

ABSTRACT
The multifunctional HIV-1 accessory protein Vif counters the antiviral activities of APOBEC3G (A3G) and APOBEC3F (A3F), and some Vifs counter stable alleles of APOBEC3H (A3H). Studies in humanized mice have shown that HIV-1 lacking Vif expression is not viable. Here, we look at the relative contributions of the three APOBEC3s to viral extinction. Inoculation of bone marrow/liver/thymus (BLT) mice with CCR5-tropic HIV-1JRCSF (JRCSF) expressing a vif gene inactive for A3G but not A3F degradation activity (JRCSFvifH42/43D) displayed either no or delayed replication. JRCSF expressing a vif gene mutated to inactivate A3F degradation but not A3G degradation (JRCSFvifW79S) always replicated to high viral loads with variable delays. JRCSF with vif mutated to lack both A3G and A3F degradation activities (JRCSFvifH42/43DW79S) failed to replicate, mimicking JRCSF without Vif expression (JRCSFΔvif). JRCSF and JRCSFvifH42/43D, but not JRCSFvifW79S or JRCSFvifH42/43DW79S, degraded APOBEC3D. With one exception, JRCSFs expressing mutant Vifs that replicated acquired enforced vif mutations. These mutations partially restored A3G or A3F degradation activity and fully replaced JRCSFvifH42/43D or JRCSFvifW79S by 10 weeks. Surprisingly, induced mutations temporally lagged behind high levels of virus in blood. In the exceptional case, JRCSFvifH42/43D replicated after a prolonged delay with no mutations in vif but instead a V27I mutation in the RNase H coding sequence. JRCSFvifH42/43D infections exhibited massive GG/AG mutations in pol viral DNA, but in viral RNA, there were no fixed mutations in the Gag or reverse transcriptase coding sequence. A3H did not contribute to viral extinction but, in combination with A3F, could delay JRCSF replication. A3H was also found to hypermutate viral DNA.
IMPORTANCE Vif degradation of A3G and A3F enhances viral fitness, as virus with even a partially restored capacity for degradation outgrows JRCSFvifH42/43D and JRCSFvifW79S. Unexpectedly, fixation of mutations that replaced H42/43D or W79S in viral RNA lagged behind the appearance of high viral loads. In one exceptional JRCSFvifH42/43D infection, vif was unchanged but replication proceeded after a long delay. These results suggest that Vif binds and inhibits the non-cytosine deaminase activities of intact A3G and intact A3F, allowing JRCSFvifH42/43D and JRCSFvifW79S to replicate with reduced fitness. Subsequently, enhanced Vif function is acquired by enforced mutations. In infected cells, JRCSFΔvif and JRCSFvifH42/43DW79S are exposed to active A3F and A3G and fail to replicate. JRCSFvifH42/43D Vif degrades A3F and, in some cases, overcomes A3G mutagenic activity to replicate. Vif may have evolved to inhibit A3F and A3G by stoichiometric binding and subsequently acquired the ability to target these proteins to proteasomes.
INTRODUCTION
HIV-1 Vif is an accessory protein that prevents inhibition of viral replication by the innate immune system proteins APOBEC3G (A3G) and APOBEC3F (A3F) (1–3). These HIV-1 restriction factors gain access to HIV-1 virions and deaminate cytosines in negative-strand DNA during reverse transcription of viral genomic RNA (4–6). This activity yields G-to-A mutations in HIV-1 genes, leading to loss of viral viability (4, 6). Vif prevents A3G and A3F antiviral activity by linkage to proteosomal degradation (7–9). Vif is also found inside virions, where it can potentially inhibit encapsidated A3G and A3F (10, 11). In addition to directly inhibiting the G-to-A mutagenic activity of A3G and A3F, virion-encapsidated Vif could also prevent A3G and A3F inhibition of reverse transcription, transcription elongation, and proviral integration and inactivation of the transactivation response element (12–16).
The devastating effect of human APOBEC3s on HIV-1JRCSF (JRCSF) replication in vivo in the absence of Vif has been previously demonstrated in NSG-hu humanized mice (17, 18). We also reported that JRCSF lacking Vif function cannot systemically replicate in bone marrow/liver/thymus (BLT) humanized mice and is quickly extinguished (17). In cases where we were able to recover archival sequences from viral DNA, high levels of G-to-A mutations incompatible with viral replication were present. The mutations were approximately 85% GG to AG and 15% GA to AA (17). A3G is the only APOBEC3 that preferentially mutates GG over GA sites, pointing to A3G as the dominant restricting factor for HIV-1 (4, 19). An alternate hypothesis is that any of the other APOBEC3s, all of which have dominantly GA-to-AA mutagenic activity, may contribute to the totality of the restriction but do so mainly by mechanisms not involving G-to-A mutation. Further, the identities of APOBEC3 genes other than A3G genes that could significantly contribute to HIV-1 extinction in humanized mice remain an outstanding question. The leading candidates are A3F, APOBEC3H (A3H), and APOBEC3D (A3D), in descending order of likely importance (2, 20–22). However, evidence for significant nondeaminase activity in vivo has not been forthcoming (22).
To advance understanding of the role of APOBEC3 genes in restricting HIV-1 vif− (i.e., vif defective), we infected BLT mice with JRCSF containing mutated vif genes that were defective for A3G and/or A3F degradation. We found that A3G acting alone was able to completely terminate HIV-1 infection in some cases, while in others, replication was greatly delayed but eventually produced high levels of virions in blood. A3F acting alone had relatively weak effects on viral replication, but Vif mutations partially restoring A3F degradation activity were nonetheless strongly selected. Strikingly, virus with the doubly mutated vif that was unable to degrade A3G or A3F mimicked vif− virus by failing to replicate in BLT mice. Also, a possible role for A3H in explaining our observations was addressed. A3H was not a necessary component for termination of HIV-1 infection but in combination with A3F may contribute to delays in viral replication. A case of HIV-1 hypermutation was found in which numerous GA-to-AA mutations could be attributed to A3H.
Therefore, we found that A3G strongly restricts HIV-1, if not countered by vif, but consistent restriction requires an additional factor which several lines of evidence suggest to be A3F. The major antiviral impact of A3F may be through a cytosine deaminase-independent mechanism.
MATERIALS AND METHODS
Cell culture.
TZM HeLa cells obtained from the NIH AIDS Research and Reference Reagent Program (23) and human embryonic kidney 293T cells were cultured at 37°C and 10% CO2 in Dulbecco's modified Eagle medium (DMEM) (Sigma) supplemented with 10% fetal bovine serum (SAFC Biosciences), 50 IU penicillin, 50 μg/ml streptomycin, and 2 mM l-glutamine (Cellgro). CEM-SS cells, a human T cell line, were cultured at 37°C and 5% CO2 in RPMI 1640 (Sigma) supplemented with 10% fetal bovine serum (SAFC Biosciences), 50 IU penicillin, 50 μg/ml streptomycin, 2 mM l-glutamine (Cellgro), and 1 mM sodium pyruvate (Cellgro).
Transfections.
HEK 293T cells were transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. Cells (7 × 105 per well in a 6-well plate or 5 × 106 in a 10-cm dish) were cultured in DMEM in the absence of antibiotics 24 h prior to transfection. Plasmid DNA was resuspended in Opti-MEM (Gibco) at a ratio of 1 μg of DNA to 2.5 μl of Lipofectamine 2000. Following a 20-min incubation at room temperature, the DNA-Lipofectamine 2000 mixture was added dropwise to the 293T cells.
Preparation of whole-cell lysates and Western blotting.
Mammalian expression vectors for A3G (pcDNA3.1A3GV5) and A3F (pcDNA3.1A3FV5) were obtained from B. M. Peterlin (24). These plasmids were transfected in 293T cells with JRCSF proviral clones. The transfected 293T cells were washed with cold phosphate-buffered saline (PBS) while attached to the culture dish and then lysed in 800 μl of cold lysis buffer (50 mM Tris, pH 8.0, 10% glycerol [Fisher], 100 mM NaCl [Fisher], 25 mM NaF [Sigma], 2 mM Na3VO2 [Sigma], 20 mM β-glycerophosphate [Sigma], 25 mM benzamidine [Sigma], 2 mM EDTA, pH 8.0, and 0.5% IGEPAL CA630 [Sigma]) supplemented with one mini-protease inhibitor tablet (Roche) per 10 ml of lysis buffer. The cell lysates were cleared by centrifugation at 15,000 × g for 10 min at 4°C, and the protein concentrations of the postnuclear supernatants were determined using IgG protein standards in the Bio-Rad protein assay reagent (Bio-Rad). Total protein (200 μg) from whole-cell lysates in sodium dodecyl sulfate (SDS) loading buffer was denatured for 5 min at 95°C and resolved on a 12% polyacrylamide gel for 900 V · h. The proteins were then transferred to a Hybond-C Extra nitrocellulose membrane (Amersham Biosciences) using a TransBlot semidry transfer cell (Bio-Rad) at 20 V for 42 min. The membrane was then blocked for 1 h in 10% milk in Tris-Tween-buffered saline (TTBS) (10 mM Tris, pH 8.0, 150 mM NaCl, 0.05% Tween 20 [Fisher Scientific]), incubated with primary antibody in 10% milk in TTBS for 12 to 16 h at 4°C, washed three times in TTBS, incubated with horseradish peroxidase (HRP)-conjugated secondary antibody in 10% milk in TTBS for 2 h, and finally washed three times in TTBS. The membrane was incubated in the luminol substrate buffer (100 mM Tris, pH 8.8, 2.5 mM luminol [Fluka], 400 μM p-coumaric acid [Sigma], and 5.4 mM H2O2 [Sigma]) for 5 min and then exposed to film for an appropriate amount of time to visualize expression of Vif, A3G, or A3F and glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Wild-type JRCSF Vif effectively depleted A3G and A3F. A3D (pCS2Apobec3DEHA) degradation activity was assayed as described above, with the exception that the SDS loading buffer contained 6 M urea (25).
Antibodies.
Rabbit anti-Vif (number 2221), obtained from the NIH AIDS Research and Reference Reagent Program (26), was used at 1:1,000. Mouse anti-V5 (Invitrogen number R960-25) was used at 1:7,500. Mouse anti-hemagglutinin (HA) (Abcam number 1818) was used at 1:1,000. Rabbit anti-GAPDH (clone 14C10; Cell Signaling number 2118L) was used at 1:3,000. Goat anti-rabbit HRP (Cell Signaling number 7074S) was used at 1:10,000. Goat anti-mouse HRP (Invitrogen number 62-6520) was used at 1:10,000.
Generation of vif-defective HIV-1.
All experiments were performed using JRCSF obtained from the NIH AIDS Research and Reference Reagent Program (27). JRCSFΔvif was previously described (17). The following JRCSF proviral clones with mutations in vif were created by site-directed mutagenesis: JRCSFvifH42/43D, JRCSFvifW79S, JRCSFvifH42/43DW79S, JRCSFvifW79F, JRCSFvifW79Y, JRCSFvifH42/43N, and JRCSFvifH42N/H43G. The H42/43D mutation was based on the report by Mehle et al. that mutating H42/H43 to N42/N43 reduced A3G degradation (28). We mutated the histidines to aspartates, as they were potentially more disruptive of function (29). For a mutation that blocked A3F degradation, we mutated tryptophan 79 to serine (JRCSFvifW79S) based on reports that mutation of this residue to alanine resulted in a specific block in the capacity of the mutant protein to degrade A3F (30, 31). Mutation of W79 to serine followed the suggestion of Yu et al. that the hydrophilic serine may be more detrimental to function than alanine (9).
Production of HIV-1 stocks and titration.
HIV-1 stocks were generated by transfecting proviral DNA into 293T cells using Lipofectamine 2000 (Invitrogen) as indicated above. The virus-containing culture supernatant was collected and cleared of cellular debris, first by centrifugation at 3,000 rpm for 20 min and then by filtering through a 0.45-μm filter (Millipore). Infectious titers of JRCSF and JRCSF vif variants were determined by using TZM-bl cells (23). Infected cells turn blue upon staining with X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) (Sigma). Blue cells were counted by microscopy, and the counts were used to calculate the titer, which was defined as tissue culture infectious units per milliliter (TCIU).
Viral cultures.
CCR5-expressing CEM-SS cells were used to propagate both wild-type and vif-defective JRCSF (17). Cells (1 × 106) were infected in a 24-well plate with 5 × 104 TCIU of virus in complete RPMI containing 4 μg/ml Polybrene at 37°C and 5% CO2 for 4 h. The cells were then washed extensively with PBS and cultured at 37°C and 5% CO2 in complete RPMI containing 0.5 μg/ml puromycin to maintain selection of CCR5-expressing cells. The cell cultures were passaged every 3 days, and a sample of the culture supernatant was collected for quantitation of the amount of viral capsid protein by p24 enzyme-linked immunosorbent assay (ELISA).
Generation and infection of BLT humanized mice.
BLT mice were prepared as previously described (32–43). Briefly, autologous human CD34+ cells isolated from fetal liver (Advanced Bioscience Resources, Alameda, CA) were transplanted into thymus/liver-implanted NOD/SCID interleukin 2γ−/− (IL-2γ−/−) mice (Jackson Laboratories, Bar Harbor, ME). Human CD45+ cells in the peripheral blood of these mice were monitored periodically by flow cytometry (FACSCanto; BD Biosciences). The mice were maintained at the Division of Laboratory Animal Medicine, University of North Carolina at Chapel Hill (UNC-CH), in accordance with protocols approved by the UNC-CH Institutional Animal Care and Use Committee. The mice were inoculated intravenously with either wild-type JRCSF or vif-defective JRCSF by tail vein injection.
Flow cytometry.
Peripheral blood obtained from the humanized mice (50 μl) was first blocked with murine IgG (Sigma) and then stained with mouse anti-human CD45 (clone 2D1; 557873; Becton-Dickinson) to exclude murine cells from the analysis. Human lymphocytes were identified by their human CD45+ expression and further characterized by staining with mouse anti-human CD3 (clone HIT3a; 555573; Becton-Dickinson). Human CD3+ cells were further analyzed for CD4+ and CD8+ expression with mouse anti-human CD4 (clone RPA-T4; 555749; Becton-Dickinson) or CD8 antibodies (clone SK1; 347314; Becton-Dickinson). Data were collected using a FACSCanto instrument and analyzed using FACS Diva software (Becton-Dickinson).
Peripheral blood analysis.
Peripheral blood was collected from humanized mice from the retro-orbital vein using EDTA-containing capillary tubes (Drummond Scientific). Whole blood was centrifuged, and the plasma was removed for analysis of viral RNA. The volume of plasma removed was replaced with solution B (1 liter PBS [Sigma], 5 g bovine serum albumin [BSA] [Sigma], 50 U/ml penicillin [Sigma], 50 mg/ml streptomycin [Sigma], 1% citrate phosphate dextrose [Sigma]). The cellular portion of the blood was used for flow cytometric analysis as previously described or for genomic-DNA isolation. Peripheral blood to be used for DNA isolation was resuspended in 1 ml of a hypotonic red blood cell lysis buffer (500 ml distilled H2O [dH2O], 4.15 g NH4Cl [Sigma], 0.5 g KHCO3 [Sigma], and 0.019 g EDTA [Sigma]). Following a 10-min incubation at room temperature, the lysed blood was centrifuged at 10,000 rpm for 1 min, and the lysis buffer was removed by aspiration. The resulting cell pellet was stored at −80°C until DNA extraction.
Plasma viral load analysis.
Plasma viremia was determined by quantitating the copy number of cell-free viral RNA. Plasma (20 μl) from the humanized mice was used to isolate viral RNA using the QIAamp Viral RNA minikit (Qiagen) according to the manufacturer's protocol. Viral RNA was then quantified using a one-step reverse transcriptase protocol. RNA isolated from the plasma (5 μl) was used as the template with the TaqMan RNA-to-Ct 1-step kit (Applied Biosystems). Primers for the real-time PCR were directed against a conserved region of gag and were previously published (44).
Tissue harvest.
The spleen, lymph nodes, and thymus were harvested and disrupted by grinding the tissue through a 70-μm-pore-size cell strainer (Falcon) with the plunger of a 3-ml syringe, and the cells were washed through the strainer with cold solution B. Red blood cells from the spleen were lysed with hypotonic lysis buffer, and cells from all the tissues were pelleted by centrifugation at 1,500 rpm for 5 min at 4°C.
Mononuclear cells were isolated from the bone marrow by crushing the long bones from the hind legs using a mortar and pestle, collecting the cells by washing with cold solution B, and filtering the cells through a 70-μm cell strainer. Red blood cells were lysed with hypotonic lysis buffer, and the cells were pelleted by centrifugation at 1,500 rpm for 5 min at 4°C. Mononuclear cells were isolated from the liver by grinding the tissue through a 70-μm cell strainer and pelleting the cells by centrifugation at 1,500 rpm for 5 min at 4°C. The resulting cell pellet was then resuspended in 40% Percoll, underlayered with 70% Percoll, and centrifuged at 2,400 rpm for 20 min at room temperature. Mononuclear cells were collected from the interface and washed with cold solution B. Cells were isolated from the lungs by first mincing the tissue with scissors and digesting the tissue in 2.5 ml RPMI containing 6 mg collagenase D (Roche) and 50 μg DNase I (Roche) for 30 min at 37°C. The digested tissue was then ground through a 70-μm cell strainer with a 3-ml syringe plunger, washed with cold solution B, and centrifuged at 1,500 rpm for 5 min at 4°C. As with the liver, lung mononuclear cells were isolated by resuspending the cell pellet in 40% Percoll, underlayering with 70% Percoll, and centrifugation at 2,400 rpm for 20 min at room temperature, and the cells were collected from the interface and washed with cold solution B.
Live cells from all tissues were counted by trypan blue exclusion and pelleted for genomic DNA isolation by centrifugation at 10,000 rpm for 1 min or pelleted by centrifugation at 1,500 rpm for 5 min, resuspended in freezing medium (90% heat-inactivated fetal bovine serum [FBS], 10% dimethyl sulfoxide [Fisher]), frozen live in a cryo-freezing container at −80°C, and then stored in liquid nitrogen.
Genomic-DNA isolation and nested PCR of viral DNA.
Genomic DNA was isolated from frozen peripheral blood cell pellets by resuspending a pellet in 0.01 M Tris, pH 7.4, containing recombinant PCR grade proteinase K (Roche) and incubating it at 56°C for 1 h. The proteinase K was then heat inactivated at 95°C for 20 min, and the mixture was frozen at −20°C. After thawing, the samples were centrifuged for 1 min at 14,000 rpm, and the DNA-containing supernatant was transferred to a new, unautoclaved Eppendorf tube.
Genomic DNA from mononuclear cells (5 × 105 to 5 × 106) from animal tissues was prepared using QIAamp DNA blood minicolumns (Qiagen) according to the manufacturer's protocol. All PCRs were performed using the Expand High Fidelity PCR System (Roche). A 1.7-kb region of the JRCSF genome from nucleotides 617 to 2358, encompassing gag, was amplified. The primers for the outer and inner reactions are listed in Table 1. The reaction conditions were as follows: a one-time denaturation at 95°C for 2 min, followed by 35 cycles of 95°C for 15 s, 55°C for 30 s, and 72°C for 1 min 45 s. Amplification concluded with an elongation step at 72°C for 7 min. PCR was performed on a 1.2-kb region of the viral genome from nucleotides 2493 to 4023 including the reverse transcriptase region of pol. The primers for the outer and inner reactions are listed in Table 1. The reaction conditions were as follows: a one-time denaturation at 95°C for 5 min, followed by 30 cycles of 95°C for 1 min, 55°C for 1 min, and 68°C for 1 min. Amplification concluded with an elongation step at 72°C for 7 min. A 3.4-kb region of the viral genome from nucleotides 1877 to 5227 including the RNase H region of pol was also performed. The primers for the outer and inner reactions are listed in Table 1. The touchdown PCR conditions were as follows: a one-time denaturation at 95°C for 2 min; 10 cycles of 95°C for 15 s and 30 s at 65°C and then subtracting 1°C each subsequent cycle; and 68°C for 3.5 min, followed by 25 cycles of 95°C for 15 s, 55°C for 30 s, and 68°C for 3.5 min. Amplification concluded with an elongation step at 68°C for 10 min. A 1.4-kb region from nucleotides 4941 through 6399 of the HIV-1JRCSF genome that included vif was also amplified with the primers listed in Table 1. The reaction conditions were as follows: a one-time denaturation at 95°C for 5 min, followed by 30 cycles of 95°C for 30 s, 55°C for 30 s, and 72°C for 1.5 min. Amplification concluded with an elongation step at 72°C for 7 min.
TABLE 1.
Nested-PCR primer sequences
| Region amplified | Primer | Primer sequence |
|---|---|---|
| HIV-1 JRCSF 617–2358 | Fwd gag Outer | 5′-CTCAATAAAGCTTGCCTTGAGTGC-3′ |
| Rev gag Outer | 5′-CTTCCAATTATGTTGACAGGTGTAGG-3′ | |
| Fwd gag Inner | 5′-GTGTGGAAAATCTCTAGCAGTGGC-3′ | |
| Rev gag Inner | 5′-CTGTATCATCTGCTCCTGTATCTAATAGAGC-3′ | |
| HIV-1 JRCSF 1877–5227 | Fwd pol Outer | 5′-GATGACAGCATGTCAGGGAG-3′ |
| Rev pol Outer | 5′-GGTCAGGGTCTACTTGTGTGC-3′ | |
| Fwd pol Inner | 5′-TGGCTGAAGCAATGAGCCAAG-3′ | |
| Rev pol Inner | 5′-GTGGGATTTGTACTTCTGAAC-3′ | |
| HIV-1 JRCSF 2493–4023 | Fwd RT Outer | 5′-GCTCTATTAGATACAGGAGC-3′ |
| Rev RT Outer | 5′-CCTAATGCATATTGTGAGTCTG-3′ | |
| Fwd RT Inner | 5′-GTAGGACCTACACCTGTCAAC-3′ | |
| Rev RT Inner | 5′-CCTGCAAAGCTAGGTGAATTGC-3′ | |
| HIV-1 JRCSF 4941–6399 | Fwd vif Outer | 5′-CAGGGACAGCAGAGATCC-3′ |
| Rev vif Outer | 5′-GTGGGTACACAGGCATGTGTGG-3′ | |
| Fwd vif Inner | 5′-ATTTGGAAAAGGACCAGCAAAGC-3′ | |
| Rev vif Inner | 5′-GATGCACAAAATAGAGTGGTGG-3′ |
Amplification of cell-free viral RNA.
Viral RNA was isolated from the plasma of JRCSF-exposed humanized mice using a QIAamp viral minikit according to the manufacturer's recommended protocol (Qiagen number 52904). Purified viral RNA was then reverse transcribed into cDNA with the reverse vif outer primer (Table 1) using SuperScript III reverse transcriptase (Invitrogen number 18080-044) according to the manufacturer's recommended protocol. The viral RNA was then digested using RNase H (Invitrogen number 18021-014), and the cDNA was used as a template in nested PCRs for vif, gag, and pol. The PCR products were sequenced with inner primers used for amplification. Individual clones from the amplified viral RNA were sequenced following TA cloning (Invitrogen).
APOBEC3H coding sequence amplification.
Cellular RNA from frozen pellets of peripheral blood mononuclear cells was extracted, and PCR amplification and sequencing of the A3H mRNA coding sequence was performed by the method of Ooms et al. (45).
DNA sequencing and analysis.
Amplified viral DNA was sequenced using the inner nested-PCR primers as sequencing primers. All chromatographs of the amplified DNA sequences were visually analyzed, and the nucleotide sequences were aligned against the corresponding proviral DNA sequence used to generate the viruses using the Highlighter program (http://www.hiv.lanl.gov/content/sequence/HIGHLIGHT/highlighter.html), which can identify G-to-A mutations indicative of APOBEC mutagenesis.
Highlighter output uses a graphical representation of DNA sequences for comparison. To assess the effects of G-to-A transition mutations on the viral protein sequence, amplified DNA sequences were translated using the online ExPASy translation tool (http://au.expasy.org/tools/dna.html).
Statistics.
Data are presented as means ± standard errors of the mean (SEM).
Nucleotide sequence accession numbers.
Sequences were deposited in GenBank with the following accession numbers: KU900139 to KU900143, KU926704, KU900138, and KU887644.
RESULTS
We previously reported that JRCSF with an inactivating deletion in vif (JRCSFΔvif) failed to replicate in BLT humanized mice (17). Here, we investigated if the anti-HIV-1 activity of A3G is sufficient to extinguish HIV-1 replication or if other APOBEC3s have important roles. A JRCSF proviral clone was constructed with a vif gene mutated at histidines 42 and 43, which are known to be critical for A3G degradation. The histidines were replaced with aspartates (JRCSFvifH42/43D) (28). We also mutated tryptophan 79, previously demonstrated to be critical for A3F degradation, to serine (JRCSFvifW79S) (30, 31). The doubly defective JRCSFvifH42/43DW79S was also constructed (see Materials and Methods). The three viruses were produced, and all three had the full replicative capacity of JRCSF in vitro (Fig. 1A). As shown in Fig. 1B, the mutated Vifs were found to be stably expressed and to be defective for A3G degradation but not A3F (H42/43D) or defective for A3F degradation but not A3G (W79S). VifH42/43DW79S failed to degrade both A3G and A3F. We also tested the abilities of the mutated Vifs to degrade A3D. Vif and VifH42/43D effectively degraded A3D, but VifW79S did not. VifH42/43DW79S showed an intermediate phenotype (Fig. 1C).
FIG 1.

Mutations in Vif that disrupt degradation of APOBEC3G or APOBEC3F do not affect virus replication in vitro. (A) Viruses were produced by transfection of 293T cell with proviral clones. CCR5-expressing CEM-SS cells were infected with wild-type JRCSF or JRCSF with point mutations in Vif that disrupt degradation of either APOBEC3G, APOBEC3F, or both. The culture supernatant was monitored longitudinally by ELISA for HIV p24gag protein as a measure of virus replication. (B) HEK-293T cells were cotransfected with the proviral clones in panel A and an expression vector for either V5-tagged APOBEC3G or APOBEC3F. The cell lysates were resolved by SDS-PAGE and probed with anti-V5 to assess APOBEC3 degradation in the presence of the designated Vif mutants. Western blots for Vif and GAPDH were conducted as controls. (C) Same as panel B except that HA-tagged APOBEC3D degradation was probed with anti-HA.
Replication of JRCSFvifH42/43D, JRCSFvifW79S, and JRCSFvifH42/43DW79S in BLT humanized mice.
In Fig. 2A, top, the time course of viral replication following intravenous injection of 90,000 TCIU of JRCSF or JRCSFvifH42/43D is shown. Seven wild-type-JRCSF-inoculated mice are plotted in aggregate (means ± SEM). In two JRCSFvifH42/43D mice (G1 and G2), viral RNA was not detected in peripheral blood for at least 8 weeks. The effective degradation of A3F and A3D by VifH42/43D makes it unlikely that these two APOBEC3s were involved in the observed viral extinction. In the other two mice (G3 and G4), virus replicated, but the appearance of viral RNA in blood was greatly delayed.
FIG 2.

Absolute restriction of HIV in vivo requires both APOBEC3G and APOBEC3F. (A) (Top) The plasma of BLT humanized mice infected with JRCSFvifH42/43D (blue lines) was monitored longitudinally for the presence of HIV RNA. Wild-type JRCSF RNA is presented in aggregate (n = 7) (black diamonds). (Bottom) Nested PCR to detect HIV DNA was performed on genomic-DNA extracts from each tissue listed from the infected humanized mice. +, tissues positive for HIV DNA; −, tissues where HIV DNA was not detected. (B) (Top) Viral RNA was present in the plasma of 4/4 mice infected with JRCSFvifW79S (red lines) at levels comparable to those of wild-type JRCSF (n = 7) (black diamonds). (Bottom) Nested PCR to detect HIV DNA was performed on genomic DNA extracted from each tissue listed from the infected BLT humanized mice. +, tissues positive for HIV DNA; −, tissues where HIV DNA was not detected. (C) (Top) Plasma viral RNA was not observed in 4/4 mice infected with JRCSFvifH42/43DW79S (green lines) for up to 8 weeks postexposure, in contrast to infection with wild-type JRCSF (black diamonds). (Bottom) Viral DNA was not detected in any tissue of JRCSFvifH42/43DW79S-inoculated BLT humanized mice (−). (D) Sequencing of viral DNA amplified from the tissues of mice infected with JRCSFvifH42/43D (G3 and G4) revealed heavy G-to-A mutation at GG sites (blue bar). Few G-to-A mutations were present at GA sites in viral DNA from mice infected with JRCSFvifH42/43D or JRCSFvifW79S, and no mutations were observed in WT1 JRCSF DNA. The data are presented as means ± SEM.
Upon sacrifice, viral DNA (reverse transcriptase coding sequence, nucleotides 2562 to 3881; GenBank accession number, M38429) was found in multiple organs of a representative JRCSF (WT1) mouse, G3, and G4 (Fig. 2A, bottom). G1 which gave no viral replication, was reinfected with JRCSF, and normal replication was observed (not shown). G2 also had no viral replication, and viral DNA could be amplified from only one tissue. Therefore, the replication of JRCSF with a vif gene defective for A3G degradation but not A3F degradation is severely restricted but not consistently extinguished. These variable results are in contrast to JRCSF with a fully defective vif (JRCSFΔvif), which fails to replicate in BLT mice at all (17).
Figure 2B, top, shows the results of the same experiment performed with JRCSFvifW79S. In all cases, there was robust replication, but one mouse (F1) had delayed replication, with 104 copies of viral RNA at 2 weeks. By 4 weeks, all JRCSFvifW79S mice had 105 or more copies of viral RNA, and by 6 weeks, all the mice had viral loads of 106 copies or more. Viral DNA from F1 to F4 was present in nearly all tissues tested (Fig. 2B, bottom). JRCSF defective for both A3G and A3F degradation (JRCSFvifH42/43DW79S) was also inoculated into BLT humanized mice (Fig. 2C). No viral RNA was detected in plasma for 6 to 8 weeks postinjection (Fig. 2C, top), and no evidence of viral DNA in tissues was found (Fig. 2C, bottom). This result mimicked our previously reported results with JRCSFΔvif and strongly implies that the combined activities of A3G and A3F rapidly extinguish viral replication (17).
Prominence of GG-to-AG mutations in archival viral DNA.
The JRCSF reverse transcriptase coding sequence was amplified from multiple organs of G3, G4, and F1 to F4 (Fig. 2A, bottom, and B, bottom). The percentage of GG sites mutated to AG and the percentage of GA sites mutated to AA sites were determined. High levels of GG-to-AG mutations were noted for G3 and G4, with 34% of GG sites but less than 1% of GA sites mutated (Fig. 2D, H42/43D). The near absence of GA-to-AA mutations suggested that A3G is highly specific for GG-to-AG mutations in vivo (4, 17). The four JRCSFvifW79S-inoculated mice had less than 1% possible GA-to-AA mutations (Fig. 2D, W79S). This finding raised the possibility that cytosine deaminase activity is minimally responsible for the antiviral effect of A3F apparent in the extinction of JRCSFvifH42/43DW79S replication. However, we could not assess GA-to-AA mutations in JRCSFvifH42/43DW79S mice, as no viral RNA or DNA was amplified from FG1 to FG4. The effectiveness of the combined actions of A3G and A3F may result from a two-pronged restriction of JRCSF by a cytosine deaminase activity and a non-cytosine deaminase activity.
Enforced mutation of defective vif.
The mechanism of viral escape from the antiviral activities of A3G (G3 and G4) or A3F (F1 to F4) was investigated by sequencing vif in viral RNA from blood drawn at 10 weeks. vif genes from the terminal time point of the course of G3 and G4 infection are aligned in Fig. 3A. Vif from G3 had the aspartate codon (GAT) for amino acid position 42 replaced with the asparagine codon (AAT) (Fig. 3A). The original aspartate codon (GAC) for amino acid position 43 was altered to RRC. No other mutations were noted in G3 Vif. To resolve the ambiguity at position 43, we cloned the PCR products from G3. In 10 clones, we found seven asparagine (AAC) and three glycine (GGC) codons. Hence, the H42/43D mutation was replaced with N42/N43 or N42/G43. Remarkably, there were no mutations in vif from G4.
FIG 3.

In vivo enforced mutation of vif in JRCSFvifH42/43D and JRCSFvifW79S. (A) HIV vif sequences amplified from plasma virions at terminal time points of JRCSFvifH42/43D infection show mutations changing the aspartate codons in mouse G3 to asparagine at position 42 and to either asparagine or glycine at codon 43. The H42/43D mutation in mouse G4 remained intact throughout the infection. (B) HIV vif sequence from the terminal time point of 4 mice infected with JRCSFvifW79S showing that the serine at codon 79 was changed to either tyrosine or phenylalanine. A single GA-to-AA mutation (nucleotide 56) was observed in mouse F3, while a point mutation (changing nucleotide 134 from A to T) was present in a portion of the HIV sequences in mouse F4. *, positions of sequence identity to JRCSF.
In Fig. 3B, vif sequence from the viral RNA of the F1 mouse had the serine codon at position 79 mutated to tyrosine (TCC to TAC) and F2 to F4 had phenylalanine at position 79 (TCC to TTC). Two other Vif mutations were found: R19K (AGA to AAA) in F3 and E45V (GAA to GWA) in F4. R19K, like S79F, first appeared at week 6 and was dominant at week 8 (not shown). E45V did not appear in F4 viral RNA until after F79 had replaced S79 (not shown). Unlike the conserved amino acid residues at positions 42 (H), 43 (H), and 79 (W), amino acid sites 19 and 45 are polymorphic (46).
If W79F, W79Y, H42N/H43N, and H42N/H43G were positively selected, then these mutations should restore at least partial A3F or A3G degradation activity to Vif. To test this hypothesis, we constructed proviral clones with Vif mutated to Y79, F79, N42/N43, and N42/G43 and performed A3G and A3F degradation assays in transfected 293T cells as shown in Fig. 1B. As demonstrated in Fig. 4A, VifW79Y and VifW79F had A3F degradation activities intermediate between those of wild-type Vif and the fully defective VifW79S. Both VifW79Y and VifW79F effectively degraded A3G. Contrary to a previous report on HIV-1NL4–3 Vif (28), we found that JRCSF Vif with the N42/N43 mutation (from G3) showed nearly wild-type A3G degradation activity, while VifN42/G43 had an intermediate level of activity (Fig. 4B). The results of these in vitro assays support the conclusion that JRCSFs with mutated Vifs overcame initial delays in replication by acquiring partial or near-wild-type APOBEC3 degradation activities. However, mouse G4 was an exception to these conclusions. Viral loads for G4 were low for 6 weeks but subsequently increased and nearly matched wild-type viral loads by 10 weeks (Fig. 2A, top). Strikingly, there was no evidence of mutations in vif from G4 (Fig. 3A).
FIG 4.

Mutations that arose in vivo partially restored Vif function. (A) Proviral DNAs with the designated Vif mutations at codon 79 and an expression vector for either V5-tagged APOBEC3G or APOBEC3F were cotransfected into HEK-293T cells. The cell lysates were resolved by SDS-PAGE and probed with anti-V5 to assess degradation of APOBEC3G or APOBEC3F in the presence of the Vif mutants, revealing that F or Y at amino acid 79 partially restored Vif degradation of APOBEC3F. (B) HEK-293T cells were cotransfected with an expression vector for either V5-tagged APOBEC3G or APOBEC3F and proviral DNA with the designated Vif mutations at codons 42 and 43. Cell lysates were resolved by SDS-PAGE and probed with anti-V5 to assess degradation of APOBEC3G or APOBEC3F in the presence of the Vif mutants, revealing that the N at position 42 with an N or G at amino acid 43 partially restored Vif degradation of APOBEC3G.
Enforced mutation and viral load are not temporally correlated.
To more rigorously test the hypothesis that the ability of JRCSFvifH42/42D to reach wild-type viral loads in G3 at 10 weeks was dependent on the N42/N43 and N42/G43 mutations, we sequenced vif genes from plasma virions at earlier time points. If the mutations to N42/N43 and N42/G43 accounted for high levels of replication, then the changes in vif sequences should correspond temporally with increased viral loads. Instead, we found that despite a viral load of over 105 copies of viral RNA per ml of blood at 4 and 6 weeks, plasma viral RNA still had the D42/D43 double mutation (Fig. 2A and 5A). By 8 weeks with a viral load at wild-type levels, the original H42/43D mutation had begun to disappear. G at the first position in codon 42 was almost completely replaced by A, giving an asparagine codon. G was still the dominant nucleotide at codon position 1 for amino acid 43, but in the second codon position, G had largely replaced A. These changes indicated that the D43 codon had mostly mutated to GGC, or glycine. However, VifN42/G43 was not particularly effective in degrading A3G in vitro (Fig. 4B). Finally, at 10 weeks, N42/N43 (7/10) was in the majority at the original D42/D43 site and N42/G43 (3/10) was in the minority (Fig. 5A). Therefore, closer analysis indicated that the appearance of in vivo-generated mutations at H42/43D lags behind the ability of the virus to replicate. Hence, the hypothesis that active replication is strictly driven by restoration of A3G degradation activity is not supported. Our observations raise the possibility that Vif is sometimes able to maintain viral viability despite loss of the A3G degradation function. Mutations restoring A3G degradation activity may facilitate the virus reaching wild-type levels of replication.
FIG 5.

Evolution of the APOBEC3 disrupting mutations in the vif gene in vivo. (A) Sequences of codons 41 to 44 of vif amplified from the plasma RNA in two viremic mice infected with JRCSFvifH42/43D. In mouse G3, Vif was subjected to positive selection to replace the aspartic acid residues at positions 42 and 43. In mouse G4, no mutation of the vif gene at amino acid 42 or 43 was observed. G, black; A, green; T, red; C, blue. (B) Sequence of codons 78 to 80 of the vif gene amplified from plasma RNA in 4 mice infected with JRCSFvifW79S revealing replacement of the W79S mutation from the initial infecting virus. In all 4 mice, the serine residue at amino acid 79 was converted to either phenylalanine or tyrosine. wpi, weeks postinfection.
Similarly, for F1 to F4, the dominant or exclusive codon at 4 weeks for amino acid position 79 is TCC, for serine (Fig. 5B). At this time point, the four viral loads are higher than 105 copies of viral RNA per ml of blood (Fig. 2B). Not until 6 to 8 weeks had codon 79 mutated to TAC (tyrosine) or TTC (phenylalanine) when viral loads were at wild-type levels. Therefore, it is evident that the mutations restoring partial A3F degradation activities to Vif do not precede but in fact lag behind active viral replication.
Relevance of APOBEC3H.
One potentially confounding variable for the extinction of viral infection in G1, G2, and FG1 to FG4 mice was the presence or absence of an active A3H allele (Fig. 2A and C). A3H is highly polymorphic, with stable/active alleles and unstable/inactive alleles (20, 45, 47–49). Primary CD4+ T cells homozygous and heterozygous for active/stable alleles of A3H are strikingly resistant to infection by HIV-1 expressing a Vif that is specifically defective for A3H degradation (20, 45). It is important to note that A3H is not countered by JRCSF Vif, since the Vif has amino acids I39 and N48 (Table 2). Vifs with F39 and H48 are highly active for A3H degradation, but mutation of one of these residues makes Vif inactive for A3H degradation while maintaining A3G and A3F degradation activity (47, 50). Specifically, B-1 and B-3 Vifs (I39 and H48) are inactive for A3H degradation, as is NL4-3 Vif (F39 and N48), strongly suggesting that JRCSF Vif (I39 and N48) is also nonfunctional for A3H degradation (20, 47, 50). JRCSF Vif is fully active for A3G, A3F, and A3D degradation despite the presence of I39 and N48 (Fig. 1B and C). Therefore, if A3H can restrict HIV-1 replication in vivo, then the presence of an active A3H allele in combination with an active A3G could account for the absence of JRCSF replication in G1, G2, and FG1 to FG4 (Fig. 2A and C) (2, 20, 45). To determine if A3H was necessary for JRCSF extinction, we sequenced A3H mRNA from human tissue in the cohorts of mice that were used in the experiments shown in Fig. 2. There were seven cohorts, each with a common human tissue used for reconstitution of the mice. Table 3 groups the mice into cohorts and documents the A3H haplotypes (20). We found haplotypes I, III, IV, and V. Of these haplotypes, only haplotype V encodes an active A3H.
TABLE 2.
Amino acids 38 through 49 of Vifs with or without A3H degradation activitya
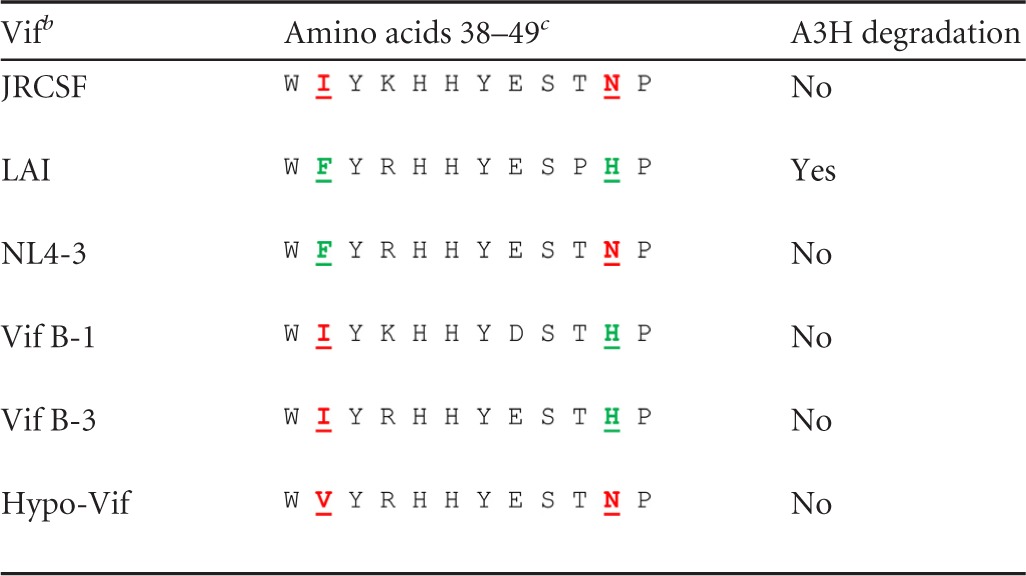
All Vifs degrade A3G and A3F.
b LAI, HIV-1LAI; NL4-3, HIV-1NL4-3. Vif B-1 and B-3 are subtype B Vifs.
c The critical residues for activity are phenylalanine 39 and histidine 48. Both a phenylalanine at position 39 and a histidine at position 48 are required for A3H degradation activity (LAI). Isoleucine at position 39 with histidine at position 48 is inactive (B-1 and B-3), and phenylalanine at position 39 and asparagine at position 48 is inactive (NL4-3). JRCSF has isoleucine at position 39 and asparagine at position 48. B-1 and B-3 are from Binka et al. (47), and Hypo-Vif is from Refsland et al. (20). Conserved residues are italic and underlined; mutated residues are boldface and underlined.
TABLE 3.
Cohorts and A3H haplotypesa
| Cohort | Mice | Amino acidb encoded at position: |
Haplotype | Activity | ||||
|---|---|---|---|---|---|---|---|---|
| 15 | 18 | 105 | 121 | 178 | ||||
| 1 | G3, G4, F1 | N | R | G | K | E | I | No |
| N | R | R | D | E | V | Yes | ||
| 2 | F2, F3, F4 | Δ | L | R | D | E | IV | No |
| N | R | R | D | E | V | Yes | ||
| 3 | F5, F6, G8, FG2, FG3 | N | R | G | K | E | I | No |
| Δ | R | R | D | E | III | No | ||
| 4 | G1 | N | R | G | K | E | I | No |
| N | R | G | K | E | I | No | ||
| 5 | G2 | N | R | R | D | E | V | Yes |
| Δ | L | R | D | E | IV | No | ||
| 6 | G5, FG1 | Δ | R | R | D | E | III | No |
| Δ | L | R | D | E | IV | No | ||
| 7 | F7, G6, G7, FG4 | N | R | R | D | E | V | Yes |
| N | R | G | K | E | I | No | ||
RNA encoding APOBEC3H from BLT mouse peripheral blood mononuclear cells (PBMCs) was amplified and sequenced. The amino acids encoded at positions 15, 18, 105, 121, and 178 were determined. The seven haplotypes for A3H are as follows: I, NRGKE; II, NRRDD; III, ΔRRDE; IV, ΔLRDE; V, NRRDE; VI, ΔLGKE; and VII, NRRKD. Active forms of A3H have asparagine at position 15 and arginine at position 105. Deletion of the codon for N15 or glycine at position 105 renders the protein unstable and hence inactive. Only four (I, III, IV, and V) of the seven possible haplotypes were present in the seven cohorts. Haplotype V represented the only active A3G allele. Functional A3H was present only in the heterozygous condition. Cohort 7 had two codons for T43, ACG and ACA.
Δ, 3-base deletion of the N15 codon.
The JRCSFvifH42/43D-inoculated mouse G1 was homozygous for defective A3H alleles, but nonetheless, infection was aborted (Fig. 2A). Given that JRCSFvifH42/43D Vif effectively degrades A3D and A3F, the extinction of viral replication in G1 appeared to be the result of A3G activity. G3 and G4 were heterozygous for the active A3H haplotype V. G3 and G4 had delayed but ultimately full blown systemic infections despite the presence of an active A3H gene. Therefore, extinction of JRCSFvifH42/43D can occur in the absence of active A3H (G1), and JRCSFvifH42/43D replication can occur in the presence of an active A3H gene (G3 and G4). These results rule out a consistently important role for A3H in viral extinction, though possible effects of A3H on viral replication could be present but not discernible because of the greater impact of A3G.
The JRCSFvifH42/43DW79S-inoculated mice also support the conclusion that A3H is minimally involved in viral extinction. FG1 to FG3 mice have no A3H activity (Table 3). Therefore, A3H activity is not required for consistent viral extinction, since FG1 to FG3 mice exhibit no viral replication. JRCSFvifW79S-infected mice (F1 to F4) were all heterozygous for the active V allele of A3H, and only F1 had delayed HIV-1 replication (Fig. 2B). Therefore, it is not possible to draw a firm conclusion regarding the role of A3F or the active alleles of A3H other than that the combination of A3F and A3H activities is much weaker than the combination of A3F and A3G activities.
Negative effects of A3F and A3H on JRCSF replication.
Figure 2 provides evidence that A3F can enhance the restriction of HIV-1 by A3G to consistently block viral replication. A role for A3H is less clear. In this regard, results with primary CD4+ T cells suggest that diminished HIV-1 replication should be observable with JRCSF replication in cohorts that express active A3H alleles (20, 45). However, multiple cohorts of mice have been infected with JRCSF by three different groups, and no evidence for individual cohorts that are highly resistant to infection has been noted (17, 18, 36). While this evidence suggests only weak anti-HIV-1 effects for A3H, the population frequencies of A3H active alleles can be low and are highly dependent on geography (51, 52). It is possible that none of the cohorts from these three reports expressed an active allele of A3H. Therefore, we compared JRCSF replication in five cohorts of mice with one of the cohorts heterozygous for active A3H. In Fig. 6, WT1 is a BLT mouse with A3H haplotypes I and V, and no difference in the viral load time course was observed relative to four other cohorts. The dramatic restriction of HIV-1 by active A3H alleles seen in primary CD4+ T cells by Refsland et al. (20) and Ooms et al. (45) may reflect the ex vivo activation of primary T cells, which are inactive in BLT mice (39, 53).
FIG 6.
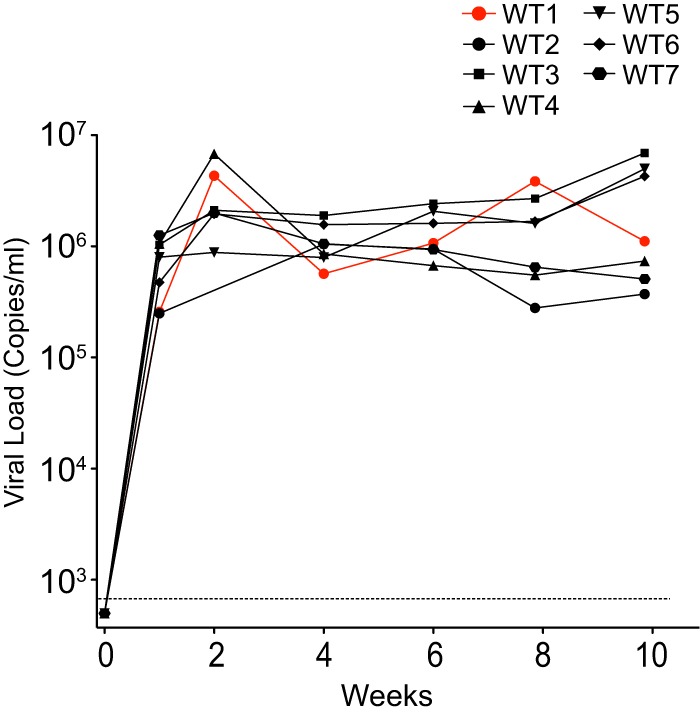
Robust replication of JRCSF in a mouse cohort heterozygous for A3H haplotype V. Individual plots of the wild-type JRCSF viral load versus time are presented. The seven mice are from Fig. 2 (WT), where JRCSF replication is presented in aggregate. WT1 has A3H haplotypes I and V, but viral replication is not diminished. WT2 to WT7 are derived from four separate cohorts.
To further investigate possible inhibitory effects of A3F and A3H on HIV-1 replication, we inoculated BLT mice with a 10-fold-lower initial dose of JRCSF (9,000 TCIU) to possibly enhance small effects, in contrast to the 90,000 TCIU employed to determine the ability of APOBEC3s to extinguish replication. At the lower dose, JRCSFvifH42/43D failed to replicate in three of four mice, and replication was greatly delayed in the fourth mouse (Fig. 7). Of note, two mice (G5 and G8) in which replication was not detected had two inactive A3H alleles (Table 3). G6 and G7 were heterozygous for the active A3H allele V (Table 3). No viral replication was observed for G6, but delayed replication was observed with G7 (Fig. 7). Again, the combination of A3G and A3H did not uniformly restrict JRCSF.
FIG 7.
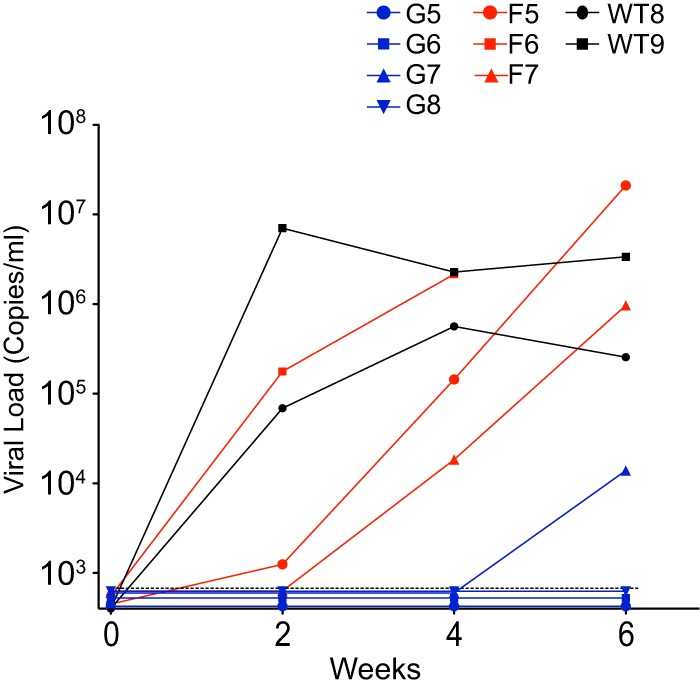
JRCSF, JRCSFvifW79S, and JRCSFvifH42/43D viral load time courses following a low-dose inoculation. Nine individual viral load time courses are presented following a 10-fold-reduced inoculation of JRCSF (9,000 TCIU) compared to Fig. 2. Two mice were inoculated with JRCSF (WT8 and WT9), three were inoculated with JRCSFvifW79S (F5, F6, and F7), and four mice were inoculated with JRCSFvifH42/43D (G5, G6, G7, and G8). WT8 and WT9 are homozygous A3H inactive (cohorts 3 and 6, respectively), F5 and F6 are homozygous A3H inactive (both cohort 3), F7 is heterozygous A3H active (cohort 7), G5 and G8 are homozygous A3H inactive (cohorts 6 and 3, respectively), and G6 and G7 are heterozygous A3H active (both cohort 7) (Table 3).
Two of the three JRCSFvifW79S mice infected with 9,000 TCIU (F6 and F7) were clearly delayed relative to JRCSF, with replication in mouse F7 the most retarded. Interestingly, mice F5 and F6 were defective for A3H while F7 was heterozygous for A3H allele V (Table 3). These results confirm the conclusion that A3G is the major restrictor of HIV-1 infection, with A3F capable of delaying but not fully restricting JRCSF. Our results also suggest that an effect of A3H is difficult to detect in the presence of active A3G but may be seen when A3H and A3F are acting in concert (F7).
A3G and A3H hypermutation in viral DNA.
Mouse G2 was inoculated with JRCSFvif42/43D and had no detectable HIV-1 RNA in blood, and viral DNA was amplified only from lymph nodes (Fig. 2A, bottom). G2 was from cohort 5 and was heterozygous for the active haplotype V allele of A3H (Table 3). The reverse transcriptase coding sequence (1,320 bp) was determined and exhibited massive G-to-A mutation (Fig. 8A). The sequence of the PCR product lacked ambiguities and may have been amplified from a discrete template. Based on results from G3 and G4, we expected extensive GG-to-AG mutation but only minor GA-to-AA mutation (Fig. 2D). On the other hand, if A3H was significantly mutating JRCSFvifH42/43D, then a definite GA-to-AA signature should be observable (2). In Fig. 8B, a relatively high level of GA-to-AA mutation was observed, accounting for 19% of the 78 total observed G-to-A mutations (61 at GG, 15 at GA, and 2 at GT). A3H is likely responsible for these GA-to-AA mutations, because JRCSFvifH42/43D effectively degrades A3F and A3D (Fig. 1B and C). It is possible that G2 is an example of HIV-1 hypermutation that was known prior to the discovery of the antiviral role of A3G (1, 54).
FIG 8.

HIV DNA amplified from an extinguished infection is hypermutated. (A) Highlighter sequence analysis of the reverse transcriptase region of HIV pol. HIV DNA amplified from the lymph nodes of mouse G2 was G-to-A hypermutated (green lines). HIV-1JRCSF nucleotide numbers are indicated at the bottom. (B) Alignment of wild-type JRCSF and hypermutated JRCSF reverse transcriptase sequence from panel A identifying G-to-A mutations at both GG and GA sites. GG-to-AG mutations are indicated in blue, GA-to-AA mutations are shown in red, and G-to-A mutations present at GT sites are shown in orange.
It should be noted that the massive mutation of DNA presented in Fig. 8B represents a nearly total mutation of the DNA by A3G. There are 32 guanosine doublets in the RT coding sequence (excluding GGG and GGGG). Twenty-four are in the context of GGA, and all 24 were mutated to AGA. Two are in the context of GGT, and both were mutated to AGT. Six were in the context of GGC, and none of them has a G-to-A change. The triplet GGG occurs 14 times (excluding GGGG). Eight were in the context of GGGA, and five of these were mutated to AAGA, two were mutated to AAAA (likely representing combined actions of A3G and A3H), and one was mutated to GAGA. Three sites were GGGT, and all three were mutated to AAGT. Interestingly, three sites were GGGC, and all three were mutated to AGGC. There were five quadruplets mutated to AAAGA, GAAGA, GAAGA, GAAGT, and AAGGC. These results suggest that in vivo a 5′ G mutation to a CC doublet in minus-strand DNA (GCC) blocks deamination (4). Taking this inhibitory effect into account, there were 61 G-to-A mutations out of 65 feasible GG targets (94%). A3H activity was much less exhaustive than A3G activity. There were 102 GA sites in the reverse transcriptase coding sequence, and 15 were mutated (15%).
The sanctity of viral RNA sequence.
The massive potential of A3G to mutate the minus strand of HIV-1 emphasizes the importance of Vif to the virus. We accordingly asked if any of this massive mutational potential resulted in fixed sequence changes in replicating viruses other than in vif. We determined the sequences of gag and the coding sequences for reverse transcriptase and RNase H from the terminal bleed of virion RNA of G3, G4, and F1 to F4. There were no fixed mutations or even clear cases of polymorphism in the viral genomic RNA sequence of gag or the reverse transcriptase coding sequence (not shown). In the case of RNase H sequences, there were no mutations evident for G3 or F1 to F4, but G4 had a nonsynonymous point mutation changing valine 27 to isoleucine (Fig. 9). G3 and F1 to F4 had enforced mutations in vif (Fig. 3), but G4 did not, raising the possibility that RNase H may be a site of extra Vif mutations with impacts on the APOBEC3 axis (15, 55).
FIG 9.

Mutations in HIV RNase H are present only in virus from mouse G4. HIV-1 RNase H sequence amplified from the plasma of viremic humanized mice infected with either JRCSFvifH42/43D or JRCSFvifW79S showed that only virus from mouse G4 had mutations in RNase H. One G-to-A mutation was present in RNase H from mouse G4 (highlighted in orange) that resulted in a valine-to-isoleucine amino acid substitution.
DISCUSSION
A3G is clearly the dominant APOBEC3 in countering HIV-1 infection (2, 17, 22). However, unlike vif− JRCSF (JRCSFΔvif), JRCSFvifH42/43D infection is not always extinguished in BLT humanized mice. This result demonstrates that A3G activity is not sufficient to consistently extinguish HIV-1 infection. An important role in extinguishing HIV-1 is likely played by A3F, since JRCSFvifH42/43DW79S was not able to replicate.
In vivo involvement of A3F and A3D.
Considerable in vitro evidence exists for an anti-HIV-1 role for A3F (2, 22). The in vivo role of this APOBEC3 is less clear. Sato et al. (22) infected NSG-hu mice with chimeric NL4-3 virus, converted to R5 tropism by replacing env V3 of NL4-3 with JRCSF V3 to give NLCSFV3 (56). NLCSFV3-4A has Vif amino acids 14 to 17 (DRMR) all mutated to alanine and is defective in A3F degradation (57). The virus replicated slowly, and viral loads at 6 weeks were reduced by 80% relative to those of the wild type, NLCSFV3. While these findings strongly suggest a significant impact on HIV-1 replication by A3F, the interpretation of the result is not straightforward because the tetra-alanine mutation may impact other Vif activities in addition to A3F degradation (58).
A complication regarding A3F's antiviral effect is the ability of NLCSFV3-4A to diversify its genomic RNA. G-to-A mutations were found by Sato et al. in 91 single-genome amplicons of env at the level of 1.6% of total G sites (22). Wild-type NLCSFV3 and A3G degradation-defective NLCSFV3-5A (amino acids 40 to 44, YRHHY to AAAAA) had only 0.21% and 0.26% of G sites mutated to A. The inability of NLCSFV3-5A to diversify viral sequence was attributed to the high propensity of A3G to introduce lethal mutations, thereby preventing mutated virus from replicating. If greater viral diversity is beneficial to the virus, then A3F may have to be considered a proviral factor, not an antiviral factor, particularly given its weak effect on viral replication relative to A3G. We consider this scenario unlikely, since HIV-1 could readily mutate reverse transcriptase to have a higher error rate if a greater level of viral diversity was beneficial (59).
Our mutational approach to A3F degradation was different from that of Sato et al. (22) in that we mutated only a single residue. This meant that unlike NLCSFV3-4A, JRCSFvifW79S could be selected against by enforced mutation. Indeed, JRCSFvifW79S reverted to the aromatic residue phenylalanine or tyrosine, which partially restored A3F degradation function. The partial reversion to wild-type activity straightforwardly defines A3F as an antiviral factor, as virus with A3F degradation activity has greater fitness and outgrows JRCSFvifW79S. We also found support for A3F as an important antiviral factor in the observation that JRCSFvifH42/43DW79S was not able to infect BLT mice. Extinction of JRCSFvifH42/43D infection (G1, G2, G5, G6, and G8, but not G3, G4, and G7) may be stochastic. Under our experimental infection conditions, viral survival is possible, but not certain. A negative impact on viral replication by A3F would limit the opportunity the virus has to mutate H42/43D. In this scenario, extinction of the virus occurs soon after inoculation or not at all.
Wild-type JRCSF Vif and Vif mutated to H42/43D degraded not only A3F, but A3D as well. This rules out a role for A3D in extinction of JRCSFvifH42/43D in mice G1, G2, G5, G6, and G8. In the case of JRCSFvifH42/43DW79S, A3D would not have been degraded, and there is the possibility that it contributed, along with A3F, to viral extinction in FG1 to FG4. Arguing against such a role are the findings that in humanized mice, A3D is expressed at lower levels than A3F, and in vitro studies demonstrating weaker inhibition of HIV-1 by A3D than A3F (2, 20, 22, 45, 60). For these reasons, any antiviral impact of A3D would be swamped by that of A3F.
In vivo involvement of A3H.
We also considered the possibility that A3H could contribute to viral extinction in the cases where inoculated JRCSFvifH42/43D failed to replicate. It is important to note that, unlike A3G and A3F, A3H would not be degraded by JRCSF Vif because of amino acids I39 and N48 (20, 47, 50). This situation could result in a combined effect of A3G and A3H, leading to viral destruction in JRCSFvifH42/43D and JRCSFvifH42/43DW79S inoculations. A3H haplotypes were determined in seven mouse cohorts, and of the four haplotypes present (I, III, IV, and V) (Table 3), only A3H V produces a stable protein. Mice G5 and G8 were inoculated with 9,000 TCIU of JRCSFvifH42/43D and displayed no viral replication (Fig. 7). In neither case was an active A3H allele present. The mice inoculated with 90,000 TCIU of JRCSFvifH42/43D that did not exhibit viral replication, G1 and G2, had haplotypes I/I and IV/V, respectively. Therefore, viral replication in G1 was robustly suppressed without a contribution of A3H (or A3F) activity, and there was not a consistent relationship between the presence of A3H activity and suppression of viral replication (Fig. 2A). Similarly, A3H is not the necessary factor that causes JRCSFvifH42/43DW79S to consistently fail to replicate. FG1 to FG3 were all homozygous for inactive A3H alleles, yet no viral replication was observed (Table 3 and Fig. 2C). Additional evidence for the suggestion that A3H has a minimal impact on JRCSF restriction is the fact that wild-type JRCSF replication is not reduced in BLT mice heterozygous for A3H haplotype V (Fig. 6). Hence, A3G is clearly the dominant restrictor of HIV-1, with A3F likely playing a critical role. We did find evidence of a small inhibitory effect of A3H on JRCSFvifW79S. A humanized mouse (F7) with a low-dose inoculation that was heterozygous for an active allele of A3H displayed a greater delay in viral replication (Fig. 7) than two JRCSFvifW79S mice that were homozygous for inactive A3H alleles (Fig. 7, F5 and F6).
A role for A3H in hypermutation.
Evidence for GA-to-AA mutations indicative of A3F and/or A3H cytosine deaminase activity was minimal in WT1, G3, G4, and F1 to F4 viral DNA (Fig. 2D), but one JRCSFvifH42/43D infection (G2) had 15% of the GA sites mutated to AA in the reverse transcriptase coding sequence from lymph node DNA. JRCSFvifH42/43D has A3F degradation activity, which strongly implicates A3H as the mutagenic factor. GG-to-AG mutations were also numerous, to the remarkable extent of 94% of potential sites. This extreme level of G-to-A mutations suggests an abnormal level of cytosine deaminase activity, which may be related to the hypermutation of viral DNA found in HIV-1-positive individuals (1, 5).
In vivo evidence for Vif activity not involving degradation of A3F or A3G.
A3G mutation of GG to AG is effective in inactivating virus prior to integration, and A3F's GA-to-AA mutations are fewer and relatively ineffective (61). Thus, it seems unlikely that GA-to-AA mutation plays a decisive role in the equivalent susceptibilities of JRCSFvif42/43DW79S and JRCSFΔvif to extinction. The notion that extinction occurs early after inoculation or not at all is supported by the fact that plasma viral RNA was not detected in G1, G2, and FG1 to FG4 despite an inoculum of 90,000 TCIU. When virus was able to overcome mutations in vif (G3, G4, and F1 to F4), viral loads ultimately reached 106 copies of viral RNA. It is also important to note that appearance of vif mutations in vivo that partially restored A3G or A3F degradation lagged behind high levels of virus or, in the case of G4, failed to appear at all. Non-cytosine deaminase Vif activities not inhibited by the H42/43D or W79S mutations may account for these observations. There are multiple anti-APOBEC3 activities other than APOBEC3 degradation, including A3G and A3F inhibition of reverse transcription, transcription elongation, and proviral integration and inactivation of the transactivation response element (12–16). The greater potency of A3F than A3G in preventing viral integration is an attractive possible mechanism for the antiviral activity of A3F (15). Early forms of Vif may have exhibited the ability to directly inhibit encapsidated APOBEC3s prior to acquiring the function of targeting APOBEC3s to proteasomes.
ACKNOWLEDGMENTS
This work was supported in part by funds from the National Institute of Allergy and Infectious Diseases (grants AI033331, AI096937, and AI96138 [J.V.G.]) and by the UNC Center for AIDS Research (grant P30 AI50410).
We thank Nathaniel Schramm for insightful comments and suggestions, as well as current and past members of the Garcia laboratory and veterinary technicians at the UNC Division of Laboratory Animal Medicine for their assistance with different aspects of this research.
REFERENCES
- 1.Albin JS, Harris RS. 2010. Interactions of host APOBEC3 restriction factors with HIV-1 in vivo: implications for therapeutics. Expert Rev Mol Med 12:e4. doi: 10.1017/S1462399409001343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hultquist JF, Lengyel JA, Refsland EW, LaRue RS, Lackey L, Brown WL, Harris RS. 2011. Human and rhesus APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H demonstrate a conserved capacity to restrict Vif-deficient HIV-1. J Virol 85:11220–11234. doi: 10.1128/JVI.05238-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sheehy AM, Erthal J. 2012. APOBEC3 versus retroviruses, immunity versus invasion: clash of the titans. Mol Biol Int 2012:974924. doi: 10.1155/2012/974924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harris RS, Bishop KN, Sheehy AM, Craig HM, Petersen-Mahrt SK, Watt IN, Neuberger MS, Malim MH. 2003. DNA deamination mediates innate immunity to retroviral infection. Cell 113:803–809. doi: 10.1016/S0092-8674(03)00423-9. [DOI] [PubMed] [Google Scholar]
- 5.Liddament MT, Brown WL, Schumacher AJ, Harris RS. 2004. APOBEC3F properties and hypermutation preferences indicate activity against HIV-1 in vivo. Curr Biol 14:1385–1391. doi: 10.1016/j.cub.2004.06.050. [DOI] [PubMed] [Google Scholar]
- 6.Wiegand HL, Doehle BP, Bogerd HP, Cullen BR. 2004. A second human antiretroviral factor, APOBEC3F, is suppressed by the HIV-1 and HIV-2 Vif proteins. EMBO J 23:2451–2458. doi: 10.1038/sj.emboj.7600246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kobayashi M, Takaori-Kondo A, Miyauchi Y, Iwai K, Uchiyama T. 2005. Ubiquitination of APOBEC3G by an HIV-1 Vif-Cullin5-Elongin B-Elongin C complex is essential for Vif function. J Biol Chem 280:18573–18578. doi: 10.1074/jbc.C500082200. [DOI] [PubMed] [Google Scholar]
- 8.Xiao Z, Ehrlich E, Yu Y, Luo K, Wang T, Tian C, Yu XF. 2006. Assembly of HIV-1 Vif-Cul5 E3 ubiquitin ligase through a novel zinc-binding domain-stabilized hydrophobic interface in Vif. Virology 349:290–299. doi: 10.1016/j.virol.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 9.Yu Y, Xiao Z, Ehrlich ES, Yu X, Yu XF. 2004. Selective assembly of HIV-1 Vif-Cul5-ElonginB-ElonginC E3 ubiquitin ligase complex through a novel SOCS box and upstream cysteines. Genes Dev 18:2867–2872. doi: 10.1101/gad.1250204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Britan-Rosich E, Nowarski R, Kotler M. 2011. Multifaceted counter-APOBEC3G mechanisms employed by HIV-1 Vif. J Mol Biol 410:1065–1076. doi: 10.1016/j.jmb.2011.03.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Feng Y, Baig TT, Love RP, Chelico L. 2014. Suppression of APOBEC3-mediated restriction of HIV-1 by Vif. Front Microbiol 5:450. doi: 10.3389/fmicb.2014.00450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bishop KN, Holmes RK, Malim MH. 2006. Antiviral potency of APOBEC proteins does not correlate with cytidine deamination. J Virol 80:8450–8458. doi: 10.1128/JVI.00839-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gillick K, Pollpeter D, Phalora P, Kim EY, Wolinsky SM, Malim MH. 2013. Suppression of HIV-1 infection by APOBEC3 proteins in primary human CD4(+) T cells is associated with inhibition of processive reverse transcription as well as excessive cytidine deamination. J Virol 87:1508–1517. doi: 10.1128/JVI.02587-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Holmes RK, Koning FA, Bishop KN, Malim MH. 2007. APOBEC3F can inhibit the accumulation of HIV-1 reverse transcription products in the absence of hypermutation. Comparisons with APOBEC3G. J Biol Chem 282:2587–2595. [DOI] [PubMed] [Google Scholar]
- 15.Mbisa JL, Bu W, Pathak VK. 2010. APOBEC3F and APOBEC3G inhibit HIV-1 DNA integration by different mechanisms. J Virol 84:5250–5259. doi: 10.1128/JVI.02358-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nowarski R, Prabhu P, Kenig E, Smith Y, Britan-Rosich E, Kotler M. 2014. APOBEC3G inhibits HIV-1 RNA elongation by inactivating the viral trans-activation response element. J Mol Biol 426:2840–2853. doi: 10.1016/j.jmb.2014.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krisko JF, Martinez-Torres F, Foster JL, Garcia JV. 2013. HIV restriction by APOBEC3 in humanized mice. PLoS Pathog 9:e1003242. doi: 10.1371/journal.ppat.1003242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sato K, Izumi T, Misawa N, Kobayashi T, Yamashita Y, Ohmichi M, Ito M, Takaori-Kondo A, Koyanagi Y. 2010. Remarkable lethal G-to-A mutations in vif-proficient HIV-1 provirus by individual APOBEC3 proteins in humanized mice. J Virol 84:9546–9556. doi: 10.1128/JVI.00823-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Refsland EW, Hultquist JF, Harris RS. 2012. Endogenous origins of HIV-1 G-to-A hypermutation and restriction in the nonpermissive T cell line CEM2n. PLoS Pathog 8:e1002800. doi: 10.1371/journal.ppat.1002800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Refsland EW, Hultquist JF, Luengas EM, Ikeda T, Shaban NM, Law EK, Brown WL, Reilly C, Emerman M, Harris RS. 2014. Natural polymorphisms in human APOBEC3H and HIV-1 Vif combine in primary T lymphocytes to affect viral G-to-A mutation levels and infectivity. PLoS Genet 10:e1004761. doi: 10.1371/journal.pgen.1004761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Refsland EW, Stenglein MD, Shindo K, Albin JS, Brown WL, Harris RS. 2010. Quantitative profiling of the full APOBEC3 mRNA repertoire in lymphocytes and tissues: implications for HIV-1 restriction. Nucleic Acids Res 38:4274–4284. doi: 10.1093/nar/gkq174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sato K, Takeuchi JS, Misawa N, Izumi T, Kobayashi T, Kimura Y, Iwami S, Takaori-Kondo A, Hu WS, Aihara K, Ito M, An DS, Pathak VK, Koyanagi Y. 2014. APOBEC3D and APOBEC3F potently promote HIV-1 diversification and evolution in humanized mouse model. PLoS Pathog 10:e1004453. doi: 10.1371/journal.ppat.1004453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Platt EJ, Wehrly K, Kuhmann SE, Chesebro B, Kabat D. 1998. Effects of CCR5 and CD4 cell surface concentrations on infections by macrophagetropic isolates of human immunodeficiency virus type 1. J Virol 72:2855–2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zheng YH, Irwin D, Kurosu T, Tokunaga K, Sata T, Peterlin BM. 2004. Human APOBEC3F is another host factor that blocks human immunodeficiency virus type 1 replication. J Virol 78:6073–6076. doi: 10.1128/JVI.78.11.6073-6076.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Duggal NK, Malik HS, Emerman M. 2011. The breadth of antiviral activity of Apobec3DE in chimpanzees has been driven by positive selection. J Virol 85:11361–11371. doi: 10.1128/JVI.05046-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goncalves J, Jallepalli P, Gabuzda DH. 1994. Subcellular localization of the Vif protein of human immunodeficiency virus type 1. J Virol 68:704–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koyanagi Y, Miles S, Mitsuyasu RT, Merrill JE, Vinters HV, Chen IS. 1987. Dual infection of the central nervous system by AIDS viruses with distinct cellular tropisms. Science 236:819–822. doi: 10.1126/science.3646751. [DOI] [PubMed] [Google Scholar]
- 28.Mehle A, Wilson H, Zhang C, Brazier AJ, McPike M, Pery E, Gabuzda D. 2007. Identification of an APOBEC3G binding site in human immunodeficiency virus type 1 Vif and inhibitors of Vif-APOBEC3G binding. J Virol 81:13235–13241. doi: 10.1128/JVI.00204-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Henikoff S, Henikoff JG. 1992. Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci U S A 89:10915–10919. doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nagao T, Yamashita T, Miyake A, Uchiyama T, Nomaguchi M, Adachi A. 2010. Different interaction between HIV-1 Vif and its cellular target proteins APOBEC3G/APOBEC3F. J Med Invest 57:89–94. doi: 10.2152/jmi.57.89. [DOI] [PubMed] [Google Scholar]
- 31.Yamashita T, Kamada K, Hatcho K, Adachi A, Nomaguchi M. 2008. Identification of amino acid residues in HIV-1 Vif critical for binding and exclusion of APOBEC3G/F. Microbes Infect 10:1142–1149. doi: 10.1016/j.micinf.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 32.Denton PW, Estes JD, Sun Z, Othieno FA, Wei BL, Wege AK, Powell DA, Payne D, Haase AT, Garcia JV. 2008. Antiretroviral pre-exposure prophylaxis prevents vaginal transmission of HIV-1 in humanized BLT mice. PLoS Med 5:e16. doi: 10.1371/journal.pmed.0050016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Denton PW, Krisko JF, Powell DA, Mathias M, Kwak YT, Martinez-Torres F, Zou W, Payne DA, Estes JD, Garcia JV. 2010. Systemic administration of antiretrovirals prior to exposure prevents rectal and intravenous HIV-1 transmission in humanized BLT mice. PLoS One 5:e8829. doi: 10.1371/journal.pone.0008829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Denton PW, Olesen R, Choudhary SK, Archin NM, Wahl A, Swanson MD, Chateau M, Nochi T, Krisko JF, Spagnuolo RA, Margolis DM, Garcia JV. 2012. Generation of HIV latency in humanized BLT mice. J Virol 86:630–634. doi: 10.1128/JVI.06120-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Denton PW, Othieno F, Martinez-Torres F, Zou W, Krisko JF, Fleming E, Zein S, Powell DA, Wahl A, Kwak YT, Welch BD, Kay MS, Payne DA, Gallay P, Appella E, Estes JD, Lu M, Garcia JV. 2011. One percent tenofovir applied topically to humanized BLT mice and used according to the CAPRISA 004 experimental design demonstrates partial protection from vaginal HIV infection, validating the BLT model for evaluation of new microbicide candidates. J Virol 85:7582–7593. doi: 10.1128/JVI.00537-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dudek TE, No DC, Seung E, Vrbanac VD, Fadda L, Bhoumik P, Boutwell CL, Power KA, Gladden AD, Battis L, Mellors EF, Tivey TR, Gao X, Altfeld M, Luster AD, Tager AM, Allen TM. 2012. Rapid evolution of HIV-1 to functional CD8+ T cell responses in humanized BLT mice. Sci Transl Med 4:143ra98. doi: 10.1126/scitranslmed.3003984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim SS, Peer D, Kumar P, Subramanya S, Wu H, Asthana D, Habiro K, Yang YG, Manjunath N, Shimaoka M, Shankar P. 2010. RNAi-mediated CCR5 silencing by LFA-1-targeted nanoparticles prevents HIV infection in BLT mice. Mol Ther 18:370–376. doi: 10.1038/mt.2009.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lan P, Tonomura N, Shimizu A, Wang S, Yang YG. 2006. Reconstitution of a functional human immune system in immunodeficient mice through combined human fetal thymus/liver and CD34+ cell transplantation. Blood 108:487–492. doi: 10.1182/blood-2005-11-4388. [DOI] [PubMed] [Google Scholar]
- 39.Long BR, Stoddart CA. 2012. Alpha interferon and HIV infection cause activation of human T cells in NSG-BLT mice. J Virol 86:3327–3336. doi: 10.1128/JVI.06676-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Melkus MW, Estes JD, Padgett-Thomas A, Gatlin J, Denton PW, Othieno FA, Wege AK, Haase AT, Garcia JV. 2006. Humanized mice mount specific adaptive and innate immune responses to EBV and TSST-1. Nat Med 12:1316–1322. doi: 10.1038/nm1431. [DOI] [PubMed] [Google Scholar]
- 41.Rajesh D, Zhou Y, Jankowska-Gan E, Roenneburg DA, Dart ML, Torrealba J, Burlingham WJ. 2010. Th1 and Th17 immunocompetence in humanized NOD/SCID/IL2rgammanull mice. Hum Immunol 71:551–559. doi: 10.1016/j.humimm.2010.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sun Z, Denton PW, Estes JD, Othieno FA, Wei BL, Wege AK, Melkus MW, Padgett-Thomas A, Zupancic M, Haase AT, Garcia JV. 2007. Intrarectal transmission, systemic infection, and CD4+ T cell depletion in humanized mice infected with HIV-1. J Exp Med 204:705–714. doi: 10.1084/jem.20062411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zou W, Denton PW, Watkins RL, Krisko JF, Nochi T, Foster JL, Garcia JV. 2012. Nef functions in BLT mice to enhance HIV-1 replication and deplete CD4+CD8+ thymocytes. Retrovirology 9:44. doi: 10.1186/1742-4690-9-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Palmer S, Wiegand AP, Maldarelli F, Bazmi H, Mican JM, Polis M, Dewar RL, Planta A, Liu S, Metcalf JA, Mellors JW, Coffin JM. 2003. New real-time reverse transcriptase-initiated PCR assay with single-copy sensitivity for human immunodeficiency virus type 1 RNA in plasma. J Clin Microbiol 41:4531–4536. doi: 10.1128/JCM.41.10.4531-4536.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ooms M, Brayton B, Letko M, Maio SM, Pilcher CD, Hecht FM, Barbour JD, Simon V. 2013. HIV-1 Vif adaptation to human APOBEC3H haplotypes. Cell Host Microbe 14:411–421. doi: 10.1016/j.chom.2013.09.006. [DOI] [PubMed] [Google Scholar]
- 46.Kikuchi T, Iwabu Y, Tada T, Kawana-Tachikawa A, Koga M, Hosoya N, Nomura S, Brumme ZL, Jessen H, Pereyra F, Trocha A, Walker BD, Iwamoto A, Tokunaga K, Miura T. 2015. Anti-APOBEC3G activity of HIV-1 Vif protein is attenuated in elite controllers. J Virol 89:4992–5001. doi: 10.1128/JVI.03464-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Binka M, Ooms M, Steward M, Simon V. 2012. The activity spectrum of Vif from multiple HIV-1 subtypes against APOBEC3G, APOBEC3F, and APOBEC3H. J Virol 86:49–59. doi: 10.1128/JVI.06082-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Duggal NK, Fu W, Akey JM, Emerman M. 2013. Identification and antiviral activity of common polymorphisms in the APOBEC3 locus in human populations. Virology 443:329–337. doi: 10.1016/j.virol.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li MM, Wu LI, Emerman M. 2010. The range of human APOBEC3H sensitivity to lentiviral Vif proteins. J Virol 84:88–95. doi: 10.1128/JVI.01344-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ooms M, Letko M, Binka M, Simon V. 2013. The resistance of human APOBEC3H to HIV-1 NL4-3 molecular clone is determined by a single amino acid in Vif. PLoS One 8:e57744. doi: 10.1371/journal.pone.0057744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.OhAinle M, Kerns JA, Li MM, Malik HS, Emerman M. 2008. Antiretroelement activity of APOBEC3H was lost twice in recent human evolution. Cell Host Microbe 4:249–259. doi: 10.1016/j.chom.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang X, Abudu A, Son S, Dang Y, Venta PJ, Zheng YH. 2011. Analysis of human APOBEC3H haplotypes and anti-human immunodeficiency virus type 1 activity. J Virol 85:3142–3152. doi: 10.1128/JVI.02049-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Watkins RL, Foster JL, Garcia JV. 2015. In vivo analysis of Nef's role in HIV-1 replication, systemic T cell activation and CD4(+) T cell loss. Retrovirology 12:61. doi: 10.1186/s12977-015-0187-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vartanian JP, Meyerhans A, Asjo B, Wain-Hobson S. 1991. Selection, recombination, and G-A hypermutation of human immunodeficiency virus type 1 genomes. J Virol 65:1779–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mbisa JL, Barr R, Thomas JA, Vandegraaff N, Dorweiler IJ, Svarovskaia ES, Brown WL, Mansky LM, Gorelick RJ, Harris RS, Engelman A, Pathak VK. 2007. Human immunodeficiency virus type 1 cDNAs produced in the presence of APOBEC3G exhibit defects in plus-strand DNA transfer and integration. J Virol 81:7099–7110. doi: 10.1128/JVI.00272-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Suzuki Y, Koyanagi Y, Tanaka Y, Murakami T, Misawa N, Maeda N, Kimura T, Shida H, Hoxie JA, O'Brien WA, Yamamoto N. 1999. Determinant in human immunodeficiency virus type 1 for efficient replication under cytokine-induced CD4(+) T-helper 1 (Th1)- and Th2-type conditions. J Virol 73:316–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Russell RA, Pathak VK. 2007. Identification of two distinct human immunodeficiency virus type 1 Vif determinants critical for interactions with human APOBEC3G and APOBEC3F. J Virol 81:8201–8210. doi: 10.1128/JVI.00395-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Albin JS, Brown WL, Harris RS. 2014. Catalytic activity of APOBEC3F is required for efficient restriction of Vif-deficient human immunodeficiency virus. Virology 450-451:49–54. doi: 10.1016/j.virol.2013.11.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mansky LM, Temin HM. 1995. Lower in vivo mutation rate of human immunodeficiency virus type 1 than that predicted from the fidelity of purified reverse transcriptase. J Virol 69:5087–5094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chaipan C, Smith JL, Hu WS, Pathak VK. 2013. APOBEC3G restricts HIV-1 to a greater extent than APOBEC3F and APOBEC3DE in human primary CD4+ T cells and macrophages. J Virol 87:444–453. doi: 10.1128/JVI.00676-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Belanger K, Langlois MA. 2015. Comparative analysis of the gene-inactivating potential of retroviral restriction factors APOBEC3F and APOBEC3G. J Gen Virol 96:2878–2887. doi: 10.1099/vir.0.000214. [DOI] [PubMed] [Google Scholar]