

ABSTRACT
Poxviridae are viruses with a large linear double-stranded DNA genome coding for up to 250 open reading frames and a fully cytoplasmic replication. The double-stranded DNA genome is covalently circularized at both ends. Similar structures of covalently linked extremities of the linear DNA genome are found in the African swine fever virus (asfarvirus) and in the Phycodnaviridae. We are studying the machinery which replicates this peculiar genome structure. From our work with vaccinia virus, we give first insights into the overall structure and function of the essential poxvirus virus helicase-primase D5 and show that the active helicase domain of D5 builds a hexameric ring structure. This hexamer has ATPase and, more generally, nucleoside triphosphatase activities that are indistinguishable from the activities of full-length D5 and that are independent of the nature of the base. In addition, hexameric helicase domains bind tightly to single- and double-stranded DNA. Still, the monomeric D5 helicase construct truncated within the D5N domain leads to a well-defined structure, but it does not have ATPase or DNA-binding activity. This shows that the full D5N domain has to be present for hexamerization. This allowed us to assign a function to the D5N domain which is present not only in D5 but also in other viruses of the nucleocytoplasmic large DNA virus (NCLDV) clade. The primase domain and the helicase domain were structurally analyzed via a combination of small-angle X-ray scattering and, when appropriate, electron microscopy, leading to consistent low-resolution models of the different proteins.
IMPORTANCE Since the beginning of the 1980s, research on the vaccinia virus replication mechanism has basically stalled due to the absence of structural information. As a result, this important class of pathogens is less well understood than most other viruses. This lack of information concerns in general viruses of the NCLDV clade, which use a superfamily 3 helicase for replication, as do poxviruses. Here we provide for the first time information about the domain structure and DNA-binding activity of D5, the poxvirus helicase-primase. This result not only refines the current model of the poxvirus replication fork but also will lead in the long run to a structural basis for antiviral drug design.
INTRODUCTION
Viruses of the family Poxviridae have a large linear double-stranded DNA (dsDNA) genome coding for up to 250 open reading frames. The double-stranded DNA genome is covalently circularized at both ends and is fully replicated in the cytoplasm. Similar structures of covalently linked extremities of the linear DNA genome are found in the African swine fever virus (asfarvirus) (1) and in the Phycodnaviridae (reviewed in reference 2). Like poxviruses, these viruses are also members of the nucleocytoplasmic large DNA virus (NCLDV) clade (3).
We are working on vaccinia virus, which was used for the vaccination campaign that led to the eradication of smallpox. It is a convenient and rather safe system for the study of general poxvirus functions. With an amino acid sequence identity of 98% between vaccinia virus and smallpox virus, the proteins of the DNA replication machinery are virtually identical between the two viruses.
The replication machinery is mainly composed of five essential proteins, which are highly conserved within the poxvirus family (reviewed in reference 4): the three proteins that form the DNA polymerase holoenzyme, E9, A20, and D4; the single-stranded DNA-binding protein I3; and the helicase-primase D5.
Here we focus on the helicase-primase D5. The D5 protein is 95 kDa and has an N-terminal archaeoeukaryotic primase (AEP) domain displaying an RNA recognition motif (RRM) type of fold (residues 1 to 240) followed by a cysteine cluster region (residues 240 to 345), which is associated with several primase domains of the NCLDV clade (5). They are followed by a D5N domain (residues 340 to 460), which is always associated with D5-type helicases, and, finally, at the C terminus, a superfamily 3 (SF3) helicase domain (residues 460 to 785) (6), which contains the classical Walker A- or P-loop motif, the Walker B motif, motif C, and a motif characteristic of the AAA+ ATPase family (7).
Despite the presence of the 4 domains (Fig. 1A), which participate in a modular architecture in a number of proteins with a helicase and/or primase function within the NCLDV clade, mimiviruses (8), as well as the P4 bacteriophages (3, 5), the only known high-resolution structures concern the primase domain (reviewed in reference 5). For the SF3 helicase domains, a ring-like structure, often with a 6-fold symmetry like that for D5, can be inferred (6, 9). Information on the arrangement of the individual domains is still lacking.
FIG 1.

Different constructs of D5. (A) Domain organization of D5 and the different constructs used. Black lines, the constructs (residues 301 to 785 and residues 412 to 785) used by Boyle and coworkers (11). (B) ATPase activity of the different proteins. (C) Nucleotide preference of D5323–735 and controls. NTP, nucleoside triphosphate. (D) Refractive index increments (black) are plotted as a function of the elution time, together with the corresponding mass determination from MALLS (gray).
D5 shows intrinsic nucleoside triphosphatase (NTPase) activity, which is independent of any nucleic acid cofactors and requires oligomerization (10, 11). The helicase activity remains elusive, whereas the primase activity has been demonstrated (12).
As a member of the SF3 family, it can be inferred that D5 moves in 3′-to-5′ direction on its substrate DNA strand (6). Recently, a role of D5 in genome uncoating, a process that liberates the genome after entry into a new host, has been shown. Here D5 localizes to incoming cores as well as to virus factories and I3-positive prereplication sites (13). Additionally, D5 possibly interacts with A20, but this interaction is supposed to be rather transient (14). Given D5's involvement in genome uncoating and replication, it could be a promising antiviral target with a strong therapeutic potential.
Here we define the oligomerization domain of D5 responsible for the formation of an active hexameric ring structure with nucleoside triphosphatase activity independent of the nature of the nucleotide. Different hexameric and monomeric domains of D5 have been studied by small-angle X-ray scattering (SAXS) and, when appropriate, electron microscopy (EM), leading to first insights into the structural arrangement. Finally, we show that the oligomeric helicase domain tightly binds single-stranded DNA (ssDNA) and dsDNA.
MATERIALS AND METHODS
Cloning and expression.
The full-length D5 construct was cloned as a His-tagged tobacco etch virus (TEV) protease-cleavable construct and expressed in SF21 cells (9).
The following constructs were cloned into the pProEx HTb vector (Life Technologies): D5 from amino acids 323 to 785 (D5323–785; primers 5′-GCGCCATGGG TAATAAACTG TTTAATATTG CAC-3′ and 5′-ATGCAAGCTT TTACGGAGAT GAAATATCCT CTATGA-3′), D5381–785 (primers 5′-GCGCCATGGC TAGCGAATTA CTCTGTCCGA G-3′, 5′-ATGCAAGCTT TTACGGAGAT GAAATATCCT CTATGA-3′), and D51–335 (primers 5′-CGCCATGGCG ATGGATGCGG CTATTAGAGG-3′, 5′-AGCCAAGCTT AGTCTAAAAT TCTTTGTGCA ATATT-3′). The constructs were expressed in Escherichia coli BL21 Star(DE3) (Novagene), in which expression was induced with 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside), at 18°C overnight.
Purification.
The insect cell pellet expressing full-length D5 was resuspended in 10 volumes of lysis buffer (50 mM Tris-HCl [pH 7], 150 mM NaCl, 5 mM MgCl2, 10 mM β-mercaptoethanol, 10% glycerol) with cOmplete protease inhibitor cocktail (Roche) and 1 μl/10 ml Benzonase (250 U/μl; Merck-Millipore). Cells were lysed by using a Potter S homogenizer (B. Braun Biotech). Pellets from E. coli bacteria expressing the D5323–785 and D5381–785 constructs were resuspended in the same lysis buffer but were lysed by sonication.
The lysates were clarified by centrifugation at 50,000 × g for 30 min at 4°C. D5 and its deletion mutants were loaded onto nickel affinity columns (HIS-select; Sigma-Aldrich). The columns were washed with 10 column volumes (CV) lysis buffer, 10 CV washing buffer (50 mM Tris-HCl [pH 7], 1 M NaCl, 10 mM β-mercaptoethanol, 10% glycerol), and 10 CV imidazole washing buffer (50 mM Tris-HCl [pH 7], 150 mM NaCl, 10 mM β-mercaptoethanol, 10% glycerol, 30 mM imidazole). The proteins were eluted with elution buffer (20 mM Tris-HCl [pH 7], 150 mM NaCl, 10 mM β-mercaptoethanol, 10% glycerol, 200 mM imidazole) and concentrated using Amicon centrifugal filter units (Millipore), prior to buffer exchange back to the lysis buffer on Econo-Pac 10DG columns (Bio-Rad). TEV digestions were carried out overnight at room temperature (RT), and the cleaved proteins were collected in the flowthrough of a second Ni column purification. For the D5323–785 and D5381–785 proteins, the samples were injected onto a Superose 6 or Superdex 200 column (GE Healthcare) that had been equilibrated with gel filtration buffer I (20 mM Tris-HCl [pH 7], 150 mM NaCl, 10% glycerol, 1 mM dithiothreitol [DTT]). D5323–785 was further purified using a Mono Q column (Bio-Rad), and the purified protein was concentrated and rechromatographed on a Superose 6 column.
Cells expressing the D51–335 construct were lysed via sonication in lysis buffer II (50 mM Tris-HCl [pH 8], 150 mM NaCl, 10 mM β-mercaptoethanol, cOmplete protease inhibitor cocktail) and purified over an Ni column (25 CV wash with lysis buffer II and elution buffer, consisting of 50 mM Tris-HCl [pH 8], 150 mM NaCl, 200 mM imidazole, 10 mM β-mercaptoethanol). After TEV cleavage, the protein was run over a second Ni column as described above, before a last purification step on a Superdex 200 column in gel filtration buffer II (20 mM Tris-HCl [pH 8], 150 mM NaCl, 1 mM DTT).
Peak fractions were analyzed by SDS-PAGE, and the gels were stained with InstantBlue (Expedeon).
NTPase activity assay.
Five hundred nanograms of D5 or a deletion construct was mixed in reaction buffer (50 mM bis-Tris propane [pH 7.6], 150 mM NaCl, 5 mM MgCl2, 3% glycerol, 0.3 mg/ml bovine serum albumin) with 3 mM nucleoside triphosphates, and the mixture was incubated at 37°C for 1 h. A malachite green phosphate assay kit (BioAssay Systems) was used in a 96-well plate format for colorimetric phosphate determination at 620 nm following the instructions in the manufacturer's manual. Color reactions were measured with a Tecan Sunrise plate reader.
Size exclusion chromatography (SEC) combined with detection by multiangle laser light scattering (MALLS) and refractometry (RI).
Purified D5 mutants were analyzed using a Superdex 200 or Superose 6 column (GE Healthcare) that had been equilibrated in the gel filtration buffers (described above) at a flow rate of 0.5 ml/min. Fifty microliters of a protein solution at concentrations ranging from 3 to 9 mg/ml was injected. A Dawn-Heleos II detector (Wyatt Technology) with a laser emitting at 690 nm was used. The protein concentration was determined by measuring the differential refractive index with an Optilab T-rEX detector (Wyatt Technology), using a refractive index increment (dn/dc) of 0.185 ml/g. Weight-averaged molecular masses were calculated with the ASTRA program (Wyatt Technology) as described previously (15).
Collection of SAXS data at ESRF.
Experiments were performed at European Synchrotron Radiation Facility (ESRF; Grenoble, France) BioSAXS beamline BM29 (16). To obtain the measurements, 30 μl of protein solution at three different concentrations for each sample alternated with buffer measurements for background subtraction and the robotic sample handling available at the beamline were used (17). The experiments were programmed using the ISPyB BioSAXS interface (18), and the sequence was triggered via the BsxCuBE control software. Ten individual frames for every exposure, each of which was 2 s in duration, were collected using a Pilatus 1M detector (Dectris). Individual frames were processed automatically and independently within the EDNA framework, yielding radially averaged curves of normalized intensity versus the magnitude of the scattering vector (s), which is equal to 4πsinθ/λ, where θ is half the scattering angle. The calculation of an average scattering curve for each exposure series used data reduction within EDNA, which is based on the automatic data processing tools of the ATSAS package (19), in order to combine individual frames; frames affected by radiation damage were excluded. Data obtained from different sample concentrations were merged and analyzed further using the tools of the ATSAS package (19) and PRIMUS software (20). Forward scattering intensity (I0) and the radius of gyration (Rg) were calculated using the Guinier approximation (21). The hydrated particle volume was computed using the Porod invariant (22), and the maximum particle size (Dmax) was determined from the pair distribution function computed by the GNOM program (23). Forty ab initio models were calculated using the DAMMIF program (24) and averaged, aligned, and compared using the DAMAVER program (25). Rigid-body modeling of the complex was done with CORAL software (19). The theoretical scattering of atomic structures was calculated with CRYSOL software (26). The EM2DAM program was used in order to convert EM electron density maps to pseudoatomic models. The theoretical scattering curve was calculated and scaled with a least-squares minimization to the experimental data using CRYSOL. Graphs were prepared with Microsoft Office Excel software, and structures were represented using the PyMOL program (Schrödinger).
Electron microscopy of D5323–785.
About 4 μl of the D5323–785 protein sample was applied to a mica sheet covered with a film of evaporated carbon. The carbon film was then floated off the mica in glycerol-free buffer, retrieved, and placed onto a 400-mesh copper electron microscopy grid. The sample was subsequently stained with 4 μl of 2% sodium silicotungstate (SST) and dried. Imaging was performed on an FEI F20 transmission electron microscope operating at 200 kV. Images were recorded on a 4k×4k Eagle digital camera with a pixel size of 2.17 Å. A total of 25,703 particles were semiautomatically selected from 50 charge-coupled-device frames with EMAN software (27). Subframes were corrected for the contrast transfer function with the CTFFIND3 program (28) and sorted with reference-free two-dimensional class averaging in the RELION program (29). Low-resolution initial models were generated with the RIco program (30) using C6 symmetry. Three-dimensional refinement was performed by maximum likelihood analysis in RELION without imposed symmetry. The resulting model was made symmetric in RELION using C6 symmetry. The contour level was adjusted in the Chimera program (31) in order to match the volume of the protein, assuming a specific volume of 1.21 Å3/Da.
Fluorescence anisotropy measurements.
Fluorescence spectroscopy was used to determine the dissociation constant (Kd) and the stoichiometry involving D5 and DNA oligomers. 6-Carboxyfluorescein-5′-TTGCAGCACATCCCCCTTTC-3′ DNA was used as the ssDNA oligomer or was used together with a fully complementary DNA as a duplex. The ssDNA oligomer and the ssDNA oligomer with a fully complementary DNA were dissolved in gel filtration buffer I and used at concentrations of 13 nM and 22 nM, respectively, for the determination of the Kd and 500 nM and 840 nM, respectively, for the determination of the stoichiometry. Protein was successively added in order to obtain the concentrations indicated above. When the equilibrium was obtained, the total fluorescence intensity and fluorescence anisotropy were measured at 515 nm at room temperature in a fluorimeter (Photon Technology International) equipped with polarizers using an excitation wavelength of 495 nm. Total fluorescence intensity was corrected for the volume change due to the addition of the protein stock, and the observed fluorescence anisotropy was corrected for the change in fluorescence intensity according to equation 1 (32):
| (1) |
where fb represents the fractional concentration of bound DNA; r represents the observed anisotropy; rf and rb represent the anisotropy of free and bound DNA, respectively; and If and Ib represent the fluorescence intensity of free and bound DNA, respectively. Using KaleidaGraph software (Synergy Software), dissociation constants were derived by fitting the binding isotherms to a 1:1 binding model of DNA and protein unit according to equation 2:
| (2) |
where L represents the total concentration of labeled DNA, and P represents the concentration of the (hexameric) protein partner.
SPR.
Surface plasmon resonance (SPR) experiments were carried out on a Biacore 3000 instrument (GE Healthcare) using CM5 sensor chips with immobilized streptavidin to which the 5′-end-biotinylated primer 5′-CCG AAT CAG GAA GAT AAC AGC GGT TTA GCC-3′ was bound directly or after annealing to the complementary sequence as described previously (33). Experiments were performed in the appropriate gel filtration buffers described above at a flow rate of 15 μl/min. Serial dilutions of the D5 deletion mutants were injected onto the streptavidin-bound DNA for 9 min (association phase), followed by a 15-min dissociation phase. Surfaces were regenerated using 0.05% SDS. Signals from which the background signal was subtracted were exported from the Biologic software (GE Healthcare) and analyzed with the LibreOffice Calc spreadsheet (www.libreoffice.org) using the Solver function for curve fitting.
RESULTS
ATPase activity requires hexamerization.
D5 is thought to display primase and helicase activities. The primase activity has been demonstrated (12), whereas the helicase activity has not; only an ATPase activity independent of the presence or absence of DNA could be shown (10, 11). We sought to attribute these activities more precisely to the different domains of D5. We searched for deletion constructs that could be produced in E. coli and that showed ATPase activity similar to that of the full-length protein, which required the baculovirus system for recombinant expression. Three constructs were well expressed: a construct containing the primase and almost all of the Cys cluster domain (residues 1 to 335) and two constructs (construct 1, D5 residues 323 to 785 [D5323–785]; construct 2, D5 residues 381 to 785 [D5381–785]) containing the helicase domain. Construct 1 also contained the entire D5N domain, which was partially truncated in construct 2 (Fig. 1A). We tested the three proteins for ATPase activity. While the purified D51–335 and the D5381–785 proteins did not show any, D5323–785 showed activity similar to that of the D5 full-length protein expressed in insect cells (Fig. 1B). We checked whether the D5323–785 construct had the same lack of nucleotide preference described previously for full-length D5 (10), and indeed, no significant difference between ATP, CTP, GTP, and UTP was observed, confirming the previous findings (Fig. 1C).
We sought to understand why D5323–785 was active but the D5381–785 construct was not. Hence, we determined the mass of all three proteins by MALLS-RI (Fig. 1D) and obtained molecular masses of 39.5 kDa for D51–335 (theoretical molecular mass, 39.0 kDa), 48.7 kDa for D5381–785 (theoretical molecular mass, 46.7 kDa), and 349 kDa for D5323–785 (theoretical molecular mass, 53.5 kDa), indicating that the last construct forms a hexamer (theoretical molecular mass, 321 kDa) like full-length D5 does (9). As a consequence, residues 323 to 380 of the D5N domain must be important for hexamer formation.
In conclusion, the D5323–785 construct is a hexameric oligomer with ATPase activity similar to that of the native protein. On the other hand, when primase and the Cys cluster domain are expressed together, as in D51–335, the construct is monomeric, excluding a role in hexamerization of the domains located N terminally of D5N.
Analysis of the monomeric and hexameric helicase fragments via a combined SAXS-electron microscopy study.
Having shown the monodisperse character of D51–335 containing the primase domain, we used SAXS to characterize its structure further (Fig. 2A; Table 1). The ab initio model indicates an elongated structure with a bulkier middle part (Fig. 2B).
FIG 2.
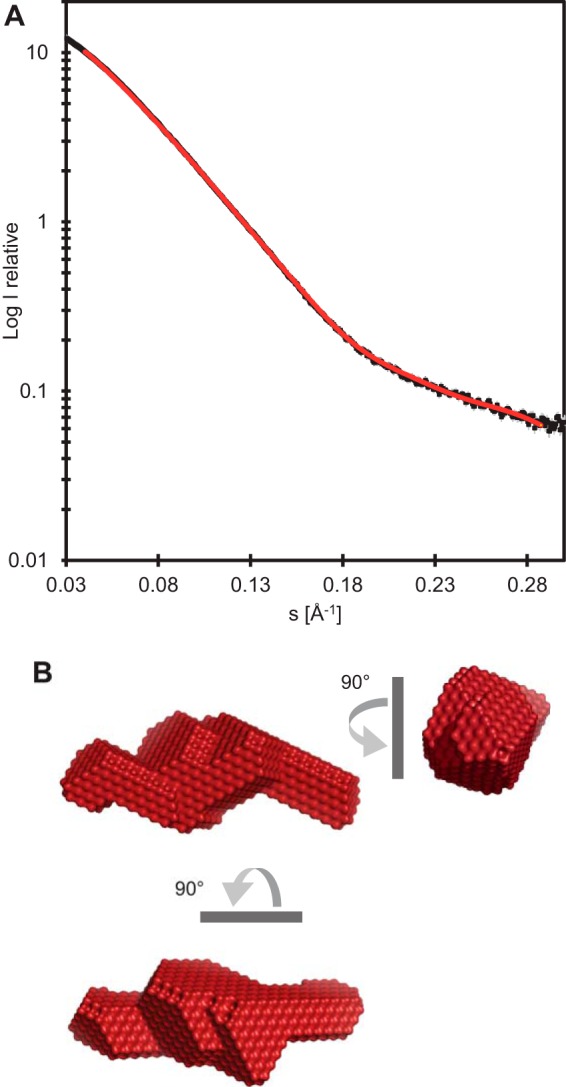
SAXS of D51–335. (A) An experimental scattering curve (black) and the curve (red) calculated for the model shown in panel B.
TABLE 1.
SAXS-derived parametersa
| Construct | Rg from Guinier plot (nm) | Reciprocal space Rg (nm)b | Dmax (nm)b | Porod vol (103 Å3) | Molecular mass (kDa) calculated from: |
|
|---|---|---|---|---|---|---|
| Porod volc | Sequence | |||||
| D51–335 | 2.9 | 2.9 | 11 | 65 | 39 | 39.0 |
| D5323–785 | 4.8 | 4.7 | 14.5 | 570 | 345 | 53.5 × 6 (321) |
| D5381–785 | 2.8 | 2.8 | 9 | 80 | 48 | 46.7 |
Absolute uncertainties for Rg from the Guinier plot, the reciprocal space Rg, and Dmax were ±0.05 nm, ±0.1 nm, and ±0.4 nm. The relative uncertainties of the Porod volume and the molecular mass from the Porod volume were 5%.
Calculated from the P(r) function.
Calculated according to reference 20.
Monodisperse samples of the D5381–785 and D5323–785 proteins containing the helicase domain were also analyzed by SAXS (Fig. 3A to D; Table 1). The modeling of the monomeric D5381–785 construct showed an elongated molecule with a more massive domain on one side (Fig. 3B). The analysis of the D5323–785 data gave a molecular mass estimate (from Porod analysis) of 345 kDa, which is in agreement with the expected mass of 321 kDa (Table 1). On the basis of the MALLS-RI data, which indicated the presence of a hexamer, as well as the observation of 6-fold symmetry for full-length D5 (9), ab initio modeling was undertaken with no imposed symmetry (χ2 = 0.88) and with C6 symmetry (χ2 = 1.0). As the overall fit to the scattering data for both reconstructions was comparable (Fig. 3A), C6 symmetry can be assumed. As a consequence, the model corresponds to a hexagonal cone-like structure with a central channel which appears to be partially obstructed (Fig. 3C and D). An examination of individual models before averaging shows that this obstruction is likely an artifact of the averaging process.
FIG 3.

Models of the helicase domain containing fragments D5381–785 and D5323–735 obtained from SAXS. (A) SAXS scattered intensity (I) of D5381–785 and D5323–735 and calculated intensity as a function of the magnitude of the scattering vector (s) for the models of both proteins with or without the use of C6 symmetry. The scattering curve calculated for SV40 Lta (PDB accession number 1N25) is also shown. Intensities are on an arbitrary scale. (B) Views of the bead model of a D5381–785 monomer (magenta). (C) Model of the D5323–735 hexamer (blue). (D) Cuts through the hexamer model showing the internal cavity. (E, F) Assembly of 6 monomeric models into a hexameric model of D5323–735 (E) and a comparison with the model of the hexamer shown in panel C (F). (G) Overlay of the models of hexameric D5323–735 (blue) and SV40 Lta (cartoon representation in green).
Based on these observations, we sought to reconstruct the hexamer from the monomeric model of the helicase (Fig. 3E), imposing 6-fold symmetry. The ab initio model of the monomer subunit could be fitted manually into the envelope of the hexamer (Fig. 3E and F). Furthermore, we compared the reconstruction obtained with C6 symmetry to known structures of SF3 helicases. The replicative helicase simian virus 40 (SV40) large tumor antigen (Lta; PDB accession number 1N25 [34]) appears to be the most similar, as it fits well the envelope (Fig. 3G) and scattering curve (Fig. 3A).
Although the monomeric protein fragments were too small for analysis by electron microscopy, this technique could be used in order to confirm the findings for the D5323–785 hexamers, which showed a clear 6-fold symmetry (Fig. 4A). Analysis of the reconstructed D5323–785 oligomer showed that the six protein subunits form a ring with tight interactions on one side, whereas on the other side, the subunits are not connected and leave a large opening along the 6-fold axis (Fig. 4B). A superposition of the electron microscopy map and the high-resolution structure of the SV40 Lta shows a good fit without further adjustments and allowed us to assign the handedness of the EM model (Fig. 4C). When the EM envelope was contoured at a level that gave the same volume as the SAXS ab initio model, there was not only a good agreement between the two models (Fig. 4D) but also a good match between the experimental scattering curve and the one calculated from the EM envelope (Fig. 4E). The discrepancy at about 0.07 Å−1 results from the smooth surface and abrupt transition between the protein and the solvent of the EM model.
FIG 4.

Reconstruction of D5323–735 from negative-stain EM. (A) (Left) Reference-free class averages of the views from negative-stain EM; (right) the best-matched projections from the reconstructed model. (B) Top, side, and bottom views of the EM reconstruction, from top to bottom, respectively. The contour level of the envelope has been set to correspond to the expected volume of the D5323–735 hexamer. (C) Comparison of the EM envelope (cyan) with the crystallographic model of the SV40 Lta hexamer (cartoon representation in green). (D) Comparison of the EM envelope (cyan) contoured at the contour level that gives the same volume as the ab initio bead model derived from SAXS (blue), which is superposed. (E) Theoretical scattering curve (blue) based on the envelope shown in panel D compared with the experimental scattering curve from SAXS (black).
The D5 helicase domain binds DNA tightly.
Despite advances concerning the overall structure of D5, classical helicase assays with blunt-end or forked DNA substrates performed by us (data not shown) or Boyle and coworkers (11) could not show detectable activity. We speculated that D5323–785 might still bind DNA. Using 6-carboxyfluorescein-labeled 20-mer ssDNA or dsDNA, a change of fluorescence anisotropy upon interaction with D5323–785 was observed. The Kd of the hexameric D5323–785 for ssDNA was about 1.7 ± 0.4 nM, and that for dsDNA was 2.8 ± 0.6 nM (Fig. 5A). DNA binding could be confirmed by surface plasmon resonance using biotinylated DNA oligomers immobilized on streptavidin. Here, ssDNA was bound with a Kd of 1.2 nM and dsDNA was bound with a Kd of 4.4 nM (Fig. 5C and D). Both methods indicated a strong binding of D5323–785 to DNA in the low-nanomolar range, whereas ssDNA appeared to be slightly preferred over dsDNA. Fluorescence anisotropy as well as SPR measurements using the monomeric helicase construct D5381–785 or the primase domain (D51–335) did not indicate any binding under our experimental conditions (data not shown). A titration experiment at higher DNA concentrations indicated the presence of one bound single-stranded or double-stranded DNA oligomer per D5323–785 hexamer (Fig. 5B).
FIG 5.
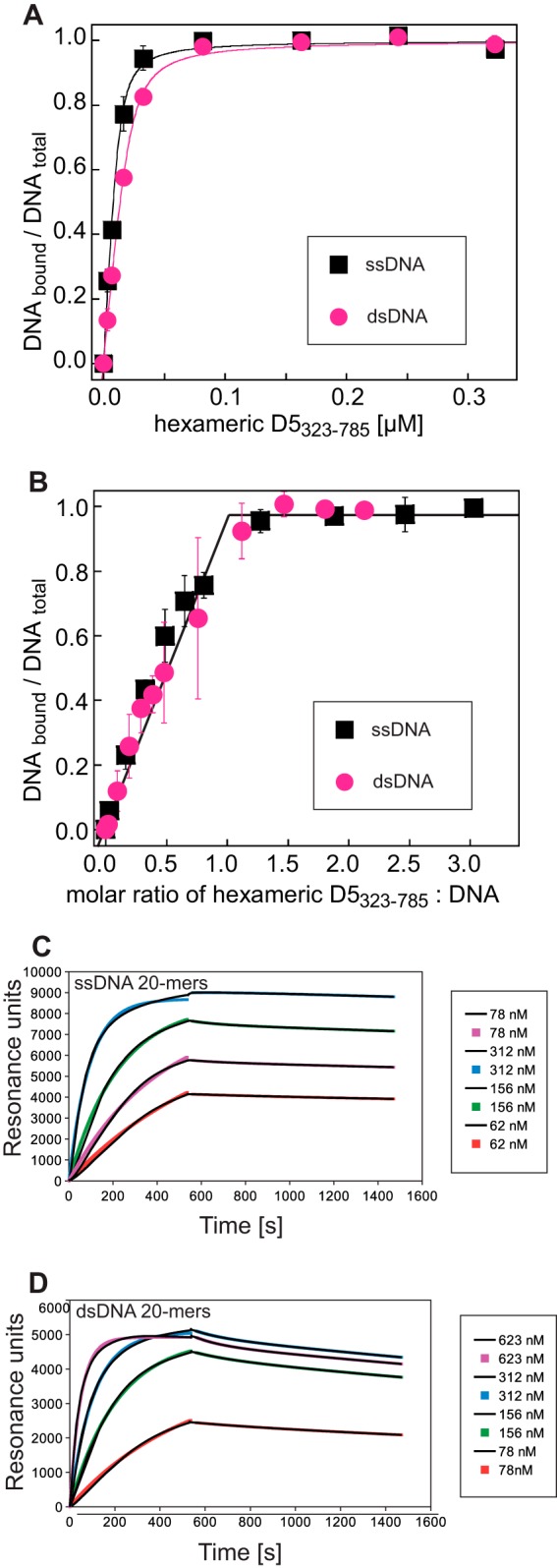
DNA binding of D5323–735 (A) Determination of the Kd of fluorescently labeled ssDNA or dsDNA oligomers using fluorescence anisotropy. (B) Determination of the binding stoichiometry. The break of the linear increase of the signal during titration indicates a stoichiometry of one DNA oligomer per hexamer. Fluorescence anisotropy measurements were performed in triplicate. Error bars indicate the standard deviation of the mean values. (C) Sensograms of an SPR experiment using biotinylated ssDNA 20-mers immobilized on a streptavidin-coated chip. Different concentrations of D5323–735 were flowed over the chip. Association phases were fitted with single exponentials. A biexponential fit was used to describe the dissociation phase, which was, however, clearly dominated by one exponential term used for the determination of the rate constant. Fitted curves use colored symbols, whereas the experimental curves are shown in black. (D) Curves corresponding to those in panel C for an experiment using immobilized dsDNA 20-mers.
DISCUSSION
The design of deletion constructs on the basis of secondary structure predictions allowed us to express separately a primase protein (D51–335), a monomeric helicase protein (D5381–785), and a hexameric helicase (D5323–785). The fact that the full D5N domain (Fig. 1A) had to be present for oligomerization assigned a first function to this domain. The hexameric helicase construct showed a nonspecific nucleoside triphosphatase activity similar to that of full-length D5 (11), proving that this activity is indeed due to the helicase domain and not the primase domain. The constructs (Fig. 1A) used by Boyle and coworkers (11) are cut within the same functional domains as our constructs and start at residue 301 in the Cys cluster domain (instead of residue 323 in our study) and at residue 412 in the D5N domain (instead of residue 381 in this study). They showed a similar behavior in ATPase assays and oligomerization, although at the time of the previous study the exact nature of the oligomer had not been addressed.
For the first time, we showed the tight binding of the hexameric construct but not the monomeric construct or the primase domain to short single-stranded or double-stranded DNA oligomers, despite the previously observed absence of an influence on ATP hydrolysis (10, 35). Although ATPase and DNA-binding activities were detected, the helicase activity remains elusive.
Structural information on D5 has been very limited, despite the presence of modular domains; the AEP primase domain, the Cys cluster domain, the D5N domain, and the SF3 helicase domain are shared with a number of viruses, in particular, members of the NCLDV clade (5). In general, with the exception of papillomavirus E1, helicases with a ring-like structure and a 3′-to-5′ directionality have been studied less than the well-characterized enzymes with opposite polarity, comprising bacteriophage T7 and bacterial replicative helicases.
Using SAXS and, for the hexameric D5323–785, EM, we obtained models of the different protein fragments, and these were consistent between the techniques used. The primase construct shows an elongated shape with a bulkier middle part (Fig. 2B). This middle part could correspond to the palm fold domain of primases, whereas the smaller extension could correspond to the N-terminal domain of AEP primases and the longer one could correspond to the Cys cluster domain (5), as judged from their sizes. The EM reconstruction of the hexameric helicase construct shows a hub with a central opening; turbine blades are attached to the hub, and these blades are probably free on the other side (Fig. 3 and 4B). The hexamer structure (Fig. 3C to E) can be built from the monomer reconstruction (Fig. 3B), which shows a bulky domain probably corresponding to the SF3 helicase domain and an extension on one side which, we believe, corresponds to the D5N domain. The fit of SV40 Lta into the hexamer envelopes from the SAXS and EM reconstructions (Fig. 3G and 4E) allowed us to orient the model. The helicase domain would be located on the wider side of the cone, whereas the N-terminally situated zinc-binding D1 domain (34) of Lta would be located on top of the cone close to the 6-fold axis. D5 does not have a direct equivalent of this domain. Instead, it has the D5N domain (Fig. 1), which is probably located at the same position. A superposition with the EM reconstruction shows that the helicase domains form the blades of the turbine (Fig. 4). A comparison of the envelope with the bovine papillomavirus E1 structure in complex with DNA (PDB accession number 2GXA [36]; data not shown) and the recent analysis of the DNA path through E1 (37) suggest an orientation of the DNA strand within the central channel of the hexamer in which the 5′ end is on the side of the D5N domain and the 3′ end is oriented toward the open side (Fig. 6). The polarity of SF3 helicases positions them on the leading strand, where they move ahead of the polymerase in the same 3′-to-5′ direction (Fig. 6). As a consequence, the model of the replication fork initially proposed by Sèle and coworkers (9) could be refined. Only the primase domain must still be placed in the model, but this requires a structure determination of full-length D5.
FIG 6.
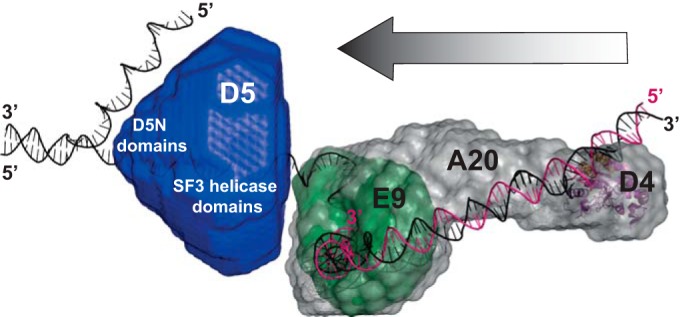
Proposed model of the poxvirus replication fork with our proposed domain structure and orientation of D5323–735. The model of the polymerase holoenzyme composed of E9, A20, and D4 is based on the model of Sèle et al. (9). Primase domains are omitted, as they cannot be positioned yet.
One of the unanswered questions is why, despite tight DNA binding, the helicase activity of D5 could not be shown in contrast to, for example, that of the papillomavirus E1 protein (38) or SV40 Lta (39). We propose that the DNA binding is different from a functional engagement of the DNA required for helicase activity. In this context, the general helicase loading mechanisms have to be considered. They involve an assembly of a dimeric precursor around the dsDNA at the origin of replication initiated by the binding of dimeric origin-binding domains, such as for the SF3 helicases SV40 Lta and papillomavirus E1, or a lateral ring opening, which has been shown for the eukaryotic MCM complex (reviewed in reference 40). However, as we never observed isolated subunits of the hexameric protein fragment either for the full-length protein or for D5381–785, this speaks against an assembly onto the viral DNA. So, a mechanism of lateral opening in order to load the DNA strand is more probable. From the current structural analysis, the possibility that the hexameric structure may be sufficiently flexible to allow a transient lateral opening of the hexameric ring cannot be excluded. This might require either very specific physicochemical conditions present only in vivo; a protein with a loader function, which still needs to be identified; or the particular telomere structure of the poxvirus genome (41), which has been shown to be necessary for efficient replication (42).
ACKNOWLEDGMENTS
We acknowledge financing of the project by French grant REPLIPOX ANR-13-BSV8-0014 and by research grants from the Service de Santé des Armées and the Délégation Générale pour l'Armement. This work used the platforms of the Grenoble Instruct Center (ISBG; UMS 3518 CNRS-CEA-UGA-EMBL) with support from FRISBI (ANR-10-INSB-05-02) and GRAL (ANR-10-LABX-49-01) within the Grenoble Partnership for Structural Biology (PSB). The electron microscopy facility is supported by the European Regional Development Fund, the French Rhône-Alpes Region, the Fondation de Recherche Médicale, and the Groupement d'Intérêt Scientifique IBISA.
Special thanks go to I. Gutsche for initial EM data, F. C. A. Gerard for her help with MALLS-RI, and P. Fender for help with the Biacore data. We thank the ESRF for SAXS beam time and thank M. Brennich for support. We further wish to acknowledge F. Garzoni from the Berger Group at EMBL Grenoble for his kind help in the setup of the MultiBac expression system.
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.González A, Talavera A, Almendral JM, Viñuela E. 1986. Hairpin loop structure of African swine fever virus DNA. Nucleic Acids Res 14:6835–6844. doi: 10.1093/nar/14.17.6835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Van Etten JL, Graves MV, Müller DG, Boland W, Delaroque N. 2002. Phycodnaviridae—large DNA algal viruses. Arch Virol 147:1479–1516. doi: 10.1007/s00705-002-0822-6. [DOI] [PubMed] [Google Scholar]
- 3.Iyer LM, Balaji S, Koonin EV, Aravind L. 2006. Evolutionary genomics of nucleo-cytoplasmic large DNA viruses. Virus Res 117:156–184. doi: 10.1016/j.virusres.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 4.Moss B. 2013. Poxvirus DNA replication. Cold Spring Harb Perspect Biol 5:a010199. doi: 10.1101/cshperspect.a010199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iyer LM, Koonin EV, Leipe DD, Aravind L. 2005. Origin and evolution of the archaeo-eukaryotic primase superfamily and related palm-domain proteins: structural insights and new members. Nucleic Acids Res 33:3875–3896. doi: 10.1093/nar/gki702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Singleton MR, Dillingham MS, Wigley DB. 2007. Structure and mechanism of helicases and nucleic acid translocases. Annu Rev Biochem 76:23–50. doi: 10.1146/annurev.biochem.76.052305.115300. [DOI] [PubMed] [Google Scholar]
- 7.Neuwald AF, Aravind L, Spouge JL, Koonin EV. 1999. AAA+: a class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res 9:27–43. [PubMed] [Google Scholar]
- 8.Raoult D, Audic S, Robert C, Abergel C, Renesto P, Ogata H, La Scola B, Suzan M, Claverie J-M. 2004. The 1.2-megabase genome sequence of mimivirus. Science 306:1344–1350. doi: 10.1126/science.1101485. [DOI] [PubMed] [Google Scholar]
- 9.Sèle C, Gabel F, Gutsche I, Ivanov I, Burmeister WP, Iseni F, Tarbouriech N. 2013. Low-resolution structure of vaccinia virus DNA replication machinery. J Virol 87:1679–1689. doi: 10.1128/JVI.01533-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Evans E, Klemperer N, Ghosh R, Traktman P. 1995. The vaccinia virus D5 protein, which is required for DNA replication, is a nucleic acid-independent nucleoside triphosphatase. J Virol 69:5353–5361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boyle KA, Arps L, Traktman P. 2007. Biochemical and genetic analysis of the vaccinia virus d5 protein: multimerization-dependent ATPase activity is required to support viral DNA replication. J Virol 81:844–859. doi: 10.1128/JVI.02217-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Silva FS, Lewis W, Berglund P, Koonin EV, Moss B. 2007. Poxvirus DNA primase. Proc Natl Acad Sci U S A 104:18724–18729. doi: 10.1073/pnas.0709276104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kilcher S, Schmidt FI, Schneider C, Kopf M, Helenius A, Mercer J. 2014. siRNA screen of early poxvirus genes identifies the AAA+ ATPase D5 as the virus genome-uncoating factor. Cell Host Microbe 15:103–112. doi: 10.1016/j.chom.2013.12.008. [DOI] [PubMed] [Google Scholar]
- 14.McCraith S, Holtzman T, Moss B, Fields S. 2000. Genome-wide analysis of vaccinia virus protein-protein interactions. Proc Natl Acad Sci U S A 97:4879–4884. doi: 10.1073/pnas.080078197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gerard FC, Ribeiro Ede A Jr, Leyrat C, Ivanov I, Blondel D, Longhi S, Ruigrok RW, Jamin M. 2009. Modular organization of rabies virus phosphoprotein. J Mol Biol 388:978–996. doi: 10.1016/j.jmb.2009.03.061. [DOI] [PubMed] [Google Scholar]
- 16.Pernot P, Round A, Barrett R, De Maria Antolinos A, Gobbo A, Gordon E, Huet J, Kieffer J, Lentini M, Mattenet M, Morawe C, Mueller-Dieckmann C, Ohlsson S, Schmid W, Surr J, Theveneau P, Zerrad L, McSweeney S. 2013. Upgraded ESRF BM29 beamline for SAXS on macromolecules in solution. J Synchrotron Radiat 20:660–664. doi: 10.1107/S0909049513010431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Round A, Felisaz F, Fodinger L, Gobbo A, Huet J, Villard C, Blanchet CE, Pernot P, McSweeney S, Roessle M, Svergun DI, Cipriani F. 2015. BioSAXS sample changer: a robotic sample changer for rapid and reliable high-throughput X-ray solution scattering experiments. Acta Crystallogr D Biol Crystallogr 71:67–75. doi: 10.1107/S1399004714026959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Maria Antolinos A, Pernot P, Brennich ME, Kieffer J, Bowler MW, Delageniere S, Ohlsson S, Malbet Monaco S, Ashton A, Franke D, Svergun D, McSweeney S, Gordon E, Round A. 2015. ISPyB for BioSAXS, the gateway to user autonomy in solution scattering experiments. Acta Crystallogr D Biol Crystallogr 71:76–85. doi: 10.1107/S1399004714019609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Petoukhov MV, Franke D, Shkumatov AV, Tria G, Kikhney AG, Gajda M, Gorba C, Mertens HDT, Konarev PV, Svergun DI. 2012. New developments in the ATSAS program package for small-angle scattering data analysis. J Appl Crystallogr 45:342–350. doi: 10.1107/S0021889812007662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Konarev PV, Volkov VV, Sokolova AV, Koch MHJ, Svergun DI. 2003. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J Appl Crystallogr 36:1277–1282. doi: 10.1107/S0021889803012779. [DOI] [Google Scholar]
- 21.Guinier A. 1938. La diffusion des rayons X sous faibles angles, appliquée à l'étude de fines particules et de suspensions colloïdales. C R Hebd Seances Acad Sci 206:1374. [Google Scholar]
- 22.Porod G. 1982. General theory. In Glatter O, Kratky O (ed), Small-angle X-ray scattering. Academic Press, London, United Kingdom. [Google Scholar]
- 23.Svergun DI. 1992. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J Appl Crystallogr 25:495–503. doi: 10.1107/S0021889892001663. [DOI] [Google Scholar]
- 24.Franke D, Svergun DI. 2009. DAMMIF, a program for rapid ab-initio shape determination in small-angle. J Appl Crystallogr 42:342–346. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Volkov VV, Svergun DI. 2003. Uniqueness of ab-initio shape determination in small-angle scattering. J Appl Crystallogr 36:860–864. doi: 10.1107/S0021889803000268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Svergun DI, Barberato C, Koch MHJ. 1995. CRYSOL—a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J Appl Crystallogr 28:768–773. doi: 10.1107/S0021889895007047. [DOI] [Google Scholar]
- 27.Ludtke SJ, Baldwin PR, Chiu W. 1999. EMAN: semiautomated software for high-resolution single-particle reconstructions. J Struct Biol 128:82–97. doi: 10.1006/jsbi.1999.4174. [DOI] [PubMed] [Google Scholar]
- 28.Mindell JA, Grigorieff N. 2003. Accurate determination of local defocus and specimen tilt in electron microscopy. J Struct Biol 142:334–347. doi: 10.1016/S1047-8477(03)00069-8. [DOI] [PubMed] [Google Scholar]
- 29.Scheres SHW. 2012. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol 180:519–530. doi: 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Estrozi LF, Navaza J. 2010. Ab initio high-resolution single-particle 3D reconstructions: the symmetry adapted functions way. J Struct Biol 172:253–260. doi: 10.1016/j.jsb.2010.06.023. [DOI] [PubMed] [Google Scholar]
- 31.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 32.Lakowicz JR. 2006. Principles of fluorescence spectroscopy, 3rd ed Springer, New York, NY. [Google Scholar]
- 33.Burmeister WP, Tarbouriech N, Fender P, Contesto-Richefeu C, Peyrefitte CN, Iseni F. 2015. Crystal structure of the vaccinia virus uracil-DNA glycosylase in complex with DNA. J Biol Chem 290:17923–17934. doi: 10.1074/jbc.M115.648352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li D, Zhao R, Lilyestrom W, Gai D, Zhang R, DeCaprio JA, Fanning E, Jochimiak A, Szakonyi G, Chen XS. 2003. Structure of the replicative helicase of the oncoprotein SV40 large tumour antigen. Nature 423:512–518. doi: 10.1038/nature01691. [DOI] [PubMed] [Google Scholar]
- 35.Sèle C. 2011. Caractérisation structurale des interactions moléculaires au sein du complexe de réplication du virus de la vaccine. Ph.D. dissertation. Université de Grenoble, Grenoble, France. [Google Scholar]
- 36.Enemark EJ, Joshua-Tor L. 2006. Mechanism of DNA translocation in a replicative hexameric helicase. Nature 442:270–275. doi: 10.1038/nature04943. [DOI] [PubMed] [Google Scholar]
- 37.Chaban Y, Stead JA, Ryzhenkova K, Whelan F, Lamber EP, Antson A, Sanders CM, Orlova EV. 2015. Structural basis for DNA strand separation by a hexameric replicative helicase. Nucleic Acids Res 43:8551–8563. doi: 10.1093/nar/gkv778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang L, Mohr I, Fouts E, Lim DA, Nohaile M, Botchan M. 1993. The E1 protein of bovine papilloma virus 1 is an ATP-dependent DNA helicase. Proc Natl Acad Sci U S A 90:5086–5090. doi: 10.1073/pnas.90.11.5086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stahl H, Dröge P, Knippers R. 1986. DNA helicase activity of SV40 large tumor antigen. EMBO J 5:1939–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Soultanas P. 2012. Loading mechanisms of ring helicases at replication origins. Mol Microbiol 84:6–16. doi: 10.1111/j.1365-2958.2012.08012.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baroudy BM, Venkatesan S, Moss B. 1982. Incompletely base-paired flip-flop terminal loops link the two DNA strands of the vaccinia virus genome into one uninterrupted polynucleotide chain. Cell 28:315–324. doi: 10.1016/0092-8674(82)90349-X. [DOI] [PubMed] [Google Scholar]
- 42.Du S, Traktman P. 1996. Vaccinia virus DNA replication: two hundred base pairs of telomeric sequence confer optimal replication efficiency on minichromosome templates. Proc Natl Acad Sci U S A 93:9693–9698. doi: 10.1073/pnas.93.18.9693. [DOI] [PMC free article] [PubMed] [Google Scholar]