

Abstract
Determining how viruses infect new hosts via receptor-binding mechanisms is important for understanding virus emergence. We studied the binding kinetics of canine parvovirus (CPV) variants isolated from raccoons—a newly recognized CPV host—to different carnivore transferrin receptors (TfRs) using single-particle tracking. Our data suggest that CPV may utilize adhesion-strengthening mechanisms during TfR binding and that a single mutation in the viral capsid at VP2 position 300 can profoundly alter receptor binding and infectivity.
TEXT
Canine parvovirus (CPV) is a pathogen of dogs that emerged and caused a pandemic of disease in the 1970s and is >99% identical in nucleotide sequence to feline panleukopenia virus (FPV), a parvovirus that infects cats and other carnivore hosts but not dogs (1–3). Although the emergence of CPV has been presumed to be the result of a direct transfer of FPV or a similar virus from domestic cats to dogs, we recently demonstrated that CPV exists endemically in sylvatic cycles in North America involving a number of wild carnivore hosts, most notably raccoons (4, 5). These recent findings, along with the lack of isolation or detection of intermediate viruses between FPV and CPV from domestic animals, suggest that parvoviruses transfer frequently between domestic and wild carnivores and that the events preceding the pandemic emergence of CPV were more complex than previously believed (6).
Although raccoons have long been known to be susceptible to FPV infection (7), they have only recently been identified as an important host for viruses that are closely related to CPV (4, 8). While CPVs from dogs, wolves, and coyotes all contain a Gly at capsid (VP2) position 300, CPVs from raccoons contain an Asp at that position, suggesting that this mutation is important for the adaptation of CPV to raccoons and possibly other wild carnivore hosts (4, 6). Additionally, VP2 position 300 is the most variable residue in the capsid (9–11). Since FPV and CPV capsids can bind to the transferrin receptor type 1 (TfR), in part by involving the structural region surrounding VP2 position 300 (12), the variations observed at this position appear to be selected by the unique TfR structures of individual carnivore hosts. To examine this phenomenon and to better understand the receptor-binding mechanisms involved, we used single-particle tracking (SPT) techniques to characterize the binding of raccoon-derived CPVs, containing either a 300-Asp or 300-Gly VP2 residue, to dog and raccoon TfRs.
The virus studied here was the prototype CPV isolated from raccoons (CPV/Raccoon/VA/118-A/07, GenBank sequence accession number JN867610), which contains an Asp at VP2 position 300 and cannot be propagated in dog cells (4, 6). We refer to this virus as Rac118-300D. However, a single point mutation of the VP2 300-Asp (codon GAT) to a Gly (codon GGT) results in efficient dog cell infection, demonstrating that this capsid residue is important for determining host range (6). The virus that contains a Gly at VP2 position 300 is referred to as Rac118-300G. For the SPT studies, both the Rac118-300G and -300D viruses were propagated and purified using methods described previously (13). Infectious virions were extracted from a sucrose gradient, dialyzed in phosphate-buffered saline (PBS), and concentrated using a 100-kDa Amicon Ultra-15 centrifugal filter unit (EMD Millipore). The viral genomes were sequenced to confirm that no additional VP1/VP2 capsid mutations had occurred during propagation. The purified particles were labeled with Alexa Fluor 594 succinimidyl ester (Invitrogen) according to the manufacturer's instructions, and unbound Alexa dye was removed using Sephadex G-25 PD-10 columns (GE Healthcare Life Sciences). The Alexa dye labeling does not appear to affect the ability of the virus to bind and enter cells, based on previous studies (13), and the Asp and Gly residues do not contain free reactive amine groups for dye conjugation.
The raccoon and dog TfR ectodomains were expressed as previously described (13, 14). Briefly, the cDNA sequence of each TfR ectodomain was cloned into a pFastBac construct containing an N-terminal hexahistidine (6×His) tag and an enhanced green fluorescent protein (eGFP) fused to its C terminus. The pFastBac TfR-eGFP constructs were expressed in High Five cells (Life Technologies) using the Bac-to-Bac baculovirus expression system (Invitrogen). TfRs were purified by binding to nickel-nitrilotriacetic acid (Ni-NTA) Superflow resin (Qiagen) and eluted with 100 mM imidazole using an ÄKTA fast-performance liquid chromatography system (GE Healthcare Life Sciences). TfRs were dialyzed overnight in PBS and concentrated using a 10-kDa Amicon Ultra-15 centrifugal filter unit (EMD Millipore). Purified TfR-eGFP was loaded on top of a supported lipid bilayer (SLB), formed inside a microfluidic device as described elsewhere (15, 16). A detailed schematic view of the SPT experimental setup is shown in Fig. 1. Lipid vesicles that are needed to form SLBs were produced by mixing 1 mol% 1,2-dioleoyl-sn-glycero-3-[(N-(5-amino-1-carboxypentyl)iminodiacetic acid)succinyl] nickel salt (DGS-NTA) and 99 mol% 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) (Avanti Polar Lipids) in chloroform, drying them in vacuum, and rehydrating them in MES buffer [1 mM 2-(N-morpholino)ethanesulfonic acid, 150 mM NaCl, pH 7.00] such that the final vesicle concentration was 1 mg/ml. Lipids were extruded using 50-nm-pore filters (Whatman) to create ∼100-nm-diameter vesicles. The vesicles were loaded into the microfluidic channel to spontaneously form supported lipid bilayers (SLBs) via the vesicle-rupture method (17). After 4 h, excess vesicles were rinsed away with MES buffer. Subsequent SPT experiments were performed at 25°C.
FIG 1.
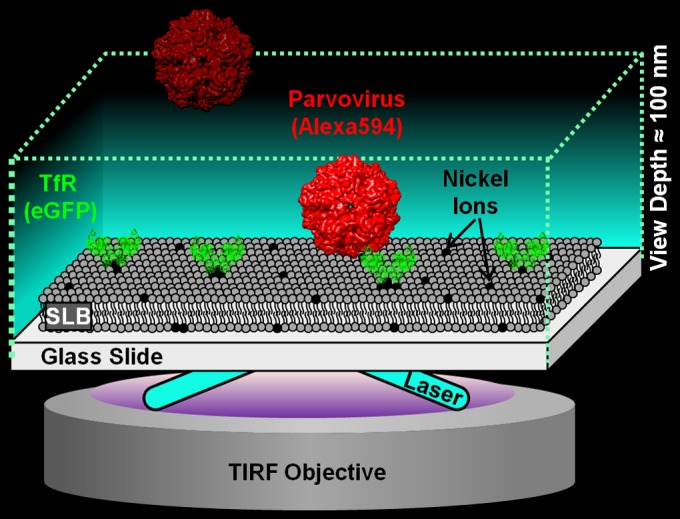
Schematic of the single-particle tracking (SPT) binding assay. The device was set up on a 100×, 1.46-numerical-aperture, oil-immersion objective in a total internal reflection fluorescence (TIRF) microscope (Carl Zeiss; Model Axio Observer Z1). The TIRF microscope used here produces an evanescent wave that illuminates only a 100-nm-deep region away from the glass surface, which is where the virus-TfR binding interaction occurs, thus ignoring virus particles floating in the bulk solution. The eGFP-labeled TfRs are tethered to the supported lipid bilayer (SLB) via nickel-His interactions. The Alexa 594-labeled virus is detected with a 561-nm laser, whereas the eGFP-labeled TfR is detected using a 488-nm laser. The CPV structure is from VIPERdB based on PDB identifier 1P5W. The TfR structure is from PDB identifier 1CX8. Note that the CPV and TfR structures are used only for illustrative purposes and deviate from the virus capsid and receptor structures used in this work.
The relative concentrations of Rac118-300D and -300G were determined by making them nonspecifically and directly adsorb onto a glass surface for 30 min. Images of the glass-adsorbed viruses were taken when the virus density, Vglass, remained fairly constant (Fig. 2A). In a separate microfluidic channel containing only the SLBs, viruses were loaded to check for nonspecific binding levels, which were negligible (Fig. 2B). Unbound viruses were rinsed away, and His-tagged dog or raccoon TfRs were loaded into the channels to bind to the DGS-NTA lipids containing nickel ions. Unbound TfRs were rinsed away, and images were taken to determine the TfR location and density, RTfR (Fig. 2C). Without changing the field of view, either Rac118-300D or -300G virus was loaded onto the SLB. As soon as the flow stopped, images were acquired at 500-ms intervals for 10 min (Fig. 2D). Using a custom image processing software made in MATLAB (MathWorks), the binding on and off times of the viruses were tracked throughout the movie.
FIG 2.

Images of the TfR-parvovirus binding experiments at different stages. (A) Images of the Alexa 594-labeled virus adsorbing to glass after 30 min of incubation. White numbers are the particle counts, Pcount. In this panel, Pcount is equal to Vglass, which is used to quantify the relative virus concentrations between virus batches. Pcount is shown instead of N or N+ because Pcount is insensitive to the binding event criteria. (B) Images of SLBs incubated with virus after a 10-min waiting period. Negligible binding events here indicate that the viruses do not readily bind to the SLBs and that the SLBs have no defects exposing the glass surface. (C) Images of the eGFP-labeled TfR attached to the SLBs, prior to loading the virus. Yellow numbers are the TfR particle counts, RTfR. (D) Images of parvovirus binding to TfR-loaded SLBs after 10 min. Image details: each image portrays an 82- by 82-μm2 physical space in 512 by 512 pixels. Uneven backgrounds were subtracted out, and all image intensities were linearly scaled, which preserves particle features.
Since binding events are extracted via visual cues from the video, we next filtered out ambiguous binding events, such as floating virions or false particles (bright pixels from shot noise that resemble particles), by setting several criteria. Based on a manual inspection of final particle tracking results, a majority of ambiguous binding events lasted at most 5 frames; hence, to remove these from the analysis, we imposed a criterion that binding events must last longer than the cutoff time of 5 frames (or 2.5 s). The cutoff time is much shorter than the lifetime of a clathrin-coated pit (∼2 min) that the virus is endocytosed into after receptor binding (13); thus, binding events that are relevant for infection are still captured. Note that changing the cutoff time or camera rate will ultimately affect the number of binding events observed and binding data, and therefore, it was important that we kept those settings the same across all trials. Additionally, only immobile particles that were colocalized with a TfR particle were tracked because the TfRs were also immobile (likely due to multiple His residues binding to DGS-NTA lipids), and immobility served as a strong visual cue for authentic virus-TfR contacts. In the cell membrane, however, the TfR is not static, and this mobility could affect viral binding avidity via the formation of multiple TfR contacts (13). The immobile TfRs in our SPT setup enable us to directly study 1:1 binding that would otherwise be challenging to study with cell membranes, which helps in understanding how relevant binding avidity is for infection.
Before describing the kinetic data, we introduce several variables: (i) N is the number of binding events, (ii) N+ (or N−) is the accumulated number of binding (or unbinding) events since the movie started, and (iii) Pcount is the number of particles in an image. Note that Pcount is not the same as N because some particles do not last long enough to meet the criteria for a binding event; hence, N is ≤Pcount. We also have two time variables: (i) t for time and (ii) tres for binding residence time of virus to receptors. Lastly, both N and N+ were normalized by dividing them by virus density (Vglass) and TfR density (RTfR), which enables a fair comparison of kinetic data across the different samples. Instead of using Vglass as a normalization factor, using the actual concentration of visible virus in the bulk solution, Vbulk, would also suffice. However, Vbulk is difficult to obtain directly. A calibration curve between Vglass and dilutions of Vbulk shows a linear relation (Fig. 3A); hence, Vbulk can be replaced by Vglass for normalization purposes.
FIG 3.
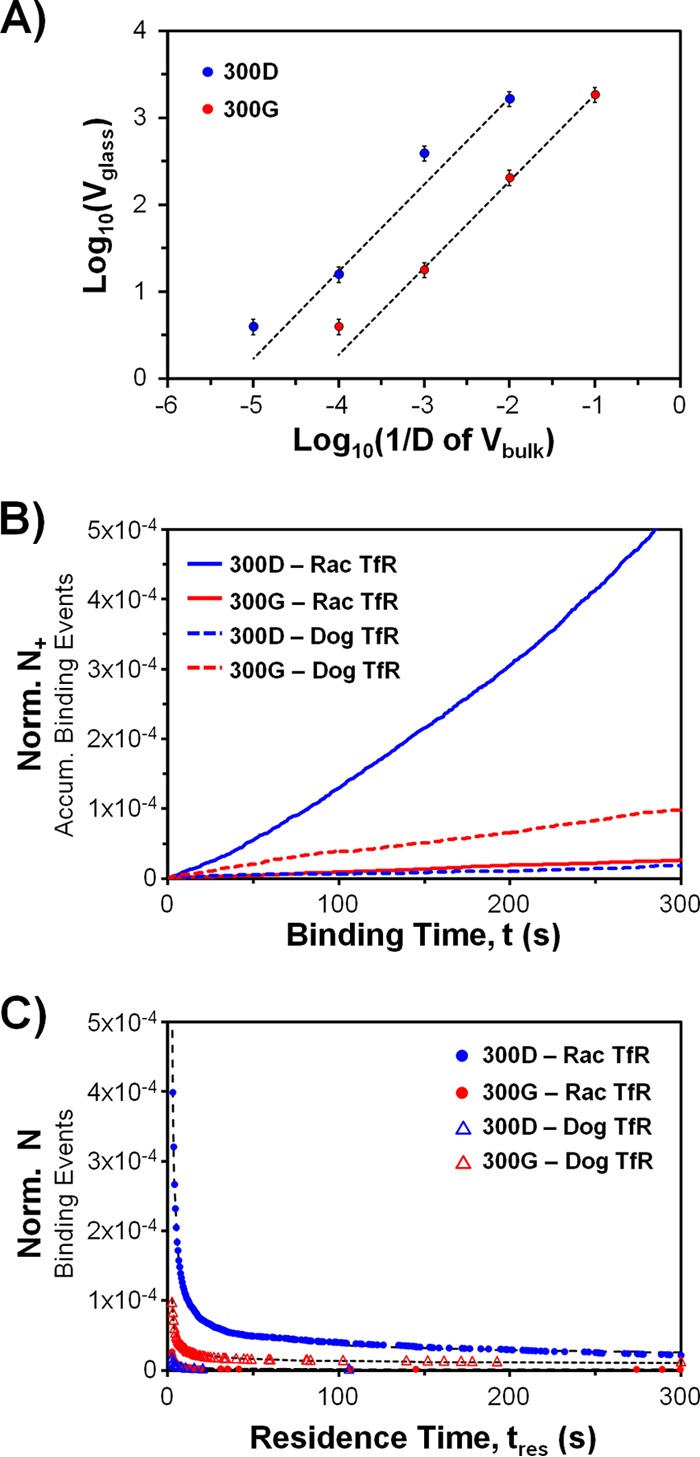
Kinetics of virus binding to raccoon or domestic dog TfR-loaded SLBs. (A) Calibration curve to show the linear relationship between Vglass and Vbulk. We measured Vglass at several dilution factors, D, of Vbulk for the Rac118-300D and -300G stock viruses. The dotted fit lines (y = mx) have R2 values greater than 0.98. The m parameters for 300D and 300G virus are 169,250 and 18,660, respectively. (B) Normalized binding rate data. Ron,norm is calculated using the linear fit of the data (Norm N+ = Ron,normt). Ron,norm values, in the order of the symbol key, are (1.6 ± 0.2) × 10−6 s−1, (9.1 ± 1.3) × 10−8 s−1, (5.9 ± 0.9) × 10−8 s−1, and (3.4 ± 0.5) × 10−7 s−1. The R2 values for linear fits are >0.98 for all cases. The error ranges were calculated via propagation of worst-case 10% errors in each Vglass and RTfR due to particle detection software limitations. The true error is expected to be less, since errors are corrected manually. (C) Normalized binding residence time distributions. The plot shows how many binding events, N, last longer than a certain residence time, tres. The data are fitted according to equation 2 (dotted lines) but in normalized form. The fit parameters (with 95% confidence intervals) and the number of binding events included in each distribution (Nbe), in the order of the symbol key, are A = 1.66 ± 0.01, B = 7.52 ± 0.01, Nbe = 2151; A = 1.91 ± 0.07, B = 5.20 ± 0.03, Nbe = 203; A = 1.57 ± 0.19, B = 3.39 ± 0.07, Nbe = 36; and A = 1.27 ± 0.03, B = 5.77 ± 0.02, Nbe = 345. The R2 values for equation 2 fits are >0.97 for all cases.
The binding frequency rate data are shown in Fig. 3B, with the normalized N+ on the y axis and the time (t) on the x axis. The slope of the plots is referred to as normalized binding frequency rate, Ron,norm (equation 1). Rac118-300D binds ∼18 times more frequently than does Rac118-300G to the raccoon TfR. Conversely, Rac118-300G binds ∼5.7 times more frequently than does Rac118-300D to the dog TfR. However, the binding rate of Rac118-300G with the dog TfR is ∼80% lower than that of Rac118-300D with the raccoon TfR. This is not an unexpected result, as previous studies have shown that a number of different CPV variants bind to very low levels to the dog TfR relative to the TfRs of other carnivore hosts (4, 14), possibly owing to the unique protein structure and/or glycosylation profile of the dog TfR (11).
| (1) |
We next analyzed the binding residence time distribution, which reflects the binding strength of the parvovirus capsid to the TfR. Biased binding events were removed using a criterion described elsewhere (18). Briefly, binding events that started since the first frame of the movie and those that started after halfway through the movie were discarded due to the inability to determine actual binding times and oversampling of short binding events, respectively. Hence, tres can be plotted only up to half the movie time. The residence time distribution was plotted as N versus tres (Fig. 3C), which shows how many binding events last longer than a certain tres time. The unbinding curves do not fit the standard 1:1 binding model, suggesting that a more complex interaction is involved. A different empirical fit equation (equation 2) was determined by testing various log-log plots. Relating equation 2 to the standard dissociation equation dN/dtres = −koffN yields equation 3 for koff, which is not a constant and varies with tres.
| (2) |
where tres ≥ binding event cutoff time (2.5 s).
| (3) |
where tres ≥ binding event cutoff time (2.5 s).
The decreasing value of koff with respect to tres (equation 3) would imply a mechanism for the virus to increase its binding strength to the receptor with longer contact times, a phenomenon generally referred to as adhesion strengthening (19–21). Adhesion strengthening can occur via either multivalent binding, coreceptor binding, or conformation changes occurring to the viral protein upon ligand binding. Multivalent binding is unlikely to occur in our SPT setup because (i) TfRs are spaced greater than 1 camera pixel (160 nm) apart, whereas the virus is only 26 nm in diameter; (ii) the TfRs are immobile and do not diffuse laterally to create multiple bonds; (iii) the planar SLB geometry restricts binding of TfR to one side of the virus; and (iv) TfRs may sterically hinder each other's access to the same virus. Whether the multivalent binding that may occur in vivo is a prerequisite for infection is unknown; rather, it may be a by-product of mobile TfRs clustering around a virus that is already strongly bound to a single TfR. However, CPV does not seem to undergo significant multivalent binding, as it appears that the engagement of the virus with TfR may potentially prevent additional binding events from occurring, although the mechanism involved is unknown (12).
A more plausible explanation for the increasing binding strength over time is that there is a complex mechanism of binding of parvovirus to the TfR (12), such that upon receptor binding, the virus capsid and/or TfR changes conformation that results in tighter binding. Our mathematical fit model and raw data agree with, but do not definitively confirm, adhesion strengthening via conformation changes that may occur when a virus binds to single TfR. Additional work is needed to confirm and potentially identify the changes to the parvovirus or TfR structure that occur as a direct result of receptor binding.
To determine if the SPT results agree with cell infection data, relative infectivity studies using raccoon uterine (Pl1Ut; ATCC CCL-74) and domestic dog (A72; ATCC CRL-1542) tumor cells were performed using procedures similar to those described elsewhere (6, 11). Briefly, cells were seeded at a density of ∼1 × 105 cells/ml in a 1.9-cm2-well format and inoculated with a multiplicity of infection of 0.4 50% tissue culture infective dose (TCID50) per cell of either Rac118-300D or -300G. At 72 h postinfection, cells were fixed in 10% formalin, triple washed with PBS, and incubated with a polyclonal rabbit anti-CPV VP1/VP2 antibody in permeabilization buffer (1× PBS, 0.5% bovine serum albumin, 0.5% Triton X-100) for 1 h. The wash step was repeated, and cells were incubated with a secondary Alexa Fluor 488-conjugated goat anti-rabbit IgG (heavy plus light chains [H+L]) antibody (Life Technologies) for 1 h, followed by a final wash in PBS. Immunofluorescence was detected using a Nikon Eclipse TE300 inverted fluorescence microscope equipped with a Hamamatsu Orca ER digital camera (Nikon Corporation). As shown in Fig. 4, there was a substantial reduction in the number of infected raccoon cells with Rac118-300G compared to Rac118-300D, whereas the reverse trend was observed with dog cells, as there was a >95% decrease in the number of infected cells with Rac118-300D in comparison to Rac118-300G. Thus, both the SPT and infectivity data confirm that although the Asp-to-Gly change at VP2 position 300 enabled the prototype raccoon-derived CPV to bind to the dog TfR and to also efficiently infect dog cells when that virus previously could not, the process of dog adaptation also reduced the virus' ability to bind to the raccoon TfR and to infect raccoon cells. These results not only further demonstrate the importance of this single capsid residue in host switching (11) but show that in this particular case, viral adaptation to a new host (dog) simultaneously led to the loss in the ability to efficiently infect the previous host of isolation (raccoon). Additionally, the unbinding kinetic data suggest that these parvoviruses may increase their binding strength during prolonged contact with their receptors, which may alter infectivity, and preliminary studies using biolayer interferometry approaches have also shown similar dissociation kinetics (unpublished data).
FIG 4.

Relative susceptibility of domestic dog and raccoon cells to infection by Rac118-300D and -300G. (A) Dog (A72) and raccoon (Pl1Ut) cells were infected with equivalent amounts of Rac118-300D or -300G and fixed, stained, and analyzed by immunofluorescence at 72 h postinfection using a polyclonal rabbit anti-CPV VP1/VP2 antibody and a goat anti-rabbit Alexa Fluor 488 secondary antibody. (B) For relative infectivity estimates, Alexa Fluor 488-positive cells were visually counted and the number of infected cells seen with the normal VP2 position 300 mutation associated with each host (i.e., Asp in raccoons, Gly in dogs) was set to 1.0. Error bars indicate standard deviations for duplicate experiments.
As the platform of the SPT binding assay illustrated here allows for the examination of a variety of TfR-parvovirus combinations along with their complex receptor-binding behaviors, future work will be aimed at applying this binding model to examine the virus unbinding kinetic data obtained from other assays and determining how single or multiple capsid or receptor mutations affect binding to the TfRs from other carnivore hosts. Such studies will give a better understanding of how viruses successfully cross species barriers to infect novel hosts.
Funding Statement
This work was performed in part at the Cornell NanoScale Facility, a member of the National Nanotechnology Infrastructure Network supported by the National Science Foundation (NSF) (grant ECCS-0335765). Research was funded by NSF grant CBET-1263701 to S.D. and National Institutes of Health (NIH) grants RO1 GM8496821 and RO1 AI092571 to C.R.P. A.B.A. is supported by a NRSA postdoctoral fellowship from the National Institute of Allergy and Infectious Diseases, NIH (F32AI100545). D.W.L. acknowledges financial support from the NSF GK12 programs, award number DGE-1045513 to Cornell's Grass Roots program and award number DGE-0841291 to Cornell's Learning Initiative for Medicine and Bioengineering (CLIMB) program. K.B.B. acknowledges funding from Cornell Engineering Learning Initiative (ELI), with donations from Genentech and Proctor & Gamble.
REFERENCES
- 1.Parrish CR, O'Connell PH, Evermann JF, Carmichael LE. 1985. Natural variation of canine parvovirus. Science 230:1046–1048. doi: 10.1126/science.4059921. [DOI] [PubMed] [Google Scholar]
- 2.Parrish CR, Have P, Foreyt WJ, Evermann JF, Senda M, Carmichael LE. 1988. The global spread and replacement of canine parvovirus strains. J Gen Virol 69:1111–1116. doi: 10.1099/0022-1317-69-5-1111. [DOI] [PubMed] [Google Scholar]
- 3.Chang S, Sgro J, Parrish C. 1992. Multiple amino acids in the capsid structure of canine parvovirus coordinately determine the canine host range and specific antigenic and hemagglutination properties. J Virol 66:6858–6867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Allison AB, Harbison CE, Pagan I, Stucker KM, Kaelber JT, Brown JD, Ruder MG, Keel MK, Dubovi EJ, Holmes EC. 2012. Role of multiple hosts in the cross-species transmission and emergence of a pandemic parvovirus. J Virol 86:865–872. doi: 10.1128/JVI.06187-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Allison AB, Kohler DJ, Fox KA, Brown JD, Gerhold RW, Shearn-Bochsler VI, Dubovi EJ, Parrish CR, Holmes EC. 2013. Frequent cross-species transmission of parvoviruses among diverse carnivore hosts. J Virol 87:2342–2347. doi: 10.1128/JVI.02428-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Allison AB, Kohler DJ, Ortega A, Hoover EA, Grove DM, Holmes EC, Parrish CR. 2014. Host-specific parvovirus evolution in nature is recapitulated by in vitro adaptation to different carnivore species. PLoS Pathog 10:e1004475. doi: 10.1371/journal.ppat.1004475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barker IK, Parrish CR. 2000. Parvovirus infections, p 131–146. In Williams ES, Barker IK (ed), Infectious diseases of wild mammals, 3rd ed Wiley-Blackwell, Hoboken, NJ. [Google Scholar]
- 8.Kapil S, Rezabek G, Germany B, Johnston L. 2010. Isolation of a virus related to canine parvovirus type 2 from a raccoon (Procyon lotor). Vet Rec 166:24–25. doi: 10.1136/vr.b5587. [DOI] [PubMed] [Google Scholar]
- 9.Qin Q, Loeffler IK, Li M, Tian K, Wei F. 2007. Sequence analysis of a canine parvovirus isolated from a red panda (Ailurus fulgens) in China. Virus Genes 34:299–302. doi: 10.1007/s11262-006-0023-6. [DOI] [PubMed] [Google Scholar]
- 10.Chen XY, Xie ZJ, Zhao ZP, Jiang SJ, Zhao HK, Zhu YL, Zhang XX. 2011. Genetic diversity of parvovirus isolates from dogs and wild animals in China. J Wildl Dis 47:1036–1039. doi: 10.7589/0090-3558-47.4.1036. [DOI] [PubMed] [Google Scholar]
- 11.Allison AB, Organtini L, Zhang S, Hafenstein SL, Holmes EC, Parrish CR. 2016. Single mutations in the VP2 300 loop region of the three-fold spike of the carnivore parvovirus capsid can determine host range. J Virol 90:753–767. doi: 10.1128/JVI.02636-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hafenstein S, Palermo LM, Kostyuchenko VA, Xiao C, Morais MC, Nelson CDS, Bowman VD, Battisti AJ, Chipman PR, Parrish CR, Rossmann MG. 2007. Asymmetric binding of transferrin receptor to parvovirus capsids. Proc Natl Acad Sci U S A 104:6585–6589. doi: 10.1073/pnas.0701574104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cureton DK, Harbison CE, Cocucci E, Parrish CR, Kirchhausen T. 2012. Limited transferrin receptor clustering allows rapid diffusion of canine parvovirus into clathrin endocytic structures. J Virol 86:5330–5340. doi: 10.1128/JVI.07194-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Palermo LM, Hafenstein SL, Parrish CR. 2006. Purified feline and canine transferrin receptors reveal complex interactions with the capsids of canine and feline parvoviruses that correspond to their host ranges. J Virol 80:8482–8492. doi: 10.1128/JVI.00683-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee D, Thapar V, Clancy P, Daniel S. 2014. Stochastic fusion simulations and experiments suggest passive and active roles of hemagglutinin during membrane fusion. Biophys J 106:843–854. doi: 10.1016/j.bpj.2013.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Costello DA, Lee DW, Drewes J, Vasquez KA, Kisler K, Wiesner U, Pollack L, Whittaker GR, Daniel S. 2012. Influenza virus-membrane fusion triggered by proton uncaging for single particle studies of fusion kinetics. Anal Chem 84:8480–8489. doi: 10.1021/ac3006473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Castellana ET, Cremer PS. 2006. Solid supported lipid bilayers: from biophysical studies to sensor design. Surf Sci Rep 61:429–444. doi: 10.1016/j.surfrep.2006.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bally M, Gunnarsson A, Svensson L, Larson G, Zhdanov VP, Höök F. 2011. Interaction of single viruslike particles with vesicles containing glycosphingolipids. Phys Rev Lett 107:188103. doi: 10.1103/PhysRevLett.107.188103. [DOI] [PubMed] [Google Scholar]
- 19.Haywood AM. 1994. Virus receptors: binding, adhesion strengthening, and changes in viral structure. J Virol 68:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barton ES, Connolly JL, Forrest JC, Chappell JD, Dermody TS. 2001. Utilization of sialic acid as a coreceptor enhances reovirus attachment by multistep adhesion strengthening. J Biol Chem 276:2200–2211. doi: 10.1074/jbc.M004680200. [DOI] [PubMed] [Google Scholar]
- 21.Stettner E, Dietrich MH, Reiss K, Dermody TS, Stehle T. 2015. Structure of serotype 1 reovirus attachment protein σ1 in complex with junctional adhesion molecule A reveals a conserved serotype-independent binding epitope. J Virol 89:6136–6140. doi: 10.1128/JVI.00433-15. [DOI] [PMC free article] [PubMed] [Google Scholar]