

Abstract
Air pollution is implicated in neurodegenerative disease risk and progression and in microglial activation, but the mechanisms are unknown. In this study, microglia remained activated 24 h after ozone (O3) exposure in rats, suggesting a persistent signal from lung to brain. Ex vivo analysis of serum from O3-treated rats revealed an augmented microglial proinflammatory response and β-amyloid 42 (Aβ42) neurotoxicity independent of traditional circulating cytokines, where macrophage-1 antigen-mediated microglia proinflammatory priming. Aged mice exhibited reduced pulmonary immune profiles and the most pronounced neuroinflammation and microglial activation in response to mixed vehicle emissions. Consistent with this premise, cluster of differentiation 36 (CD36)−/− mice exhibited impaired pulmonary immune responses concurrent with augmented neuroinflammation and microglial activation in response to O3. Further, aging glia were more sensitive to the proinflammatory effects of O3 serum. Together, these findings outline the lung–brain axis, where air pollutant exposures result in circulating, cytokine-independent signals present in serum that elevate the brain proinflammatory milieu, which is linked to the pulmonary response and is further augmented with age.—Mumaw, C. L., Levesque, S., McGraw, C., Robertson, S., Lucas, S., Stafflinger, J. E., Campen, M. J., Hall, P., Norenberg, J. P., Anderson, T., Lund, A. K., McDonald, J. D., Ottens, A. K., Block, M. L. Microglial priming through the lung–brain axis: the role of air pollution–induced circulating factors.
Keywords: neuroinflammation, inhaled pollutants, glia
Increasing evidence implicates air pollution exposures in central nervous system (CNS) diseases (1, 2), where living in conditions with elevated levels of urban air pollution is associated with decreased cognitive function in the elderly (3–5), increased autism risk (6–9), elevated Alzheimer’s disease (AD) risk (10), increased Parkinson’s disease (PD) risk (11), accelerated disease progression to the first hospital admission in neurodegenerative diseases (12), elevated AD-like neuropathology in humans (13–15), Parkinson’s disease–like neuropathology in humans (16, 17), and increased stroke incidence (18, 19). Although the detailed mechanisms are poorly understood, there is increasing support for the hypothesis that neuroinflammation and microglial activation is the common underlying mechanism through which air pollution exerts its effects in these diverse CNS conditions.
Outdoor air pollution is a complex chemical mixture derived from multiple sources, including engine emissions, coal combustion, biomass burning, and secondary photochemical products, such as ground level ozone (O3). In the United States alone, it is estimated that >88 million people are exposed to levels of air pollution above safety standards (20). Ground-level O3 is a widely prevalent air pollutant linked to deleterious health effects worldwide (21), including recent findings implicating O3 in increased AD (10) and PD (11) risk. O3 is formed when precursors, such as oxides of nitrogen or volatile organics from car emissions and other sources of pollutants, undergo photochemical reactions (22). Inhaled O3 is highly reactive and does not transfer to the bloodstream or the brain, as O3 rapidly reacts with protein and lipid components of the airway surfactant, to generate a mix of secondary and tertiary reactants implicated in the biologic effects of O3 (23, 24). How inhaled O3 affects the brain remains a major uncertainty. The biologic health effects of ground-level O3 are of significant concern, as levels in certain urban U.S. regions have remained high, despite efforts to reduce precursors, and O3 levels are expected to climb worldwide with the predicted climate change (25, 26).
Essential to CNS health, microglia serve many functions in the brain, as they are the resident innate immune cells, sentinels surveying the CNS environment (27), and “electricians” regulating synaptic communication (28). As sentinels, microglia respond to a vast repertoire of stimuli, including cellular damage, environmental toxins, and pathogens (29). Neuropathology and CNS disease are believed to occur when the microglial proinflammatory response is exacerbated and unregulated (29, 30), which is linked to microglial priming (31). Microglia have long been implicated in the neuronal damage that occurs in many CNS diseases and conditions, including AD (32), PD (29), and autism (33), supporting the potential of a common underlying neuroinflammatory mechanism. Although most microglial function is beneficial, little is known about how the microglia are reprogrammed to a heightened proinflammatory phenotype (priming) to become deleterious in CNS disease.
Recent reports have indicated that ambient air pollutants are also a common environmental source of chronic neuroinflammation and microglial activation. Consistent with human reports (13), animal studies have revealed that exposure to diverse forms of outdoor air pollution by inhalation, such as urban particulate matter (34, 35), O3 (36), diesel exhaust (37–39), and manganese (40, 41), results in elevated cytokines and oxidative stress in the brain. More specifically, several inhaled air pollutants are shown to activate microglia with direct effects in vitro (38, 42, 43), in animal models (38, 44), and in human (13) studies.
Given the high prevalence and chronic nature of human air pollution exposure, combined with the wide-reaching impact that urban air pollution may have on neurodegenerative disease, it is critical to understand the mechanisms through which inhaled pollutants affect the brain. In the current study, we began to explore the lung–brain axis and addressed whether circulating factors, independent of traditional cytokines, produced in response to inhaled pollutants, can regulate microglial function. More specifically, we tested the ability of inhaled O3 (which does not translocate beyond the lung to the brain parenchyma) to persistently activate microglia in vivo, explore the serum cytokine signature at the time of microglia activation, reveal the impact of O3-induced serum to initiate or prime the microglial proinflammatory response and associated neurotoxicity ex vivo, and began to explore the effect of aging on the lung–brain axis.
MATERIALS AND METHODS
Reagents
LPS (strain O111:B4) was purchased from EMD Chemicals (Gibbstown, NJ, USA). Cell culture reagents were obtained from Thermo Scientific–Invitrogen (Carlsbad, CA, USA). The polyclonal antibody against the ionized calcium-binding adapter molecule 1 (IBA1) microglial marker was purchased from Wako (Richmond, VA, USA). The biotinylated goat anti-rabbit secondary antibody was purchased from Vector Laboratories (Burlingame, CA, USA). The monoclonal macrophage-1 antigen (MAC1) inhibitor antibody was purchased from Lifespan Biosciences (Seattle, WA, USA), and the normal mouse isotype IgG control antibody was purchased from Medical and Biologic Research Laboratories (Woburn, MA, USA). ELISA kits were purchased from R&D Systems (Minneapolis, MN, USA) and Thermo Scientific–Invitrogen. Aβ1-42 was purchased from American Peptide Company (Sunnyvale, CA, USA). All cell culture reagents were purchased from Thermo Scientific–Invitrogen, and all other reagents were procured from Sigma-Aldrich (St. Louis, MO, USA).
Animals
Mature, 8-wk-old male Sprague-Dawley rats (Charles River Laboratories, Raleigh, NC, USA) provide a significant volume of serum for analysis, are well-characterized models for O3 research (45) and were used for O3 exposures in serum studies. Timed-pregnant (gestational day 12) adult female Fisher 344 rats were purchased from Charles River Laboratories, to provide postnatal day (P)1–P3 pups for primary rat microglia and embryonic day (E)17 embryos for rat cortical cultures, as these are characterized models routinely used to investigate the role of microglia in CNS disease (46). C57BL/6 mice have an average lifespan of ∼24 months, are well documented for the in vivo study of neuroimmune effects in CNS disease, and were therefore used as a model to study the aging effects of air pollution in the brain. Young (8-wk-old, sexually mature) and aged (18-mo-old, entering the later quarter of the lifespan) male C57BL/6 mice were used for the in vivo mixed vehicle exhaust (MVE) exposures to study aging and CNS effects. Young adult (8 wk) and aging (10 mo, approximately midway through the lifespan) male C57BL/6 mice (Charles River Laboratories) were used to generate the adult hippocampal mixed glia cultures. Rats and mice were housed at least 5 d before exposures for proper conditioning. Eight mature 10-wk-old female C57BL/6 [cluster of differentiation 36 (CD36)+/+] and CD36−/− mice were also used. CD36−/− mice are on a C57BL/6 background and have an attenuated peripheral macrophage immune response (47), including that females show impaired pulmonary immune response to O3 (23). CD36−/− mice were kindly provided by Dr. Maria Febbraio (University of Alberta, Edmonton, AB, Canada). The mice were acclimated for 1 wk before the start of the experiments. Mice and rats were housed in an Association for Assessment and Accreditation of Laboratory Animal Care–accredited housing facility, maintained at 20–24°C and on a 12 h light–dark schedule. All experiments were conducted with International Care and Use Committee approval and the Guidelines for the Care and Use of Laboratory Animals (National Institutes of Health, Bethesda, MD, USA) were strictly followed.
O3 exposure
O3 was generated via an OREC silent arc discharge O3 generator (Osmonics, Phoenix, AZ, USA). The O3 concentration was continuously monitored with a photometric O3 analyzer (TG-501,; GrayWolf Sensing Solutions, Shelton, CT, USA), and temperature was maintained at 21 ± 2°C. The rats and mice were randomly assigned to a group and were exposed to either filtered air (FA) or 1 part per million (ppm) O3 for 4 h. The measured chemical characteristics of the O3 exposures are shown in Table 1. Rodents are insensitive to O3 toxicity, owing to their complex nasal turbinates, lung morphologic differences, and high urate and ascorbate concentrations in the airway surfactant. For this reason, a factor of 3 is accepted practice for extrapolating concentrations from rodents to primates. O3 concentrations of 0.2–0.3 ppm are frequently achieved in areas of high air pollution, where 1 ppm is considered the equivalent in rodent experiments.
TABLE 1.
O3 constructions
| Exposure atmosphere composition ( ppm) | Control | O3 |
|---|---|---|
| O3 | 0.0 ± 0.0 | 1.02 ± 0.03 |
| Oxides of nitrogen | 0.0 ± 0.0 | 0.0 ± 0.0 |
During exposures, rats and mice were singly housed within a sealed chamber (Biospherix, Parish, NY, USA) without bedding to avoid decomposition of the O3. Food, but not water, was withheld during the 4 h exposure period to preclude ingestion of ozonation products. The exposure chambers were also analyzed for oxides of nitrogen contaminants, which were invariably below levels of detection (10 ppb). Brain tissue and whole blood were obtained 24 h after the 4 h O3 exposure, from rats and mice under isoflurane anesthesia. Blood was centrifuged at 10,000 g for 5 min to isolate serum, which was stored at −80°C until use. One hemisphere was fixed in 4% paraformaldehyde, and the other hemisphere was snap-frozen and stored at −80°C.
MVE exposure
Mice were exposed 6 h/d for 50 d to either 300 μg particulate matter (PM)/m3 , which was a combination of roughly 50 μg PM/m3 gasoline engine emissions mixed with 250 μg PM/m3 diesel engine emissions mixed in a preexposure chamber upstream of the animal-exposure chamber (48). MVE exposure was developed to model an urban high-traffic environment, complete with particulate and gaseous copollutants, with a Yammar (Osaka, Japan) diesel engine generator operated under constant load, combined with emissions from a gasoline-powered 4.3 L engine operated on a transient engine cycle (48). Control mice were exposed to FA. The measured chemical characteristics of the MVE exposures are shown in Table 2. The concentration used is high for urban regions of the United States, but not atypical for many industrial urban regions worldwide. Brain tissue was obtained 24 h after the last exposure. One hemisphere was fixed in 4% paraformaldehyde, and the other hemisphere was snap-frozen and stored at −80°C.
TABLE 2.
Mixed vehicle emissions chemical characteristics
| Exposure atmosphere composition | Units | Control | MVE |
|---|---|---|---|
| Filter sample, chamber | µg/m³ | 10.0 ± 8.0 | 328.0 ± 73.0 |
| Elemental carbon | µg/m³ | 0.4 | 248.3 ± 3.1 |
| Organic carbon | µg/m³ | 5.2 ± 0.1 | 37.7 ± 2.2 |
| Ammonium | ng/m³ | 0.0 | 2080.4 ± 23.3 |
| Sulfate | ng/m³ | 137.3 ± 0.03 | 3613.2 ± 13.5 |
| Nitrate | ng/m³ | 63.3 ± 0.13 | 2448.6 ± 34.7 |
| Elements (metals) | ng/m³ | 253.7 ± 10.5 | 2541.9 ± 34.1 |
| Alkanes | µg/m³ | 2.2 ± 0.1 | 29.7 ± 1.7 |
| Carbonyl | µg/m³ | 24.7 ± 1.8 | 58.8 ± 3.5 |
| Polycyclic aromatic hydrocarbon (PAH) | µg/m³ | 1.4 ± 0.1 | 21.9 ± 1.2 |
| Nitro-PAH | ng/m³ | 0.4 ± 0.1 | 22.3 ± 1.8 |
| Polar (acids) | µg/m³ | 3.1 ± 0.3 | 21.7 ± 1.6 |
| Hopane and steranes | ng/m³ | 2.2 ± 0.2 | 37.2 ± 2.9 |
| Volatile organic carbon (VOC) | µg/m³ | 92.6 ± 6.81 | 6653.5 ± 406.3 |
Primary rat microglia cells
Primary enriched microglia cultures were prepared from the whole brains of 1-d-old Fisher 344 rat pups (49). The cells were treated 24 h after the enriched microglia were seeded. Cultures were treated with LPS (5 ng/ml) or medium alone (control) in treatment medium containing 2% FA or O3 serum. Immunocytochemistry revealed less than 1% astrocyte or neuron contamination.
Rat microglial cell line
The highly aggressively proliferating immortalized (HAPI) rat microglia cells were maintained at 37°C in DMEM supplemented with 10% fetal bovine serum, 50 U/ml penicillin, and 50 μg/ml streptomycin in a humidified incubator with 5% CO2/95% air. Only passages 3–7 were used for the study. The cells were seeded in maintenance medium for 24 h and later treated with LPS (5 ng/ml) or medium alone (control) in treatment medium containing 2% FA or O3+ serum from rats.
Assessment of pulmonary inflammation by imaging and lavage
Single-photon emission computed tomography/computed tomography (SPECT/CT) studies were performed with a multipinhole small-animal imaging camera (NanoSPECT/CT) (50). The mice were injected intravenously with a previously described [111In]-labeled radioligand for lymphocyte function-associated antigen 1 (LFA-1) (50), ∼45 min before SPECT/CT imaging of the thorax. Imaging studies were performed on prone anesthetized mice (1.5–2.0% isoflurane) on a temperature-controlled bed. Image analyses (crosstalk removal, reconstruction, and concatenation) were performed with InVivoScope (inviCro, LLC, Boston, MA, USA). Data are presented as a percentage of the injected dose, normalized to thoracic volume and time after injection. Bronchiole alveolar lavage was performed on rats and mice after euthanasia (23).
Cortical neuron–glia cultures
Rat cortical neuron–glia cultures were prepared according to a published protocol (46). Cultures were treated 7 d after seeding with aggregated β-amyloid 42 (Aβ42) (2 μM) or medium alone (control) in treatment medium containing 2% fetal bovine serum (FBS), in addition to 2% FA or O3 serum from rats. Cell survival was assessed by MTT assay.
Adult hippocampus mixed glia cultures
The hippocampus from adult male C57BL/6 mice at 8 wk (young adult) or 10 mo (aged) was dissected and cultured (51) in an effort to assess the responses of mature and aging glia. In brief, tissue was minced in 10% medium (DMEM:F12 50:50 containing 10% FBS, 15 mM HEPES, 2 mM glutamine, and penicillin/streptomycin). Tissues were digested in 0.5% trypsin for 10 min at 37°C and then resuspended in 20% DMEM:F12 (50:50). The cells were then further dissociated via gentle mechanical trituration in 10% DMEM:F12 (50:50), resuspended in 20% DMEM:F12 (50:50) , filtered through a 70 μm filter, and immediately seeded (5 × 105/well) in poly d-lysine (20 μg/ml)-precoated 24-well plates. Two days after seeding, the medium was removed and replenished with 500 μl fresh 20% DMEM:F12 (50:50). The 20% DMEM:F12 (50:50) was replaced 2×/wk thereafter. Cultures were treated 4 wk after seeding with LPS (5 ng/ml) or medium alone (control) in treatment medium containing 2% vol/vol FA or O3+ serum. The final cultures consisted of ∼55% astrocytes and 45% microglia, as determined by immunohistochemistry.
Quantitative RT-PCR
Total RNA was extracted from the mouse brain cortex with Trizol (Thermo Scientific–Invitrogen), according to the manufacturer’s instructions. The RNA was treated with Ambion DNase I (Thermo Scientific–Invitrogen), which was subsequently removed with an RNeasy RNA cleanup kit (Qiagen, Germantown, MD, USA). Reverse transcription of RNA (0.3 - 1.0 µg/sample) was performed with iScript Reverse Transcription Supermix (Bio-Rad, Hercules, CA,) according to the manufacturer’s instructions. SsoFast Evagreen Supermix (Bio-Rad) and 500 nM forward and reverse primers were used to perform quantitative PCR on a CFX96 (Bio-Rad) real-time PCR detection system, per the manufacturer’s instructions. Cycling parameters were 1 cycle at 95°C for 5 min, 40 cycles of 95°C (5 s) and 56°C (5 s), followed by a melting curve measurement consisting of 5 s 0.5°C incremental increases from 65 to 95°C. The primer sequences are listed in Table 3.
TABLE 3.
Primer sequences
| Gene |
Primer sequence, 5′–3′ |
|
|---|---|---|
| Forward | Reverse | |
| GAPDH | AACTTTGGCATTGTGGAAGG | ATCCACAGTCTTCTGGGTGG |
| IL-1b | TGAAGAAGAGCCCATCCTCTGTGA | GGTCCGACAGCACGAGGCTT |
| TNFa | GCCCACGTCGTAGCAAACCACC | CCCATCGGCTGGCACCACTA |
Immunohistochemistry
The right hemisphere of the brain was fixed in 4% paraformaldehyde for 2 d and cryoprotected in 30% sucrose. Coronal sections (40 µm) were collected with a freezing-stage microtome (Microm HM 450; Thermo Scientific, Waltham, MA, USA). Free-floating brain slices were treated with 1% H2O2, washed twice for 10 min with PBS, incubated 20 min with a blocking solution (PBS containing 1% bovine serum albumin, 4% goat serum, ,0.4% Triton X-100), and incubated overnight at 4°C with primary IBA1 antibody (Wako), diluted 1:1000 in antibody diluent (Dako, Carpinteria, CA, USA). Slices were then washed twice in PBS, incubated with biotinylated anti-rabbit antibody (Vector Laboratories) for 1 h, washed twice in PBS, and incubated with the Vectastain ABC Kit (Vector Laboratories) reagents, according to the manufacturer’s instructions. Staining was visualized with 3,3′-diaminobenzidine and urea-H2O2 tablets (Sigma-Aldrich). Images were captured with a BX51 microscope (Olympus America, Center Valley, PA, USA).
Cytokine ELISAs
The production and release of IL-6, TNFα, and IL-1β into serum and medium were measured with a commercial ELISA kit from R&D Systems (52). Chemokine (C-C motif) ligand 2 (CCL2; R&D Systems) and CCL11 (LSBio, Seattle, WA, USA) in serum were measured with commercially available ELISA kits, per the manufacturer’s instructions.
H2O2 assay
Levels of H2O2 production in cell culture were determined as has been described (38). Results were calculated as catalase-inhibitable fluorescence and reported as a percentage of control values.
Nitrite assay
As an indicator of NO production, the amount of nitrite accumulated in culture supernatant was determined with a colorimetric assay using Griess reagent [1% sulfanilamide, 2.5% H3PO4, and 0.1% N-(1-naphthyl) ethylenediamine dihydrochloride] (49). The sample nitrite concentration was determined from a sodium nitrite standard curve, with a lower limit of detection of 1.2 μM.
Cell survival assay
Cell survival was measured using thiazole blue (MTT) to evaluate metabolic viability of cells, as previously described (46).
Statistical analysis
Data are expressed as representative images, raw values, the percentage of control or LPS, or the difference from control, where control values were set to either 100 or 0%, accordingly. The treatment groups are expressed as the means ± sem, and statistical significance was assessed with a 1- or 2-way ANOVA, followed by Bonferroni’s post hoc analysis (Prism, ver. 6.0; GraphPad, La Jolla, CA, USA).
RESULTS
O3 exposure causes persistent microglial activation in vivo
O3 does not translocate to the brain parenchyma to directly induce effects in the brain, and the CNS response to pulmonary O3 exposure/injury is therefore poorly understood. As sentinels, microglia are activated, and their function is regulated by events in the periphery (52, 53). We sought to determine whether microglia respond to short-term O3 inhalation and the associated mild lung pathology (45). The time point of 24 h after exposure was chosen based on prior studies, when extrapulmonary effects of O3 in the cardiovascular system are evident (23). Histopathological evidence revealed that, despite the inability of O3 to enter the brain parenchyma, microglia were activated in rats after short-term O3 inhalation (4 h), and microglia morphology remained changed for 24 h after exposure compared to FA (sham)-exposed controls (Fig. 1). This persistent response of the microglia to O3 (24 h after stimulation) supports an active ongoing signal from the lung to the brain with a single exposure.
Figure 1.
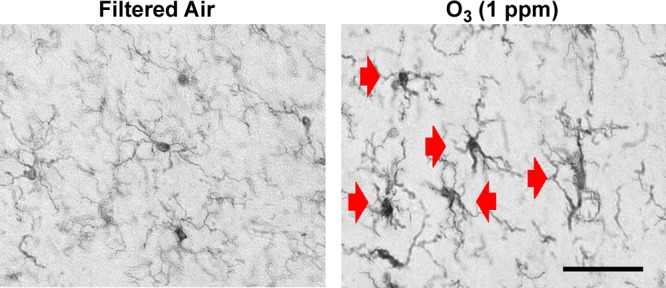
Changes in microglia morphology persist 24 hours after short-term O3 exposure. Young adult male rats were exposed to FA or O3 (1 ppm) for 4 h, and changes in microglia morphology were assessed 24 h later. Three coronal sections (40 µm) per animal were stained with the IBA1 antibody. Red arrows: activated microglia morphology (n = 3). Magnification, ×40; scale bar, 50 μm.
O3-induced circulating factors prime microglia ex vivo
In an effort to discern whether an inflammatory signal was present in the blood after O3 inhalation, to which microglia may respond, ex vivo assays testing the bioactivity of serum collected from rats 24 h after the 4 h inhalation exposure (FA or 1 ppm O3) were performed on microglial proinflammatory and neurotoxicity responses. O3 serum from exposed rats, diluted 1:50 in medium, was unable to initiate TNFα production (Fig. 2A, B) and produced only a low-level reactive oxygen species (ROS) response with serum alone (Fig. 2C) in microglia cultures. However, serum from O3-exposed rats enhanced LPS (5 ng/ml)-induced TNFα in the HAPI microglia cell line (Fig. 2A) and rat primary microglia cultures, as compared with serum from FA-exposed rats (Fig. 2B). O3 serum also augmented H2O2 production in primary rat microglia (Fig. 2C). In regard to the potential impact on ongoing neurodegenerative disease processes, such as AD, O3 serum amplified aggregated Aβ42-induced H2O2 in primary microglia cultures (Fig. 3A) and neurotoxicity in mixed cortical neuron–glia cultures (Fig. 3B). O3 serum was not directly toxic to microglia, but O3 serum attenuated LPS-induced microglial toxicity, as measured by MTT (Fig. 2D). LPS-induced microglial cell death has been implicated in the attenuation of the microglial proinflammatory response (54) and our current data suggest that O3 impairs this mechanism. Serum from O3-exposed rats had no effects on LPS-induced nitrite measurements (data not shown). Together, these data indicate that O3 serum from a single 4 h exposure carries a bioactive signal initiating a low-grade ROS response, priming microglia to be sensitive to additional proinflammatory stimuli and augmenting ongoing AD-like toxicity.
Figure 2.

O3 serum primes the microglial proinflammatory response to LPS ex vivo. Young adult male rats were exposed to FA or O3 (1 ppm) for 4 h, and serum was collected 24 h later, during the time microglia had shown activated morphology. Ex vivo serum bioactivity was assessed using cultures treated with 2% rat serum acquired from FA- or O3-exposed rats. A, B) Although O3 serum did not initiate TNFα production, O3 serum enhanced LPS-induced TNFα production in the 3 h supernatant of the HAPI rat microglial cell line (A) and rat primary microglia cultures (B). C) O3 serum caused a slight but insignificant elevation of H2O2 at 3 h after treatment and amplified LPS-induced H2O2 in primary rat microglia cultures. D) O3 serum failed to cause any microglia toxicity alone at 3 h after treatment in HAPI rat microglia cell lines and significantly reduced LPS-induced toxicity, as measured by MTT (n = 3–6). Values are reported as the mean or the mean percentage of control ± sem. *P < 0.05 vs. control; †P < 0.05, O3 serum vs. FA serum.
Figure 3.

O3 serum primes the microglial ROS response and neurotoxicity to Aβ42 ex vivo. Young adult male rats were exposed to FA or O3 (1 ppm) for 4 h, and serum was collected 24 h later, during the time microglia had shown activated morphology. Ex vivo serum bioactivity was assessed in cultures treated with 2% rat serum acquired from FA- or O3-exposed rats. A) O3 serum caused a slight but insignificant elevation of H2O2 and amplified Aβ42-induced H2O2 in primary microglia cultures. B) O3 serum amplified Aβ42-induced neurotoxicity in rat cortical neuron–glia cultures, showing that the serum may augment AD-like neuropathology (n = 3). Values are reported as mean percentage of control ± sem. *P < 0.05 vs. control; †P < 0.05, O3 serum vs. FA serum.
Traditional circulating cytokines are absent from bioactive O3 serum
To begin to identify how pulmonary pathology communicate with the brain, we focused on circulating cytokines that are routinely accepted as essential for transfer of inflammation from the periphery to the brain and CNS effects. Analysis of serum CCL2, CCL11, TNFα, IL-6, and IL-1β levels by ELISA indicated that these traditional circulating cytokines were not modified by O3 exposure in bioactive serum samples (Supplementary Table S1), supporting that microglial activation and modulation occur through an alternate mechanism. This finding is consistent with results of work exploring how air pollution affects the cardiovascular system, where postexposure serum from animals and humans exposed to various pollutants (O3, NO2, and diesel emissions) carries a bioactive signal that induces proinflammatory responses in endothelial cells that are independent from the “usual suspect” cytokines, such as IL-1β, IL-6, TNFα, and monocyte chemotactic protein-1 (23). Thus, the data indicate that the circulating bioactive signals present in O3 serum that prime and activate microglia are independent from traditional cytokine triggers and need extensive further investigation.
MAC1 is critical for O3 serum-induced microglial priming
To explore how microglia may identify this serum signal, we first assessed whether the MAC1 surface receptor, which is frequently implicated in deleterious microglia responses (29), played a role in how O3 serum primes the microglial proinflammatory response. The ability of MAC1 blocking antibodies (20 μg/ml) to attenuate the O3-induced priming of the LPS-induced TNFα response was compared to the effects of the IgG control antibodies (20 μg/ml). As expected (55), MAC1 blocking antibodies reduced the LPS-induced TNFα response in the presence of FA serum (Fig. 4). However, O3 amplified only the TNFα response to LPS in the presence of the IgG control antibody and failed to augment the TNFα response in the presence of the MAC1 blocking antibody. Thus, these data suggest that MAC1is key to the mechanism of how O3 serum augments the microglial response to LPS.
Figure 4.

MAC1 mediates O3 serum priming in microglia ex vivo. Young adult male rats were exposed to FA or O3 (1 ppm) for 4 h and serum was collected 24 h later. The ability of O3 serum to prime microglia and augment LPS-induced TNFα production in the presence of the control IgG antibody or the MAC1 blocking antibody was determined. Microglia cells were pretreated with the MAC1 inhibitor antibody (20 μg/ml) or mouse IgG control antibody (20 μg/ml) for 30 min followed by LPS (10 ng/ml) treatment. Supernatant was collected 3 h later, and TNFα protein was assessed by ELISA. Values are reported as means ± sem. *P < 0.05, difference in LPS-induced TNFα response from IgG antibody control; †P < 0.05, O3 serum augmentation of LPS-induced TNFα response is only significant in the IgG antibody control (n = 3).
Aging impairs the pulmonary cellular immune response and thoracic inflammation
To explore how pulmonary responses to pollutants affect the lung–brain axis, we assessed neuroinflammation in 2 models with altered lung innate immune responses to inhaled toxicants. Aging is well known to alter the pulmonary effects of urban air pollution (56), but mechanisms underlying the role of aging are poorly understood. We explored the effects of MVE exposure because the vulnerability of the aging human CNS has been clearly linked to near-roadway traffic exposures (57, 58). Older (18 mo) mice were exposed alongside younger (2 mo) mice to 300 μg of PM/m3 of MVE, a combination of gasoline and diesel engine emissions, for 6 h/d for 50 d. Pulmonary inflammation was assessed by conventional bronchoalveolar lavage and by a novel radioligand for activated LFA-1 ([111In]-DANBIRT). In 2-mo-old mice, the total lavageable cells showed a slight increase that was not significant (Fig. 5A), but lavageable neutrophils were significantly elevated, as expected (Fig. 5B), in response to MVE exposure. This neutrophilic response to MVE was absent in the 18-mo-old mice, however, although these older mice may have had elevated baseline airway macrophage counts (Fig. 5A, B). SPECT/CT imaging of LFA-1 also confirmed a thoracic inflammatory response in young mice (Fig. 5C, D) that was absent in aged mice. Thus, the data indicate that aging may impart an impaired pulmonary immune response to MVE.
Figure 5.

The aging lung. Aging attenuates the pulmonary immune response to mixed vehicle exhaust and augments thoracic inflammation. Young adult (2 mo) and aged (18 mo) male C57BL/6 mice were exposed to 300 μg PM/m3 MVE or FA 6 h/d for a month. A) Total cells in the bronchoalveolar lavage fluid were not significantly altered by exposure in either group. B) Two-month-old mice displayed a significant increase in BAL fluid neutrophils, and 18-mo-old mice exhibited no neutrophilic response to MVE. *P < 0.05. C) LFA-1 SPECT/CT images showed consistent increases in inflammatory patterns in young mice in response to MVE exposure, with no significant response in the 18-mo-old mice. D) Representative SPECT/CT images of [111In]-NorBIRT radioligand for LFA-1 (n = 3). Values are reported as means ± SEM. * P < 0.05, MVE exposure vs. FA.
Aged CNS tissue is sensitive to the proinflammatory effects of air pollutants
In an effort to explore whether aging augments the CNS response to urban air pollution, we assessed neuroinflammation and changes in microglia morphology in response to 50 d inhalation exposure to MVE in the 2- and 18-mo-old mice. Aged cortical tissue exhibited the highest TNFα mRNA expression (Fig. 6A) and changes in microglia morphology (Fig. 6B). Notably, MVE is composed of multiple components with the potential to travel to the brain parenchyma, such as PM, metals, and polyaromatic hydrocarbons (PAHs). To discern whether aging CNS tissue may be more vulnerable to the effects of air pollution–induced circulating factors, we compared the proinflammatory sensitivity of hippocampal mixed-glia cultures obtained from aging (10 mo) and young adult (2 mo) mice to O3 serum priming of the LPS-induced TNFα response. Data are expressed as the percentage increase over FA values in LPS-induced TNFα production. O3 serum from young adult rats augmented TNFα production in young adult mouse mixed glia hippocampal cultures by 18%, whereas aged cultures showed a 48% increase with O3 serum (Fig. 6C), indicating that the aging mixed glia cells are hypersensitive to the proinflammatory priming effects of O3 serum.
Figure 6.

The aging brain. Aging augments neuroinflammation in response to air pollutants. Young adult (2 mo) and aged (18 mo) male C57BL/6 mice were exposed to either 300 μg PM/m3 MVE or FA 6 h/d for 50 d. A) MVE-induced neuroinflammation is highest in aged mice. Cortical tissue was collected, and TNFα mRNA levels were assessed with quantitative RT-PCR (n = 3). Expression was normalized to GAPDH by using the 2−ΔΔCt method and is reported as the mean ± sem. *P < 0.05, MVE vs. FA. B) MVE-induced activated microglia morphology is more pronounced in aged mice. Three coronal sections (40 µm) per animal were stained with the IBA1 antibody. Magnification, ×40; scale bar, 50 μm. Red arrows depict activated microglia morphology (n = 3). C) The aging brain is more sensitive to O3 serum The ability of O3 serum collected 24 h after a 4 h O3 exposure to prime microglia and augment LPS-induced TNFα production in mixed glia cultures from young adult (2 mo) and aged (10 mo) rats was determined. Supernatant was collected 3 h after LPS treatment, and TNFα protein was assessed by ELISA. Data are reported as the percentage increase from mean filtered LPS values ± sem (n = 3). †P < 0.05, O3 serum demonstrated significantly greater augmentation of the LPS-induced TNFα response in aged cultures.
Thus, although the pulmonary cellular immune response to inhaled pollutants appears attenuated by aging, the aged CNS tissue is more sensitive to the proinflammatory effects of inhaled pollutants, including an increased sensitivity of aging glia to the circulating signals triggered by O3 exposure.
Young adult CD36−/− mice have augmented O3-induced neuroinflammation
To further explore the role of impaired pulmonary immune responses in air pollution–induced neuroinflammation, CD36−/− mice were used. CD36−/− mice fail to express this scavenger receptor globally and exhibit minimal pulmonary cellular immune response to O3, as compared to wild-type mice (23). CD36−/− mice also demonstrate a blunted vascular impairment after O3 inhalation, although this outcome relates to a lack of endothelial CD36, which is necessary to respond to altered serum components; serum from CD36−/− mice exposed to O3 still exhibited bioactivity ex vivo on aortic rings from wild-type mice (23). In the current study, at 24 h after 4 h exposure to O3, CD36+/+ mice showed no neural O3 effects, but O3 inhalation causes a significant increase in frontal lobe TNFα (Fig. 7A), frontal lobe IL-1β (Fig. 7B), and cortical microglial morphology change in the CD36−/− mice (Fig. 7C). These data indicate that despite significantly muted pulmonary inflammatory responses, CD36−/− mice have an augmented neuroimmune response to O3 inhalation, further supporting that an impaired pulmonary immune response to airborne pollutants may be linked to augmented neuroinflammation.
Figure 7.

CD36−/− mice have augmented O3-induced neuroinflammation and microglia activation. Young adult female CD36+/+ and CD36−/− mice were exposed to FA or O3 (1 ppm) for 4 h, and brain tissue was collected 24 h later. A, B) TNFα (A) and IL-1β (B) mRNA levels were assessed with quantitative RT-PCR. Values were normalized to GAPDH by the 2−ΔΔCt method and are reported as means ± sem. *P < 0.05 vs. FA control. C) O3-induced activated microglia morphology is more pronounced in the cortex of CD36−/− mice. Three coronal sections (40 µm) per animal were stained with the IBA1 antibody (n = 3). Magnification, ×40; scale bar, 50 μm.
DISCUSSION
The mechanisms underlying how inhaled air pollution triggers microglial activation to affect CNS disease are complex and poorly understood. We propose a new route of microglial activation and begin to outline the lung–brain axis, where the pulmonary interactions with air pollutants, such as O3, results in circulating signals that are detectable by microglia, resulting in a persistent microglial response; are independent from traditional circulating cytokines; reprogram microglia to be more sensitive to additional proinflammatory stimuli (priming); augment ongoing Aβ42 neurotoxicity; and have a greater proinflammatory priming response in aging CNS tissue. These findings provide much-needed insight into the mechanisms through which air pollutants may enhance risk for and augment the progression of aging associated neurodegenerative diseases, such as AD.
The concept of a lung–brain axis where lung damage/pathology regulates CNS function is supported by the fact that individuals with chronic obstructive pulmonary disease (59–61) and asthma (62, 63) show increased risk for development of dementia, which includes AD. This premise is further bolstered by recent findings pointing to a role for inhaled air pollution in CNS disease (10–12), including gases such as O3, where direct effects are limited to the airways with minimal, if any, direct translocation of pollutants to the rest of the body. As such, we first confirmed that microglia, the first responders in the brain, would detect O3 exposure in vivo, where a single 4 h exposure was shown to induce a persistent change in microglia morphology 24 h after the instigating stimulus (Fig. 1). These findings support that direct interaction of inhaled air pollutants with the brain is unnecessary to activate microglia.
As CNS sentinels, microglia readily detect events in the periphery, such as systemic LPS administration (52) and intestinal ischemia–reperfusion injury (53), where the microglial response to peripheral injury has been shown to augment ongoing neuropathology (53). To begin to address how peripheral responses to air pollution regulate microglia function, we used an ex vivo approach with serum collected from O3-treated rats to determine whether a bioactive signal was present in the circulation at the time of persistent microglial activation in the brain. Data revealed that, although microglial cultures treated with dilute serum alone exhibited no response, O3 serum augmented the TNFα and H2O2 response of microglia to LPS, a known trigger of the M1 innate immune response (Fig. 2). Further, O3 serum augmented aggregated Aβ42-induced superoxide production and neurotoxicity (Fig. 3), supporting that O3-induced circulating factors may augment ongoing AD-like neuropathology. Taken together, these findings indicate that O3 causes a circulating bioactive signal that primes the microglial proinflammatory response and associated neurotoxicity. These findings notably support a lung–brain axis where the instigating interaction with the toxicant first occurs in the airways, but the source of these CNS bioactive circulating factors may not originate only from the lung, but may include input from the liver, cardiovascular system, gut, and multiple other potentially affected organs (64). Further, the mechanisms underlying the CNS effects of air pollution are clearly multifaceted and likely include additional pathways, including autonomic nervous system effects, the hypothalamic–pituitary–adrenal axis (64), and the direct effects of the pollutants themselves, involving the translocation of the particle and chemical components of air pollution to the brain (2).
Although the specific serum components that enter the brain parenchyma are undetermined, blood–brain barrier permeability is notably impaired in aging (65), by neurodegenerative diseases (66), and by air pollution exposure (67), which facilitates entry of aberrant circulating factors into the brain and optimizes the opportunity of serum factors to interact with microglia. In fact, peripheral circulating cytokines are the first candidates implicated in how peripheral damage regulates the brain (52), particularly microglial priming (68). Thus, although our findings were consistent with previous work investigating how air pollution affects the cardiovascular system (23) and an exhaustive analysis of potential cytokines/chemokines was not used, it was surprising that traditional circulating cytokines (CCL2, CCL11, TNFα, IL-6, and IL-1β) were not elevated in the O3 bioactive serum. Although it remains possible that other untested proinflammatory factors, such as other ILs, IFNs, or prostaglandin Es (PGEs) may still mediate the serum bioactivity, the current study implicated MAC1 as what is likely one of many key microglial receptors the mediate the proinflammatory priming effects of O3 (Fig. 4), further supporting the role of a nontraditional ligand that is independent of cytokines. The augmentation of O3-induced neuroinflammation in the CD36−/− mice show that this receptor may not be a key component of the receptor complex in microglia that recognizes the O3-induced circulating signal. At present, the identity of these circulating signals responsible for transducing pulmonary pathology to CNS and microglial effects remain unknown and are the key focus of ongoing inquiry.
Aging is the predominant risk factor for the development of AD and reports implicate traffic-related air pollutant exposures in cognitive decline in the elderly (57, 58), but the mechanisms are unclear. Using the MVE model of traffic-related exposures, we demonstrated that the brains of aged mice have more pronounced neuroinflammation and changes in microglia morphology (Fig. 6). Pulmonary immune responses were reduced concomitant with this augmented neuroinflammation (Fig. 5), indicating that elevated lung inflammation may not reliably predict CNS effects and further suggests the possibility that an impaired immune response in the lung associated with aging could potentiate CNS effects. This possibility is further supported by our findings in CD36−/− mice, which have an impaired pulmonary immune response (23) where we have shown that CD36−/− mice exhibit augmented O3-induced neuroinflammation and microglial activation (Fig. 7). In addition, bioactive O3 serum from young adult rats was shown to have the greatest amplifying effects on LPS-induced TNFα production in hippocampal mixed glia cultures from aged mice, indicating that aging CNS tissue is more vulnerable to the circulating factors in response to O3. Thus, the lung–brain axis may be critical for age-related vulnerability to the CNS effects of air pollution.
In summary, this work reveals the role of the lung–brain axis (Fig. 8) in how inhaled pollutants regulate the proinflammatory milieu in the brain. More specifically, we demonstrate that inhalation of gaseous pollutants, such as O3, that are unable to reach the brain, instigate a circulating signal that activates and reprograms microglia to a primed state, which can augment neurotoxicity. This bioactive circulating signal does not appear to be cytokine in nature, but rather the proinflammatory microglial priming was regulated by ligands to the MAC1 receptor. In addition, age was identified as a key factor for conferring vulnerability to augmented air pollution–induced neuroinflammation, impaired pulmonary immune response to air pollutants, and glial cell sensitivity to the proinflammatory priming effects of air pollution–induced circulating factors. An impaired pulmonary immune response was revealed as an important pathology associated with an exacerbated neuroimmune response to air pollutants. Continuing research elucidating these mechanisms remains of pressing scientific concern for future efforts, not only to limit the impact of environmental exposures, but to identify novel targets and pathways for treatment of CNS disease.
Figure 8.

The lung–brain axis. Circulating factors in response to air pollutants prime microglia. Increasing evidence supports the conclusions that pulmonary damage through disease and exposure to air pollutants may regulate the proinflammatory milieu in the brain and potentially influence CNS disease and damage through the lung–brain axis. Outlining the lung–brain axis, the findings from the current study support the conclusions that after exposure to air pollutants such as ground-level O3, there is a pulmonary immune response, where impaired function (due to genetic modification or aging) is pathology associated with augmented neuroinflammation and microglia responses (A); brain regulating serum factors independent of circulating cytokines are present in circulation (B); and these circulating factors signal and are detected by microglia in the brain to result in a primed proinflammatory phenotype measured by enhanced sensitivity to additional proinflammatory stimuli (C). These findings have significant implications for how air pollution may affect the brain to augment CNS disease and how aging may confer vulnerability to the CNS effects of air pollution.
Supplementary Material
Acknowledgments
This research was supported by the U.S. National Institutes of Health/National Institute of Environmental Health Sciences [ES016951 (to M.L.B.) and ES014639 (to M.J.C.)]. The authors declare no conflicts of interest.
Glossary
- AD
Alzheimer’s disease
- Aβ42
β-amyloid 42
- CCL2
chemokine (C-C motif) ligand 2
- CCL11
chemokine (C-C motif) ligand 11
- CD36
cluster of differentiation 36
- FA
filtered air
- FBS
fetal bovine serum
- HAPI
highly aggressively proliferating immortalized
- IBA1
ionized calcium-binding adapter molecule 1
- LFA-1
lymphocyte function-associated antigen 1
- MAC1
macrophage-1 antigen
- MVE
mixed vehicle exhaust
- PAH
polycyclic aromatic hydrocarbon
- PD
Parkinson’s disease
- ppm
parts per million
- PM
particulate matter
- ROS
reactive oxygen species
- SPECT/CT
single-photon emission computed tomography/computed tomography
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
REFERENCES
- 1.Block M. L., Elder A., Auten R. L., Bilbo S. D., Chen H., Chen J. C., Cory-Slechta D. A., Costa D., Diaz-Sanchez D., Dorman D. C., Gold D. R., Gray K., Jeng H. A., Kaufman J. D., Kleinman M. T., Kirshner A., Lawler C., Miller D. S., Nadadur S. S., Ritz B., Semmens E. O., Tonelli L. H., Veronesi B., Wright R. O., Wright R. J. (2012) The outdoor air pollution and brain health workshop. Neurotoxicology 33, 972–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Block M. L., Calderón-Garcidueñas L. (2009) Air pollution: mechanisms of neuroinflammation and CNS disease. Trends Neurosci. 32, 506–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Power M. C., Weisskopf M. G., Alexeeff S. E., Wright R. O., Coull B. A., Spiro A. III, Schwartz J. (2013) Modification by hemochromatosis gene polymorphisms of the association between traffic-related air pollution and cognition in older men: a cohort study. Environ. Health 12, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wellenius G. A., Boyle L. D., Coull B. A., Milberg W. P., Gryparis A., Schwartz J., Mittleman M. A., Lipsitz L. A. (2012) Residential proximity to nearest major roadway and cognitive function in community-dwelling seniors: results from the MOBILIZE Boston Study. J. Am. Geriatr. Soc. 60, 2075–2080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Power M. C., Weisskopf M. G., Alexeeff S. E., Coull B. A., Spiro A. III, Schwartz J. (2011) Traffic-related air pollution and cognitive function in a cohort of older men. Environ. Health Perspect. 119, 682–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roberts A. L., Lyall K., Hart J. E., Laden F., Just A. C., Bobb J. F., Koenen K. C., Ascherio A., Weisskopf M. G. (2013) Perinatal air pollutant exposures and autism spectrum disorder in the children of Nurses’ Health Study II participants. Environ. Health Perspect. 121, 978–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Becerra T. A., Wilhelm M., Olsen J., Cockburn M., Ritz B. (2013) Ambient air pollution and autism in Los Angeles County, California. Environ. Health Perspect. 121, 380–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Volk H. E., Lurmann F., Penfold B., Hertz-Picciotto I., McConnell R. (2013) Traffic-related air pollution, particulate matter, and autism. JAMA Psychiatry 70, 71–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Volk H. E., Hertz-Picciotto I., Delwiche L., Lurmann F., McConnell R. (2011) Residential proximity to freeways and autism in the CHARGE study. Environ. Health Perspect. 119, 873–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jung C. R., Lin Y. T., Hwang B. F. (2015) Ozone, particulate matter, and newly diagnosed Alzheimer’s disease: a population-based cohort study in Taiwan. J. Alzheimers Dis. 44, 573–584 [DOI] [PubMed] [Google Scholar]
- 11.Kirrane E. F., Bowman C., Davis J. A., Hoppin J. A., Blair A., Chen H., Patel M. M., Sandler D. P., Tanner C. M., Vinikoor-Imler L., Ward M. H., Luben T. J., Kamel F. (2015) Associations of ozone and PM2.5 concentrations with Parkinson’s disease among participants in the agricultural health study. J. Occup. Environ. Med. 57, 509–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kioumourtzoglou M. A., Schwartz J. D., Weisskopf M. G., Melly S. J., Wang Y., Dominici F., Zanobetti A. (2016) Long-term PM exposure and neurological hospital admissions in the northeastern united states. Environ. Health Perspect. 124, 23–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Calderón-Garcidueñas L., Solt A. C., Henríquez-Roldán C., Torres-Jardón R., Nuse B., Herritt L., Villarreal-Calderón R., Osnaya N., Stone I., García R., Brooks D. M., González-Maciel A., Reynoso-Robles R., Delgado-Chávez R., Reed W. (2008) Long-term air pollution exposure is associated with neuroinflammation, an altered innate immune response, disruption of the blood-brain barrier, ultrafine particulate deposition, and accumulation of amyloid beta-42 and alpha-synuclein in children and young adults. Toxicol. Pathol. 36, 289–310 [DOI] [PubMed] [Google Scholar]
- 14.Calderón-Garcidueñas L., Reed W., Maronpot R. R., Henríquez-Roldán C., Delgado-Chavez R., Calderón-Garcidueñas A., Dragustinovis I., Franco-Lira M., Aragón-Flores M., Solt A. C., Altenburg M., Torres-Jardón R., Swenberg J. A. (2004) Brain inflammation and Alzheimer’s-like pathology in individuals exposed to severe air pollution. Toxicol. Pathol. 32, 650–658 [DOI] [PubMed] [Google Scholar]
- 15.Calderón-Garcidueñas L., Kavanaugh M., Block M., D’Angiulli A., Delgado-Chávez R., Torres-Jardón R., González-Maciel A., Reynoso-Robles R., Osnaya N., Villarreal-Calderon R., Guo R., Hua Z., Zhu H., Perry G., Diaz P. (2012) Neuroinflammation, hyperphosphorylated tau, diffuse amyloid plaques, and down-regulation of the cellular prion protein in air pollution exposed children and young adults. J. Alzheimers Dis. 28, 93–107 [DOI] [PubMed] [Google Scholar]
- 16.Calderón-Garcidueñas L., Franco-Lira M., Mora-Tiscareño A., Medina-Cortina H., Torres-Jardón R., Kavanaugh M. (2013) Early Alzheimer’s and Parkinson’s disease pathology in urban children: friend versus foe responses—it is time to face the evidence. BioMed Res. Int. 2013, 161687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Calderón-Garcidueñas L., D’Angiulli A., Kulesza R. J., Torres-Jardón R., Osnaya N., Romero L., Keefe S., Herritt L., Brooks D. M., Avila-Ramirez J., Delgado-Chávez R., Medina-Cortina H., González-González L. O. (2011) Air pollution is associated with brainstem auditory nuclei pathology and delayed brainstem auditory evoked potentials. Int. J. Dev. Neurosci. 29, 365–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bedada G. B., Smith C. J., Tyrrell P. J., Hirst A. A., Agius R. (2012) Short-term effects of ambient particulates and gaseous pollutants on the incidence of transient ischaemic attack and minor stroke: a case-crossover study. Environ. Health 11, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wellenius G. A., Burger M. R., Coull B. A., Schwartz J., Suh H. H., Koutrakis P., Schlaug G., Gold D. R., Mittleman M. A. (2012) Ambient air pollution and the risk of acute ischemic stroke. Arch. Intern. Med. 172, 229–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mauderly J. L., Burnett R. T., Castillejos M., Ozkaynak H., Samet J. M., Stieb D. M., Vedal S., Wyzga R. E. (2010) Is the air pollution health research community prepared to support a multipollutant air quality management framework? Inhal. Toxicol. 22(Suppl 1), 1–19 [DOI] [PubMed] [Google Scholar]
- 21.World Health Organization. (2002) The World Health Report 2002: Reducing Risks, Promoting Healthy Life. Available at http://www.who.int/whr/2002/en/
- 22.Cole M. P., Freeman B. A. (2009) Promotion of cardiovascular disease by exposure to the air pollutant ozone. Am. J. Physiol. Lung Cell. Mol. Physiol. 297, L205–L208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Robertson S., Colombo E. S., Lucas S. N., Hall P. R., Febbraio M., Paffett M. L., Campen M. J. (2013) CD36 mediates endothelial dysfunction downstream of circulating factors induced by O3 exposure. Toxicol. Sci. 134, 304–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Postlethwait E. M., Cueto R., Velsor L. W., Pryor W. A. (1998) O3-induced formation of bioactive lipids: estimated surface concentrations and lining layer effects. Am. J. Physiol. 274, L1006–L1016 [DOI] [PubMed] [Google Scholar]
- 25.Fiore A. M., Naik V., Spracklen D. V., Steiner A., Unger N., Prather M., Bergmann D., Cameron-Smith P. J., Cionni I., Collins W. J., Dalsøren S., Eyring V., Folberth G. A., Ginoux P., Horowitz L. W., Josse B., Lamarque J. F., MacKenzie I. A., Nagashima T., O’Connor F. M., Righi M., Rumbold S. T., Shindell D. T., Skeie R. B., Sudo K., Szopa S., Takemura T., Zeng G. (2012) Global air quality and climate. Chem. Soc. Rev. 41, 6663–6683 [DOI] [PubMed] [Google Scholar]
- 26.Bell M. L., Davis D. L., Cifuentes L. A., Krupnick A. J., Morgenstern R. D., Thurston G. D. (2008) Ancillary human health benefits of improved air quality resulting from climate change mitigation. Environ. Health 7, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hickman S. E., Kingery N. D., Ohsumi T. K., Borowsky M. L., Wang L. C., Means T. K., El Khoury J. (2013) The microglial sensome revealed by direct RNA sequencing. Nat. Neurosci. 16, 1896–1905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sun Q., Wu W., Hu Y. C., Li H., Zhang D., Li S., Li W., Li W. D., Ma B., Zhu J. H., Zhou M. L., Hang C. H. (2014) Early release of high-mobility group box 1 (HMGB1) from neurons in experimental subarachnoid hemorrhage in vivo and in vitro. J. Neuroinflammation 11, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Block M. L., Zecca L., Hong J. S. (2007) Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat. Rev. Neurosci. 8, 57–69 [DOI] [PubMed] [Google Scholar]
- 30.Heneka M. T., Kummer M. P., Latz E. (2014) Innate immune activation in neurodegenerative disease. Nat. Rev. Immunol. 14, 463–477 [DOI] [PubMed] [Google Scholar]
- 31.Perry V. H., Holmes C. (2014) Microglial priming in neurodegenerative disease. Nat. Rev. Neurol. 10, 217–224 [DOI] [PubMed] [Google Scholar]
- 32.Heppner F. L., Ransohoff R. M., Becher B. (2015) Immune attack: the role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 16, 358–372 [DOI] [PubMed] [Google Scholar]
- 33.Takano T. (2015) Role of microglia in autism: recent advances. Dev. Neurosci. 37, 195–202 [DOI] [PubMed] [Google Scholar]
- 34.Campbell A., Araujo J. A., Li H., Sioutas C., Kleinman M. (2009) Particulate matter induced enhancement of inflammatory markers in the brains of apolipoprotein E knockout mice. J. Nanosci. Nanotechnol. 9, 5099–5104 [DOI] [PubMed] [Google Scholar]
- 35.Campbell A., Oldham M., Becaria A., Bondy S. C., Meacher D., Sioutas C., Misra C., Mendez L. B., Kleinman M. (2005) Particulate matter in polluted air may increase biomarkers of inflammation in mouse brain. Neurotoxicology 26, 133–140 [DOI] [PubMed] [Google Scholar]
- 36.Santiago-López D., Bautista-Martínez J. A., Reyes-Hernandez C. I., Aguilar-Martínez M., Rivas-Arancibia S. (2010) Oxidative stress, progressive damage in the substantia nigra and plasma dopamine oxidation, in rats chronically exposed to ozone. Toxicol. Lett. 197, 193–200 [DOI] [PubMed] [Google Scholar]
- 37.Gerlofs-Nijland M. E., van Berlo D., Cassee F. R., Schins R. P., Wang K., Campbell A. (2010) Effect of prolonged exposure to diesel engine exhaust on proinflammatory markers in different regions of the rat brain. Part. Fibre Toxicol. 7, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Levesque S., Taetzsch T., Lull M. E., Kodavanti U., Stadler K., Wagner A., Johnson J. A., Duke L., Kodavanti P., Surace M. J., Block M. L. (2011) Diesel exhaust activates and primes microglia: air pollution, neuroinflammation, and regulation of dopaminergic neurotoxicity. Environ. Health Perspect. 119, 1149–1155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Levesque S., Surace M. J., McDonald J., Block M. L. (2011) Air pollution & the brain: subchronic diesel exhaust exposure causes neuroinflammation and elevates early markers of neurodegenerative disease. J. Neuroinflammation 8, 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Antonini J. M., Sriram K., Benkovic S. A., Roberts J. R., Stone S., Chen B. T., Schwegler-Berry D., Jefferson A. M., Billig B. K., Felton C. M., Hammer M. A., Ma F., Frazer D. G., O’Callaghan J. P., Miller D. B. (2009) Mild steel welding fume causes manganese accumulation and subtle neuroinflammatory changes but not overt neuronal damage in discrete brain regions of rats after short-term inhalation exposure. Neurotoxicology 30, 915–925 [DOI] [PubMed] [Google Scholar]
- 41.Elder A., Gelein R., Silva V., Feikert T., Opanashuk L., Carter J., Potter R., Maynard A., Ito Y., Finkelstein J., Oberdörster G. (2006) Translocation of inhaled ultrafine manganese oxide particles to the central nervous system. Environ. Health Perspect. 114, 1172–1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sama P., Long T. C., Hester S., Tajuba J., Parker J., Chen L. C., Veronesi B. (2007) The cellular and genomic response of an immortalized microglia cell line (BV2) to concentrated ambient particulate matter. Inhal. Toxicol. 19, 1079–1087 [DOI] [PubMed] [Google Scholar]
- 43.Campbell A., Daher N., Solaimani P., Mendoza K., Sioutas C. (2014) Human brain derived cells respond in a type-specific manner after exposure to urban particulate matter (PM). Toxicol. In Vitro 28, 1290–1295 [DOI] [PubMed] [Google Scholar]
- 44.Bolton J. L., Smith S. H., Huff N. C., Gilmour M. I., Foster W. M., Auten R. L., Bilbo S. D. (2012) Prenatal air pollution exposure induces neuroinflammation and predisposes offspring to weight gain in adulthood in a sex-specific manner. FASEB J. 26, 4743–4754 [DOI] [PubMed] [Google Scholar]
- 45.Paffett M. L., Zychowski K. E., Sheppard L., Robertson S., Weaver J. M., Lucas S. N., Campen M. J. (2015) Ozone inhalation impairs coronary artery dilation via intracellular oxidative stress: evidence for serum-borne factors as drivers of systemic toxicity. Toxicol. Sci. 146, 244–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Qin L., Liu Y., Cooper C., Liu B., Wilson B., Hong J. S. (2002) Microglia enhance beta-amyloid peptide-induced toxicity in cortical and mesencephalic neurons by producing reactive oxygen species. J. Neurochem. 83, 973–983 [DOI] [PubMed] [Google Scholar]
- 47.Febbraio M., Abumrad N. A., Hajjar D. P., Sharma K., Cheng W., Pearce S. F., Silverstein R. L. (1999) A null mutation in murine CD36 reveals an important role in fatty acid and lipoprotein metabolism. J. Biol. Chem. 274, 19055–19062 [DOI] [PubMed] [Google Scholar]
- 48.Lund A. K., Lucero J., Harman M., Madden M. C., McDonald J. D., Seagrave J. C., Campen M. J. (2011) The oxidized low-density lipoprotein receptor mediates vascular effects of inhaled vehicle emissions. Am. J. Respir. Crit. Care Med. 184, 82–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Block M. L., Wu X., Pei Z., Li G., Wang T., Qin L., Wilson B., Yang J., Hong J. S., Veronesi B. (2004) Nanometer size diesel exhaust particles are selectively toxic to dopaminergic neurons: the role of microglia, phagocytosis, and NADPH oxidase. FASEB J. 18, 1618–1620 [DOI] [PubMed] [Google Scholar]
- 50.Poria R. B., Norenberg J. P., Anderson T. L., Erion J., Wagner C. R., Arterburn J. B., Larson R. S. (2006) Characterization of a radiolabeled small molecule targeting leukocyte function-associated antigen-1 expression in lymphoma and leukemia. Cancer Biother. Radiopharm. 21, 418–426 [DOI] [PubMed] [Google Scholar]
- 51.Rozovsky I., Finch C. E., Morgan T. E. (1998) Age-related activation of microglia and astrocytes: in vitro studies show persistent phenotypes of aging, increased proliferation, and resistance to down-regulation. Neurobiol. Aging 19, 97–103 [DOI] [PubMed] [Google Scholar]
- 52.Qin L., Wu X., Block M. L., Liu Y., Breese G. R., Hong J. S., Knapp D. J., Crews F. T. (2007) Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 55, 453–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhou J., Huang W. Q., Li C., Wu G. Y., Li Y. S., Wen S. H., Lei W. L., Liu K. X. (2012) Intestinal ischemia/reperfusion enhances microglial activation and induces cerebral injury and memory dysfunction in rats. Crit. Care Med. 40, 2438–2448 [DOI] [PubMed] [Google Scholar]
- 54.Liu B., Wang K., Gao H. M., Mandavilli B., Wang J. Y., Hong J. S. (2001) Molecular consequences of activated microglia in the brain: overactivation induces apoptosis. J. Neurochem. 77, 182–189 [DOI] [PubMed] [Google Scholar]
- 55.Pei Z., Pang H., Qian L., Yang S., Wang T., Zhang W., Wu X., Dallas S., Wilson B., Reece J. M., Miller D. S., Hong J. S., Block M. L. (2007) MAC1 mediates LPS-induced production of superoxide by microglia: the role of pattern recognition receptors in dopaminergic neurotoxicity. Glia 55, 1362–1373 [DOI] [PubMed] [Google Scholar]
- 56.Geriatric Study in Europe on Health Effects of Air Quality in Nursing Homes Group (2012) Adverse respiratory effects of outdoor air pollution in the elderly. Int. J. Tuberc. Lung Dis. 16, 1149–1161 [DOI] [PubMed] [Google Scholar]
- 57.Weuve J., Puett R. C., Schwartz J., Yanosky J. D., Laden F., Grodstein F. (2012) Exposure to particulate air pollution and cognitive decline in older women. Arch. Intern. Med. 172, 219–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen J. C., Schwartz J. (2009) Neurobehavioral effects of ambient air pollution on cognitive performance in US adults. Neurotoxicology 30, 231–239 [DOI] [PubMed] [Google Scholar]
- 59.Liao K. M., Ho C. H., Ko S. C., Li C. Y. (2015) Increased risk of dementia in patients with chronic obstructive pulmonary disease. Medicine (Baltimore) 94, e930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dodd J. W. (2015) Lung disease as a determinant of cognitive decline and dementia. Alzheimers Res. Ther. 7, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liao W. C., Lin C. L., Chang S. N., Tu C. Y., Kao C. H. (2015) The association between chronic obstructive pulmonary disease and dementia: a population-based retrospective cohort study. Eur. J. Neurol. 22, 334–340 [DOI] [PubMed] [Google Scholar]
- 62.Peng Y. H., Wu B. R., Su C. H., Liao W. C., Muo C. H., Hsia T. C., Kao C. H. (2015) Adult asthma increases dementia risk: a nationwide cohort study. J. Epidemiol. Community Health 69, 123–128 [DOI] [PubMed] [Google Scholar]
- 63.Chen M. H., Li C. T., Tsai C. F., Lin W. C., Chang W. H., Chen T. J., Pan T. L., Su T. P., Bai Y. M. (2014) Risk of dementia among patients with asthma: a nationwide longitudinal study. J. Am. Med. Dir. Assoc. 15, 763–767 [DOI] [PubMed] [Google Scholar]
- 64.Thomson E. M., Vladisavljevic D., Mohottalage S., Kumarathasan P., Vincent R. (2013) Mapping acute systemic effects of inhaled particulate matter and ozone: multiorgan gene expression and glucocorticoid activity. Toxicol. Sci. 135, 169–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Montagne A., Barnes S. R., Sweeney M. D., Halliday M. R., Sagare A. P., Zhao Z., Toga A. W., Jacobs R. E., Liu C. Y., Amezcua L., Harrington M. G., Chui H. C., Law M., Zlokovic B. V. (2015) Blood-brain barrier breakdown in the aging human hippocampus. Neuron 85, 296–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Da Fonseca A. C., Matias D., Garcia C., Amaral R., Geraldo L. H., Freitas C., Lima F. R. (2014) The impact of microglial activation on blood-brain barrier in brain diseases. Front. Cell. Neurosci. 8, 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Oppenheim H. A., Lucero J., Guyot A. C., Herbert L. M., McDonald J. D., Mabondzo A., Lund A. K. (2013) Exposure to vehicle emissions results in altered blood brain barrier permeability and expression of matrix metalloproteinases and tight junction proteins in mice. Part. Fibre Toxicol. 10, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Perry V. H., Cunningham C., Holmes C. (2007) Systemic infections and inflammation affect chronic neurodegeneration. Nat. Rev. Immunol. 7, 161–167 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.