fukuda S, Hatakeyama S. 2008. Tripartite motif protein 32 facilitates cell growth and migration via degradation of Abl-interactor 2. Cancer Res 68:5572–5580. doi: 10.1158/0008-5472.CAN-07-6231. [DOI] [PubMed] [Google Scholar]
- 21.Gack MU, Shin YC, Joo CH, Urano T, Liang C, Sun L, Takeuchi O, Akira S, Chen Z, Inoue S, Jung JU. 2007. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 446:916–920. doi: 10.1038/nature05732. [DOI] [PubMed] [Google Scholar]
- 22.Di Pietro A, Kajaste-Rudnitski A, Oteiza A, Nicora L, Towers GJ, Mechti N, Vicenzi E. 2013. TRIM22 inhibits influenza A virus infection by targeting the viral nucleoprotein for degradation. J Virol 87:4523–4533. doi: 10.1128/JVI.02548-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shen Y, Li NL, Wang J, Liu B, Lester S, Li K. 2012. TRIM56 is an essential component of the TLR3 antiviral signaling pathway. J Biol Chem 287:36404–36413. doi: 10.1074/jbc.M112.397075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang J, Liu B, Wang N, Lee YM, Liu C, Li K. 2011. TRIM56 is a virus- and interferon-inducible E3 ubiquitin ligase that restricts pestivirus infection. J Virol 85:3733–3745. doi: 10.1128/JVI.02546-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu B, Li NL, Wang J, Shi PY, Wang T, Miller MA, Li K. 2014. Overlapping and distinct molecular determinants dictating the antiviral activities of TRIM56 against flaviviruses and coronavirus. J Virol 88:13821–13835. doi: 10.1128/JVI.02505-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsuchida T, Zou J, Saitoh T, Kumar H, Abe T, Matsuura Y, Kawai T, Akira S. 2010. The ubiquitin ligase TRIM56 regulates innate immune responses to intracellular double-stranded DNA. Immunity 33:765–776. doi: 10.1016/j.immuni.2010.10.013. [DOI] [PubMed] [Google Scholar]
- 27.Wang P, Song W, Mok BW, Zhao P, Qin K, Lai A, Smith GJ, Zhang J, Lin T, Guan Y, Chen H. 2009. Nuclear factor 90 negatively regulates influenza virus replication by interacting with viral nucleoprotein. J Virol 83:7850–7861. doi: 10.1128/JVI.00735-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bao X, Liu T, Shan Y, Li K, Garofalo RP, Casola A. 2008. Human metapneumovirus glycoprotein G inhibits innate immune responses. PLoS Pathog 4:e1000077. doi: 10.1371/journal.ppat.1000077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Elbahesh H, Cline T, Baranovich T, Govorkova EA, Schultz-Cherry S, Russell CJ. 2014. Novel roles of focal adhesion kinase in cytoplasmic entry and replication of influenza A viruses. J Virol 88:6714–6728. doi: 10.1128/JVI.00530-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Balish AL, Katz JM, Klimov AI. 2013. Influenza: propagation, quantification, and storage. Curr Protoc Microbiol Chapter 15:Unit 15G.1. doi: 10.1002/9780471729259.mc15g01s29. [DOI] [PubMed] [Google Scholar]
- 31.Liu S, Qiu C, Miao R, Zhou J, Lee A, Liu B, Lester SN, Fu W, Zhu L, Zhang L, Xu J, Fan D, Li K, Fu M, Wang T. 2013. MCPIP1 restricts HIV infection and is rapidly degraded in activated CD4+ T cells. Proc Natl Acad Sci U S A 110:19083–19088. doi: 10.1073/pnas.1316208110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang N, Dong Q, Li J, Jangra RK, Fan M, Brasier AR, Lemon SM, Pfeffer LM, Li K. 2010. Viral induction of the zinc-finger antiviral protein is IRF3-dependent but NF-kappa B-independent. J Biol Chem 285:6080–6090. doi: 10.1074/jbc.M109.054486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen G, Liu CH, Zhou L, Krug RM. 2014. Cellular DDX21 RNA helicase inhibits influenza A virus replication but is counteracted by the viral NS1 protein. Cell Host Microbe 15:484–493. doi: 10.1016/j.chom.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marklund JK, Ye Q, Dong J, Tao YJ, Krug RM. 2012. Sequence in the influenza A virus nucleoprotein required for viral polymerase binding and RNA synthesis. J Virol 86:7292–7297. doi: 10.1128/JVI.00014-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li K, Chen Z, Kato N, Gale M Jr, Lemon SM. 2005. Distinct poly(I-C) and virus-activated signaling pathways leading to interferon-beta production in hepatocytes. J Biol Chem 280:16739–16747. doi: 10.1074/jbc.M414139200. [DOI] [PubMed] [Google Scholar]
- 36.Chase G, Deng T, Fodor E, Leung BW, Mayer D, Schwemmle M, Brownlee G. 2008. Hsp90 inhibitors reduce influenza virus replication in cell culture. Virology 377:431–439. doi: 10.1016/j.virol.2008.04.040. [DOI] [PubMed] [Google Scholar]
- 37.Chou YY, Heaton NS, Gao Q, Palese P, Singer RH, Lionnet T. 2013. Colocalization of different influenza viral RNA segments in the cytoplasm before viral budding as shown by single-molecule sensitivity FISH analysis. PLoS Pathog 9:e1003358. doi: 10.1371/journal.ppat.1003358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gack MU, Albrecht RA, Urano T, Inn KS, Huang IC, Carnero E, Farzan M, Inoue S, Jung JU, García-Sastre A. 2009. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe 5:439–449. doi: 10.1016/j.chom.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Le Goffic R, Pothlichet J, Vitour D, Fujita T, Meurs E, Chignard M, Si-Tahar M. 2007. Cutting edge: influenza A virus activates TLR3-dependent inflammatory and RIG-I-dependent antiviral responses in human lung epithelial cells. J Immunol 178:3368–3372. doi: 10.4049/jimmunol.178.6.3368. [DOI] [PubMed] [Google Scholar]
- 40.Barber GN. 2011. Innate immune DNA sensing pathways: STING, AIMII and the regulation of interferon production and inflammatory responses. Curr Opin Immunol 23:10–20. doi: 10.1016/j.coi.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fu B, Wang L, Ding H, Schwamborn JC, Li S, Dorf ME. 2015. TRIM32 senses and restricts influenza A virus by ubiquitination of PB1 polymerase. PLoS Pathog 11:e1004960. doi: 10.1371/journal.ppat.1004960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Edwards TA, Wilkinson BD, Wharton RP, Aggarwal AK. 2003. Model of the brain tumor-Pumilio translation repressor complex. Genes Dev 17:2508–2513. doi: 10.1101/gad.1119403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Slack FJ, Ruvkun G. 1998. A novel repeat domain that is often associated with RING finger and B-box motifs. Trends Biochem Sci 23:474–475. doi: 10.1016/S0968-0004(98)01299-7. [DOI] [PubMed] [Google Scholar]
- 44.Loedige I, Gaidatzis D, Sack R, Meister G, Filipowicz W. 2013. The mammalian TRIM-NHL protein TRIM71/LIN-41 is a repressor of mRNA function. Nucleic Acids Res 41:518–532. doi: 10.1093/nar/gks1032. [DOI] [PMC free article] [PubMed] [Google Scholar]