

ABSTRACT
Alphaviruses are small enveloped RNA viruses that infect cells via clathrin-mediated endocytosis and low-pH-triggered fusion in the early endosome. Using a small interfering RNA (siRNA) screen in human cells, we previously identified TSPAN9 as a host factor that promotes infection by the alphaviruses Sindbis virus (SINV), Semliki Forest virus (SFV), and chikungunya virus (CHIKV). Depletion of TSPAN9 specifically decreases SFV membrane fusion in endosomes. TSPAN9 is a member of the tetraspanin family of multipass membrane proteins, but its cellular function is currently unknown. Here we used U-2 OS cells stably overexpressing TSPAN9 to show that TSPAN9 is localized at the plasma membrane and in early and late endosomes. Internalized SFV particles colocalized with TSPAN9 in vesicles early during infection. Depletion of TSPAN9 led to reductions in the amounts of the late endosomal proteins LAMP1 and CD63 and an increase in the amount of LAMP2. However, TSPAN9 depletion did not alter the delivery of SFV to early endosomes or change their pH or protease activity. Comparative studies showed that TSPAN9 depletion strongly inhibited infection by several viruses that fuse in early endosomes (SFV, SINV, CHIKV, and vesicular stomatitis virus [VSV]), while viruses that fuse in the late endosome (recombinant VSV-Lassa and VSV-Junin), including an SFV point mutant with a lower pH threshold for fusion (SFV E2 T12I), were relatively resistant. Our data suggest that TSPAN9 modulates the early endosome compartment to make it more permissive for membrane fusion of early-penetrating viruses.
IMPORTANCE Alphaviruses are spread by mosquitoes and can cause serious human diseases such as arthritis and encephalitis. Recent outbreaks of CHIKV infection are responsible for millions of cases of acute illness and long-term complications. There are no vaccines or antiviral treatments for these important human pathogens. Alphaviruses infect host cells by utilizing the endocytic machinery of the cell and fusing their membrane with that of the endosome. Although the mechanism of virus-membrane fusion is well studied, we still know relatively little about the host cell proteins that are involved in alphavirus entry. Here we characterized the role of the host membrane protein TSPAN9 in alphavirus infection. TSPAN9 was localized to early endosomes containing internalized alphavirus, and depletion of TSPAN9 inhibited virus fusion with the early endosome membrane. In contrast, infection of viruses that enter through the late endosome was relatively resistant to TSPAN9 depletion, suggesting an important role for TSPAN9 in the early endosome.
INTRODUCTION
Alphaviruses are a genus of viruses that are generally transmitted by mosquito vectors and can infect a variety of hosts, including small and large mammals, birds, and humans (reviewed in reference 1). A number of alphaviruses cause human illnesses, including fever, encephalitis, and polyarthritis. For example, in humans, the alphavirus chikungunya virus (CHIKV) causes painful arthritis with symptoms that can persist for years and can also cause neurological complications and neonatal encephalitis (2). CHIKV reemerged in 2004 to cause outbreaks of millions of cases in countries around the Indian ocean (3, 4) and spread to the Americas in late 2013, where, to date, it has caused an estimated 1.2 million human cases (5, 6). There are currently no effective vaccines or treatments for human alphavirus infections.
Alphaviruses are small, enveloped, highly organized viruses that have a plus-sense RNA genome (1). The genome is enclosed in a capsid protein shell that is surrounded by a lipid membrane containing a lattice of the E1 and E2 membrane glycoproteins. To infect cells, alphaviruses bind to cell surface receptors via the E2 protein and are internalized by clathrin-mediated endocytosis (reviewed in reference 7). The virus is delivered to the early endosome compartment, where the low-pH environment triggers the dissociation of the E2-E1 heterodimer and conformational changes in the E1 membrane fusion protein. E1 inserts its hydrophobic fusion loop into the endosome membrane, trimerizes, and refolds into a hairpin-like conformation with the fusion loop and the transmembrane domain at the same end of the molecule (8). These conformational changes drive fusion between the viral and endosome membranes and thus release the nucleocapsid into the cytoplasm. Although we have considerable insights into the functions of the viral proteins during entry, we have relatively little information about cellular proteins that are involved in the alphavirus entry process.
Using a whole-genome small interfering RNA (siRNA) screen, we previously identified TSPAN9 as a human host factor that promotes alphavirus infection (9). TSPAN9, a member of the tetraspanin protein family, is a small protein of ∼27 kDa with four transmembrane domains and both the N and C termini in the cytoplasm (10). It is conserved from human to mosquito and expressed in all assayed tissues and cell lines. Depletion of TSPAN9 inhibited infection by several alphaviruses (Semliki Forest virus [SFV], Sindbis virus [SINV], and CHIKV) and by the rhabdovirus vesicular stomatitis virus (VSV) (9). All of these viruses infect cells by endocytosis and fusion in the early endosome (7, 11–13). Depletion of TSPAN9 caused little or no effect on alphavirus binding, internalization, or low-pH-triggered E1 conformational changes. However, the fusion of internalized virus with the endosome membrane was strongly inhibited. While tetraspanin proteins have been reported to be involved in a wide variety of cellular processes (14, 15), the specific cellular functions of TSPAN9 are undefined.
Here we set out to characterize the mechanism by which TSPAN9 promotes alphavirus infection. Expression studies localized TSPAN9 to the plasma membrane and to the early and late endosome compartments. Internalized SFV colocalized with TSPAN9 in early endosomes. TSPAN9 depletion did not alter the delivery of internalized SFV to the early endosome, even though virus fusion and infection were strongly inhibited. In contrast, infection by viruses that enter from the late endosome was relatively unimpaired. These results suggest that TSPAN9 is specifically required for the fusion-permissive functions of the early endosome.
MATERIALS AND METHODS
Cell lines and viruses.
U-2 OS cells were maintained at 37°C in McCoy's 5A medium with 10% fetal bovine serum (FBS), 100 U penicillin/ml, 100 μg streptomycin/ml, and 2 mM l-glutamine. The alphaviruses used in this study were SINV pE2R1-GFP/2A (referred to as SINV-GFP) (a kind gift from Hans Heidner) (9, 16), a well-characterized plaque-purified SFV stock referred to here as M1K (17), wild-type (WT) SFV derived from the pSP6-SFV4 infectious clone (18), and the SFV E2 T12I mutant (17, 19) constructed by using this infectious clone. VSV expressing the green fluorescent protein (GFP) reporter (VSV-GFP) (20, 21) and recombinant VSV missing the VSV G protein but expressing the GFP reporter plus the glycoprotein from Lassa virus (rVSV-Lassa) (22) or Junin virus (rVSV-Junin) were kind gifts of Kartik Chandran and Sean Whelan, respectively.
Antibodies.
The polyclonal rabbit anti-E1/E2 antibody and monoclonal antibody (MAb) E1a-1 to the acid conformation of E1 were previously described (23). The following MAbs to endocytosis pathway proteins were obtained from the Developmental Studies Hybridoma Bank (DSHB) created by the NICHD of the NIH and maintained at the University of Iowa Department of Biology, Iowa City, IA: anti-LAMP1 (clone H4A3), anti-LAMP2 (clone H4B4), anti-CD63 (clone H5C6), and anti-mannose-6-phosphate receptor (M6PR) (clone 22d4). Anti-EEA1 MAb was obtained from BD Biosciences (San Jose, CA), anti-lysobisphosphatidic acid (LBPA) MAb was obtained from Echelon (Salt Lake City, UT), and anti-cathepsin B polyclonal antibody (PAb) was obtained from Athens Research & Technology (Athens, GA).
siRNA depletion of cellular proteins.
The following siRNAs were used in this study: a nontargeting (NT) siRNA (Ambion Silencer negative-control siRNA 7), a transfection control siRNA that targets RPL27A and causes cell death (Ambion siRNA 9257) (24), TSPAN9 siRNA (Ambion siRNA 18435) (9), LAMP1 siRNA (Ambion siRNA 144036) (25), and CD63 siRNA (Ambion siRNA 10318) (26). For infection assays, siRNAs were added to Corning black polystyrene 384-well plates and used to reverse transfect U-2 OS cells (3,000 cells/well) at a final concentration of 37.5 nM siRNA (NT, TSPAN9, CD63, or RPL27A siRNA) or 100 nM siRNA (LAMP1 siRNA) with 0.375% Lipofectamine RNAiMAX (Invitrogen) in medium without antibiotics for 48 h. For microscopy assays, U-2 OS cells seeded at 4 × 104 cells/well on 8-well Lab-Tek chambered cover glass (Thermo Scientific) were transfected with 22.5 nM siRNA and 0.15% RNAiMax and cultured for 48 h.
FusionRed TSPAN9 construct and cells.
The fluorescent FusionRed (FR) protein (Evrogen) (a kind gift from Erik Snapp) (27) was fused to the N terminus of TSPAN9 by amplifying the two proteins separately by PCR. TSPAN9 was amplified by using forward primer MCK 1002 (5′-ATGGCCAGGGGCTGCCTCTGCTGC) and reverse primer MCK 667 (5′-CCGAATTCTCATGCGTCGTACTTCTTACCAG). FusionRed was amplified by using forward primer MCK 1000 (5′-CGGCTAGCATGGTGTCCGAGCTGATTAAGG) and reverse primer MCK 1001 (5′-GCAGCAGAGGCAGCCCCTGGCCATTTTCCCTCCATCGCCTGCCCC). The two PCR products were gel purified, combined into one reaction mixture, and amplified by using forward primer MCK 1000 and reverse primer MCK 667 to yield the final full-length product. The PCR product was digested with NheI and EcoRI and ligated into pcDNA3.1 to generate plasmid pcDNA-FR-TSPAN9. U-2 OS cells were transfected with pcDNA-FR-TSPAN9 by using Lipofectamine 2000 as recommended by the manufacturer. Clonal cell lines were selected by limiting dilution in the presence of 1 mg/ml G418. Clone 15 was used in this study and was selected by immunofluorescence for FR-TSPAN9 expression.
Transient overexpression of LAMP2.
A plasmid containing the gene for human LAMP2B (HsCD00325256) was obtained from the plasmid repository at Harvard University and was cloned by PCR amplification into pcDNA 3.1 to create plasmid pcDNA-hLAMP2. U-2 OS cells were transiently transfected with pcDNA or pcDNA-hLAMP2 by using Lipofectamine 2000 according to the manufacturer's protocols. At 24 h posttransfection, cells were infected with SFV M1K (multiplicity of infection [MOI] of 0.03) for 12 h at 37°C and then washed and fixed with 4% paraformaldehyde (PFA). Cells were stained with polyclonal anti-E1/E2 and anti-LAMP2 (DSHB clone H4B4) followed by goat anti-rabbit Alexa 488 and goat-anti mouse Alexa 568 secondary antibodies. Nuclei were stained with Hoechst dye.
DQ Red-BSA degradation assay.
U-2 OS cells were transfected on 8-well chambered cover glass as described above. The cells were incubated for 5 h with BODIPY-labeled bovine serum albumin (BSA) (DQ Red-BSA; Molecular Probes) at a final concentration of 200 μg/ml in U-2 OS medium. Cells were subsequently washed three times with phosphate-buffered saline (PBS), fixed with 4% PFA, and imaged by confocal microscopy.
Protease inhibitor assay.
U-2 OS cells were transfected as described above on a 384-well plate. Cells were treated with 5 μM E64d and 100 μM leupeptin for 5 h before infection with SINV-GFP at an MOI of 1 for 24 h and with SFV at an MOI of 0.01 for 12 h. Protease inhibitor treatment was continued during virus infection. Cells were then washed with PBS and fixed with 4% PFA, and SFV-infected cells were stained with polyclonal anti-E1/E2 and goat anti-rabbit Alexa 488 antibodies. Infected cells were imaged by fluorescence microscopy.
LysoSensor green assay.
U-2 OS cells on 8-well chambered cover glass were transfected with NT and TSPAN9 siRNAs as described above. Cells were washed with PBS before incubation with LysoSensor (LysoSensor Green DND-189; Invitrogen) at a final concentration of 1 μM in U-2 OS growth medium at 37°C for 1 h. Cells were then washed four times with PBS, fixed with 4% PFA, and permeabilized by treatment with 0.01% Triton X-100 and 1% BSA in PBS for 30 min. Cells were incubated with EEA1 or LAMP2 primary antibody for 2 h at room temperature (RT), followed by incubation with goat anti-mouse Alexa 594 secondary antibody for 30 min, and imaged by confocal microscopy on the same day.
Primary infection experiments.
U-2 OS cells on 384-well plates were transfected with NT and TSPAN9 siRNAs as described above and infected with SINV-GFP (MOI = 10), SFV M1K (MOI = 0.1), rVSV-Lassa (MOI = 0.5), rVSV-Junin (MOI = 2), WT SFV (MOI = 0.2), or the SFV T12I mutant (MOI = 0.2) for 1 h at 37°C. The media were then replaced with media containing 30 mM NH4Cl to block secondary infection, and incubation was continued overnight at 28°C. Cells were washed with PBS and fixed with 4% PFA. SFV-infected cells were permeabilized with 0.1% Triton X-100 and stained with anti-E1/E2 polyclonal antibody, followed by goat anti-rabbit Alexa 488 secondary antibody. All other viruses express GFP in the cell cytoplasm. Nuclei were stained with Hoechst dye. Cells were imaged by fluorescence microscopy, and infected cells and nuclei were counted by using a customized pipeline in Cell Profiler v. 2.0 r11710 cell image analysis software (Broad Institute) (28).
Colocalization of virus with cellular markers.
U-2 OS cells on an 8-well chambered cover glass were transfected as described above. Purified SFV (0.2 μg/well) in Rmed (RPMI without bicarbonate plus 0.2% BSA and 10 mM HEPES), pH 6.8, was bound to U-2 OS cells for 60 min on ice. The cells were incubated at 37°C in Rmed (pH 7.4) for 20 min to permit endocytosis, washed on ice three times with PBS, and fixed with 4% PFA. Cell surface virus was stained with anti-SFV E1/E2 PAb and goat anti-rabbit Ig Alexa Fluor 647. Cells were then permeabilized with 0.1% Triton X-100, intracellular virus was stained with anti-SFV E1/E2 rabbit PAb and goat anti-rabbit Alexa Fluor 594, and endosome protein markers were stained with the indicated MAbs and goat anti-mouse Alexa 488. Nuclei were stained with Hoechst dye.
Confocal microscopy and image analysis.
z-stack images were captured by using a Leica SP5 confocal microscope. The fluorescence signal was quantitated by using Image J by measuring the integrated density of fluorescence (area × intensity) per 50 cells and normalized to the value obtained with the NT control. For colocalization studies, fluorescence signals were quantitated and assessed for colocalization by using a customized pipeline in Cell Profiler v. 2.0 r11710 cell image analysis software (Broad Institute) (28).
Statistical analysis.
Microsoft Excel was used to calculate statistical significance by the two-tailed unpaired Student t test. Significance is indicated in the figures.
RESULTS
Intracellular localization of TSPAN9.
Our previous studies identified TSPAN9 as a host factor that promotes alphavirus infection (9). Depletion of TSPAN9 specifically decreased alphavirus membrane fusion, which normally occurs in the early endosome. TSPAN9 is a member of the tetraspanin family of membrane proteins, but little is known about its normal cellular function. While a polyclonal antibody to TSPAN9 is available, we found that the endogenous levels of TSPAN9 are too low to be detected in U-2 OS and other tested cell lines. To define the cellular localization of TSPAN9 under various conditions, we therefore generated a U-2 OS cell line expressing TSPAN9 with the FusionRed fluorescent protein at its N terminus (FR-TSPAN9). Confocal microscopy showed that FR-TSPAN9 was expressed at the cell surface and in vesicles that colocalized with markers of early endosomes, late endosomes, and lysosomes (Fig. 1A). This localization was very similar to that observed in cells overexpressing untagged TSPAN9 and stained with the polyclonal antibody (data not shown) (9). We compared the localization of FR-TSPAN9 in cells incubated with SFV for 20 min and observed no change in the protein's colocalization with cellular marker proteins (Fig. 1B, gray versus black bars). Thus, endocytosis of alphavirus particles did not modify the normal pattern of TSPAN9 localization.
FIG 1.

Intracellular localization of FR-TSPAN9. (A) U-2 OS cells stably expressing FR-TSPAN9 were stained with MAbs to the indicated proteins of the endosomal pathway, followed by anti-mouse Alexa 488 secondary antibody (green). FR-TSPAN9 is shown in red. A single confocal slice from a representative experiment of three is shown. (B) U-2 OS cells stably expressing FR-TSPAN9 were incubated with or without 0.2 μg SFV/well for 20 min at 37°C and then fixed and stained for the indicated proteins as described above for panel A. Colocalization between FR-TSPAN9 and endosome proteins was quantitated by using Cell Profiler in the presence and absence of SFV internalization. Data represent the means ± standard errors of the means of data from three independent experiments. No significant differences were observed between the results with and without SFV.
We next examined whether SFV was internalized into endocytic vesicles that also contained TSPAN9. Cells expressing FR-TSPAN9 were incubated with SFV for 20 min at 37°C to allow uptake. Internalized virus was then stained with MAb E1a-1, which is specific for the acid conformation of the SFV E1 protein (9, 23). Approximately 70% of E1a-1-positive virus was found to colocalize with punctate spots of FR-TSPAN9 fluorescence (Fig. 2). Together, our data showed that TSPAN9 is found in vesicles of the endocytic pathway and is present in the acidic early endosomes that contain newly internalized SFV.
FIG 2.

Colocalization of FR-TSPAN9 with SFV. U-2 OS cells stably expressing FR-TSPAN9 were incubated with 0.2 μg SFV/well for 20 min at 37°C and then fixed and stained with acid conformation-specific E1 MAb E1a-1 (green). Approximately 70% of the acid-exposed SFV colocalizes with punctate FR-TSPAN9 (red). A single confocal slice from a representative experiment of three is shown.
TSPAN9 depletion does not affect SFV localization.
Our previously reported data show that depletion of TSPAN9 inhibits SFV infection by blocking a late step in entry: the fusion of internalized, acid-triggered virus with the cell membrane (9). In control cells, WT SFV fusion occurs in the early endosome once the pH reaches a threshold value of ∼6.2 (29, 30). We tested whether the trafficking of SFV to the early endosome compartment was altered in TSPAN9-depleted cells. Control or depleted cells were allowed to internalize SFV for 20 min, and the localization of intracellular virus was then compared to that of various markers of endocytic compartments. In control cells, >70% of internalized SFV particles colocalized with EEA1, a marker of the early endosome compartment (Fig. 3). We also observed that ∼30% of internalized SFV colocalized with M6PR, consistent with the known localization of this receptor to the early endosome as well as to compartments including late endosomes and the trans-Golgi network (31). There was little colocalization of internalized SFV with the late endosome/lysosome markers CD63, LAMP1, LAMP2, and LBPA. This colocalization pattern was not significantly changed by TSPAN9 depletion; SFV still colocalized mainly with EEA1-positive early endosomes after 20 min of uptake. Thus, TSPAN9 depletion does not alter SFV delivery to the normal site of membrane fusion.
FIG 3.
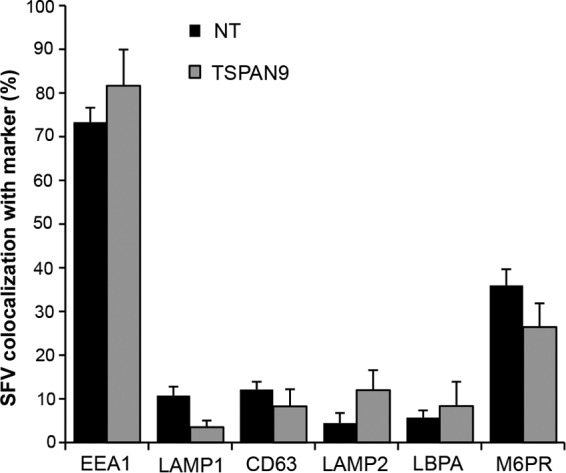
Effect of TSPAN9 depletion on the intracellular localization of SFV. U-2 OS cells were transfected with NT or TSPAN9 siRNA and cultured for 48 h. SFV was bound to cells for 1 h on ice, followed by warming to 37°C for 20 min before fixation. SFV was stained with anti-E1/E2 PAb before and after permeabilization, under conditions that differentiate virus remaining on the cell surface from internalized virus (9), and endosomal proteins were stained with the indicated MAbs. Colocalization of internalized virus and cellular proteins was quantitated by using Cell Profiler and expressed as a percentage of the total internalized SFV puncta. Data represent the means ± standard errors of the means of data from three independent experiments. No significant differences between the NT and TSPAN9 siRNA samples were observed.
TSPAN9 depletion alters the levels of late endosome proteins.
While performing the imaging studies shown in Fig. 3, we observed that the staining of some of the endosomal proteins appeared to be altered by TSPAN9 depletion. We therefore compared the immunofluorescence stainings of various endosomal proteins in control and TSPAN9-depleted cells. Quantitation of fluorescence images showed that TSPAN9 depletion did not significantly change the staining of the early endosome proteins Rab5 and EEA1 (Fig. 4A). However, fluorescence of the late endosome proteins CD63 and LAMP1 was significantly decreased while that of LAMP2 was significantly increased by TSPAN9 depletion (Fig. 4A). In contrast, M6PR, which transports proteases between the trans-Golgi network and endosomes (31), and cathepsin B, a protease found in late endosomes and lysosomes, showed modest but not significant increases in staining levels, and the late endosome lipid LBPA was unchanged (Fig. 4A). To determine whether the changes in LAMP1 and LAMP2 fluorescence resulted from changes in protein distribution versus changes in the amount of protein, we quantitated the cellular protein amounts by Western blot analysis of control and TSPAN9-depleted cells (Fig. 4B). In agreement with the immunofluorescence data, no difference was observed in the amounts of EEA1, while the amount of LAMP1 protein was significantly decreased and that of LAMP2 was significantly increased.
FIG 4.
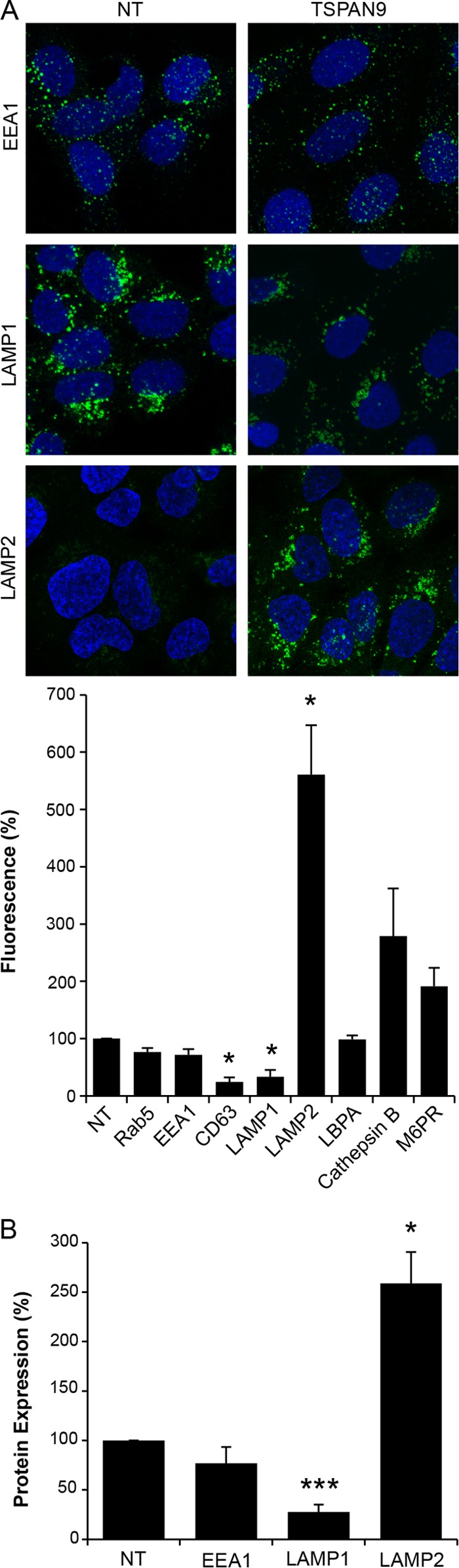
Quantitation of endosomal proteins after TSPAN9 depletion. U-2 OS cells were transfected with NT or TSPAN9 siRNA for 48 h before fixation. Endosomal proteins were stained with the indicated MAbs. (A) Representative confocal extended-focus images from three experiments. The total integrated density of fluorescence per image was quantitated by using Image J and normalized to the number of cells per image. Fifty cells were counted in each of three independent experiments (bar graph). (B) U-2 OS cells transfected as described above for panel A were lysed and subjected to SDS-PAGE and Western blotting with the indicated antibodies. Protein amounts were quantitated and normalized to tubulin levels as the loading control. The bar graphs in panels A and B represent the means ± standard errors of the means for three independent experiments (*, P < 0.05; ***, P < 0.001).
To test if the observed decrease in the amount of CD63 or LAMP1 or the increase in the amount of LAMP2 was responsible for the inhibition of virus infection in TSPAN9-depleted cells, we depleted CD63 or LAMP1 by using siRNA in U-2 OS cells (Fig. 5A). We found no significant difference in infection by SFV or SINV-GFP in cells depleted of either CD63 or LAMP1 (Fig. 5B). Next, we overexpressed LAMP2B in U-2 OS cells (Fig. 5C) but found no significant difference in the ability of SFV to infect these cells (Fig. 5D). These results suggest that the inhibition of alphavirus infection is caused by the lack of TSPAN9 rather than the depletion of LAMP1 or CD63 or the increase in the amount of LAMP2.
FIG 5.
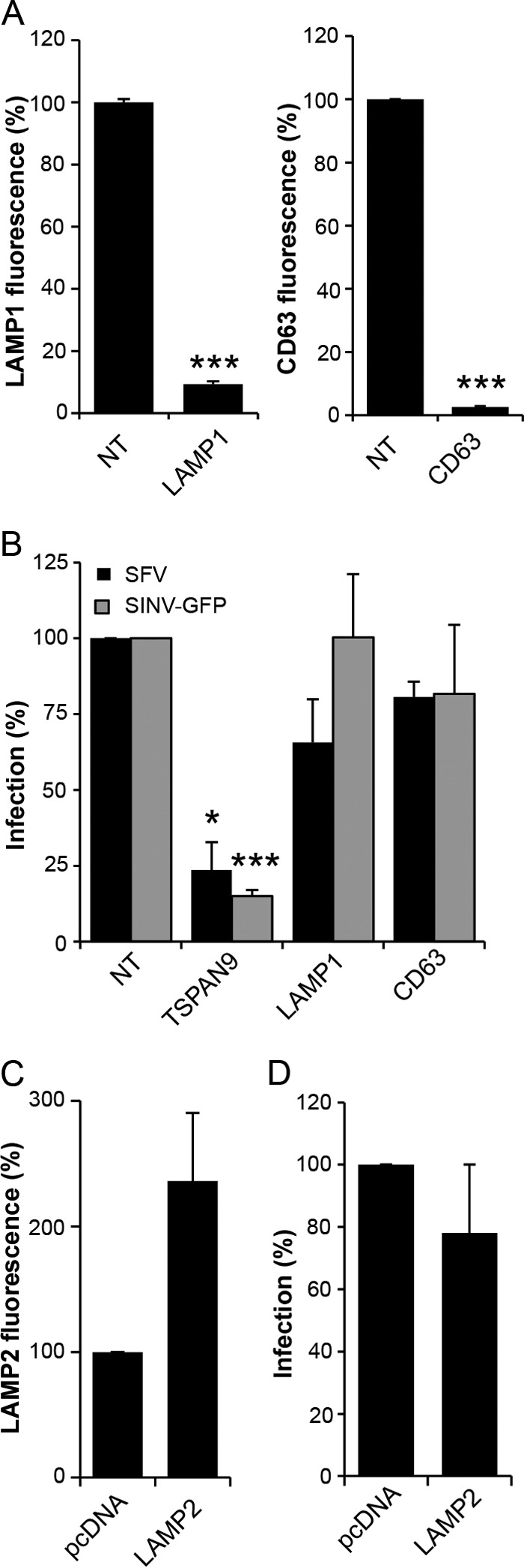
Effect of depletion of LAMP1 and CD63 or overexpression of LAMP2. (A) U-2 OS cells were transfected with NT, LAMP1, or CD63 siRNA; cultured for 48 h; and stained with anti-LAMP1 antibody (left) or anti-CD63 antibody (right). The total integrated density of fluorescence per image was quantitated by using Image J and normalized to the number of cells per image. Fifty cells were quantitated under each condition in each of three independent experiments. (B) U-2 OS cells transfected as described above for panel A were cultured for 48 h and then infected with SFV (MOI = 0.1) or SINV-GFP (MOI = 10) for 1 h before the addition of 20 mM NH4Cl and incubation for 14 h (primary infection). Cells were then fixed, and SFV-infected cells were stained with PAb against E1/E2. Infected cells and nuclei were counted by using Cell Profiler to determine the percentage of infected cells, and values were normalized to those for the NT control. (C) U-2 OS cells were transfected with pcDNA or pcDNA-hLAMP2 for 24 h and stained with anti-LAMP2 MAb. The total integrated density of fluorescence per image was quantitated by using Image J and normalized to the number of cells per image. Fifty cells were quantitated under each condition in each of three independent experiments. (D) U-2 OS cells were transfected with pcDNA or pcDNA-hLAMP2 for 24 h before infection with SFV and staining as described above for panel B. Infected cells and nuclei were counted by using Cell Profiler, and values were normalized to values for the pcDNA control. Data in all bar graphs represent the means ± standard errors of the means for three independent experiments (*, P < 0.05; ***, P < 0.001).
Increased protease activity is not involved in inhibition of alphavirus infection.
We observed an ∼2-fold increase in the fluorescence staining of cathepsin B and M6PR in TSPAN9-depleted cells (Fig. 4A). It was possible that this produced an increased protease level in early endosomes, thus inhibiting alphavirus infection. However, there was little colocalization of cathepsin B with internalized, acid-exposed SFV in either control or TSPAN9-depleted cells, suggesting that this protease did not relocalize to SFV-containing early endosomes (Fig. 6A). Moreover, we did not observe increased degradation of BODIPY-labeled BSA (DQ-BSA) after depletion of TSPAN9 (Fig. 6B). To test if treatment with protease inhibitors would rescue virus infection, control and TSPAN9-depleted cells were preincubated with the panspecific cathepsin inhibitor E64d and the serine/thiol protease inhibitor leupeptin for 5 h before virus addition. Infection by SFV and SINV-GFP was still strongly inhibited in TSPAN9-depleted cells in the presence of protease inhibitors (Fig. 6C). In contrast, the same protease inhibitor conditions inhibited the cleavage of fluorescent DQ-BSA by >90% (Fig. 6D). Together, our results suggest that the inhibition of alphavirus infection in TSPAN9-depleted cells is not due to increased endosomal protease activity.
FIG 6.

Effect of TSPAN9 depletion on endosomal proteases. (A) U-2 OS cells were transfected with NT or TSPAN9 siRNA and cultured for 48 h, and 0.2 μg SFV/well was bound to cells for 1 h on ice, followed by warming to 37°C for 20 min. Cells were then fixed, permeabilized, and stained with SFV MAb E1a-1 and anti-cathepsin B PAb. Colocalization of SFV and cathepsin B staining was quantitated by using Cell Profiler. (B) U-2 OS cells were transfected as described above for panel A and incubated with 200 μg/ml DQ-BSA for 5 h at 37°C. The total integrated density of fluorescence per image was quantitated by using Image J and normalized to the number of cells per image for a total of 50 cells per experiment. (C) U-2 OS cells transfected as described above for panel A were pretreated with 5 μM E64d and 100 μM leupeptin for 5 h before the addition of SFV (MOI = 0.01) or SINV-GFP (MOI = 1). Infected cells were incubated for 12 h postinfection (SFV) or 24 h postinfection (SINV-GFP), and infection was quantitated by GFP expression or staining with SFV anti-E1/E2 PAb. (D) U-2 OS cells were treated with protease inhibitors as described above for panel C, incubated with 200 μg/ml DQ-BSA, and quantitated as described above for panel B. Bar graphs represent the means ± standard errors of the means for three independent experiments (**, P < 0.01; ***, P < 0.001).
TSPAN9 depletion does not affect early endosome pH or cellular cholesterol levels.
Our previously reported data show that endosomes in TSPAN9-depleted cells become acidified, as detected by LysoTracker staining and reactivity of internalized SFV with an E1 acid conformation-specific MAb (9). However, these assays would not detect an increase in early endosome acidity, which could inactivate the virus before fusion. To test for dysregulation of early endosome pH, we stained cells with LysoSensor DND-189 (pKa of 5.2), which becomes fluorescent only in highly acidic environments, and analyzed its colocalization with EEA1 and LAMP1/LAMP2. In control cells, LysoSensor DND-189 strongly colocalized with LAMP1/LAMP2 and not with EEA1, and this pattern was not changed by TSPAN9 depletion (Fig. 7A).
FIG 7.
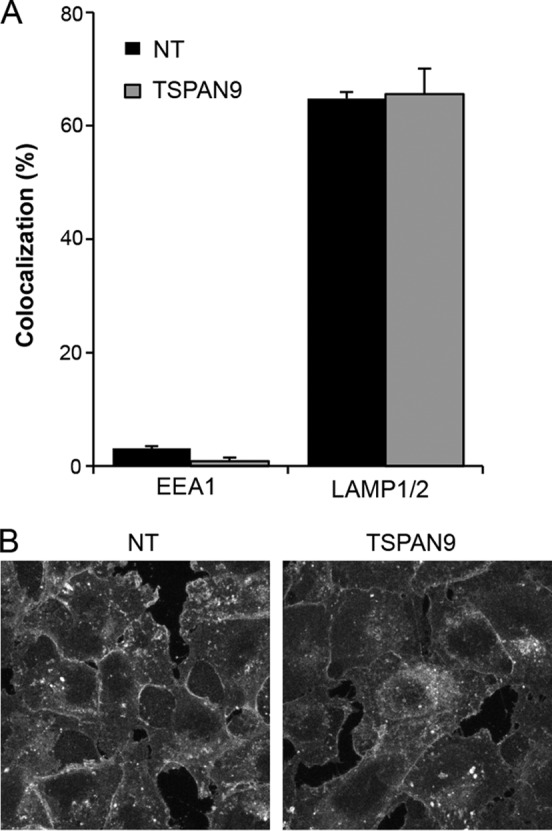
Effect of TSPAN9 depletion on endosomal pH and cholesterol levels. (A) U-2 OS cells were transfected with NT or TSPAN9 siRNA, cultured for 48 h, and then incubated with LysoSensor Green DND-189 (pKa of ∼5.2) for 1 h at 37°C before fixation. Cells were then permeabilized and stained with either a MAb against EEA1 or MAbs against LAMP1 and LAMP2. Colocalization of LysoSensor and the indicated endosomal proteins was quantitated by using Cell Profiler. Bar graphs represent the means ± standard errors of the means of data from three independent experiments. No significant differences were observed between the NT and TSPAN9 siRNA samples. (B) Control or TSPAN9-depleted U-2 OS cells prepared as described above for panel A were fixed and stained with filipin (50 μg/ml). A single confocal slice from a representative experiment of three is shown.
Alphavirus fusion and infection are strongly dependent on the presence of cholesterol in the endosome membrane (32). Since several tetraspanin proteins interact directly with cholesterol (33), we examined the cholesterol contents of control and TSPAN9-depleted cells. Filipin staining of cellular cholesterol did not decrease or show an altered distribution when TSPAN9 was depleted (Fig. 7B). Thus, neither the pH of early endosomes nor cellular cholesterol levels appear responsible for the inhibition of virus infection in TSPAN9-depleted cells.
TSPAN9 depletion inhibits early- but not late-fusing viruses.
TSPAN9 depletion inhibits infection by the three alphaviruses SINV, SFV, and CHIKV and the rhabdovirus VSV. In contrast, depletion had no significant effect on infection by dengue virus (DENV) (9). While the alphaviruses and VSV are known to fuse in the early endosome compartment (7, 11–13), DENV fuses in the late endosome (34, 35). The arenaviruses Lassa virus and Junin virus are two other well-characterized late-entering viruses (22, 36, 37). We used recombinant VSV containing the GPs from these viruses to test their ability to infect TSPAN9-depleted cells. TSPAN9 depletion did not significantly inhibit infection by either rVSV-Lassa GP or rVSV-Junin GP (Fig. 8A).
FIG 8.
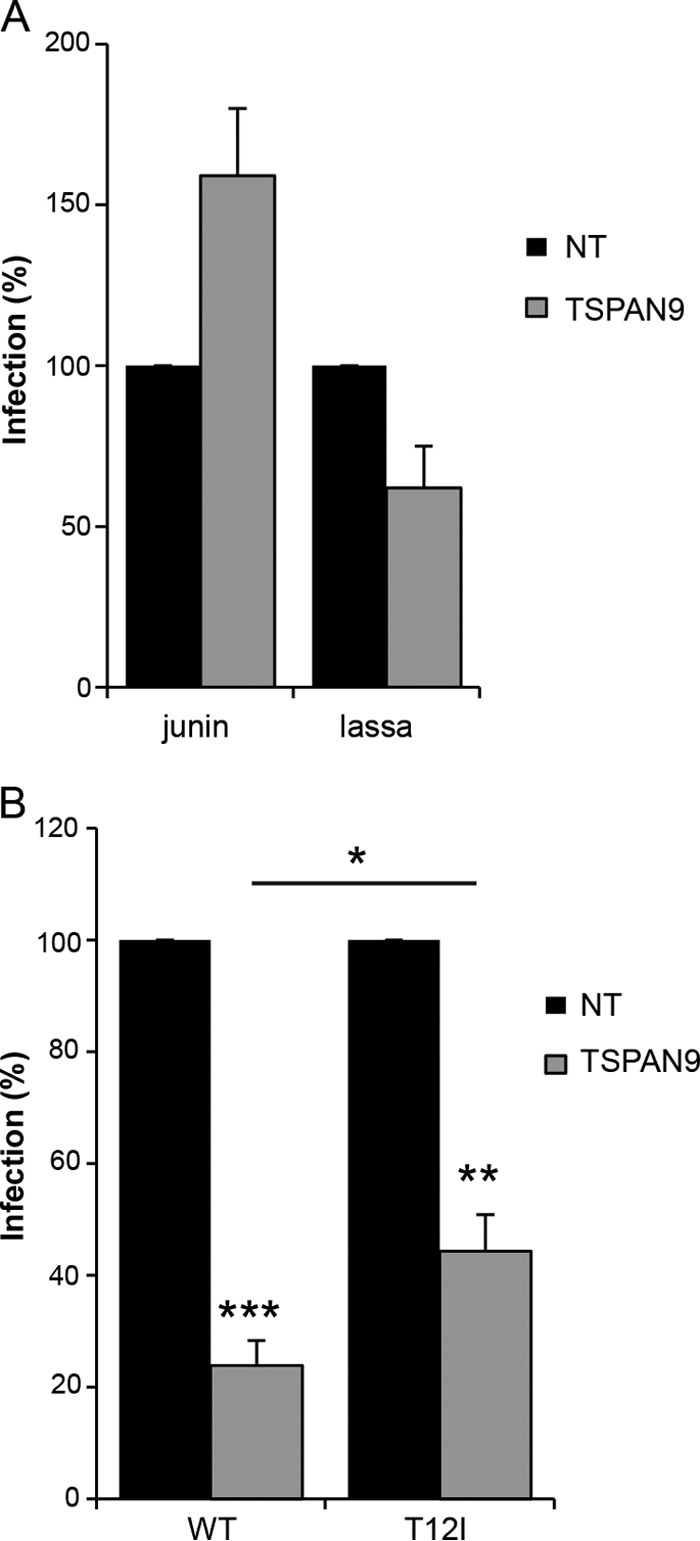
Effect of TSPAN9 depletion on viruses entering from late endosomes. U-2 OS cells were transfected with NT or TSPAN9 siRNA, cultured for 48 h, and infected as described in the text. (A) Cells were infected with rVSV-Lassa (MOI = 0.5) or rVSV-Junin (MOI = 2) for 2 h before the addition of NH4Cl and incubation for 14 h (primary infection). GFP-positive cells and nuclei were counted by immunofluorescence. Bar graphs represent the means ± standard errors of the means of data from three independent experiments. No significant differences were observed between the NT and TSPAN9 siRNA samples. (B) Cells were infected with SFV or the SFV T12I mutant (MOI = 0.2) for 1 h before the addition of NH4Cl and incubation for 14 h (primary infection). Infected cells were stained with anti-E1/E2 PAb and counted by immunofluorescence analysis. Bar graphs represent the means ± standard errors of the means of data from three independent experiments (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
We then took advantage of an SFV mutant, fus-1, which has a lower threshold for fusion of pH ∼5.0 than the WT SFV threshold of pH ∼6.2 (17) and therefore enters from a late endosome compartment (29, 30). The key mutation in the fus-1 mutant is a change of E2 residue 12 from threonine to isoleucine (17). A virus constructed with this single point mutation, T12I, shows the same pH profile as that of the fus-1 mutant (19). We compared primary infection by WT SFV to that of the E2 T12I mutant (Fig. 8B). While both viruses were inhibited by depletion of TSPAN9, the T12I point mutation significantly rescued infection. Together, our results indicate that TSPAN9 promotes virus fusion in the early endosome, while viruses that have adapted to fuse later in the endosomal pathway are less dependent on TSPAN9.
DISCUSSION
When we identified TSPAN9 as a host factor in alphavirus infection (9), almost no information was available on this protein, with a single report on its presence in tetraspanin-enriched microdomains on platelets (38) and no studies of its cellular functions. We therefore aimed to obtain information on both the localization of TSPAN9 in cells and its role in alphavirus membrane fusion. A requirement for a direct TSPAN9-virus interaction during fusion appeared unlikely based on our finding that low-pH-triggered fusion of SFV and SINV with the cell plasma membrane was not altered by TSPAN9 depletion (9) and previous reports that alphaviruses fuse efficiently with protein-free liposomes at low pH (e.g., see references 39 and 40). Thus, it seemed most probable that TSPAN9 promoted virus fusion by effects on the endosome compartment.
Our results showed that TSPAN9 is localized to both the cell surface and vesicular compartments containing markers of early and late endosomes/lysosomes. This localization was not altered by the addition of purified SFV to cells, and after a 20-min incubation, the majority of internalized virus was detected in TSPAN9-positive compartments that were also positive for the early endosome marker EEA1. Depletion of TSPAN9 did not alter the delivery of SFV to the early endosome compartment (as defined by EEA1) but strongly inhibited its fusion. We tested several properties of the early endosome compartment that potentially could explain its decreased permissiveness for alphavirus fusion. No changes in its pH, cholesterol levels, or protease content were detected, suggesting that other mechanisms are responsible for inhibition of fusion.
TSPAN9 depletion produced unexpectedly strong effects on the levels of late endosome proteins, with significant decreases in the amounts of both CD63 and LAMP1 and a striking increase in the amount of LAMP2. It seems likely that, as described previously by other investigators (41), the observed increase in the amount of LAMP2 acts to compensate for the loss of LAMP1. LAMP1 and LAMP2 are highly glycosylated membrane proteins that are the most abundant proteins in the lysosomal membrane, where part of their role is to protect the limiting membrane from lysosomal hydrolases (42). The LAMP2A isoform has a well-defined role as the receptor for chaperone-mediated autophagy (43). The tetraspanin protein CD63 (also termed LAMP3) has been implicated in the regulation of protein transport, but its specific intracellular function is not well defined (44). While TSPAN9 depletion caused significant changes in the levels of late endosome proteins, we found that newly internalized SFV did not colocalize with CD63, LAMP1, or LAMP2, in keeping with the entry of alphaviruses via early endosomes. In addition, direct depletion of CD63 or LAMP1 or overexpression of LAMP2 did not inhibit SFV infection. Thus, our results suggest that changes in the levels of these late endosome proteins are unlikely to be directly responsible for the inhibition of alphavirus fusion in early endosomes. However, these alterations may well reflect the effect of TSPAN9 depletion on membrane domains or protein interactions in the endosomal compartment, which produce a less fusion-permissive early endosome environment, and the detected changes in later endosome compartments.
Our previously reported results show that DENV infection is not inhibited by TSPAN9 depletion. DENV fuses in the late endosome compartment (34, 35), and we demonstrated here that the late-penetrating viruses rVSV-Lassa and rVSV-Junin were also unaffected by TSPAN9 depletion. A point mutation in the SFV E2 protein, T12I, shifts the pH of fusion by changing the pH of E2-E1 dimer dissociation (17, 19). The mutant virus has a threshold of fusion of pH ∼5.0, compared with the WT threshold of pH ∼6.2, and therefore fuses in a later endosome compartment (29, 30). The T12I mutation conferred a substantial rescue of SFV infection in TSPAN9-depleted cells. The resistance of late-entering viruses to TSPAN9 depletion may mean that the key, fusion-inhibiting effect of depletion is specifically localized to the early endosome. Alternatively, our results may indicate that the key effect of TSPAN9 depletion occurs throughout the endosomal pathway but that the fusion mechanisms of early- and late-entering viruses differ in their sensitivity to this inhibition.
What might the role of TSPAN9 in the endosomal compartment be? Tetraspanin family proteins are known to create tetraspanin-enriched microdomains consisting of oligomers of tetraspanins and their cis-interacting partner proteins (14). Such domains are thought to play roles in cell adhesion, signaling, membrane compartmentalization of proteins, and cell-cell fusion, among other roles. Tetraspanin proteins have been implicated in sperm-egg fusion, and the depletion of the tetraspanin CD9 decreased the ability of sperm and egg cells to fuse (45–47). The exact function of CD9 in sperm-egg fusion is not clear, but it was suggested that its ability to recruit partner proteins promotes a state of “enhanced fusion competency” (14). It is possible that TSPAN9 interacts with important microdomain partners in the early endosome and that its depletion causes the loss of these interactors, thus either directly affecting fusion or more indirectly producing an unfavorable change in the endosome lipid composition. It is also possible that TSPAN9 normally acts to sequester a fusion inhibitor from the endosome and, thus, its loss leads to the endosomal delivery of the inhibitor.
Tetraspanin proteins are also associated with the entry of other viruses. For example, CD81 acts as one of the coreceptors for hepatitis C virus infection (48) and is also involved in the fusion of influenza virus in endosomes (49). Similar to our findings with TSPAN9 and alphavirus fusion, depletion of CD81 inhibited the fusion of influenza virus with endosomes, as measured by a lipid-mixing assay. The mechanism of this inhibition is not yet understood, but in contrast to alphaviruses, influenza virus fuses later in the endosome pathway. Together, the limited data suggest that tetraspanin proteins can play important although as-yet-incompletely understood roles in the endosome and can affect the entry and fusion of a variety of viruses. Our results demonstrate that TSPAN9 is an endosomal tetraspanin whose depletion has a pronounced effect on the entry of several viruses that fuse in the early endosome. Future studies will provide more information on the functional roles of the relatively poorly understood tetraspanin protein family during virus infection.
ACKNOWLEDGMENTS
We thank Ana Maria Cuervo for reagents and advice on LAMP1 and LAMP2, Sean Whelan and Kartik Chandran for recombinant VSVs, Youqing Xiang for technical assistance, and the Kielian laboratory and Kartik Chandran and his laboratory for helpful discussions.
This work was supported by a grant to M.K. from the National Institute of General Medicine (R01 GM057454) and by Cancer Center Core Support grant NIH/NCI P30 CA13330.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medicine or the National Institutes of Health.
REFERENCES
- 1.Kuhn RJ. 2013. Togaviridae, p 629–650. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed, vol 1 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Schwartz O, Albert ML. 2010. Biology and pathogenesis of chikungunya virus. Nat Rev Microbiol 8:491–500. doi: 10.1038/nrmicro2368. [DOI] [PubMed] [Google Scholar]
- 3.Enserink M. 2007. Infectious diseases. Chikungunya: no longer a third world disease. Science 318:1860–1861. doi: 10.1126/science.318.5858.1860. [DOI] [PubMed] [Google Scholar]
- 4.Morrison TE. 2014. Reemergence of chikungunya virus. J Virol 88:11644–11647. doi: 10.1128/JVI.01432-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johansson MA. 2015. Chikungunya on the move. Trends Parasitol 31:43–45. doi: 10.1016/j.pt.2014.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Enserink M. 2014. Crippling virus set to conquer Western Hemisphere. Science 344:678–679. doi: 10.1126/science.344.6185.678. [DOI] [PubMed] [Google Scholar]
- 7.Kielian M, Chanel-Vos C, Liao M. 2010. Alphavirus entry and membrane fusion. Viruses 2:796–825. doi: 10.3390/v2040796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gibbons DL, Vaney M-C, Roussel A, Vigouroux A, Reilly B, Lepault J, Kielian M, Rey FA. 2004. Conformational change and protein-protein interactions of the fusion protein of Semliki Forest virus. Nature 427:320–325. doi: 10.1038/nature02239. [DOI] [PubMed] [Google Scholar]
- 9.Ooi YS, Stiles KM, Liu CY, Taylor GM, Kielian M. 2013. Genome-wide RNAi screen identifies novel host proteins required for alphavirus entry. PLoS Pathog 9:e1003835. doi: 10.1371/journal.ppat.1003835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Serru V, Dessen P, Boucheix C, Rubinstein E. 2000. Sequence and expression of seven new tetraspans. Biochim Biophys Acta 1478:159–163. doi: 10.1016/S0167-4838(00)00022-4. [DOI] [PubMed] [Google Scholar]
- 11.Sieczkarski SB, Whittaker GR. 2003. Differential requirements of Rab5 and Rab7 for endocytosis of influenza and other enveloped viruses. Traffic 4:333–343. doi: 10.1034/j.1600-0854.2003.00090.x. [DOI] [PubMed] [Google Scholar]
- 12.Mire CE, White JM, Whitt MA. 2010. A spatio-temporal analysis of matrix protein and nucleocapsid trafficking during vesicular stomatitis virus uncoating. PLoS Pathog 6:e1000994. doi: 10.1371/journal.ppat.1000994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johannsdottir HK, Mancini R, Kartenbeck J, Amato L, Helenius A. 2009. Host cell factors and functions involved in vesicular stomatitis virus entry. J Virol 83:440–453. doi: 10.1128/JVI.01864-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hemler ME. 2005. Tetraspanin functions and associated microdomains. Nat Rev Mol Cell Biol 6:801–811. doi: 10.1038/nrm1736. [DOI] [PubMed] [Google Scholar]
- 15.Charrin S, Jouannet S, Boucheix C, Rubinstein E. 2014. Tetraspanins at a glance. J Cell Sci 127:3641–3648. doi: 10.1242/jcs.154906. [DOI] [PubMed] [Google Scholar]
- 16.Thomas JM, Klimstra WB, Ryman KD, Heidner HW. 2003. Sindbis virus vectors designed to express a foreign protein as a cleavable component of the viral structural polyprotein. J Virol 77:5598–5606. doi: 10.1128/JVI.77.10.5598-5606.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Glomb-Reinmund S, Kielian M. 1998. fus-1, a pH-shift mutant of Semliki Forest virus, acts by altering spike subunit interactions via a mutation in the E2 subunit. J Virol 72:4281–4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liljeström P, Lusa S, Huylebroeck D, Garoff H. 1991. In vitro mutagenesis of a full-length cDNA clone of Semliki Forest virus: the small 6,000-molecular-weight membrane protein modulates virus release. J Virol 65:4107–4113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang X, Kielian M. 2005. An interaction site of the envelope proteins of Semliki Forest virus that is preserved after proteolytic activation. Virology 337:344–352. doi: 10.1016/j.virol.2005.04.021. [DOI] [PubMed] [Google Scholar]
- 20.Whelan SP, Ball LA, Barr JN, Wertz GT. 1995. Efficient recovery of infectious vesicular stomatitis virus entirely from cDNA clones. Proc Natl Acad Sci U S A 92:8388–8392. doi: 10.1073/pnas.92.18.8388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chandran K, Sullivan NJ, Felbor U, Whelan SP, Cunningham JM. 2005. Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection. Science 308:1643–1645. doi: 10.1126/science.1110656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jae LT, Raaben M, Herbert AS, Kuehne AI, Wirchnianski AS, Soh TK, Stubbs SH, Janssen H, Damme M, Saftig P, Whelan SP, Dye JM, Brummelkamp TR. 2014. Virus entry. Lassa virus entry requires a trigger-induced receptor switch. Science 344:1506–1510. doi: 10.1126/science.1252480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kielian M, Jungerwirth S, Sayad KU, DeCandido S. 1990. Biosynthesis, maturation, and acid-activation of the Semliki Forest virus fusion protein. J Virol 64:4614–4624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wild T, Horvath P, Wyler E, Widmann B, Badertscher L, Zemp I, Kozak K, Csucs G, Lund E, Kutay U. 2010. A protein inventory of human ribosome biogenesis reveals an essential function of exportin 5 in 60S subunit export. PLoS Biol 8:e1000522. doi: 10.1371/journal.pbio.1000522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Madan R, Rastogi R, Parashuraman S, Mukhopadhyay A. 2012. Salmonella acquires lysosome-associated membrane protein 1 (LAMP1) on phagosomes from Golgi via SipC protein-mediated recruitment of host Syntaxin6. J Biol Chem 287:5574–5587. doi: 10.1074/jbc.M111.286120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tham TN, Gouin E, Rubinstein E, Boucheix C, Cossart P, Pizarro-Cerda J. 2010. Tetraspanin CD81 is required for Listeria monocytogenes invasion. Infect Immun 78:204–209. doi: 10.1128/IAI.00661-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shemiakina II, Ermakova GV, Cranfill PJ, Baird MA, Evans RA, Souslova EA, Staroverov DB, Gorokhovatsky AY, Putintseva EV, Gorodnicheva TV, Chepurnykh TV, Strukova L, Lukyanov S, Zaraisky AG, Davidson MW, Chudakov DM, Shcherbo D. 2012. A monomeric red fluorescent protein with low cytotoxicity. Nat Commun 3:1204. doi: 10.1038/ncomms2208. [DOI] [PubMed] [Google Scholar]
- 28.Kamentsky L, Jones TR, Fraser A, Bray MA, Logan DJ, Madden KL, Ljosa V, Rueden C, Eliceiri KW, Carpenter AE. 2011. Improved structure, function and compatibility for CellProfiler: modular high-throughput image analysis software. Bioinformatics 27:1179–1180. doi: 10.1093/bioinformatics/btr095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schmid SL, Fuchs R, Kielian M, Helenius A, Mellman I. 1989. Acidification of endosome subpopulations in wild-type Chinese hamster ovary cells and temperature-sensitive acidification-defective mutants. J Cell Biol 108:1291–1300. doi: 10.1083/jcb.108.4.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kielian MC, Marsh M, Helenius A. 1986. Kinetics of endosome acidification detected by mutant and wild-type Semliki Forest virus. EMBO J 5:3103–3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ghosh P, Dahms NM, Kornfeld S. 2003. Mannose 6-phosphate receptors: new twists in the tale. Nat Rev Mol Cell Biol 4:202–212. doi: 10.1038/nrm1050. [DOI] [PubMed] [Google Scholar]
- 32.Phalen T, Kielian M. 1991. Cholesterol is required for infection by Semliki Forest virus. J Cell Biol 112:615–623. doi: 10.1083/jcb.112.4.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Charrin S, Manie S, Thiele C, Billard M, Gerlier D, Boucheix C, Rubinstein E. 2003. A physical and functional link between cholesterol and tetraspanins. Eur J Immunol 33:2479–2489. doi: 10.1002/eji.200323884. [DOI] [PubMed] [Google Scholar]
- 34.van der Schaar HM, Rust MJ, Chen C, van der Ende-Metselaar H, Wilschut J, Zhuang X, Smit JM. 2008. Dissecting the cell entry pathway of dengue virus by single-particle tracking in living cells. PLoS Pathog 4:e1000244. doi: 10.1371/journal.ppat.1000244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zaitseva E, Yang ST, Melikov K, Pourmal S, Chernomordik LV. 2010. Dengue virus ensures its fusion in late endosomes using compartment-specific lipids. PLoS Pathog 6:e1001131. doi: 10.1371/journal.ppat.1001131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martinez MG, Forlenza MB, Candurra NA. 2009. Involvement of cellular proteins in Junin arenavirus entry. Biotechnol J 4:866–870. doi: 10.1002/biot.200800357. [DOI] [PubMed] [Google Scholar]
- 37.Lozach PY, Huotari J, Helenius A. 2011. Late-penetrating viruses. Curr Opin Virol 1:35–43. doi: 10.1016/j.coviro.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 38.Protty MB, Watkins NA, Colombo D, Thomas SG, Heath VL, Herbert JM, Bicknell R, Senis YA, Ashman LK, Berditchevski F, Ouwehand WH, Watson SP, Tomlinson MG. 2009. Identification of Tspan9 as a novel platelet tetraspanin and the collagen receptor GPVI as a component of tetraspanin microdomains. Biochem J 417:391–400. doi: 10.1042/BJ20081126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smit JM, Bittman R, Wilschut J. 1999. Low-pH-dependent fusion of Sindbis virus with receptor-free cholesterol- and sphingolipid-containing liposomes. J Virol 73:8476–8484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.White J, Helenius A. 1980. pH-dependent fusion between the Semliki Forest virus membrane and liposomes. Proc Natl Acad Sci U S A 77:3273–3277. doi: 10.1073/pnas.77.6.3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Andrejewski N, Punnonen EL, Guhde G, Tanaka Y, Lullmann-Rauch R, Hartmann D, von Figura K, Saftig P. 1999. Normal lysosomal morphology and function in LAMP-1-deficient mice. J Biol Chem 274:12692–12701. doi: 10.1074/jbc.274.18.12692. [DOI] [PubMed] [Google Scholar]
- 42.Eskelinen EL. 2006. Roles of LAMP-1 and LAMP-2 in lysosome biogenesis and autophagy. Mol Aspects Med 27:495–502. doi: 10.1016/j.mam.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 43.Cuervo AM, Dice JF. 1996. A receptor for the selective uptake and degradation of proteins by lysosomes. Science 273:501–503. doi: 10.1126/science.273.5274.501. [DOI] [PubMed] [Google Scholar]
- 44.Pols MS, Klumperman J. 2009. Trafficking and function of the tetraspanin CD63. Exp Cell Res 315:1584–1592. doi: 10.1016/j.yexcr.2008.09.020. [DOI] [PubMed] [Google Scholar]
- 45.Kaji K, Oda S, Shikano T, Ohnuki T, Uematsu Y, Sakagami J, Tada N, Miyazaki S, Kudo A. 2000. The gamete fusion process is defective in eggs of Cd9-deficient mice. Nat Genet 24:279–282. doi: 10.1038/73502. [DOI] [PubMed] [Google Scholar]
- 46.Le Naour F, Rubinstein E, Jasmin C, Prenant M, Boucheix C. 2000. Severely reduced female fertility in CD9-deficient mice. Science 287:319–321. doi: 10.1126/science.287.5451.319. [DOI] [PubMed] [Google Scholar]
- 47.Miyado K, Yamada G, Yamada S, Hasuwa H, Nakamura Y, Ryu F, Suzuki K, Kosai K, Inoue K, Ogura A, Okabe M, Mekada E. 2000. Requirement of CD9 on the egg plasma membrane for fertilization. Science 287:321–324. doi: 10.1126/science.287.5451.321. [DOI] [PubMed] [Google Scholar]
- 48.Pileri P, Uematsu Y, Campagnoli S, Galli G, Falugi F, Petracca R, Weiner AJ, Houghton M, Rosa D, Grandi G, Abrignani S. 1998. Binding of hepatitis C virus to CD81. Science 282:938–941. doi: 10.1126/science.282.5390.938. [DOI] [PubMed] [Google Scholar]
- 49.He J, Sun E, Bujny MV, Kim D, Davidson MW, Zhuang X. 2013. Dual function of CD81 in influenza virus uncoating and budding. PLoS Pathog 9:e1003701. doi: 10.1371/journal.ppat.1003701. [DOI] [PMC free article] [PubMed] [Google Scholar]