

ABSTRACT
In contrast to many viruses, human cytomegalovirus (HCMV) is unable to productively infect most cancer-derived cell lines. The mechanisms of this restriction are unclear. To explore this issue, we tested whether defined oncogenic alleles, including the simian virus 40 (SV40) T antigen (TAg) and oncogenic H-Ras, inhibit HCMV infection. We found that expression of SV40 TAg blocks HCMV infection in human fibroblasts, whereas the replication of a related herpesvirus, herpes simplex virus 1 (HSV-1), was not impacted. The earliest restriction of HCMV infection involves a block of viral entry, as TAg expression prevented the nuclear delivery of viral DNA and pp65. Subsequently, we found that TAg expression reduces the abundance of platelet-derived growth factor receptor α (PDGFRα), a host protein important for HCMV entry. Viral entry into TAg-immortalized fibroblasts could largely be rescued by PDGFRα overexpression. Similarly, PDGFRα overexpression in HeLa cells markedly increased the levels of HCMV gene expression and DNA replication. However, the robust production of viral progeny was not restored by PDGFRα overexpression in either HeLa cells or TAg-immortalized fibroblasts, suggesting additional restrictions associated with transformation and TAg expression. In TAg-expressing fibroblasts, expression of the immediate early 2 (IE2) protein was not rescued to the same extent as that of the immediate early 1 (IE1) protein, suggesting that TAg expression impacts the accumulation of major immediate early (MIE) transcripts. Transduction of IE2 largely rescued HCMV gene expression in TAg-expressing fibroblasts but did not rescue the production of infectious virions. Collectively, our data indicate that oncogenic alleles induce multiple restrictions to HCMV replication.
IMPORTANCE HCMV cannot replicate in most cancerous cells, yet the causes of this restriction are not clear. The mechanisms that restrict viral replication in cancerous cells represent viral vulnerabilities that can potentially be exploited therapeutically in other contexts. Here we found that SV40 T antigen-mediated transformation inhibits HCMV infection at multiple points in the viral life cycle, including through inhibition of proper viral entry, normal expression of immediate early genes, and viral DNA replication. Our results suggest that the SV40 T antigen could be a valuable tool to dissect cellular activities that are important for successful infection, thereby potentially informing novel antiviral development strategies. This is an important consideration, given that HCMV is a leading cause of birth defects and causes severe infection in immunocompromised individuals.
INTRODUCTION
Human cytomegalovirus (HCMV) is a ubiquitous opportunistic betaherpesvirus that infects ∼50 to 70% of the global population. While infection of healthy individuals is frequently resolved without severe complications, HCMV poses a major threat to immunocompromised individuals, such as AIDS patients and organ transplant recipients (1, 2). Further, HCMV is a leading cause of birth defects, affecting approximately 5,000 newborns in the United States every year (3). In the immunocompetent host, an efficient antiviral immune response limits viral infection, and HCMV enters a latent phase in hematopoietic progenitor cells, which is characterized by silencing of the major immediate early (MIE) promoter and the subsequent limitation of viral gene expression (4).
HCMV is somewhat unique in comparison to many viruses that can be propagated in vitro, in that it is incapable of productively infecting most commonly used transformed cell lines (1, 5). In contrast to other herpesviruses, such as the herpes simplex viruses (HSVs), HCMV must be propagated in nontransformed cells, most commonly, in primary fibroblasts. One known contributing factor is the inability of laboratory-adapted strains of HCMV to infect a broad range of cell types, including epithelial cells, whereas clinical isolates of HCMV exhibit a wide tropic range and are typically capable of infecting fibroblasts, macrophages, and epithelial and endothelial cells (1). Continued propagation of laboratory strains in fibroblasts results in the inability to productively infect epithelial and endothelial cells. This tropic defect results from the accumulation of mutations in the UL128-131 region (6, 7). This region encodes proteins that form the gH/gL/UL128-131 complex, which is essential for viral entry into epithelial and endothelial cells (6–10). While repair of this region enables entry into transformed epithelial cell lines, such as HeLa cells, production of viral progeny is still minimal (6), suggesting additional host cell restriction mechanisms. Further, fibroblastic cells are also unable to support productive HCMV infection (5). The mechanisms of this viral cellular restriction are unclear.
To explore how oncogenic signaling might limit HCMV replication, we employed a genetically defined model of tumorigenesis, which consists of primary human foreskin fibroblasts, life-extended telomerase-expressing fibroblasts, and life-extended fibroblasts that express the simian virus 40 (SV40) T antigen (TAg) either alone or in conjunction with expression of an oncogenic H-Ras G12V allele (11). This genetically defined model enables the deconvolution of the genetic complexity associated with transformation and therefore permits study of the mechanisms of oncogenic viral restriction. We found that the expression of the SV40 TAg severely attenuated HCMV replication but not herpes simplex virus replication. This restriction involved inhibition of the nuclear delivery of viral DNA and the pp65 tegument protein, suggesting that TAg expression interferes with viral entry. TAg expression was found to substantially downregulate platelet-derived growth factor receptor α (PDGFRα), a cell surface protein that plays a major role during viral entry (12). Viral entry could largely be rescued by PDGFRα overexpression, whereas viral gene expression and DNA replication were only partially rescued, and the production of infectious viral progeny was not rescued at all. In the presence of increased amounts of PDGFRα, expression of the SV40 TAg inhibited the accumulation of the immediate early 2 (IE2) protein, the major viral transcriptional activator. Transduction of IE2 further rescued viral gene expression but not the production of infectious HCMV. Combined, these results indicate that oncogenic signaling induces several blocks to the HCMV life cycle, including the reduced expression of an important host entry factor and the downregulation of viral gene expression.
MATERIALS AND METHODS
Cell culture and viral infection.
H-Ras-transformed BJ fibroblasts, telomerase-SV40 TAg-immortalized BJ fibroblasts, and primary BJ fibroblasts (ATCC CRL-2522) were courteously provided by Robert Weinberg (Whitehead Institute for Biomedical Research). Telomerase-expressing BJ fibroblasts were created via lentiviral transduction (see below). Primary fibroblasts and their telomerase-expressing, immortalized, and transformed derivatives were cultured in Dulbecco's modified Eagle medium (DMEM; Invitrogen) supplemented with 10% fetal bovine serum, 4.5 g/liter glucose, and 1% penicillin-streptomycin (Pen-Strep; Life Technologies). Primary fibroblasts and their telomerase-expressing, immortalized, and transformed derivatives were seeded at a density of 50,000 cells per well of a standard 12-well plate 24 h before viral infection. Unless indicated otherwise, the strain utilized in all studies was BADwt, derived from a bacterial artificial chromosome (BAC) clone of the HCMV AD169 laboratory strain (13). BADsubUL21.5, which expresses enhanced green fluorescent protein (EGFP) driven by the SV40 early promoter, replicates with kinetics similar to those of the wild-type virus (14). The HCMV TB40/e strain, which expresses EGFP driven by the SV40 early promoter, was provided by Eain Murphy (15). Viral genomes were labeled with bromodeoxyuridine (BrdU) as previously described (16). Briefly, MRC-5 fibroblasts were infected with AD169 at a multiplicity of infection (MOI) of 0.05. When the cells exhibited an ∼90% cytopathic effect (CPE) (at ∼5 days postinfection [dpi]), fresh medium containing 10 μM BrdU was added. Forty-eight hours later, the medium was spiked with an additional 10 μM BrdU for an additional 24 h before harvesting to obtain a viral stock. This stock was designated BrdU-AD169 and was previously found to replicate with kinetics similar to those of nonlabeled wild-type virus (16).
Cells were infected with HSV-1 (KOS) or HCMV at an MOI of 3.0 for 1.5 h in the absence of serum. The viral inoculum was then removed and replaced with fresh serum-free medium. To measure the production of infectious viral progeny, infected cells (at 5 dpi, unless otherwise specified) were scraped, sonicated, centrifuged at 3,000 rpm for 5 min, and stored in −80°C before a plaque-forming assay was performed on MRC-5 human fibroblasts (for HCMV) or Vero cells (for HSV).
Cloning.
Human telomerase (hTERT) cDNA was amplified by PCR from pWZL-Blast-Flag-HA-hTERT (Addgene plasmid 22396) using the following primers: forward primer 5′-GGAACCAATTCAGTCGACTGGGATCCCGTCCTGCTGCGCACGTG-3′ and reverse primer 5′-TTTGTACAAGAAAGCTGGGTTCTAGATCAGTCCAGGATGGTCTTGAAGTCTG-3′. hTERT cDNA was then cloned via Gibson assembly (17) into the BamHI and XbaI sites of pLenti CMV/TO/Hygro (Addgene plasmid 17484). To generate pLenti6/CMV/PDGFRα, human PDGFRα (isoform X1) was amplified from cDNA synthesized with SuperScript II reverse transcriptase (Invitrogen) from total RNA purified from primary BJ fibroblasts (TRIzol, Invitrogen). The following primers were used: forward primer 5′-CATAGAAGACACCGACTCTAGAGGGATCCATGGGGACTTCCCATCCGGC-3′ and reverse primer 5′-GAACCGCGGGCCCTCTAGACCTCGAGTTACAGGAAGCTGTCTTCCACCAG-3′. The PDGFRα cDNA was then cloned into the BamHI and XhoI sites of pLenti6/CMV/V5-D-TOPO (Invitrogen) via Gibson assembly. To generate pLenti CMV/TO/MIE, the HCMV MIE gene was amplified by PCR from a BAC plasmid of the HCMV TB40/e strain using the following primers: forward primer 5′-GGAACCAATTCAGTCGACTGGGATCCAGAGCTCGTTTAGTGAAC-3′ and reverse primer 5′-TTTGTACAAGAAAGCTGGGTTTACTGAGATTTGTTCCTCAGG-3′. The MIE gene was then cloned into the BamHI and XbaI sites of the pLenti CMV/TO Puro vector (a gift from Eric Campeau; Addgene plasmid 22262) via Gibson assembly.
Lentiviral transfection and transduction.
293T cells were seeded at 2 × 106 cells per 10-cm dish and grown for 24 h. For the generation of pseudotyped lentivirus, each 10-cm dish of 293T cells was transfected with 2.6 μg lentiviral vector, 2.4 μg PAX2, and 0.25 μg vesicular stomatitis virus G glycoprotein using the Fugene 6 reagent (Promega). Twenty-four hours later, the medium was removed and replaced with 4 ml fresh medium, and the medium was collected after an additional 24 h and filtered through a 0.45-μm-pore-size filter prior to transduction. The fibroblasts were transduced with lentivirus in the presence of 5 μg/ml Polybrene and incubated overnight. The lentivirus-containing medium was then removed and replaced with fresh DMEM. At 72 h after transduction, the cells were placed under selection with antibiotics. Cells transduced with pLenti CMV/TO/Hygro/hTERT were grown in 200 μg/ml hygromycin B (Invitrogen) for 1 week, and the expression of hTERT was confirmed by quantitative PCR (qPCR). Cells transduced with pLenti6/CMV/PDGFRα were selected in 10 μg/ml blasticidin (Invitrogen) for 4 days. TAg-expressing fibroblasts transduced with pLenti CMV/TO/MIE were not drug selected, but the robust expression of IE2 was verified by Western blotting analysis.
Immunoblotting.
For Western blotting, cells were washed with phosphate-buffered saline (PBS) and lysed in 1× radioimmunoprecipitation assay buffer (50 mM Tris, 150 mM NaCl, 0.1% SDS, 1% Triton X-100, 0.5% sodium deoxycholate, a cocktail of protease inhibitors [EDTA free; cOmplete; Roche]). Lysates were sonicated and centrifuged at 14,000 × g for 5 min to pellet insoluble material. The protein concentration was measured by a Bradford protein assay (Bio-Rad). Supernatants were mixed with 4× loading buffer (200 mM Tris [pH 7.0], 8% SDS, 20% 2-mercaptoethanol, 11% sucrose), boiled, briefly centrifuged, run on an ∼10% polyacrylamide gel, and transferred to nitrocellulose in Tris-glycine transfer buffer. The blots were then stained with Ponceau S to visualize protein bands and ensure equal protein loading. The membranes were blocked in 5% milk in Tris-buffered saline–Tween 20 (TBST), followed by incubation in primary antibodies. After subsequent washes, the blots were treated with secondary antibodies, visualized using an enhanced chemiluminescence (ECL) system (Bio-Rad), and imaged using a Molecular Imager Gel Doc XR+ system (Bio-Rad). The antibodies used were specific for H-Ras (Santa Cruz Biotechnology Inc.), SV40 small T antigen and large T antigen (Santa Cruz Biotechnology Inc.), GAPDH (glyceraldehyde-3-phosphate dehydrogenase; Cell Signaling Technology), IE1/IE2 (18), UL26 (19), pp28 (20), pp65 (21), UL44 (Virusys) (46), and PDGFRα (Santa Cruz Biotechnology Inc.).
Immunofluorescence.
For analysis of pp65 localization, cells were grown on glass coverslips. At 4 h postinfection (hpi), the cells were washed once with PBS, fixed with 2% paraformaldehyde in PBS for 20 min, washed three times with PBS, permeabilized by treatment with 0.1% Triton X-100 and 0.1% SDS for 15 min, and washed twice with PBS containing 0.1% Tween 20. The cells were subsequently blocked by incubation overnight in PBS containing 2% bovine serum albumin (BSA), 5% goat serum, 5% human serum, and 0.3% Triton X-100. The cells were then incubated with primary antibody to pp65, diluted in 0.05% Tween 20 in PBS for 1 h, washed with PBS containing 0.1% Tween 20 three times, incubated with Alexa Fluor 488-conjugated anti-mouse immunoglobulin secondary (Invitrogen) antibody for 1 h, and washed with PBS containing 0.1% Tween 20 three times. Coverslips were mounted in SlowFade Gold antifade reagent (Molecular Probes) and 4′,6′-diamidino-2-phenylindole (DAPI). Confocal images were captured with an FV1000 Olympus laser scanning confocal microscope. All images were captured under identical confocal settings.
Flow cytometry.
Cells were serum starved for 4 days and infected with HCMV. At 24 hpi, cells were harvested in PBS, fixed in 70% ethanol, and stained with 1 μg/ml DAPI in 0.1% Triton X-100. Flow cytometric determination of the DNA content was conducted using an LSRII flow cytometer, and the data were processed with FlowJo (v10) software.
Quantitative PCR.
Cytoplasmic and nuclear fractions were obtained via a nuclear and cytoplasmic extraction kit (G-Biosciences). Viral DNA was harvested (at 5 dpi, unless otherwise specified) in lysis buffer (100 mM NaCl, 100 mM Tris-HCl, 25 mM, EDTA, 0.5% SDS, 0.1 mg/ml proteinase K, 40 mg/ml RNase A). qPCR was performed using Fast SYBR green master mix (Applied Biosystems), a model 7500 Fast real-time PCR system (Applied Biosystems), and Fast 7500 software (Applied Biosystems) according to the manufacturer's instructions. For quantifying viral DNA, a standard curve was generated using the BADwt plasmid (AD169) as the template and a primer set targeting pp65 (forward primer, 5′-CAGGAAGATTTGCTGCCCGTTCAT-3′; reverse primer, 5′-GGCTTTACGGTGTTGTGTCCCAAA-3′). To measure viral gene expression, relative quantities were normalized to GAPDH levels using the ΔCT threshold cycle (CT) method and primers targeting the following: IE1 (forward primer, 5′-CCATGTCCACTCGAACCTTAAT-3′; reverse primer, 5′-TGAACAAGTGACCGAGGATTG-3′), IE2 (forward primer, 5′-CCCTTCACGATTCCCAGTATG-3′; reverse primer, 5′-CTCATGATTGCGGGTGTAGAT-3′), UL37 (forward primer, 5′-CCGAGTTCTCACCGTCAATTA-3′; reverse primer, 5′-CTCTCCCGCCTTGGTTAAG-3′), and GAPDH (forward primer, 5′-GGTGTGAACCATGAGAAGTATGA-3′; reverse primer, 5′-GAGTCCTTCCACGATACCAAAG-3′).
Statistical analysis.
For all the graphs shown in the figures, the data are represented as means ± standard deviations. Statistical analysis was performed using a two-sided paired Student's t test.
RESULTS
Expression of oncogenic proteins inhibits HCMV infection.
To gain insight into the mechanisms of oncogenic restriction of HCMV infection, we examined the impact of oncogenic SV40 TAg and H-Ras G12V expression on HCMV replication in a previously defined stepwise model of transformation (11), consisting of parental human primary foreskin fibroblasts (P), telomerase-expressing fibroblasts (P-ht), telomerase-SV40 TAg-expressing fibroblasts (TAg cells), and Ras-transformed fibroblasts (TAgR cells) (Fig. 1A). Consistent with the ability of HSV-1 to grow in a large variety of tumor-derived cell lines, expression of the SV40 TAg did not impact HSV-1 titers (Fig. 1B). In contrast, consistent with the inability of HCMV to productively infect most transformed cell lines, HCMV infection was attenuated by about 100-fold in the immortalized and the Ras-transformed fibroblasts (Fig. 1C). This attenuation was not a result of the expression of human telomerase, which did not affect the production of viral progeny (Fig. 1D). Given that it has been shown that HCMV cannot replicate well in S-phase cells (22), we assessed whether differences in the cell cycle could be playing a role in the observed reduction in virus production. Toward this end, we synchronized the cell cycle of the primary fibroblasts, the TAg cells, and the TAgR cells through serum starvation. Despite similar percentages of cells in each stage of the cell cycle, the TAg and TAgR cells still exhibited a substantial defect in HCMV replication relative to the primary fibroblasts (Fig. 1E), suggesting that the cell cycle differences between the cells were not playing a major role.
FIG 1.

The expression of SV40 TAg blocks HCMV replication. (A) The protein levels of SV40 large T antigen (LT) and H-Ras in primary fibroblasts (P) or their transduced derivatives were measured by Western blotting. These fibroblast lines include those expressing telomerase alone (P-ht), those expressing telomerase plus the SV40 early region (TAg), or those expressing the combination of telomerase, the SV40 early region, and H-RasV12 (TAgR). (B to D) The cells for which the results are shown in panel A were infected with herpes simplex virus (B) or HCMV AD169 (C and D) at an MOI of 3.0. The production of infectious virions at 1 dpi (B), 3 to 7 dpi (C), and 5 dpi (D) was measured by plaque assay. (C) Asterisks indicate significant differences between the values for primary cells and those for TAg-expressing cells (*, P < 0.05; **, P < 0.01; ****, P < 0.001). Number signs indicate significant differences between the values for primary cells and those for TAgR cells (#, P < 0.05; ##, P < 0.01). (E) The cells for which the results are shown in panel A were maintained in serum-free medium for 96 h and infected with HCMV AD169 at an MOI of 3.0. Cell cycle analysis was performed by analyzing the cellular DNA content by flow cytometry at 24 hpi. Asterisks indicate significant differences between the values for primary cells and those for TAg-expressing cells (P < 0.001). (F) The cells for which the results are shown in panel A were infected with HCMV (AD169, MOI = 3.0), and the protein levels of IE1, UL26, pp28, and GAPDH were measured by Western blotting at 4, 24, 48, and 72 hpi. The numbers to the left of the gels in panels A and F are molecular masses (in kilodaltons).
We next set out to investigate whether the expression of these oncogenic alleles affects HCMV gene expression. Compared to the level of accumulation of HCMV proteins in primary fibroblasts, the accumulation of a variety of HCMV proteins was substantially reduced in TAg and TAgR cells over the course of infection. Viral genes from the various kinetic classes of infection, including the IE1 immediate early gene, the UL26 early gene, and the pp28 late gene, all failed to accumulate in TAg or TAgR cells from 4 hpi to 72 hpi (Fig. 1F). Together, these results indicate that the expression of SV40 TAg inhibits HCMV infection at a very early stage of infection.
Given that HCMV IE1 expression was inhibited at 4 hpi in TAg and TAgR cells (Fig. 1F), we hypothesized that HCMV entry may not occur properly in these cells. Immediately upon successful envelope fusion, the viral nucleocapsid, together with specific tegument proteins, is transported to the nucleus, where the expression of immediate early genes begins. The pp65 tegument protein is delivered to the nucleus upon initial viral envelope fusion. After examining the localization of pp65 and the viral genomes at 4 hpi, we found that pp65 and the viral genomes were localized to the nuclei of primary fibroblasts but not the nuclei of TAg or TAgR fibroblasts (Fig. 2A and C). As the quantities in Fig. 2B and D show, about 70% of the primary fibroblasts exhibited nuclear pp65 and the viral genome upon infection. In contrast, no viral genome/pp65-specific staining was evident in the nuclei of cells expressing TAg. We also quantified the abundance of viral genomes in purified cellular cytosolic and nuclear fractions. As shown in Fig. 2E and F, the majority of viral genomes were localized in the nuclei of the primary fibroblasts. In comparison, there was a substantial decrease in the amount of viral genomes in the nuclei of TAg or TAgR fibroblasts. Together, these results suggest that the entry of HCMV is blocked in T antigen-expressing fibroblasts.
FIG 2.

Expression of SV40 TAg blocks proper HCMV entry. (A) Primary fibroblasts and TAg and TAgR cells were infected with HCMV (AD169, MOI = 3.0), and the localization of pp65 was determined by measurement of the level of immunofluorescence at 4 hpi. Bars, 20 μm. (B) The number of nuclei with positive pp65 staining in cells infected as described in the legend to panel A was quantified. (C) The cells for which the results are shown in panel A were infected with BrdU-labeled AD169, and the localization of BrdU was determined by measurement of the level of immunofluorescence at 4 hpi. Bars, 20 μm. (D) The number of nuclei with positive BrdU staining in the cells for which the results are shown in panel C was quantified. (E) The indicated cells were infected as described in the legend to panel A, and the HCMV genomes in the cytosolic or the nuclear fractions were quantified at 4 hpi. (F) The abundance of the nuclear lamin A/C and GAPDH proteins in the cytosolic and nuclear fractions from the lysates generated in the assay whose results are presented in panel E was measured. The numbers to the left of the gels are molecular masses (in kilodaltons).
PDGFRα expression increases HCMV entry in TAg-expressing fibroblasts.
A number of different growth factor receptors have been reported to be important for HCMV entry, including PDGFRα and the epidermal growth factor receptor (EGFR) (12, 23). Given the observed defect in viral entry, we sought to determine whether PDGFRα levels were impacted by TAg expression. In comparison to the protein and mRNA levels of PDGFRα in primary cells, both the protein (Fig. 3A) and mRNA levels (Fig. 3B) of PDGFRα were markedly reduced in TAg and TAgR-fibroblasts. As the decreased levels of PDGFRα could attenuate viral entry in the TAg-expressing fibroblasts, we examined whether PDGFRα overexpression could increase the amount of HCMV that enters these cells. Primary and TAg and TAgR fibroblasts were transduced with a control or PDGFRα-expressing lentiviral vector (Fig. 3C). PDGFRα overexpression significantly increased pp65 nuclear localization in TAg fibroblasts (∼55%) and TAgR fibroblasts (∼20%) relative to that achieved after expression of a control vector, where no detectable nuclear pp65 was evident (Fig. 3D and E). pp65 was primarily localized in the nuclei of primary fibroblasts, irrespective of whether they were transduced with a control or PDGFRα expression vector (Fig. 3D and E). PDGFRα transduction also increased the expression of genes carried on the viral genome, enhancing virus genome-encoded EGFP expression (Fig. 3F). These results indicate that PDGFRα overexpression can substantially rescue HCMV entry in TAg-immortalized fibroblasts.
FIG 3.

PDGFRα overexpression partly rescues HCMV entry in fibroblasts expressing SV40 TAg. (A and B) The PDGFRα protein abundance (A) or mRNA level (B) in primary fibroblasts (P) or TAg or TAgR fibroblasts was measured by Western blot and qPCR, respectively. (C) Cell lysates from primary fibroblasts or TAg or TAgR fibroblasts transduced with either PDGFRα or a control vector (Vec) were subjected to Western blotting analysis of PDGFRα. (D) The localization of pp65 in primary fibroblasts or TAg or TAgR fibroblasts transduced with PDGFRα or a control vector was determined by measurement of the level of immunofluorescence at 4 hpi (AD169, MOI = 3.0). Bars, 20 μm. (E) The number of nuclei with pp65 staining in cells infected as described in the legend to panel D was quantified. (F) The indicated cells were infected with EGFP-tagged AD169 (MOI = 3.0), and EGFP fluorescence was measured at 48 hpi. Bars, 100 μm. The numbers to the left of the gels in panels A and C are molecular masses (in kilodaltons).
PDGFRα overexpression partially rescues HCMV gene expression and DNA replication but not virion production in TAg-expressing fibroblasts.
Given that PDGFRα overexpression significantly increased HCMV entry in TAg-expressing fibroblasts, we tested whether PDGFRα overexpression rescued viral gene expression, DNA replication, and the production of infectious progeny. As shown in Fig. 4A, at 4 hpi, the IE1 protein accumulated to a lesser extent in TAg and TAgR cells than in primary fibroblasts (Fig. 4A). However, by 24 hpi, PDGFRα transduction substantially increased the level of IE1 expression in TAg and TAgR fibroblasts to levels comparable to those in primary fibroblasts (Fig. 4A). However, the levels of IE2, the major viral transactivator, were rescued to a lesser extent. Substantially reduced levels of IE2 accumulated in PDGFRα-expressing TAg and TAgR fibroblasts at all times postinfection (Fig. 4A). The accumulation of downstream viral proteins, such as UL26 and pp28, was partially rescued by PDGFRα overexpression, but this occurred only to a small extent, consistent with the observed reduction in IE2 levels. In addition, PDGFRα overexpression only partially rescued viral DNA replication in TAg-expressing cells (Fig. 4B) and did not rescue virion production in TAg-expressing cells (Fig. 4C). Notably, the expression of PDGFRα reduced virion production in primary fibroblasts and TAgR-expressing fibroblasts (Fig. 4C), suggesting that its constitutive expression may inhibit the production of infectious virus. Host proteins that are involved in viral entry could potentially complicate viral egress strategies, e.g., through interactions with viral receptors prior to egress, which could block subsequent infection. A viral strategy to prevent this would be to reduce the expression of the host proteins involved in entry. Consistent with this possibility, we found that PDGFRα expression was acutely downregulated by HCMV infection (Fig. 4D and E). Combined, our results indicate that while PDGFRα expression can substantially rescue HCMV entry and IE1 expression in TAg-expressing fibroblasts, its expression does not restore either wild-type levels of viral gene expression or wild-type levels of production of viral progeny.
FIG 4.
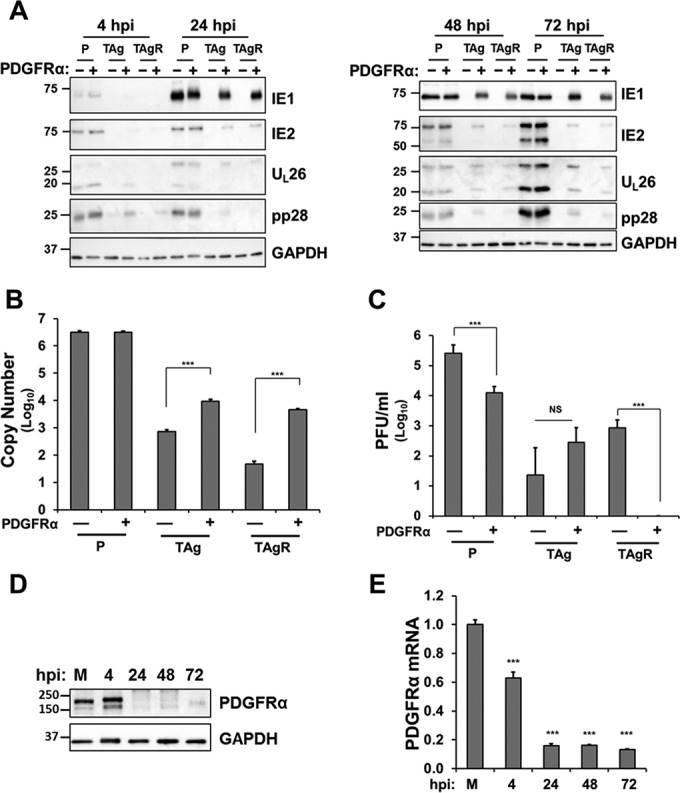
PDGFRα overexpression partly rescues HCMV gene expression and DNA replication but not virion production in TAg-expressing fibroblasts. (A) Primary fibroblasts (P) or TAg or TAgR fibroblasts transduced with PDGFRα cDNA or a control vector were infected with HCMV (AD169, MOI = 3.0). Cell lysates were harvested at 4, 24, 48, and 72 hpi and subjected to Western blotting analysis for IE1, IE2, UL26, pp28, and GAPDH. (B) The viral DNA abundance in cells infected as described in the legend to panel A was quantified and is displayed as the copy number per nanogram of input DNA on a log10 scale. Asterisks indicate significant differences between control and PDGFRα-transduced cells (***, P < 0.001). (C) Production of infectious virions was measured by plaque assay in the cells infected as described in the legend to panel A and harvested at 120 hpi. Asterisks indicate significant differences between control cells and PDGFRα-transduced cells (***, P < 0.001; NS, not significant). (D and E) Primary fibroblasts mock infected or infected with HCMV (AD169, MOI = 3.0) were harvested at the indicated times and analyzed for the abundance of the PDGFRα protein by Western blotting (D) or PDGFRα mRNA by qPCR (E). Asterisks indicate significant differences between values for mock-infected and HCMV-infected cells (***, P < 0.001). The numbers to the left of the gels in panels A and D are molecular masses (in kilodaltons).
PDGFRα and TAg expression in fibroblasts results in the differential accumulation of major immediate early transcripts.
The observation that PDGFRα expression largely rescued IE1 but not IE2 levels suggests that TAg expression may differentially impact the accumulation of HCMV immediate early transcripts. To explore this issue, we employed gene-specific qPCR primers to examine the amounts of various immediate early transcripts, including IE1, IE2, and UL37, during infection. At 4 hpi, PDGFRα expression increased the IE1-specific mRNA abundance by over 200-fold in TAg cells and 180-fold in TAgR cells (Fig. 5A and Table 1). At 24 hpi, PDGFRα expression increased the IE1 mRNA abundance by 119-fold and 63-fold in TAg and TAgR cells, respectively (Fig. 5A and Table 1). IE2-specific mRNA was also induced upon PDGFRα expression, with ∼30-fold and 40-fold increases in PDGFRα-expressing TAg and TAgR cells, respectively, at 4 hpi, and ∼50-fold increases in both PDGFRα-expressing TAg and TAgR cells at 24 hpi (Fig. 5B and Table 1). We also examined whether PDGFRα expression could rescue the expression of another immediate early gene, UL37. As shown in Fig. 5C, PDGFRα transduction increased the level of UL37 mRNA expression at both 4 hpi and 24 hpi in TAg-expressing cells. As viral entry was not completely rescued by PDGFRα transduction in TAg- and TAgR-expressing cells (Fig. 3E), it follows that the expression of these immediate early genes would not be fully rescued to levels typical of those seen in infected primary fibroblasts. Collectively, these results suggest that PDGFRα transduction substantially increases the expression of various viral immediate early genes.
FIG 5.

PDGFRα and TAg expression in fibroblasts results in the differential accumulation of major immediate early transcripts. (A to C) Primary fibroblasts (P) or TAg or TAgR fibroblasts transduced with PDGFRα cDNA or a control vector were infected with HCMV (AD169, MOI = 3.0) and harvested for analysis of mRNA by qPCR. The abundances of IE1 (A), IE2 (B), and UL37 (C) mRNA were plotted after normalization to GAPDH mRNA levels. Asterisks indicate significant differences between control cells and PDGFRα-transduced cells (**, P < 0.01; ***, P < 0.001). (D and E) The fraction of IE1 or IE2 in the total sum of IE1 and IE2 was measured in cells infected as described in the legend to panel A. Asterisks indicate significant differences between the values for IE1 in the primary cells and TAg-expressing cells (*, P < 0.05). Number signs indicate significant differences between the values for IE2 in the primary cells and other cells (##, P < 0.01).
TABLE 1.
Abundances of IE1, IE2, and UL37 mRNA in primary fibroblasts and TAg and TAgR fibroblasts transduced with PDGFRα or a control vectora
| hpi | Gene | mRNA abundance in the following fibroblasts transduced with the indicated vector: |
||||
|---|---|---|---|---|---|---|
| Primary fibroblasts | TAg fibroblasts |
TAgR fibroblasts |
||||
| Control vector | PDGFRα | Control vector | PDGFRα | |||
| 4 | IE1 | 22,952 ± 185 | 6 ± 1 | 1,210 ± 47 | 4 ± 0 | 743 ± 62 |
| IE2 | 11,690 ± 123 | 10 ± 1 | 303 ± 18 | 7 ± 2 | 311 ± 44 | |
| UL37 | 181,841 ± 12,424 | 209 ± 7 | 5,371 ± 185 | 141 ± 15 | 3,718 ± 139 | |
| 24 | IE1 | 16,622 ± 435 | 54 ± 9 | 6,425 ± 282 | 45 ± 4 | 2,839 ± 322 |
| IE2 | 14,257 ± 1,318 | 6 ± 1 | 330 ± 13 | 4 ± 0 | 205 ± 22 | |
| UL37 | 263,876 ± 10,411 | 707 ± 83 | 15,156 ± 374 | 98 ± 18 | 10,946 ± 1,333 | |
The abundances of IE1, IE2, and UL37 mRNA were normalized to the level of GAPDH mRNA (which was given a value of 1), measured by qPCR at 4 hpi and 24 hpi.
The genes for both IE1 and IE2 are expressed from the major immediate early (MIE) promoter, and their respective proteins arise from the differential splicing of common transcripts (1, 24). In primary fibroblasts, the relative proportion of the IE2-specific message relative to the sum of the IE1 and IE2 messages increased from 30% at 4 hpi to ∼50% at 24 hpi (Fig. 5D and E). Notably, this transition did not occur in TAg-expressing fibroblasts. The proportion of the IE2 transcript fraction dropped from 20 to 30% at 4 hpi to below 10% at 24 hpi in TAg-expressing cells (Fig. 5D and E). These results suggest that TAg expression prevents a shift that favors IE2 mRNA accumulation over IE1 mRNA accumulation. This defect likely results in the reduced level of IE2 protein accumulation observed in Fig. 4A and potentially contributes to the defects in viral gene expression associated with TAg expression.
The impact of IE2 overexpression on HCMV infection of TAg-expressing fibroblasts.
Given the essential role that IE2 plays in the HCMV life cycle, we hypothesized that the decreased IE2 levels may be responsible for the attenuated HCMV virion production seen in PDGFRα-transduced TAg-expressing cells. To test this hypothesis, we sought to examine whether IE2 overexpression could rescue viral infection in TAg-expressing cells. Transduction of HCMV MIE into TAg-expressing fibroblasts resulted in IE2 expression levels that were similar to those in infected primary fibroblasts (Fig. 6A). MIE transduction also increased the levels of accumulation of the viral early proteins UL26 and UL44, as well as the true late protein pp28 (Fig. 6B). However, while viral DNA replication increased by ∼2-fold, viral DNA still accumulated to a level less than 10% of the level of viral DNA present in primary fibroblasts (Fig. 6C). Further, MIE transduction did not rescue infectious virion production in TAg-expressing cells (Fig. 6D). Combined, these data indicate that while IE2 overexpression can rescue the expression of the HCMV genes analyzed, IE2 overexpression is not sufficient to rescue infectious virion production.
FIG 6.

HCMV IE2 overexpression rescues HCMV gene expression but not virion production in TAg-expressing fibroblasts. (A) Lysates of primary fibroblasts infected with HCMV (MOI = 3.0) at 24 hpi PDGFRα-expressing TAg cells transduced with either the HCMV MIE gene or a control vector were subjected to Western blotting analysis of IE2. (B) Primary fibroblasts and PDGFRα-expressing TAg cells transduced with the HCMV MIE gene or a control vector were infected with HCMV (AD169, MOI = 3.0) and harvested at 4, 24, 48, and 72 hpi. The resulting lysates were subjected to Western blotting analysis of IE1, IE2, UL26, UL44, pp28, and GAPDH. The numbers to the left of the gels in panels A and B are molecular masses (in kilodaltons). (C) The viral DNA abundance in the cells infected as described in the legend to panel B was quantified and is displayed as the copy number per nanogram of input DNA on a log10 scale. (D) PDGFRα-expressing TAg cells transduced with the HCMV MIE gene or a control vector were infected with HCMV (AD169, MOI = 3.0). Production of infectious virions was measured by plaque assay at 120 hpi.
PDGFRα overexpression increases HCMV gene expression and DNA replication but not virion production in HeLa cells.
To explore whether the restrictions observed in the genetically defined fibroblast model of oncogenesis were similar in cancer-derived cells, we examined how PDGFRα overexpression affects HCMV replication in HeLa cells. Lentiviral transduction increased PDGFRα expression in HeLa cells (Fig. 7A). As shown in Fig. 7B, PDGFRα overexpression markedly increased the level of virus genome-encoded EGFP expression upon infection. Further, the levels of expression of viral genes from the various kinetic classes of infection, immediate early genes IE1 and IE2, early genes UL26 and UL44, and late genes pp28 and pp65, were increased substantially by PDGFRα overexpression in HeLa cells (Fig. 7C). The expression levels of IE1, IE2, UL26, and UL44 in PDGFRα-expressing HeLa cells were comparable to those in the primary fibroblasts (Fig. 7C). However, the late proteins pp28 and pp65 were rescued to a lesser extent (Fig. 7C). Given that these true late genes require DNA replication, we sought to examine how PDGFRα overexpression affects viral DNA replication in HeLa cells. As shown in Fig. 7D, PDGFRα overexpression partially rescued viral DNA replication, but only to about 12% of the level found in primary fibroblasts. Further, PDGFRα overexpression did not increase virion production in HeLa cells (Fig. 7E). As AD169 is defective in its ability to replicate in epithelial cells, we sought to investigate whether PDGFRα overexpression would influence the replication of an HCMV clinical strain (TB40/e) in HeLa cells. As shown in Fig. 7F, PDGFRα overexpression increased the level of virus genome-encoded EGFP expression in HeLa cells. However, PDGFRα overexpression failed to rescue TB40/e virion production in HeLa cells (Fig. 7G). With respect to PDGFRα expression, these results largely recapitulate the previous results obtained with TAg-expressing fibroblasts. Collectively, our data suggest that PDGFRα overexpression can largely rescue HCMV entry and gene expression but can only partly rescue DNA replication in tumor-derived cell lines.
FIG 7.

PDGFRα overexpression increases HCMV gene expression and DNA replication but not virion production in HeLa cells. (A) Lysates of HeLa cells transduced with PDGFRα cDNA or a control vector were subjected to Western blotting analysis of PDGFRα. (B) HeLa cells transduced with PDGFRα cDNA or a control vector were infected with EGFP-tagged AD169, and the fluorescence of EGFP was observed at 48 hpi. Bars, 100 μm. (C) Primary fibroblasts (P) or HeLa cells transduced with PDGFRα (HeLa-R) or a control vector (HeLa-V) were infected with HCMV (AD169, MOI = 3.0), harvested at 4, 24, 48, and 72 hpi, and subjected to Western blotting analysis for IE1, IE2, UL26, UL44, pp65, pp28, PDGFRα, and GAPDH. (D) HCMV DNA abundance in cells infected as described in the legend to panel C was measured and normalized to the quantity in HCMV-infected primary fibroblasts. (E) The production of infectious virions in HeLa cells transduced with PDGFRα cDNA or a control vector (AD169, MOI = 3.0, 120 hpi) was measured. (F) HeLa cells transduced with PDGFRα cDNA or a control vector were infected with EGFP-tagged TB40/e, and the fluorescence of EGFP was observed at 72 hpi. Bars, 100 μm. (G) The cells for which the results are presented in panel C were infected with EGFP-tagged TB40/e (MOI = 3.0), and virion production was measured at 5, 7, and 10 dpi. *, levels below the detection limit. The numbers to the left of the gels in panels A and C are molecular masses (in kilodaltons).
DISCUSSION
In contrast to many herpesviruses, HCMV cannot productively infect cells of most cancer-derived cell lines (1, 5). The mechanisms of this restriction are not clear. Here we show that the expression of the SV40 TAg blocks HCMV replication but not HSV replication in human fibroblasts. Specifically, TAg expression inhibited nuclear delivery of HCMV genome and the tegument protein pp65, indicative of a block of virion fusion. Further, TAg expression downregulated PDGFRα expression. This reduction in PDGFRα was found to be functionally relevant, as PDGFRα transduction partly rescued viral entry and gene expression, although it did not rescue the production of infectious viral progeny. TAg expression induced further downstream restrictions, including attenuation of the accumulation of IE2 and viral DNA replication. Transduction of IE2 further restored HCMV gene expression and increased viral DNA replication, but it did not increase the production of infectious progeny. Collectively, our results indicate that oncogenic TAg expression induces multiple blocks to successful HCMV infection.
HCMV phosphorylates and activates PDGFRα, which is required for HCMV entry and gene expression in human embryonic lung fibroblasts (12). Specifically, the HCMV gB protein physically interacts with and activates PDGFRα in human fibroblasts (12). It was later reported that PDGFRα is not the physical receptor for HCMV entry in endothelial and epithelial cells but, instead, that its increased expression promotes the entry of HCMV strains that lack gH/gL/UL128-131, key viral determinants of tropism in endothelial/epithelial cells, via aberrant pathways involving endocytosis and endosome fusion (25). Our results show that SV40 TAg expression downregulates the accumulation of PDGFRα in human foreskin fibroblasts. This is in agreement with reports indicating that the SV40 early region reduces the abundance of mRNA for growth factor receptors in fibroblasts (26, 27). The expression of PDGFRα appears to be generally important for HCMV entry into transformed cells, as we found that PDGFRα overexpression substantially increases HCMV AD169 entry, gene expression, and DNA replication in TAg-expressing fibroblasts as well as HeLa cells.
Despite promoting entry, the expression of PDGFRα did not rescue the production of infectious HCMV. Notably, PDGFRα overexpression decreased the level of production of infectious virions in primary fibroblasts (Fig. 4C). During HCMV infection in primary fibroblasts, HCMV was found to acutely downregulate PDGFRα expression as early as 24 hpi (Fig. 4D and E). Similarly, another postulated HCMV receptor, EGFR, is reportedly downregulated transcriptionally upon HCMV infection in human fetal lung fibroblasts (28). Collectively, these data suggest that the downregulation of host proteins involved in entry at the later times of infection may be important for the proper production of viral progeny. Such scenarios have been proposed for other viruses, for example, duck hepatitis B virus, which induces the degradation of its receptor, gp180 (29).
We found that the balance of IE1 and IE2 expression was altered in cells expressing TAg and PDGFRα, resulting in substantially less IE2 expression than IE1 expression. Given the role of IE2 as the major viral transcriptional activator, the viral gene expression defects associated with HCMV infection of PDGFRα-expressing TAg fibroblasts are likely a result of this reduced IE2 expression. This hypothesis is supported by the data showing that the transduction of IE2 rescues viral gene expression. However, the findings that IE2 transduction did not rescue the production of viral progeny and did not substantially rescue viral DNA replication suggest that an additional restriction(s) inhibits viral DNA replication.
Our results indicate that in primary cells, the processing of MIE transcripts that encode IE2 and IE1 shifts from a 35:65 ratio of IE2/IE1 at 4 hpi to an approximately 50:50 ratio at 24 hpi. In contrast, in TAg-expressing fibroblasts, this ratio of IE2/IE1 starts at about 20:80 at 4 hpi and shifts to a ratio of less than 10:90 at 24 hpi. It is currently unclear how the production, splicing, or processing of these transcripts may differ in primary versus TAg-expressing fibroblasts. Notably, a similar switch favoring IE1 expression at the expense of IE2 expression was observed upon inhibition of Cox-2, which blocks HCMV infection (30). While it is unclear if there is any link between our observations and those made upon Cox-2 inhibition, given the essential nature of IE2 expression for successful HCMV infection, the mechanisms that govern these events are of particular interest and warrant study in the future.
HCMV has increasingly been implicated as an etiological agent in oncogenesis. HCMV genes have been found in a variety of human carcinomas but not in matched tissue controls (31–34). HCMV nucleic acids and proteins have also been found to be associated with a high percentage of malignant gliomas and meduloblastomas (35–39). Further, many HCMV gene products have been shown to induce oncogenic activities, including the activation/deregulation of cell growth pathways and inhibition of tumor suppressor pathways (reviewed in references 40 and 41). Consistent with the activation of these oncogenic events, HCMV gene products have been found to transform rodent fibroblasts (42) and to stimulate oncogenesis and cooperate with other oncogenic mutations in in vivo models of tumorigenesis (38, 43, 44). While HCMV is clearly not sufficient to induce tumor formation in people, a hit-and-run contribution has been proposed (45). Collectively, these results suggest the possibility that abortive infection of an immortalized cell, in a manner analogous to that suggested by our observations, could potentially provide precancerous cells with viral factors/functions that could stimulate the oncogenic process. Such a scenario would be consistent with the findings that cells derived from clinical meduloblastoma samples were found to express high levels of functional viral gene products (39). Further study is necessary to explore the potential links between HCMV and oncogenesis.
In summary, we found that SV40 TAg expression induces multiple restrictions to HCMV infection: (i) TAg expression blocks HCMV entry by inhibiting the expression of the entry protein PDGFRα, (ii) TAg expression inhibits viral gene expression by preferentially downregulating IE2 expression, and (iii) TAg expression inhibits viral DNA replication upon IE2 restoration. Most viruses that are capable of in vitro propagation grow to substantially higher titers in cancer-derived cell lines. The inability to do so makes HCMV somewhat unique in relation to most viruses studied. At first pass, the failure of HCMV to replicate in transformed cells appears to have limited relevance to HCMV-associated disease. However, understanding the mechanisms through which specific oncogenic signaling events can limit HCMV infection is broadly informative, and the information gained can be applied to other infectious contexts. For example, the mechanisms responsible for the observed modulation of IE1 versus IE2 levels could be important in other types of HCMV infection, e.g., controlling viral gene expression during latency or reactivation. Once fully elucidated, these mechanisms could potentially be therapeutically targeted to modulate infectious outcomes.
REFERENCES
- 1.Mocarski ES, Shenk T, Pass RF. 2007. Cytomegaloviruses, p 2701–2757. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE PM (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Gerna G, Baldanti F, Revello MG. 2004. Pathogenesis of human cytomegalovirus infection and cellular targets. Hum Immunol 65:381–386. doi: 10.1016/j.humimm.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 3.Cannon MJ. 2009. Congenital cytomegalovirus (CMV) epidemiology and awareness. J Clin Virol 46(Suppl 4):S6–S10. doi: 10.1016/j.jcv.2009.09.002. [DOI] [PubMed] [Google Scholar]
- 4.Hahn G, Jores R, Mocarski ES. 1998. Cytomegalovirus remains latent in a common precursor of dendritic and myeloid cells. Proc Natl Acad Sci U S A 95:3937–3942. doi: 10.1073/pnas.95.7.3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith JD. 1986. Human cytomegalovirus: demonstration of permissive epithelial cells and nonpermissive fibroblastic cells in a survey of human cell lines. J Virol 60:583–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang D, Shenk T. 2005. Human cytomegalovirus UL131 open reading frame is required for epithelial cell tropism. J Virol 79:10330–10338. doi: 10.1128/JVI.79.16.10330-10338.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang D, Shenk T. 2005. Human cytomegalovirus virion protein complex required for epithelial and endothelial cell tropism. Proc Natl Acad Sci U S A 102:18153–18158. doi: 10.1073/pnas.0509201102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ryckman BJ, Chase MC, Johnson DC. 2008. HCMV gH/gL/UL128-131 interferes with virus entry into epithelial cells: evidence for cell type-specific receptors. Proc Natl Acad Sci U S A 105:14118–14123. doi: 10.1073/pnas.0804365105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ryckman BJ, Rainish BL, Chase MC, Borton JA, Nelson JA, Jarvis MA, Johnson DC. 2008. Characterization of the human cytomegalovirus gH/gL/UL128-131 complex that mediates entry into epithelial and endothelial cells. J Virol 82:60–70. doi: 10.1128/JVI.01910-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ryckman BJ, Jarvis MA, Drummond DD, Nelson JA, Johnson DC. 2006. Human cytomegalovirus entry into epithelial and endothelial cells depends on genes UL128 to UL150 and occurs by endocytosis and low-pH fusion. J Virol 80:710–722. doi: 10.1128/JVI.80.2.710-722.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hahn WC, Counter CM, Lundberg AS, Beijersbergen RL, Brooks MW, Weinberg RA. 1999. Creation of human tumour cells with defined genetic elements. Nature 400:464–468. doi: 10.1038/22780. [DOI] [PubMed] [Google Scholar]
- 12.Soroceanu L, Akhavan A, Cobbs CS. 2008. Platelet-derived growth factor-alpha receptor activation is required for human cytomegalovirus infection. Nature 455:391–395. doi: 10.1038/nature07209. [DOI] [PubMed] [Google Scholar]
- 13.Yu D, Smith GA, Enquist LW, Shenk T. 2002. Construction of a self-excisable bacterial artificial chromosome containing the human cytomegalovirus genome and mutagenesis of the diploid TRL/IRL13 gene. J Virol 76:2316–2328. doi: 10.1128/jvi.76.5.2316-2328.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang D, Bresnahan W, Shenk T. 2004. Human cytomegalovirus encodes a highly specific RANTES decoy receptor. Proc Natl Acad Sci U S A 101:16642–16647. doi: 10.1073/pnas.0407233101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.O'Connor CM, Murphy EA. 2012. A myeloid progenitor cell line capable of supporting human cytomegalovirus latency and reactivation, resulting in infectious progeny. J Virol 86:9854–9865. doi: 10.1128/JVI.01278-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rosenke K, Fortunato EA. 2004. Bromodeoxyuridine-labeled viral particles as a tool for visualization of the immediate-early events of human cytomegalovirus infection. J Virol 78:7818–7822. doi: 10.1128/JVI.78.14.7818-7822.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA III, Smith HO. 2009. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 18.Zhu H, Shen Y, Shenk T. 1995. Human cytomegalovirus IE1 and IE2 proteins block apoptosis. J Virol 69:7960–7970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Munger J, Yu D, Shenk T. 2006. UL26-deficient human cytomegalovirus produces virions with hypophosphorylated pp28 tegument protein that is unstable within newly infected cells. J Virol 80:3541–3548. doi: 10.1128/JVI.80.7.3541-3548.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Silva MC, Yu QC, Enquist L, Shenk T. 2003. Human cytomegalovirus UL99-encoded pp28 is required for the cytoplasmic envelopment of tegument-associated capsids. J Virol 77:10594–10605. doi: 10.1128/JVI.77.19.10594-10605.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Browne EP, Shenk T. 2003. Human cytomegalovirus UL83-coded pp65 virion protein inhibits antiviral gene expression in infected cells. Proc Natl Acad Sci U S A 100:11439–11444. doi: 10.1073/pnas.1534570100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fortunato EA, Sanchez V, Yen JY, Spector DH. 2002. Infection of cells with human cytomegalovirus during S phase results in a blockade to immediate-early gene expression that can be overcome by inhibition of the proteasome. J Virol 76:5369–5379. doi: 10.1128/JVI.76.11.5369-5379.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang X, Huong SM, Chiu ML, Raab-Traub N, Huang ES. 2003. Epidermal growth factor receptor is a cellular receptor for human cytomegalovirus. Nature 424:456–461. doi: 10.1038/nature01818. [DOI] [PubMed] [Google Scholar]
- 24.Stinski MF, Petrik DT. 2008. Functional roles of the human cytomegalovirus essential IE86 protein. Curr Top Microbiol Immunol 325:133–152. [DOI] [PubMed] [Google Scholar]
- 25.Vanarsdall AL, Wisner TW, Lei H, Kazlauskas A, Johnson DC. 2012. PDGF receptor-alpha does not promote HCMV entry into epithelial and endothelial cells but increased quantities stimulate entry by an abnormal pathway. PLoS Pathog 8:e1002905. doi: 10.1371/journal.ppat.1002905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bae VL, Jackson-Cook CK, Brothman AR, Maygardens SJ, Ware JL. 1994. Tumorigenicity of SV40 T antigen immortalized human prostate epithelial cells: association with decreased epidermal growth factor receptor (EGFR) expression. Int J Cancer 58:721–729. doi: 10.1002/ijc.2910580517. [DOI] [PubMed] [Google Scholar]
- 27.Wang JL, Nister M, Bongcam-Rudloff E, Ponten J, Westermark B. 1996. Suppression of platelet-derived growth factor alpha- and beta-receptor mRNA levels in human fibroblasts by SV40 T/t antigen. J Cell Physiol 166:12–21. doi:. [DOI] [PubMed] [Google Scholar]
- 28.Beutler T, Hoflich C, Stevens PA, Kruger DH, Prosch S. 2003. Downregulation of the epidermal growth factor receptor by human cytomegalovirus infection in human fetal lung fibroblasts. Am J Respir Cell Mol Biol 28:86–94. doi: 10.1165/rcmb.4881. [DOI] [PubMed] [Google Scholar]
- 29.Breiner KM, Urban S, Glass B, Schaller H. 2001. Envelope protein-mediated down-regulation of hepatitis B virus receptor in infected hepatocytes. J Virol 75:143–150. doi: 10.1128/JVI.75.1.143-150.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu H, Cong JP, Yu D, Bresnahan WA, Shenk TE. 2002. Inhibition of cyclooxygenase 2 blocks human cytomegalovirus replication. Proc Natl Acad Sci U S A 99:3932–3937. doi: 10.1073/pnas.052713799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Samanta M, Harkins L, Klemm K, Britt WJ, Cobbs CS. 2003. High prevalence of human cytomegalovirus in prostatic intraepithelial neoplasia and prostatic carcinoma. J Urol 170:998–1002. doi: 10.1097/01.ju.0000080263.46164.97. [DOI] [PubMed] [Google Scholar]
- 32.Harkins L, Volk AL, Samanta M, Mikolaenko I, Britt WJ, Bland KI, Cobbs CS. 2002. Specific localisation of human cytomegalovirus nucleic acids and proteins in human colorectal cancer. Lancet 360:1557–1563. doi: 10.1016/S0140-6736(02)11524-8. [DOI] [PubMed] [Google Scholar]
- 33.El-Shinawi M, Mohamed HT, El-Ghonaimy EA, Tantawy M, Younis A, Schneider RJ, Mohamed MM. 2013. Human cytomegalovirus infection enhances NF-kappaB/p65 signaling in inflammatory breast cancer patients. PLoS One 8:e55755. doi: 10.1371/journal.pone.0055755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Taher C, de Boniface J, Mohammad AA, Religa P, Hartman J, Yaiw KC, Frisell J, Rahbar A, Soderberg-Naucler C. 2013. High prevalence of human cytomegalovirus proteins and nucleic acids in primary breast cancer and metastatic sentinel lymph nodes. PLoS One 8:e56795. doi: 10.1371/journal.pone.0056795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cobbs CS, Harkins L, Samanta M, Gillespie GY, Bharara S, King PH, Nabors LB, Cobbs CG, Britt WJ. 2002. Human cytomegalovirus infection and expression in human malignant glioma. Cancer Res 62:3347–3350. [PubMed] [Google Scholar]
- 36.Cobbs CS, Soroceanu L, Denham S, Zhang W, Kraus MH. 2008. Modulation of oncogenic phenotype in human glioma cells by cytomegalovirus IE1-mediated mitogenicity. Cancer Res 68:724–730. doi: 10.1158/0008-5472.CAN-07-2291. [DOI] [PubMed] [Google Scholar]
- 37.Ranganathan P, Clark PA, Kuo JS, Salamat MS, Kalejta RF. 2012. Significant association of multiple human cytomegalovirus genomic loci with glioblastoma multiforme samples. J Virol 86:854–864. doi: 10.1128/JVI.06097-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Soroceanu L, Matlaf L, Bezrookove V, Harkins L, Martinez R, Greene M, Soteropoulos P, Cobbs CS. 2011. Human cytomegalovirus US28 found in glioblastoma promotes an invasive and angiogenic phenotype. Cancer Res 71:6643–6653. doi: 10.1158/0008-5472.CAN-11-0744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baryawno N, Rahbar A, Wolmer-Solberg N, Taher C, Odeberg J, Darabi A, Khan Z, Sveinbjornsson B, Fuskevag OM, Segerstrom L, Nordenskjold M, Siesjo P, Kogner P, Johnsen JI, Soderberg-Naucler C. 2011. Detection of human cytomegalovirus in medulloblastomas reveals a potential therapeutic target. J Clin Invest 121:4043–4055. doi: 10.1172/JCI57147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sanchez V, Spector DH. 2008. Subversion of cell cycle regulatory pathways. Curr Top Microbiol Immunol 325:243–262. [DOI] [PubMed] [Google Scholar]
- 41.Yurochko AD. 2008. Human cytomegalovirus modulation of signal transduction. Curr Top Microbiol Immunol 325:205–220. [DOI] [PubMed] [Google Scholar]
- 42.Doniger J, Muralidhar S, Rosenthal LJ. 1999. Human cytomegalovirus and human herpesvirus 6 genes that transform and transactivate. Clin Microbiol Rev 12:367–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Price RL, Song J, Bingmer K, Kim TH, Yi JY, Nowicki MO, Mo X, Hollon T, Murnan E, Alvarez-Breckenridge C, Fernandez S, Kaur B, Rivera A, Oglesbee M, Cook C, Chiocca EA, Kwon CH. 2013. Cytomegalovirus contributes to glioblastoma in the context of tumor suppressor mutations. Cancer Res 73:3441–3450. doi: 10.1158/0008-5472.CAN-12-3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Price RL, Bingmer K, Harkins L, Iwenofu OH, Kwon CH, Cook C, Pelloski C, Chiocca EA. 2012. Cytomegalovirus infection leads to pleomorphic rhabdomyosarcomas in Trp53+/− mice. Cancer Res 72:5669–5674. doi: 10.1158/0008-5472.CAN-12-2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shen Y, Zhu H, Shenk T. 1997. Human cytomegalovirus IE1 and IE2 proteins are mutagenic and mediate “hit-and-run” oncogenic transformation in cooperation with the adenovirus E1A proteins. Proc Natl Acad Sci U S A 94:3341–3345. doi: 10.1073/pnas.94.7.3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Strang BL, Boulant S, Coen DM. 2010. Nucleolin associates with the human cytomegalovirus DNA polymerase accessory subunit UL44 and is necessary for efficient viral replication. J Virol 84:1771–1784. doi: 10.1128/JVI.01510-09. [DOI] [PMC free article] [PubMed] [Google Scholar]