

ABSTRACT
Human bronchoalveolar fluid is known to have anti-influenza activity. It is believed to be a frontline innate defense against the virus. Several antiviral factors, including surfactant protein D, are believed to contribute to the activity. The 2009 pandemic H1N1 influenza virus was previously shown to be less sensitive to surfactant protein D. Nevertheless, whether different influenza virus strains have different sensitivities to the overall anti-influenza activity of human bronchoalveolar fluid was not known. We compared the sensitivities of 2009 pandemic H1N1, seasonal H1N1, and seasonal H3N2 influenza virus strains to inhibition by human bronchoalveolar lavage (BAL) fluid. The pandemic and seasonal H1N1 strains showed lower sensitivity to human BAL fluid than the H3N2 strains. The BAL fluid anti-influenza activity could be enhanced by oseltamivir, indicating that the viral neuraminidase (NA) activity could provide resistance to the antiviral defense. In accordance with this finding, the BAL fluid anti-influenza activity was found to be sensitive to sialidase. The oseltamivir resistance mutation H275Y rendered the pandemic H1N1 virus but not the seasonal H1N1 virus more sensitive to BAL fluid. Since only the seasonal H1N1 but not the pandemic H1N1 had compensatory mutations that allowed oseltamivir-resistant strains to maintain NA enzymatic activity and transmission fitness, the resistance to BAL fluid of the drug-resistant seasonal H1N1 virus might play a role in viral fitness.
IMPORTANCE Human airway secretion contains anti-influenza activity. Different influenza strains may vary in their susceptibilities to this antiviral activity. Here we show that the 2009 pandemic and seasonal H1N1 influenza viruses were less sensitive to human bronchoalveolar lavage (BAL) fluid than H3N2 seasonal influenza virus. The resistance to the pulmonary innate antiviral activity of the pandemic virus was determined by its neuraminidase (NA) gene, and it was shown that the NA inhibitor resistance mutation H275Y abolished this resistance of the pandemic H1N1 but not the seasonal H1N1 virus, which had compensatory mutations that maintained the fitness of drug-resistant strains. Therefore, the innate respiratory tract defense may be a barrier against NA inhibitor-resistant mutants, and evasion of this defense may play a role in the emergence and spread of drug-resistant strains.
INTRODUCTION
Despite being regarded as a mild pandemic, the H1N1 influenza virus can cause unusually severe diseases, which can rapidly progress to respiratory failure and death (1). Comparative studies in the same outbreak season showed that patients infected with the pandemic H1N1 (pdmH1N1) virus are more likely to develop severe disease and consequently die than those infected with seasonal influenza virus (2). In addition, animal experiments with mice, ferrets, and macaques have shown that the H1N1 2009 pandemic influenza virus is more virulent and replicates more efficiently in lungs than seasonal influenza virus (3, 4). These findings suggest that the pandemic virus may invade lungs more effectively.
Innate defense plays important roles in susceptibility to viral infection and pathogenesis. Soluble antiviral factors are important components of innate defense in the respiratory tract. Several soluble factors with anti-influenza activity have been identified in bronchoalveolar lavage (BAL) fluid, including surfactant protein D (SP-D), SP-A, scavenger receptor gp340, long pentraxin PTX3, L-ficolin, H-ficolin, and serum amyloid P (5–12). These factors are present in serum and respiratory secretions. Serum influenza virus inhibitors can be classified as α-, β-, and γ-inhibitors according to their physical properties and mechanisms of inhibition (13–15). Because α- and γ-inhibitors are sialylated glycoproteins or receptor analogues that compete with sialic acid in the binding of influenza virus hemagglutinin (HA), they are heat resistant and Ca2+ independent but can be destroyed by sialidase or receptor-destroying enzyme (RDE). In contrast, β-inhibitors, such as SP-D, are heat labile, require Ca2+, and can be inhibited by some monosaccharides but are resistant to RDE. They are lectins that bind influenza virus HA via carbohydrate moiety on glycosylation sites of HA and inhibit HA function, probably through steric hindrance. In addition, exosome from human bronchial epithelial cells has been shown to exhibit anti-influenza activity (16).
Among these known anti-influenza factors, SP-D is believed to be the main contributor to the BAL fluid anti-influenza activity (6). It was shown that the 2009 H1N1 pandemic virus is resistant to SP-D because it had fewer N-linked glycosylation sites on the globular head of hemagglutinin (HA) (17, 18). However, whether susceptibility to other innate antiviral factors, especially α- and γ-inhibitors, contributes to the virulence of the 2009 pandemic influenza virus is not known. The antiviral activities of α- and γ-inhibitors can be detected without interference from the β-inhibitor by performing the assay under conditions without calcium ions. We therefore assessed the sensitivities to human BAL fluid of 2009 pandemic influenza virus strains in comparison with those of seasonal influenza strains.
MATERIALS AND METHODS
Ethics statement.
The human study was approved by the Ethics Committee of the Faculty of Medicine, Siriraj Hospital (Siriraj Institutional Review Board), which is in full compliance with the Declaration of Helsinki, the Belmont Report, the CIOMS Guidelines, and the International Conference on Harmonization in Good Clinical Practice (ICH-GCP). The study was performed under protocol COA no. SI572/2009, “Sensitivity of Influenza 2009 H1N1 to Antiviral Factors in Bronchoalveolar Lavage.” All human subjects provided written informed consent.
BAL fluid samples.
BAL fluid samples were collected from 52 patients 30 to 96 years of age (mean ± [SD], = 64 ± 12 years) with a male/female ratio of 1:1.17, who underwent routine bronchoscopy and bronchoalveolar lavage for investigation of suspected lung cancer. None of the patients was being treated with any antiviral treatment. Bronchoalveolar lavage with 0.9% normal saline was performed at a nonlesional lung segment. The retrieved fluid was collected and immediately carried on ice to the laboratory. The BAL fluid samples were centrifuged at 1,500 × g at 4°C for 5 min. The supernatant was stored frozen in aliquots at −80°C.
Viral strains.
The 2009 pandemic H1N1 (pdmH1N1) influenza virus strains were isolated from patients with influenza-like illness in Thailand during 2009 to 2011 (A/Nonthaburi/102/2009, A/Thailand/104/2009, A/Thailand/MVCU-013/2009, and A/Thailand/ICRC_NSN_1/2011). The seasonal influenza strains used in this study are A/Thailand/Siriraj-03/2006(sH1N1) (A/New Caledonia/20/99-like virus), A/Thailand/Siriraj-12/2006(sH1N1) (A/New Caledonia/20/99-like virus), A/Thailand/SirirajICRC_SEA-3479/2008(sH1N1) (A/Brisbane/59/2007-like virus), A/Thailand/Siriraj-03/2004(H3N2) (A/Fujian/411/02-like virus), and A/Thailand/Siriraj-04/2003(H3N2). All of the viruses were isolated and propagated for less than 10 passages in Madin-Darby canine kidney (MDCK) cells with 1 mM l-1-tosylamido-2-phenylethyl chloromethyl ketone (TPCK)-treated trypsin (Sigma-Aldrich) in minimum essential medium (MEM) (Gibco).
Antiviral activity.
The antiviral activity in BAL fluid samples was assessed using hemagglutination inhibition (HI) and neutralization (NT) assays as previously described for the serum antibody assay (19), except the samples were not treated with sialidase prior to the assay. Briefly, for the HI assay, BAL fluid was preabsorbed with a 0.5% goose red blood cell (GRBC) suspension to eliminate nonspecific agglutinins, serially diluted in phosphate-buffered saline (PBS) with and without 5 mM CaCl2, mixed with 4 hemagglutination units of virus and 0.5% GRBC in a 96-well U-shaped microtiter plate, and incubated at 4°C for 30 min. HI titers were read as the reciprocal of the highest dilution with complete hemagglutination. For the NT assay, BAL fluid was serially diluted in MEM plus 1 mM TPCK-treated trypsin (with 1.8 mM CaCl2) and mixed with 25 50% tissue culture infectious doses (TCID50s) of virus, incubated at 37°C for 1 h, and inoculated onto MDCK cells in a 96-well tissue culture plate. Briefly, each well was seeded with 3 × 104 cells in 200 μl MEM plus 10% heat-inactivated fetal bovine serum (FBS) (Gibco) and incubated at 37°C overnight. After an overnight incubation at 37°C, the tissue culture plate was washed and fixed with 80% cold acetone in 1× PBS, and the level of viral infection was measured using an antiviral nucleoprotein monoclonal antibody and a peroxidase-conjugated secondary antibody. Optical densities were measured, and NT titers with 50% inhibition of specific signals were calculated. The BAL fluid sample was treated with receptor-destroying enzyme (RDE II; Denka Seiken) at 1:2 at 37°C overnight. RDE was inactivated at 56°C for 30 min before the HI testing. Protease treatment was done by adding 30 U of proteinase K (Sigma-Aldrich) to 1 ml of BAL fluid sample, and the mixture was incubated at 37°C overnight and inactivated at 92°C for 10 min.
Reverse genetic viruses.
The HA and NA genes of selected pdmH1N1 strains were amplified from RNA extracted from infected MDCK cells, cloned into the reverse genetic plasmid pHw2000, and sequenced. Specific mutations were introduced into the NA gene by PCR site-directed mutagenesis with DpnI digestion. Reverse genetic viruses were produced in HEK293T cells by cotransfecting pHw2000 plasmids carrying the cloned HA and NA genes of the 2009 pandemic virus and the other 6 genes from the A/Puerto Rico/8/1934(H1N1) (PR8) strain as previously described (20).
IgA and IgG depletion.
IgA and IgG in BAL fluid sample were precipitated using a lectin (jacalin) from Artocarpus integrifolia cross-linked with 4% beaded agarose (Sigma-Aldrich) (21) and Pierce protein G-agarose (Thermo Scientific), respectively. Two hundred fifty microliters of BAL fluid sample was added with 100 μl of settled agarose beads and mixed thoroughly. The mixture was rotated and incubated at 4°C overnight. The agarose beads were precipitated from sample by centrifugation at 1,000 rpm for 30 s. The supernatant was collected. To precipitate total protein in the sample, 10 μl of supernatant was added with 250 μl of absolute ethanol and mixed thoroughly, and the mixture was centrifuged at 13,000 rpm at 4°C for 1 h. The supernatant was discarded and dried. Twenty microliters of reducing loading dye was added and mixed thoroughly, and the mixture was incubated at 70°C for 10 min. The sample was run in sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and Western blotting was performed. IgA and IgG were detected using 1:2,000 goat anti-human IgA horseradish peroxidase (HRP)-labeled antibody (Sigma-Aldrich) and 1:2,000 goat anti-human IgG HRP-labeled antibody (Invitrogen) in 2% bovine serum albumin (Sigma-Aldrich), respectively. Both detected IgA and IgG antibodies were developed with DAB (3,3′-diaminobenzidine)-HRP substrate (Sigma-Aldrich). One hundred microliters of IgA- and IgG-depleted sample was further measured for the HI titer.
Animal experiment.
Ferrets of ages 6 to 18 months were screened for influenza virus HI antibody with a 2009 pandemic H1N1 strain. Three animals with no HI titer were randomly assigned to each experimental group. The animals were intratracheally inoculated with either MVCU-013/2009 or 104/2009 at the dosage of 106 TCID50s in a 1.2-ml volume. The animals were kept separately in animal isolators in a biosafety level 3 (BSL3) laboratory. The animals were observed clinically every day, and at day 10 postinfection, they were euthanized, and then lung tissue was collected and processed for pathological study and RNA extraction for viral load measurement. The tissue was cut into small pieces in TRIzol solution, and RNA was extracted using the manufacturer's protocol. Viral RNA was measured by a real-time reverse transcription-PCR (RT-PCR) using a primer pair for the influenza virus A M gene and TaqMan probes (5′-6-carboxyfluorescein [6-FAM] and 3′-Black Hole Quencher 1 [BHQ1]) according to the CDC protocol of real-time RT-PCR for influenza virus A (H1N1) (22, 23).
Statistical analysis.
HI/NT titers and NT percentages of inhibition are shown as the geometric mean with 95% confidence interval (CI) and arithmetic mean ± standard error of the mean (SEM), respectively. The comparisons of HI and NT titers and NT percentage of inhibition were tested by paired/unpaired t test (GraphPad Prism version 5.1 software).
RESULTS
H1N1 influenza viruses were less sensitive to human BAL fluid.
BAL fluid samples from 24 subjects were tested for their NT activities against two H1N1 pandemic strains (A/Nonthaburi/102/2009 and A/Thailand/104/2009), two seasonal H3N2 strains (A/Thailand/Siriraj03/2004 and A/Thailand/Siriraj04/2003), and two seasonal H1N1 strains (A/Thailand/Siriraj03/2006 and A/Thailand/Siriraj12/2006). Together as a group, pandemic and seasonal H1N1 viruses had lower BAL fluid NT titers than the H3N2 virus (t test, P < 0.0001), and there was a smaller degree of difference between NT titers against pandemic and seasonal H1N1 viruses (P = 0.0026; paired t test) (Fig. 1A). There was no difference in the demographic profiles of BAL fluid samples with high and low antiviral activities (data not shown). As the NT assay in cell culture had to be performed in the presence of Ca2+, it did not tell us whether the anti-influenza activity was Ca2+ dependent. We further tested the sensitivity to human BAL fluid of the H1N1 and H3N2 viruses by HI assay in the presence and absence of Ca2+. Similar titers were observed in reactions with and without Ca2+, indicating that the HI activity was Ca2+ independent. Similar to the NT profile, BAL fluid HI titers against the H1N1 viruses were lower than those against H3N2 virus (P < 0.0305; t test), and the HI titer of seasonal H1N1 virus was significantly higher than that of pandemic H1N1 virus (P = 0.0379; paired t test) (Fig. 1B). The presence or absence of Ca2+ did not affect the HI titers, suggesting that the anti-influenza activity was Ca2+ independent. Pandemic 2009 H1N1 strains were previously shown to be resistant to SP-D due to their hypoglycosylated HA. However, the lower sensitivity to BAL fluid of the pandemic H1N1 virus by HI assay was Ca2+ independent. This indicated that the observed resistance to BAL fluid of the H1N1 virus could not be fully explained by the previously published resistance to SP-D of 2009 pandemic H1N1 virus.
FIG 1.

(A) Antiviral activity in 24 BAL fluid samples against 2 pdmH1N1 strains (102/2009 and 104/2009) and 4 seasonal influenza virus strains: Siriraj-03/2006(sH1N1), Siriraj-12/2006(sH1N1), Siriraj-03/2004(H3N2), and Siriraj-04/2003(H3N2) using the neutralization (NT) assay. (B) The hemagglutination inhibition (HI) assay was run in duplicate with and without Ca2+. HI titers were derived from 12 BAL fluid samples. HI and NT titers are shown as the geometric mean with 95% CI (*, P < 0.05; **, P < 0.005; ***, P < 0.0005; t test). Samples that were reused in more than one experiment are numbered.
The resistance to human BAL fluid of the H1N1 viruses required NA activity.
The fact that the observed BAL fluid antiviral activity did not show Ca2+ dependency suggested that these anti-influenza factors might be sialic acid-containing molecules (13–15). As the net result of virion binding to sialic acid depends on both the binding activity of HA and the cleavage activity of NA, we asked whether NA activity contributed to the different susceptibilities to BAL fluid of the tested viruses. To test whether NA enzymatic activity contributed to the resistance to human BAL fluid, the HI assay was done in the presence of the anti-NA drug oseltamivir. All of the viral strains showed increased HI titers in the presence of oseltamivir, indicating that the NA activity could reduce the HI titers and contributed to the viral resistance to BAL fluid (Fig. 2). This indicated that sialic acid-containing factors contributed significantly to the BAL fluid anti-influenza activity. In the presence of oseltamivir, only the HI titer of pandemic H1N1 remained lower than that of H3N2 virus, whereas seasonal H1N1 and H3N2 viruses showed comparable HI titers. This suggested that, in addition to NA, HA of the pandemic H1N1 virus might also contribute to the resistance to human BAL fluid.
FIG 2.
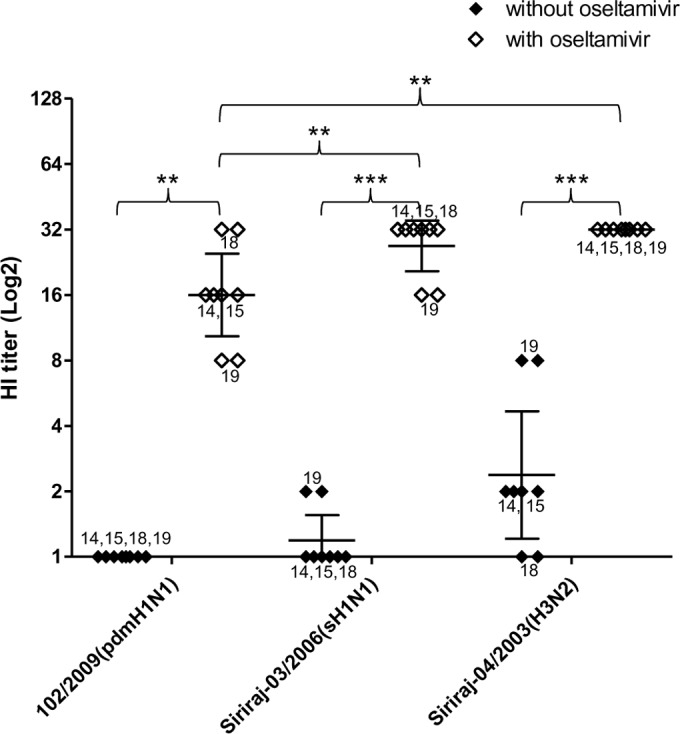
Antiviral activity in 4 BAL fluid samples against pdmH1N1 (102/2009), sH1N1 (Siriraj-03/2006), and H3N2 (Siriraj-04/2003) viruses using the hemagglutination inhibition (HI) assay. The assay was run in duplicate with and without oseltamivir. HI titers are shown as the geometric mean with 95% CI (**, P < 0.005; ***, P < 0.0001; t test).
We further expand the number of pandemic H1N1 isolates to see whether the resistance to BAL fluid was uniformly present. Of 40 tested pandemic H1N1 strains, two (A/Thailand/MVCU-013/2009 and A/Thailand/NSN_1/2011) showed HI titers comparable to those of H3N2 seasonal influenza virus (Fig. 3A). The higher sensitivity of the two pandemic H1N1 strains was further confirmed with both the HI and NT assays using more BAL fluid samples (Fig. 3B). When the HA sequences of the A/Thailand/MVCU-013/2009(H1N1) and A/Thailand/NSN_1/2011(H1N1) were compared to those of the other pandemic H1N1 strains, we did not find any common mutation. We then generated reverse genetic viruses carrying the HA gene of A/Thailand/MVCU-013/2009(H1N1) in the PR8 backbone and found that the HA gene did not cause any change in the susceptibility to BAL fluid (Fig. 3C). We then compared the NA sequences of the A/Thailand/MVCU-013/2009(H1N1) and A/Thailand/NSN_1/2011(H1N1) strains to those of the other pandemic strains and found that both the BAL fluid-sensitive strains carried an H275Y mutation in the NA sequences. In addition to the H275Y mutations, a number of other mutations were observed, including V241I and N369K in the A/Thailand/NSN_1/2011 strain, none of which was observed in the A/Thailand/MVCU-013/2009 strain. These mutations were previously shown to partially compensate for the fitness of oseltamivir-resistant H1N1 virus (24). An NA gene of a pandemic H1N1 strain, A/Thailand/104/2009, was used to generate a reverse genetic virus with the other genes from the PR8 strain. The virus carrying the pandemic NA showed a lower HI and NT titer to BAL fluid compared to a PR8-based reverse genetic virus carrying the NA gene from an H3N2 virus, A/Thailand/AW10/2010(H3N2) (Fig. 4A). We then introduced the H275Y mutation into the pandemic NA and generated a mutant reverse genetic virus. The mutant virus showed higher sensitivity to antiviral activity of BAL fluid by both the HI and NT assays (Fig. 4A and B). This indicated that the NA gene was responsible for the reduced sensitivity of the pandemic H1N1 influenza virus to the innate anti-influenza activity of human BAL fluid and that the oseltamivir resistance mutation H275Y could render the virus more susceptible to human BAL fluid. The role of NA in susceptibility to BAL fluid suggested that the BAL fluid antiviral factor might be sialic acid-containing molecules, which could bind to HA via sialic acid-HA interaction. Sialic acid was probably cleaved from these molecules by the viral NA, thereby releasing the bound virions from the antiviral factor and eliminating its antiviral activity.
FIG 3.

(A) Hemagglutination inhibition (HI) assay of one BAL fluid sample against 40 pdmH1N1 strains. (B) Neutralization curves as determined by NT. The NT activity from one sample against 2 pdmH1N1 strains (NSN_1/2011 and MVCU-013/2009) and the Siriraj-04/2003(H3N2) strain was significantly higher than that of the 104/2009(pdmH1N1) strain. The experiment was run in duplicate. The neutralization curves are shown as the arithmetic mean ± SEM (*, P < 0.05; **, P < 0.005; t test). (C) Comparisons of HI titers from 3 BAL fluid samples (run in duplicate) against 6 + 2 reverse genetic viruses carrying MVCU-013/2009 HA or 102/2009-HA and 102/2009-NA. The HI titers are shown as the geometric mean with 95% CI.
FIG 4.

Comparisons of HI titers and percentages of inhibition with or without oseltamivir against 7 + 1 reverse genetic viruses. (A) Comparisons of HI titers against reverse genetic viruses carrying the pdmH1N1 102/2009-NA wild type (RG-102wt), H275Y mutant (RG-102H275Y), and N2 (RG-N2). HI titers were derived from 4 BAL fluid samples (run in duplicate). HI titers are shown as the geometric mean with 95% CI (*, P < 0.05; ***, P < 0.0001; t test). (B) Comparisons of percentages of neutralization with or without oseltamivir. The neutralization curves were derived from one BAL fluid sample that shows high HI activity. The experiment was run in duplicate, and results are shown as the arithmetic mean ± SEM (***, P < 0.0005; t test).
An oseltamivir-resistant seasonal H1N1 strain did not show increased sensitivity to BAL fluid.
Oseltamivir-resistant 2009 pandemic H1N1 strains have not evolved to maintain transmission efficiency and have not widely spread. In contract, oseltamivir-resistant strains of the seasonal H1N1 virus have gained the ability to transmit efficiently and caused a widespread epidemic of drug-resistant influenza in 2007 to 2008 independent of drug usage. To test whether the drug resistance mutation had a similar effect on susceptibility to BAL fluid in seasonal H1N1 virus, an oseltamivir-resistant strain (A/Thailand/SirirajICRC_SEA-3479/2008) was tested. The NA of this strain contained the H275Y mutation as well as a number of known adaptive mutations: V234M, R222Q, K239E, D344N, and D354G. The strain was found to have comparable levels of sensitivity to human BAL fluid in both HI (Fig. 5A) and NT (Fig. 5B). This association between the widespread epidemic and BAL fluid resistance of the oseltamivir-resistant seasonal H1N1 virus suggested that the resistance to BAL fluid may play a role in the viral transmission efficiency.
FIG 5.

Antiviral activity against 2 sH1N1 strains, Siriraj-03/2006 (oseltamivir sensitive) and SirirajICRC_SEA-3479/2008 (oseltamivir resistant), using the (A) hemagglutination inhibition (HI) assay and (B) neutralization curves as determined by neutralization (NT) assay. HI titers were derived from 6 BAL fluid samples and are shown as the geometric mean with 95% CI. The neutralization curves were derived from one BAL fluid sample that shows high HI activity. The experiment was run in duplicate, and results are shown as the arithmetic mean ± SEM. Both the HI and NT assays were run in duplicate.
The BAL fluid antiviral activity was sensitive to sialidase.
The effect of the NA gene on the viral susceptibility to BAL fluid suggested that the BAL fluid antiviral factor was more efficiently destroyed by the NA of the pandemic virus and that the BAL fluid antiviral factor was sialic acid-containing molecules. In order to understand the nature of the anti-influenza factors, BAL fluid samples were subjected to different treatments prior to the HI assay using A/Thailand/MVCU-013/2009(H1N1) (Fig. 6). To test the effect of Ca2+, 5 mM CaCl2 and 5 mM EDTA were added to each BAL fluid dilution during HI assay. A heat treatment at 56°C for 30 min or the presence of Ca2+ had no effect on the HI titer, whereas the HI activity was abolished when the BAL fluid sample was treated with RDE. These results indicate that the main anti-influenza factor in the BAL fluid samples was neither SP-D nor the other β-inhibitors. The elimination of the antiviral activity by RDE and protease indicated that the antiviral factor was a sialylated glycoprotein.
FIG 6.
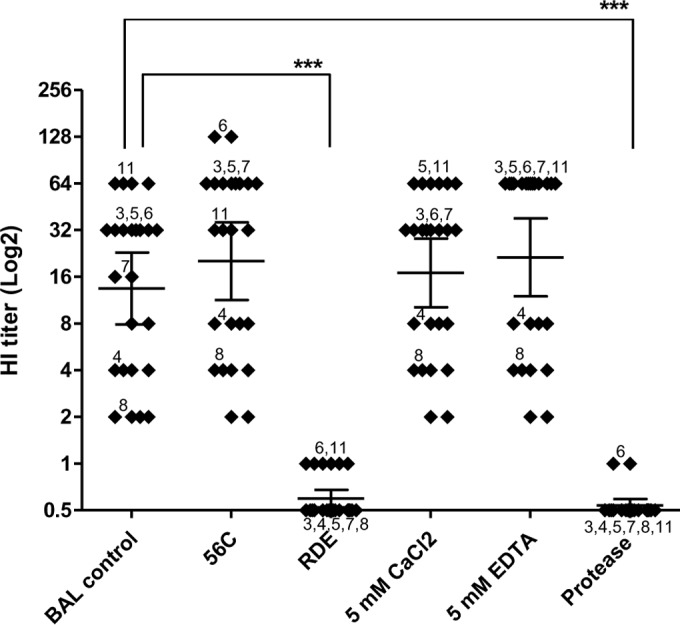
BAL fluid antiviral activity was sensitive to sialidase. Shown are HI titers from 12 BAL fluid samples against MVCU-013/20099(pdmH1N1). The BAL fluid samples were subjected to the following treatments prior to the assay: heat treatment at 56°C and exposure to the receptor-degrading enzyme (RDE), CaCl2, EDTA, and protease. HI titers are shown as the geometric mean with 95% CI (***, P < 0.001; t test).
IgA and IgG depletion.
The BAL fluid samples were collected in 2009 when the 2009 H1N1 virus was extensively circulating and should have induced specific immunity in majority of the population. It has been estimated that during the first year of circulation, 20 to 27% had been infected with the pandemic strain (25). In a serological survey in Thailand, HI antibody against the 2009 pandemic virus was detected in 52% of the tested subjects after the first wave of pandemic in July to October 2009 (26). The observed resistance of the 2009 H1N1 virus to BAL fluid was therefore unlikely to be the result of a lower level of specific immunity to this virus. To exclude the role of secretory IgA and IgG in the observed difference in viral susceptibility to BAL fluid antiviral activity, IgA and IgG were depleted from 3 selected BAL fluid specimens with medium to high HI titers by jacalin and protein G, respectively, and the IgA- and IgG-depleted BAL fluid samples were retested for HI activity. The completeness of IgA and IgG depletion was confirmed by Western blot analysis (Fig. 7A). In 2 out of 3 samples, depletion of IgA and IgG did not change the HI titers. In one sample with a very high titer against H3N2, IgA depletion did not affect the HI titer, but IgG depletion reduced the titer from 512 to 64. Nevertheless, this did not affect the overall difference among the pandemic H1N1, seasonal H1N1, and H3N2 viruses (Fig. 7B). This indicated that secretory IgA and IgG were not responsible for the higher resistance of the 2009 H1N1 pandemic virus to BAL fluid. Jacalin is a lectin that binds only to O-glycosidically linked oligosaccharides. Besides IgA, jacalin also binds to a number of plasma glycoproteins, including C1-inhibitor, hemopexin, and α2-HSG (fetuin A) (27). The persistence of the antiviral activity after jacalin treatment indicated that these glycoproteins were also not involved in the antiviral activity.
FIG 7.

IgA and IgG depletion. IgA and IgG were depleted from one BAL fluid sample using the IgA-binding lectin jacalin and protein G beads, respectively. (A) After the depletion, the samples were analyzed by Western blotting using human IgA- and IgG-specific antibodies. The IgA and IgG heavy chains have molecular masses of approximately 58 to 80 kDa. (B) HI titers of IgA- and IgG-depleted BAL fluid against Siriraj-04/2003(H3N2), Siriraj-03/2006(sH1N1), and 104/2009(pdmH1N1) were comparable to those of untreated BAL fluid. The data were derived from 3 BAL fluid samples and run in duplicate. HI titers are shown as the geometric mean with 95% CI (**, P = 0.0075; ***, P = 0.0009; t test).
Oseltamivir-resistant pandemic H1N1 virus showed lower viral loads in ferrets and increased sensitivity to ferret BAL fluid.
To ensure that the ferret is a relevant model for the human BAL fluid anti-influenza activity, two uninfected healthy ferrets were sacrificed for BAL fluid collection. The ferret BAL fluid showed similar patterns of activity with a high titer against the H3N2 virus and a low titer against the pdmH1N1 virus (Fig. 8). The oseltamivir-resistant pdmH1N1 strain MVCU-013/2009 showed a slightly higher HI titer than (Fig. 8A) and comparable NT titer to (Fig. 8B) the wild-type 104/2009 virus. The MVCU-013/2009 and 104/2009 strains were then inoculated into ferrets via the intratracheal route at a dosage of 106 TCID50s. Three ferrets were used in each group. At day 10 postinoculation, the animals were euthanized, and lung tissue samples were collected. Five lung tissue samples were collected from each animal for pathological examination and viral load determination. Pathological changes were scored 0 to 4 for each of the three findings: alveolar infiltration, peribronchiolar infiltration, and perivascular infiltration, as previously described (28). The mean total scores ± standard deviation (SD) were 6.4 ± 1.1 and 6.6 ± 1.0 for the wild-type 104/2009 strain and the MVCU-013 group, respectively (t test, P = 0.66). While lung tissue samples from all of the ferrets showed similar degrees of inflammation, the pulmonary viral loads of ferrets inoculated with the BAL fluid-susceptible strain MVCU-013/2009 and the BAL fluid-resistant strain 104/2009 were found to be 5.79 ± 0.45 and 7.67 ± 0.32 log10 copies/g of tissue (mean ± SEM), respectively (t test, P = 0.0143). This suggests that the resistance to the pulmonary innate defense could contribute to viral fitness in ferrets. On the other hand, since the viral load was measured at the late phase of infection, the difference might reflect the efficiency of viral clearance related to the pulmonary innate defense.
FIG 8.

Antiviral activity in 2 ferret BAL fluid samples against 2 pdmH1N1 influenza strains (104/2009 and MVCU-013/2009) and Siriraj-04/2003(H3N2) using the (A) HI assay and (B) neutralization (NT) assay. The HI assay was run in duplicate, and results are shown as the geometric mean with 95% CI (**, P < 0.005; t test). The neutralization experiment was run in duplicate, and results are shown as the arithmetic mean ± SEM (*, P < 0.05; t test).
DISCUSSION
It has been shown that the 2009 pandemic H1N1 viruses are resistant to SP-D owing to their HA hypoglycosylation. Certain N-linked glycosylation sites on the HA globular head have been shown to be essential for SP-D sensitivity (23), and accumulation of N-linked glycosylation on HA of seasonal influenza viruses occurred during decades of circulation in human population (29). These data suggest that the level of N-linked glycosylation on HA may be associated with the viral virulence and that seasonal influenza viruses are evolving toward lower virulence in humans. Our data suggested that in addition to this resistance to SP-D, the 2009 pandemic H1N1 virus is also more resistant to the other classes of inhibitors owing to its NA activity. This makes the virus more resistant to all types of the inhibitors.
While SP-D is calcium dependent and binds to the carbohydrate moiety on the HA head, many other innate anti-influenza molecules were shown to bind influenza virus HA via their sialic acid. These molecules are therefore calcium independent and sensitive to sialidase. These inhibitors include long pentraxin PTX3, SP-A, gp340, L-ficolin, H-ficolin, and serum amyloid P (5, 7, 8, 10–12). The BAL fluid antiviral activity shown in this study is consistent with these inhibitors and may be the result of the combined effect of these inhibitors. The NA activity of the 2009 H1N1 influenza virus was previously shown to be stronger than that of seasonal influenza viruses for both α2,3- and α2,6-linked sialic acid (30). In general, avian influenza viruses have high NA activity against α2,3-linked sialic acid but low activity against α2,6-linked sialic acid, whereas swine influenza viruses have high NA activity against α2,6-linked sialic acid but low activity against α2,3-linked sialic acid. Seasonal influenza viruses, on the other hand, have relatively lower activity against both α2,3- and α2,6-linked sialic acid (30). In contrast, the 2009 H1N1 virus was shown to have high NA activity against α2,3- and α2,6-linked sialic acid at a level comparable to those of avian and swine influenza viruses, respectively (30). This is in agreement with our data showing the role of NA in the resistance of pandemic H1N1 virus to human BAL fluid. This unique high NA activity for both types of sialic acid may help the virus to evade innate defense in multiple species and not only enhance the virulence in those species but also facilitate the interspecies transmission.
Our result on the effect of the H275Y mutation is in concordance with recently published data showing a reduced viral fitness and virulence in ferrets and a reduction in the enzymatic activity (31). The 2009 H1N1 pandemic influenza virus was a reassortant with the HA gene from the H1N1 classical swine influenza virus and the NA gene from an H1N1 avian-like swine influenza virus (32–34). The acquisition of the NA gene with increased enzymatic activity was previously shown to contribute to effective transmission in ferrets (35). It was hypothesized that the NA contributed to the effective transmission by improving the balance in HA and NA activities (36).
NA is believed to function not only to release virions from infected cells but also to help virions penetrate mucosal secretion by digesting sialic acid that can trap virions and prevent them from attaching to the target cells (37). Changes in NA activity can therefore influence the effectiveness of the virus to evade the innate antiviral activity of mucosal surface. Monitoring the changes in the NA activity may help us understand the viral evolution and its adaption to the complex virus-host interaction, which may influence the level of viral virulence and fitness.
The difference in the effects of the H275Y mutation on the BAL fluid sensitivity of pandemic and seasonal H1N1 viruses and the difference in the levels of fitness of mutant strains between the two viruses suggested that successful evasion of the respiratory innate defense may play a role in the viral transmission fitness. Oseltamivir-resistant seasonal H1N1 strains gained adaptive mutations and transmission fitness (38) and spread widely in 2007 to 2008 (39). The adaptive mutations allowed normal surface expression of NA and full activity of the enzyme (40), which helped to evade the respiratory innate anti-influenza activity. In contrast, some oseltamivir-resistant 2009 pandemic H1N1 strains emerged sporadically but have not caused large-scale spreading. Some adaptive mutations in the pandemic H1N1 NA have been reported. Nevertheless, the adaptive mutations have not caused full fitness of oseltamivir-resistant strains (24). Resistance to BAL fluid may play a role in the fitness of drug-resistant strains, and monitoring of BAL fluid resistance in drug-resistant strains may provide a biomarker for strains that are gaining fitness. The biomarker may help in surveillance and control of drug resistance.
ACKNOWLEDGMENTS
This study was supported by a research grant (61047) from the National Research Council of Thailand (NRCT) and a senior research scholar grant (RTA5780009) from the Thailand Research Fund (TRF).
We gratefully acknowledge Robert G. Webster for providing the pHW2000 plasmid. We thank Supparerk Disayabutr for clinical assistance with BAL fluid sample collection.
P.A. designed and planned the experiments, K.R., C.B., and O.S. ran the experiments, P.A. and K.R. wrote the manuscript, K.M. and J.A. performed sample collection, W.C. and W.W. performed the animal experiments, M.U. performed the pathological examination, P.P. and P.B. provided viruses, and A.J. constructed the reverse genetic virus.
REFERENCES
- 1.Perez-Padilla R, de la Rosa-Zamboni D, Ponce de Leon S, Hernandez M, Quinones-Falconi F, Bautista E, Ramirez-Venegas A, Rojas-Serrano J, Ormsby CE, Corrales A, Higuera A, Mondragon E, Cordova-Villalobos JA. 2009. Pneumonia and respiratory failure from swine-origin influenza A (H1N1) in Mexico. N Engl J Med 361:680–689. doi: 10.1056/NEJMoa0904252. [DOI] [PubMed] [Google Scholar]
- 2.Riquelme R, Torres A, Rioseco ML, Ewig S, Cilloniz C, Riquelme M, Inzunza C, Polverino E, Gomez Y, Marcos MA, Contreras C, Gabarrus A, Fasce R. 2011. Influenza pneumonia: a comparison between seasonal influenza virus and the H1N1 pandemic. Eur Respir J 38:106–111. doi: 10.1183/09031936.00125910. [DOI] [PubMed] [Google Scholar]
- 3.Itoh Y, Shinya K, Kiso M, Watanabe T, Sakoda Y, Hatta M, Muramoto Y, Tamura D, Sakai-Tagawa Y, Noda T, Sakabe S, Imai M, Hatta Y, Watanabe S, Li C, Yamada S, Fujii K, Murakami S, Imai H, Kakugawa S, Ito M, Takano R, Iwatsuki-Horimoto K, Shimojima M, Horimoto T, Goto H, Takahashi K, Makino A, Ishigaki H, Nakayama M, Okamatsu M, Takahashi K, Warshauer D, Shult PA, Saito R, Suzuki H, Furuta Y, Yamashita M, Mitamura K, Nakano K, Nakamura M, Brockman-Schneider R, Mitamura H, Yamazaki M, Sugaya N, Suresh M, Ozawa M, Neumann G, Gern J, Kida H, Ogasawara K, Kawaoka Y. 2009. In vitro and in vivo characterization of new swine-origin H1N1 influenza viruses. Nature 460:1021–1025. doi: 10.1038/nature08260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maines TR, Jayaraman A, Belser JA, Wadford DA, Pappas C, Zeng H, Gustin KM, Pearce MB, Viswanathan K, Shriver ZH, Raman R, Cox NJ, Sasisekharan R, Katz JM, Tumpey TM. 2009. Transmission and pathogenesis of swine-origin 2009 A(H1N1) influenza viruses in ferrets and mice. Science 325:484–487. doi: 10.1126/science.1177238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Benne CA, Kraaijeveld CA, van Strijp JA, Brouwer E, Harmsen M, Verhoef J, van Golde LM, van Iwaarden JF. 1995. Interactions of surfactant protein A with influenza A viruses: binding and neutralization. J Infect Dis 171:335–341. doi: 10.1093/infdis/171.2.335. [DOI] [PubMed] [Google Scholar]
- 6.Hartshorn KL, Crouch EC, White MR, Eggleton P, Tauber AI, Chang D, Sastry K. 1994. Evidence for a protective role of pulmonary surfactant protein D (SP-D) against influenza A viruses. J Clin Invest 94:311–319. doi: 10.1172/JCI117323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hartshorn KL, White MR, Mogues T, Ligtenberg T, Crouch E, Holmskov U. 2003. Lung and salivary scavenger receptor glycoprotein-340 contribute to the host defense against influenza A viruses. Am J Physiol Lung Cell Mol Physiol 285:L1066–L1076. doi: 10.1152/ajplung.00057.2003. [DOI] [PubMed] [Google Scholar]
- 8.Job ER, Bottazzi B, Gilbertson B, Edenborough KM, Brown LE, Mantovani A, Brooks AG, Reading PC. 2013. Serum amyloid P is a sialylated glycoprotein inhibitor of influenza A viruses. PLoS One 8:e59623. doi: 10.1371/journal.pone.0059623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.LeVine AM, Whitsett JA, Hartshorn KL, Crouch EC, Korfhagen TR. 2001. Surfactant protein D enhances clearance of influenza A virus from the lung in vivo. J Immunol 167:5868–5873. doi: 10.4049/jimmunol.167.10.5868. [DOI] [PubMed] [Google Scholar]
- 10.Pan Q, Chen H, Wang F, Jeza VT, Hou W, Zhao Y, Xiang T, Zhu Y, Endo Y, Fujita T, Zhang XL. 2012. L-ficolin binds to the glycoproteins hemagglutinin and neuraminidase and inhibits influenza A virus infection both in vitro and in vivo. J Innate Immun 4:312–324. doi: 10.1159/000335670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reading PC, Bozza S, Gilbertson B, Tate M, Moretti S, Job ER, Crouch EC, Brooks AG, Brown LE, Bottazzi B, Romani L, Mantovani A. 2008. Antiviral activity of the long chain pentraxin PTX3 against influenza viruses. J Immunol 180:3391–3398. doi: 10.4049/jimmunol.180.5.3391. [DOI] [PubMed] [Google Scholar]
- 12.Verma A, White M, Vathipadiekal V, Tripathi S, Mbianda J, Ieong M, Qi L, Taubenberger JK, Takahashi K, Jensenius JC, Thiel S, Hartshorn KL. 2012. Human H-ficolin inhibits replication of seasonal and pandemic influenza A viruses. J Immunol 189:2478–2487. doi: 10.4049/jimmunol.1103786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anders EM, Hartley CA, Jackson DC. 1990. Bovine and mouse serum beta inhibitors of influenza A viruses are mannose-binding lectins. Proc Natl Acad Sci U S A 87:4485–4489. doi: 10.1073/pnas.87.12.4485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krizanova O, Rathova V. 1969. Serum inhibitors of myxoviruses. Curr Top Microbiol Immunol 47:125–151. [DOI] [PubMed] [Google Scholar]
- 15.Ryan-Poirier KA, Kawaoka Y. 1993. Alpha 2-macroglobulin is the major neutralizing inhibitor of influenza A virus in pig serum. Virology 193:974–976. doi: 10.1006/viro.1993.1208. [DOI] [PubMed] [Google Scholar]
- 16.Kesimer M, Scull M, Brighton B, DeMaria G, Burns K, O'Neal W, Pickles RJ, Sheehan JK. 2009. Characterization of exosome-like vesicles released from human tracheobronchial ciliated epithelium: a possible role in innate defense. FASEB J 23:1858–1868. doi: 10.1096/fj.08-119131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Job ER, Deng YM, Tate MD, Bottazzi B, Crouch EC, Dean MM, Mantovani A, Brooks AG, Reading PC. 2010. Pandemic H1N1 influenza A viruses are resistant to the antiviral activities of innate immune proteins of the collectin and pentraxin superfamilies. J Immunol 185:4284–4291. doi: 10.4049/jimmunol.1001613. [DOI] [PubMed] [Google Scholar]
- 18.Qi L, Kash JC, Dugan VG, Jagger BW, Lau YF, Sheng ZM, Crouch EC, Hartshorn KL, Taubenberger JK. 2011. The ability of pandemic influenza virus hemagglutinins to induce lower respiratory pathology is associated with decreased surfactant protein D binding. Virology 412:426–434. doi: 10.1016/j.virol.2011.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lerdsamran H, Pittayawonganon C, Pooruk P, Mungaomklang A, Iamsirithaworn S, Thongcharoen P, Kositanont U, Auewarakul P, Chokephaibulkit K, Oota S, Pongkankham W, Silaporn P, Komolsiri S, Noisumdaeng P, Chotpitayasunondh T, Sangsajja C, Wiriyarat W, Louisirirotchanakul S, Puthavathana P. 2011. Serological response to the 2009 pandemic influenza A (H1N1) virus for disease diagnosis and estimating the infection rate in Thai population. PLoS One 6:e16164. doi: 10.1371/journal.pone.0016164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hoffmann E, Krauss S, Perez D, Webby R, Webster RG. 2002. Eight-plasmid system for rapid generation of influenza virus vaccines. Vaccine 20:3165–3170. doi: 10.1016/S0264-410X(02)00268-2. [DOI] [PubMed] [Google Scholar]
- 21.Roque-Barreira MC, Campos-Neto A. 1985. Jacalin: an IgA-binding lectin. J Immunol 134:1740–1743. [PubMed] [Google Scholar]
- 22.World Health Organization. 6 October 2009. CDC protocol of realtime RTPCR for influenza A (H1N1). World Health Organization, Geneva, Switzerland: http://www.who.int/csr/resources/publications/swineflu/realtimeptpcr/en/. [Google Scholar]
- 23.Tate MD, Brooks AG, Reading PC. 2011. Specific sites of N-linked glycosylation on the hemagglutinin of H1N1 subtype influenza A virus determine sensitivity to inhibitors of the innate immune system and virulence in mice. J Immunol 187:1884–1894. doi: 10.4049/jimmunol.1100295. [DOI] [PubMed] [Google Scholar]
- 24.Butler J, Hooper KA, Petrie S, Lee R, Maurer-Stroh S, Reh L, Guarnaccia T, Baas C, Xue L, Vitesnik S, Leang SK, McVernon J, Kelso A, Barr IG, McCaw JM, Bloom JD, Hurt AC. 2014. Estimating the fitness advantage conferred by permissive neuraminidase mutations in recent oseltamivir-resistant A(H1N1)pdm09 influenza viruses. PLoS Pathog 10:e1004065. doi: 10.1371/journal.ppat.1004065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Van Kerkhove MD, Hirve S, Koukounari A, Mounts AW. 2013. Estimating age-specific cumulative incidence for the 2009 influenza pandemic: a meta-analysis of A(H1N1)pdm09 serological studies from 19 countries. Influenza Other Respir Viruses 7:872–886. doi: 10.1111/irv.12074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Prachayangprecha S, Makkoch J, Vuthitanachot C, Vuthitanachot V, Payungporn S, Chieochansin T, Theamboonlers A, Poovorawan Y. 2011. Epidemiological and serological surveillance of human pandemic influenza A virus infections during 2009-2010 in Thailand. Jpn J Infect Dis 64:377–381. [PubMed] [Google Scholar]
- 27.Kabir S. 1998. Jacalin: a jackfruit (Artocarpus heterophyllus) seed-derived lectin of versatile applications in immunobiological research. J Immunol Methods 212:193–211. doi: 10.1016/S0022-1759(98)00021-0. [DOI] [PubMed] [Google Scholar]
- 28.Kajon AE, Gigliotti AP, Harrod KS. 2003. Acute inflammatory response and remodeling of airway epithelium after subspecies B1 human adenovirus infection of the mouse lower respiratory tract. J Med Virol 71:233–244. doi: 10.1002/jmv.10475. [DOI] [PubMed] [Google Scholar]
- 29.Vigerust DJ, Ulett KB, Boyd KL, Madsen J, Hawgood S, McCullers JA. 2007. N-linked glycosylation attenuates H3N2 influenza viruses. J Virol 81:8593–8600. doi: 10.1128/JVI.00769-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gerlach T, Kuhling L, Uhlendorff J, Laukemper V, Matrosovich T, Czudai-Matwich V, Schwalm F, Klenk HD, Matrosovich M. 2012. Characterization of the neuraminidase of the H1N1/09 pandemic influenza virus. Vaccine 30:7348–7352. doi: 10.1016/j.vaccine.2012.09.078. [DOI] [PubMed] [Google Scholar]
- 31.Wong DD, Choy KT, Chan RW, Sia SF, Chiu HP, Cheung PP, Chan MC, Peiris JS, Yen HL. 2012. Comparable fitness and transmissibility between oseltamivir-resistant pandemic 2009 and seasonal H1N1 influenza viruses with the H275Y neuraminidase mutation. J Virol 86:10558–10570. doi: 10.1128/JVI.00985-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Neumann G, Noda T, Kawaoka Y. 2009. Emergence and pandemic potential of swine-origin H1N1 influenza virus. Nature 459:931–939. doi: 10.1038/nature08157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Garten RJ, Davis CT, Russell CA, Shu B, Lindstrom S, Balish A, Sessions WM, Xu X, Skepner E, Deyde V, Okomo-Adhiambo M, Gubareva L, Barnes J, Smith CB, Emery SL, Hillman MJ, Rivailler P, Smagala J, de Graaf M, Burke DF, Fouchier RA, Pappas C, Alpuche-Aranda CM, Lopez-Gatell H, Olivera H, Lopez I, Myers CA, Faix D, Blair PJ, Yu C, Keene KM, Dotson PD Jr, Boxrud D, Sambol AR, Abid SH, St George K, Bannerman T, Moore AL, Stringer DJ, Blevins P, Demmler-Harrison GJ, Ginsberg M, Kriner P, Waterman S, Smole S, Guevara HF, Belongia EA, Clark PA, Beatrice ST, Donis R, Katz J, Finelli L, Bridges CB, Shaw M, Jernigan DB, Uyeki TM, Smith DJ, Klimov AI, Cox NJ. 2009. Antigenic and genetic characteristics of swine-origin 2009 A(H1N1) influenza viruses circulating in humans. Science 325:197–201. doi: 10.1126/science.1176225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smith GJ, Vijaykrishna D, Bahl J, Lycett SJ, Worobey M, Pybus OG, Ma SK, Cheung CL, Raghwani J, Bhatt S, Peiris JS, Guan Y, Rambaut A. 2009. Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza A epidemic. Nature 459:1122–1125. doi: 10.1038/nature08182. [DOI] [PubMed] [Google Scholar]
- 35.Yen HL, Liang CH, Wu CY, Forrest HL, Ferguson A, Choy KT, Jones J, Wong DD, Cheung PP, Hsu CH, Li OT, Yuen KM, Chan RW, Poon LL, Chan MC, Nicholls JM, Krauss S, Wong CH, Guan Y, Webster RG, Webby RJ, Peiris M. 2011. Hemagglutinin-neuraminidase balance confers respiratory-droplet transmissibility of the pandemic H1N1 influenza virus in ferrets. Proc Natl Acad Sci U S A 108:14264–14269. doi: 10.1073/pnas.1111000108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu R, Zhu X, McBride R, Nycholat CM, Yu W, Paulson JC, Wilson IA. 2012. Functional balance of the hemagglutinin and neuraminidase activities accompanies the emergence of the 2009 H1N1 influenza pandemic. J Virol 86:9221–9232. doi: 10.1128/JVI.00697-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bhatia A, Kast RE. 2007. How influenza's neuraminidase promotes virulence and creates localized lung mucosa immunodeficiency. Cell Mol Biol Lett 12:111–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hurt AC, Nor'e SS, McCaw JM, Fryer HR, Mosse J, McLean AR, Barr IG. 2010. Assessing the viral fitness of oseltamivir-resistant influenza viruses in ferrets, using a competitive-mixtures model. J Virol 84:9427–9438. doi: 10.1128/JVI.00373-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moscona A. 2009. Global transmission of oseltamivir-resistant influenza. N Engl J Med 360:953–956. doi: 10.1056/NEJMp0900648. [DOI] [PubMed] [Google Scholar]
- 40.Bloom JD, Gong LI, Baltimore D. 2010. Permissive secondary mutations enable the evolution of influenza oseltamivir resistance. Science 328:1272–1275. doi: 10.1126/science.1187816. [DOI] [PMC free article] [PubMed] [Google Scholar]