

Abstract
Recently we have reported that age-dependent decline in antioxidant levels accelerated apoptosis and skeletal muscle degeneration. Here, we demonstrate genetic ablation of the master cytoprotective transcription factor, nuclear factor (erythroid-derived-2)–like 2 (Nrf2), aggravates cardiotoxin (CTX)-induced tibialis anterior (TA) muscle damage. Disruption of Nrf2 signaling sustained the CTX-induced burden of reactive oxygen species together with compromised expression of antioxidant genes and proteins. Transcript/protein expression of phenotypic markers of muscle differentiation, namely paired box 7 (satellite cell) and early myogenic differentiation and terminal differentiation (myogenin and myosin heavy chain 2) were increased on d 2 and 4 postinjury but later returned to baseline levels on d 8 and 15 in wild-type (WT) mice. In contrast, these responses were persistently augmented in Nrf2-null mice suggesting that regulation of the regeneration-related signaling mechanisms require Nrf2 for normal functioning. Furthermore, Nrf2-null mice displayed slower regeneration marked by dysregulation of embryonic myosin heavy chain temporal expression. Histologic observations illustrated that Nrf2-null mice displayed smaller, immature TA muscle fibers compared with WT counterparts on d 15 after CTX injury. Improvement in TA muscle morphology and gain in muscle mass evident in the WT mice was not noticeable in the Nrf2-null animals. Taken together these data show that the satellite cell activation, proliferation, and differentiation requires a functional Nrf2 system for effective healing following injury.—Shelar, S. B., Narasimhan, M., Shanmugam, G., Litovsky, S. H., Gounder, S. S., Karan, G., Arulvasu, C., Kensler, T. W., Hoidal, J. R., Darley-Usmar, V. M., Rajasekaran, N. S. Disruption of nuclear factor (erythroid-derived-2)–like 2 antioxidant signaling: a mechanism for impaired activation of stem cells and delayed regeneration of skeletal muscle.
Keywords: TA muscle, Pax7, Nrf2/ARE, satellite cell, cardiotoxin
Daily activities related to normal wear and tear of muscles, along with strains associated with overload, sports, drugs, chronic pain, ischemia, neurologic dysfunctions, and other disease states, either acutely or chronically impair the normal functioning of muscle (1–4). However, being a durable tissue, adult skeletal muscle possesses an extensive regenerative capacity that allows maintenance of muscle function following injury (5). Optimal recovery is influenced by the type of muscle, severity of injury, and the nature of treatment. In most cases, muscle damage follows 3 phases: destruction, inflammation, and repair and remodeling (6, 7). Effective regeneration of single muscle fibers or the complete muscle occurs via activation and proliferation of satellite cells (8). In healthy muscles, the bulk of the muscle satellite cells remain quiescent in their sublaminal niche and are transcriptionally inactive (9, 10). Following muscle damage, satellite cells migrate either to the damaged area or to the adjacent myofibers depending on the intactness of basal lamina during the injury to repair and remodel it (9, 11). Particularly, upon muscle injury, the satellite cells egress from G0 and rapidly enter the cell cycle and proliferate (satellite cell activation) (12, 13). Furthermore, activated satellite cells are capable of inducing the transcription of Myf5 and myogenic differentiation (MyoD) to promote differentiation into myoblasts, which later fuse to form myofibers (8). Thus, any impairment in the activation and/or satellite cell number or the migration or combination of these processes is shown to impair muscle regeneration (14). Although nonsatellite cells could supply myogenic precursors and orchestrate regeneration, their contribution has been shown to be low to negligible (15–19).
The current treatment of muscle injury is often plagued by partial or incomplete functional recovery due to the slower healing as well as development of fibrosis and scars after the injury (20). Therefore, a better understanding of the biologic and pathophysiologic processes of muscle repair following injury is needed. Several pathways including redox signaling have been identified to be involved in the inefficient and/or compromised muscle regenerative ability (21–23). Particularly, redox-dependent regulation is involved in self-renewal of stem cells and myogenic differentiation (12, 24, 25). Previous findings from our laboratory have demonstrated that either ablation or age-dependent decline of the transcription factor, nuclear factor (erythroid-derived-2)–like 2 (Nrf2), accelerates skeletal muscle degeneration through induction of apoptotic and ubiquitin-mediated protein degradation pathways (26, 27). Nrf2 is a master regulator of cellular oxidative stress and is normally bound to Kelch like-ECH-associated protein 1 (Keap1) in the cytosol, but Keap1 is inactivated in response to oxidative stress allowing nascent Nrf2 to translocate into the nucleus and to bind to the antioxidant response elements in the promoter region of multiple cytoprotective/antioxidative genes (28). However, the role of Nrf2 signaling on the regulation of stem cell activation and differentiation in tibialis anterior (TA) muscle regeneration remains elusive.
Although the degenerative and the regenerative phases of the muscle repair process largely overlap among varying muscle types, the kinetics and length of each phase may vary depending on the severity of injury, the muscle injured, and the animal model interrogated (29–31). In the current study, we evaluated the role of Nrf2 in the regenerative ability of TA muscle in response to CTX, a peptide isolated from snake venom that inhibits PKC and induces the depolarization and contraction of muscular cells thereby disrupting membrane organization and leading to lysis of the cells. The myotoxin CTX is capable of producing phenotypes such as necrosis, inflammation, sarcolemma disruption, and so on that are commonly noted in muscular dystrophies and inflammatory myopathies (32). Although CTX injury model is studied in cell lines and animals to understand signal transduction alterations, membrane damages, cell death, and so on (33, 34), its effect in the background of absence of Nrf2 in TA muscle injury is not studied. Our results demonstrate that loss of Nrf2 promotes CTX-induced oxidative stress and impairs the timely activation, proliferation, and differentiation of myogenic progenitors, thus retarding the regeneration of TA muscle.
MATERIALS AND METHODS
Reagents and sources
Catalase (219010) was from Calbiochem (San Diego, CA, USA). Glutathione reductase (GSR; ab16801), NQO1 (ab34173), GCLC, GCLM (ab18445), PAX7 (ab92317), and myogenin (ab124800) were obtained from Abcam (Cambridge, MA, USA). Anti-GAPDH (51745), anti-proliferating cell nuclear antigen (2586), anti-Ki67 (912) was from Cell Signaling Technology (Danvers, MA, USA). SOD1 (ADI-SOD-100) and SOD2 (ADI-SOD-110) were obtained from Enzo Life Sciences (Farmingdale, NY, USA). Anti-laminin antibody (L9393), cardiotoxin (CTX; V9000), and all other fine chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA).
CTX-mediated TA muscle injury, regeneration, and tissue collection
Wild-type (WT) and Nrf2−/− mice (C57Bl6J male and female at 6–8 mo of age) were used to study the effect of redox imbalance on postinjury regeneration of skeletal muscle. Mice were fed with water and food ad libitum, housed under controlled temperature and humidity and 12-h light-dark cycles. The experimental protocol was approved by Institutional Animal Care and Use Committee (University of Utah and University of Alabama at Birmingham). Hind limbs of mice (n = 3–6/d per group) were shaved a day before the CTX injection, and on the day of treatment, sterile filtered 10 µM CTX in PBS was injected into TA muscle to induce muscle injury. Control animals received an equal volume of PBS. Mice were observed daily for behavioral and physical changes and visible changes at the site of injury were recorded. Mice were euthanized after CTX injury (d 0, 2, 4, 8, and 15) by CO2 inhalation and TA muscles collected for histologic and biochemical analyses. Samples for RNA isolation were immersed in 300 µl of RNAlater (Sigma-Aldrich) for 45–60 min at room temperature and frozen at −80°C until analyzed. For histochemical analysis (hematoxylin and eosin staining), tissues were stored into 10% formalin at room temperature and for the reactive oxygen species (ROS) determination, tissues were immersed into optimal cutting temperature compound and frozen at −80°C after snap freezing in liquid nitrogen.
ROS determination in TA muscle
ROS levels in TA muscle were measured after CTX-mediated muscle injury and during regeneration process by fluorescent probes 2′7′-dichlorodihydrofluorescein diacetate and dihydroethidium (DHE) that is associated with the intracellular levels of peroxides (H2O2) and superoxide (O2−.), respectively. CTX/PBS-treated TA muscles were collected at the indicated times and the tissues were cryosectioned (10 µM, transverse section) and placed on clean glass slide for fluorescence microscopy. Cryosections were stained with CM-2′7′-dichlorodihydrofluorescein diacetate (C6827; Thermo Fisher Scientific, Waltham, MA, USA) and DHE (D11347; Thermo Fisher Scientific) as described earlier (35), and fluorescence images were acquired using laser scanning confocal microscopy (Zeiss 510; Carl-Zeiss, Jena, Germany). Fluorescence intensity of the images was analyzed using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Quantitative real-time-PCR
Total RNA was extracted from ∼30 mg TA muscle tissues treated CTX/PBS. Following genomic DNA elimination, RNA was isolated using RNeasy extraction kit (Qiagen, Germantown, MD, USA) by following manufacturer’s instructions. cDNA was synthesized with 1.25 µg of RNA using Quantitect cDNA synthesis kit (Qiagen). Quantification of mRNA levels was performed using gene specific primers (1 pmol) and cDNA (50 ng) in a 10 µl final reaction mixture. Quantifast SYBR green PCR kit (Qiagen) was used for amplification and detection using Light Cycler (Roche, Basel, Switzerland). Relative mRNA levels of genes were calculated using differences in CT values among the different groups, which were normalized to mRNA levels of housekeeping gene Gapdh. Details of primer assays were provided in Table 1.
TABLE 1.
Primers used for semiquantitative and quantitative PCR
| Gene | Qiagen catalogue |
|---|---|
| Nrf2 | QT00095270 |
| Keap1 | QT00147371 |
| Gapdh | QT01658692 |
| Pax7 | QT00147728 |
| Myod1 | QT00101983 |
| Myh2 | QT01060850 |
| Gclc | QT00130543 |
| Gclm | QT00174300 |
| Nqo1 | QT00094367 |
| G6pd | QT00120750 |
| Gsr | QT01758232 |
| Sod1 | QT00165039 |
| Sod2 | QT00161707 |
Preparation of protein lysates for immunoblotting
Tissue was homogenized using cytosol extraction buffer (10 mM HEPES, 10 mM KCl, 0.1 mM EDTA, 0.5 mM MgCl2, pH 7.9) containing dithiothreitol (1 mM), phenyl methyl-sulfonyl fluoride (0.1 mM), 1% Triton, protease-phosphatase inhibitor cocktail (Roche and Sigma-Aldrich) and clarified by centrifugation at 5000 rpm for 5–6 min. Equal concentrations (10–20 µg) of protein lysates were mixed with 4× loading dye containing 5% β-mercaptoethanol (Bio-Rad, Hercules, CA, USA) followed by boiling for 5 min and subjected to SDS-PAGE separation. The proteins were then electrotransferred onto PVDF membrane (Immobilon-P; EMD Millipore, Billerica, MA, USA) and blocked with 5% (W/V) nonfat milk in 1× TBS-T for 2 h. Membranes were then incubated with respective primary antibodies (1:200 to 1:5000) in 1× TBS-T containing 1–2% bovine serum albumin for 2 h to overnight at 4°C. Following 5 × 10 min TBS-T washes, the membranes were incubated with horseradish peroxidase-conjugated anti-rabbit/anti-mouse (1:10,000 to 1:25,000) antibodies (Vector Laboratories, Burlingame, CA, USA) for 1 h, washed 5 × 10 min with 1× TBS-T, and subjected to chemiluminescence detection using ECL kit (Pierce, Rockford, IL, USA). Intensity of appropriate immunoreactive signals was visualized by autoradiography and the quantification was performed using ImageJ software.
Immunofluorescence microscopy
TA muscles frozen in optimal cutting temperature compound were cryosectioned (10 µM, transverse section) and placed on clean glass slides for fluorescence microscopy. Briefly, tissue sections were fixed in acetone permeabilized using 0.5% Triton × 100 in PBS and blocked with 10% goat serum in PBS. Sections were then probed with embryonic myosin heavy chain [anti-embryonic myosin heavy chain (MHC-emb): red, anti-laminin: green/red, paired box (Pax)7-green/red, Ki67-red and DAPI: blue/nucleus] for 2 h followed by incubation with anti-rabbit antibody conjugated with Alexa Fluor 488 and 594 for 2 h. The sections were mounted with Vectasheild mounting medium (Vector Laboratories, Burlingame, CA, USA) containing nuclear stain DAPI. Immunofluorescence images were captured using epifluorescence microscope.
Histologic analysis of TA muscles
For histologic analysis, formalin-fixed and paraffin-embedded TA muscles were cryosectioned and placed on clean glass slides. Sections were deparaffinized using xylene and ethanol, and stained with hematoxylin. Following washes and destaining with 0.3% acid ethanol, and the sections were stained with eosin for 2 min and washed again with organic solvents xylene and ethanol. The sections were dried and mounted on a coverslip using mounting medium. Images were captured using light microscopy.
In vitro model for myogenic differentiation
Satellite cell-mimetic C2C12 myoblasts (passage number <25) were cultured in proliferation medium (PM) (DMEM containing 10% fetal bovine serum, 100 U penicillin/streptomycin and 1 mM l-glutamine). At 70–80% confluence, myogenic differentiation was induced by replacing PM with differentiation medium (DMEM containing 2% horse serum and 100 U of penicillin/streptomycin). Fresh differentiation medium was replenished every 24 h, and cells were subjected to CTX treatment and experimental analysis.
Cytotoxicity assay
C2C12 cells were cultured in 96-well plate and treated with 0.1 to 4 µM CTX for 24 h, and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) (20 µl of 1 mg/ml in PBS) was added to each well and incubated for 3 h at 37°C in the incubator. The formazan crystals formed were dissolved in 100 µl DMSO containing 1% glacial acetic acid and absorbance of dissolved crystals was measured at 570 nm. The percentage viability of CTX-treated cells was calculated relative to the untreated controls.
Nrf2 knockdown using shRNA and wound-healing assay
C2C12 cells were cultured in PM and at 50% confluence; cells were transfected with either 5 µg of control or small hairpin (sh)RNA plasmids specific against Nrf2 (TR30021 and TL515053A; OriGene Technologies, Rockville, MD, USA) using Lipofectamine 3000 (Thermo Fisher Scientific) and allowed to grow for 24 h. When the cells reached 70–80% confluence they were treated with 0.5 µM of CTX for 24 h. Following treatment, cells were artificially wounded by scratching the cells in the center across the well with a sterile pipette tip cut at the bottom. After wounding, the culture medium was changed and then myoblasts were allowed to proliferate for additional 24 h with or without CTX. Light microscopy images of the wound closure were captured at 0 and 24 h. The percentage of wound healing was calculated by measuring the differences between the initial (0 h) surface area of the wound with the cell-covered areas at 24 h.
Statistical analysis
Statistical analysis was performed using Student’s t test or 1- way ANOVA followed by Student’s Newman–Keul correction.
RESULTS
Ablation of Nrf2 affects TA muscle growth in response to CTX-induced injury
Deletion of Nrf2 was confirmed in TA muscles using semiquantitative real-time PCR and RT-PCR for Nrf2 expression. As expected, Nrf2 transcript was undetectable in TA muscle of Nrf2−/− as compared with WT mice (Fig. 1A, B) with no significant changes in the expression of Keap1 (Fig. 1A, B). Among the numerous skeletal muscle injury models such as crush, freeze, and contusion, the chemical induction by CTX is widely used (5). CTX was injected intramuscularly into TA muscles of the experimental animals, and subsequent analyses was performed as indicated in Fig. 1C. The average weight of CTX and PBS-injected WT and Nrf2−/− mice TA muscles at different time points (2, 4, 8, and 15 d post-CTX injury) of muscle regeneration are presented in Fig. 1D. The weight of TA muscle normalized to the body weight of the respective animal was found to be unaltered among the control (PBS-treated) WT and Nrf2−/− animals over the time course. In contrast, 2 d after CTX administration, the weight of the TA muscle mass was significantly decreased in both WT (1.5 ± 0.1 mg) and Nrf2−/− (1.3 ± 0.05 mg) mice compared with the PBS group (2.2 ± 0.3 mg). The muscle weight was slightly increased after d 4 in WT (1.7 ± 0.09) and Nrf2−/− (1.47 ± 0.12) mice compared with that observed on d 2 after CTX treatment, but was still significantly lower in Nrf2−/− mice than in the control animals. Progressive increase in TA muscle weight was observed in WT mice after 8 (1.96 ± 0.39) and 15 (2.2 ± 0.3) d of postinjury, but the weight of TA muscle in Nrf2−/− mice was still significantly lower (1.4 ± 0.27; 1.76 ± 0.17) at the respective time points. Thus the relative inability of Nrf2−/− mice to regain muscle mass at the end of 8 and 15 d post-CTX injury is suggestive of defective skeletal muscle regeneration and/or growth when the Nrf2 signaling pathway is disrupted.
Figure 1.
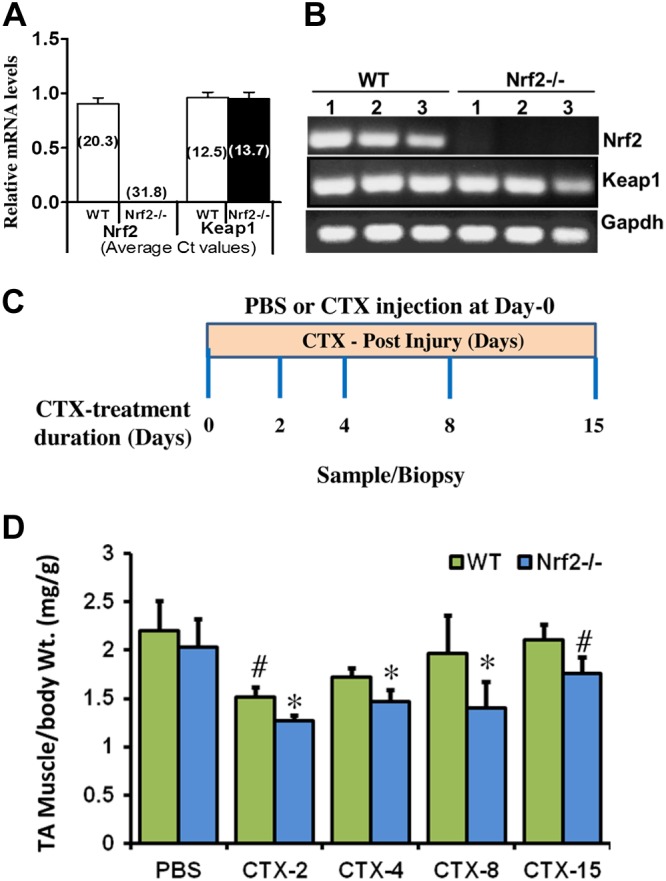
Model for CTX-induced skeletal muscle injury and regeneration. A, B) Confirmation of genotypes of WT and Nrf2−/− mice by analysis of relative mRNA levels of Nrf2 and Keap1 genes, analyzed by real-time quantitative PCR (A) and traditional PCR analysis (B) in TA muscle. C) Experimental strategy for CTX-mediated TA muscle injury and post-CTX injury regeneration of skeletal muscle regeneration. WT and Nrf2−/− test animals injected with 10 μM CTX in PBS into the TA muscle and control animals received equal volumes of PBS. D) Graphical presentation of means ± sd of TA muscle (mg)/body weight (g) of WT and Nrf2−/− mice to measure the CTX-mediated TA muscle damage and post-CTX injury regeneration at the indicated points. n = 4/d per group. Statistical significance was calculated by 1-way ANOVA. #P < 0.05 PBS vs. CTX-WT, *P < 0.05 CTX-WT vs. Nrf2−/−.
Genetic ablation of Nrf2 impairs antioxidant gene and protein expression in CTX-injured TA muscle
To assess the effect of genetic loss of Nrf2 on antioxidative genes in TA muscle, we analyzed the gene expression of NAD(P)H dehydrogenase quinone 1 (Nqo1) (Fig. 2A), glutamate-cysteine ligase, catalytic subunit (Gclc) (Fig. 2B), glucose 6-phosphate dehydrogenase (G6pd) (Fig. 2C), superoxide dismutase 1 (Sod1) (Fig. 2D), superoxide dismutase 2 (Sod2) (Fig. 2E), glutathione reductase (Gsr) (Fig. 2F), glutamate-cysteine ligase, modifier subunit (Gclm) (Fig. 2G). The gene expression of Gclc, Gclm, and Nqo1 followed the same trend, while G6pd and Gsr mRNA peaked at d 2 in WT mice after CTX injury and started declining afterward. Although Sod1 and Sod2 are also targets of Nrf2 regulation, we did not observe any significant changes in WT after CTX injury. In contrast, Nrf2−/− mice showed blunted responses to the temporal regulation of antioxidant gene expression. Along with these changes, we observed an initial increase in Nrf2 mRNA in WT mice subjected to CTX-mediated injury that peaked at d 4 and then started declining to reach normal levels at the end of d 15 post-CTX injury (Fig. 2H). To confirm whether the changes in transcription observed in antioxidant genes is reflected at the level of translational, immunoblotting analysis of these antioxidant proteins was performed. We observed that levels of antioxidant proteins such as NQO1 (Fig. 3A–D, F), GCLC (Fig. 3A–D, G), GSR (Fig. 3E, K), and catalase (Fig. 3E, L) were significantly attenuated in Nrf2−/− mice recovering from CTX injury compared with its WT counterparts. The expression of G6PD and SOD2 showed varying response in WT and Nrf2−/− mice subjected to CTX injury (Fig. 3A–D, H, J) and similar to the transcript changes, the protein levels of SOD1 were not significantly altered (Fig. 3A–D, I). Taken together, these data indicate that genes coding for enzymes involved in GSH synthesis (Gclc, Gclm) along with expression of some of the antioxidant genes and proteins were severely dampened in Nrf2−/− mice following CTX injury. Furthermore, the lower levels of electrophile response element-driven antioxidant genes observed in TA muscles of Nrf2−/− mice injured with CTX indicating, as expected, that these animals are unable to regulate critical antioxidant and redox regulatory enzymes.
Figure 2.

Down-regulation of Nrf2-regulated antioxidant gene expression in the CTX-injured regenerating TA muscles of Nrf2−/− mice. Transcript levels of Nrf2 (A), Gclc (B), Gclm (C), Nqo1 (D), G6pd (E), Gsr (F), Sod1 (G), and Sod2 (H) were determined in WT and Nrf2−/− TA muscle by quantitative RT-PCR. The relative gene expression was calculated by normalizing the mRNA levels of gene of interest with the levels of housekeeping gene, Gapdh. n = 4/d per group. Statistical significance was calculated by 1-way ANOVA. #P < 0.05 PBS vs. CTX-WT, *P < 0.05 CTX-WT vs. Nrf2−/−.
Figure 3.

Poor recovery of Nrf2-regulated antioxidant protein levels in regenerating TA muscles of Nrf2−/− mice injured with CTX. A–D) Protein levels of Nrf2-regulated antioxidants (catalase, NQO1, GCLC, G6PD, SOD1, and SOD2) were determined by Western blot analysis of tissues homogenates of regenerating TA muscles collected after CTX-mediated injury and regeneration on d 2 (A), d 4 (B), d 8 (C), and d 15 (D). E) Western blots for GSR investigating changes in its levels during post-CTX injury regeneration of TA muscle of WT and Nrf2−/−. F–L) ImageJ densitometry quantification for NQO1 (F), GCLC (G), G6PD (H), SOD1 (I), SOD2 (J), GSR (K), and catalase (L) immunoblots. n = 4/d per group. Statistical significance was calculated by 1-way ANOVA. #P < 0.05 PBS vs. CTX-WT, *P < 0.05 CTX-WT vs. Nrf2−/−.
Nrf2 loss augments the ROS accumulation upon CTX-induced muscle injury
Generation/accumulation of superoxide was found to be higher in Nrf2−/− than WT mice TA muscle 2, 4, and 8 d after CTX-induced injury (Fig. 4A, C). However, the burden of superoxide accumulation after CTX injection was comparatively decreased at all-time points in WT when compared with Nrf2 knockouts. By d 8 postinjury, the ROS signals were comparable between CTX and PBS-injected WT mice. However, the ROS signals were still higher in Nrf2−/− than those of WT mice at 15 d post-CTX injury. Next, to ensure the load of ROS (especially, H2O2) during muscle regeneration, we performed H2DCFH-DA staining in CTX-induced TA muscles of WT and Nrf2−/− mice (Fig. 4B, D). Similar to superoxide changes, CTX injury induced higher level of hydrogen peroxide in Nrf2−/− mice than those of WT mice TA muscle at all points. Thus these results clearly indicate a persistent pro-oxidant status associated with ablation of Nrf2 in CTX injury model.
Figure 4.

Nrf2-deficient mice displayed increased levels of ROS in CTX-injured regenerating TA muscle. A, B) Frozen TA muscle sections from control and CTX-treated WT and Nrf2−/− mice were loaded with fluorescent ROS probes: DHE (A) and chloromethyl derivative of 2′7′-dichlorodihydrofluorescein diacetate (CM-DCFDA) (B) were processed for fluorescent microscope (×40 magnification) analysis. C, D) The mean fluorescence intensity of DHE (red fluorescence) (C) and CM-DCFDA (green fluorescence) (D) were measured by ImageJ software; n = 4/d per group. Statistical significance was calculated by 1-way ANOVA. #P < 0.05 PBS vs. CTX-WT, *P < 0.05 CTX-WT vs. Nrf2−/−.
Role of Nrf2 in satellite cell activation and proliferation upon TA muscle injury
To investigate the role of Nrf2 on satellite cell response (activation and proliferation), we measured the protein and mRNA levels of Pax7 in normal (PBS/basal) and regenerating TA muscle of Nrf2−/− mice after CTX injection for comparison with control WT mice. Quantitative PCR analyses showed the levels of Pax7 mRNA expression were significantly increased in WT and Nrf2−/− mice on d 2 after postinjury when compared with PBS-injected control (d 0) mice (Fig. 5A); however, this response was considerably higher in the WT than the Nrf2−/− mice. Indeed, the changes on Pax7 transcript expression on d 4, 8, and 15 post-CTX injury followed a similar trend. PAX7 protein expression was significantly increased in both WT (∼100-fold) and Nrf2−/− (∼80 fold) mice 2 d after CTX injury as compared with PBS-injected WT and Nrf2−/− mice (Fig. 6). Furthermore, the PAX7 protein was constantly upregulated (∼70 fold) in regenerating WT mice even after d 4 when compared with d 0. Of note, PAX7 protein expression was significantly lower (∼25 fold) in Nrf2−/− mice after d 4 than that of 2 d postinjury. On d 8 postinjury, PAX7 expression fell to a level comparable to PBS-injected WT mice. By contrast, at d 8 post-CTX injury PAX7 protein level was considerably higher in CTX-injected Nrf2−/− mice when compared with CTX-injected WT mice. By d 15 of postinjury PAX7 expression decreased in both WT and Nrf2−/− mice to near basal levels. In summary, both transcript and protein levels of Pax7 were up-regulated in response to CTX-mediated muscle injury in WT and Nrf2−/− mice. However, Pax7 levels were comparatively higher in WT mice shortly after CTX administration/injury and resolved by d 8 in WT but not in Nrf2−/− mice.
Figure 5.

Delayed transcriptional activation of skeletal muscle regeneration markers in regenerating TA muscle of Nrf2−/− mice. Real-time quantitative PCR analyses of skeletal muscle regeneration markers Pax7 (A), Myod1 (B), and Myh2 (C) in TA muscle of PBS and CTX-treated WT and Nrf2−/− mice. Relative mRNA levels were calculated by normalizing to Gapdh expression. n = 4/d per group. Statistical significance was calculated by 1-way ANOVA. #P < 0.05 PBS vs. CTX-WT, *P < 0.05 CTX-WT vs. Nrf2−/−.
Figure 6.

Loss of Nrf2 affects the expression of skeletal muscle regeneration markers during CTX-mediated TA muscle injury and regeneration. A, C) Immunoblot analyses for PAX7 and MYOD1 (A) and myogenin (C) in TA muscle of PBS and CTX-treated WT and Nrf2−/− mice. B, D) Relative intensity of the PAX7 and MYOD1 (B) and myogenin (D) protein signals normalized to GAPDH was calculated using ImageJ software; n = 4/d per group. Statistical significance was calculated by 1-way ANOVA. #P < 0.05 PBS vs. CTX-WT, *P < 0.05 CTX-WT vs. Nrf2−/−.
Similarly, immunostaining of Pax7 showed significantly differed expression of Pax7 in regenerating TA muscle of WT and Nrf2−/− mice. Nrf2 deletion was found to delay the expression of Pax7 in post-CTX-injected TA muscle (Fig. 7A, B). Furthermore, we investigated whether the activation and proliferative potential of satellite cells were also impaired under aggravated oxidizing conditions in Nrf2 deficient mice. Proliferation markers (PCNA and Ki67) were studied in the regenerating TA muscle of WT and Nrf2−/− mice. As shown in Fig. 7C, D, the expression of PCNA was significantly protracted even at 8–15 d after CTX in regenerating TA muscle of Nrf2−/− mice, whereas in WT it started to fall back to normal at 8 d and appeared similar to control at 15 d. Ki67 was also studied by immunofluorescence staining of regenerating TA muscle. As seen in Supplemental Fig. 1, Ki67 was expressed significantly on d 2 and d 4 in TA muscle of WT and Nrf2−/− mice. However, the levels of Ki67 were significantly lower in Nrf2−/− mice. Overall, the significantly different expression of Pax7, PCNA, and Ki67 in WT and Nrf2−/− mice confirms the impaired proliferation/activation of satellite cells in regenerating TA muscles of Nrf2−/− mice.
Figure 7.

Absence of antioxidant defense hampers activation and proliferation of satellite cells in regenerating TA muscle. A) Immunofluorescence images showing Pax7+ satellite cells (green fluorescence) and the muscle cell plasma membrane protein laminin staining (red fluorescence) of TA muscles from WT (a, c, e, g, i) and Nrf2−/− (b, d, f, h, j) mice injected with PBS (a, b) and CTX (c–j) and collected on d 2 (c, d), 4 (e, f), 8 (g, h), and 15 (i, j) postinjury (×40 magnification). B) Graphs represent the mean fluorescence intensity of Pax7 expression. C) Western blot analysis of proliferation marker PCNA in the TA muscle of WT and Nrf2−/− mice. D) Densitometric analysis of PCNA expression was calculated using ImageJ software and the data are presented in a bar graph. n = 3/d per group. Statistical significance was calculated by 1-way ANOVA. #P < 0.05 PBS vs. CTX-WT, *P < 0.05 CTX-WT vs. Nrf2−/−.
Effect of Nrf2 on the expression of early and terminal muscle differentiation biomarkers in regenerating skeletal muscle
Transcription factors MyoD and Myf5 determine the differentiation potential of activated myoblasts downstream of Pax7 (36–38). We thus sought to assess the expression of these myogenic regulatory factors after injury caused by CTX administration, in Nrf2−/− and WT mice (Figs. 5 and 6). The level of MyoD was significantly increased in both Nrf2−/− and WT mice on d 2 post-CTX injury when compared with PBS-injected control animals, although to a dampened extent in Nrf2−/− mice. On d 4 post-CTX injury, elevated MyoD expression was maintained in WT mice. Whereas, the expression level of MyoD in Nrf2−/− mice was significantly decreased when compared with d 2 post-CTX injury. Eight days after CTX injury, MyoD expression declined in the WT to near baseline. In contrast, Nrf2−/− mice still exhibited substantial expression of MyoD at d 8. On d 15, although MyoD signal was absent in WT, the Nrf2−/− mice showed a modest MyoD expression (Fig. 5B). Similarly, immunoblot analysis demonstrated that MYOD protein expression was markedly increased from d 2 to 4 after CTX injury in WT and Nrf2−/− mice and returned to basal level in WT mice thereafter, but not in Nrf2−/− mice (Fig. 6A, B). These data suggested that differentiation started at least by d 2 post-CTX injury and was terminated by d 8 in WT mice, but was prolonged until d 15 in Nrf2−/− mice.
In adult skeletal muscle, the transcription factor, myogenin regulates metabolic activity and plays an active role in the terminal differentiation of muscle fibers. Thus we determined the expression of myogenin during skeletal muscle regeneration in CTX injured Nrf2−/− and WT mice (Fig. 6C, D). Immunoblot analyses indicated that myogenin expression was not detected in either WT or Nrf2−/− until d 4 after CTX induced injury, indicating that the terminal differentiation program had yet to commence. After d 8 post-CTX injury, myogenin expression was higher in WT compared with Nrf2−/− mice. This indicates that the terminal differentiation was effectively initiated at d 8 after injury in WT but not in Nrf2−/− mice. Expression of myogenin in WT mice was extensively reduced at d 15 postinjury in comparison to d 8 indicating the cessation of the terminal differentiation process. In contrast, myogenin was still robustly expressed in Nrf2−/− mice on d 15 postinjury, indicating that the terminal differentiation was incomplete and progressing in the Nrf2−/− mice. Furthermore, mRNA expression for myosin heavy chain (Myh)2, a terminal differentiation factor, was analyzed to confirm our findings (Fig. 5C). The transcript level of Myh2 was undetectable in WT and Nrf2−/− mice on d 2 postinjury compared with control animals. Thereafter, the transcription level was modestly increased in WT but not in Nrf2−/− on d 4 postinjury. On d 8 after CTX injury, the Myh2 mRNA expression was substantially increased up to 8 fold in WT with a minor increase in Nrf2−/− when compared with control animals. On d 15 postinjury, the expression of Myh2 had decreased to 5.5 fold in WT mice but still remained significantly higher than that of control animals. In contrast, the Mhy2 transcript level was further augmented on d 15 in CTX-injured Nrf2−/− mice, which were comparatively 2-fold higher than that of d 8 in the CTX-injured Nrf2−/− mice. Altogether these results indicate that the orderly and temporal pattern of expression of the classic early and terminal differentiation markers displayed by WT mice is disturbed and broadly expanded in Nrf2−/− mice, consistent with a delay in the regenerative process in response to CTX injury.
Histologic analysis of TA muscle showing impaired regeneration in Nrf2−/− mice
Next, we examined whether the genetic ablation of Nrf2 impairs the skeletal muscle regeneration after CTX injury. Effect of administration of 10 μM CTX into anterior tibialis muscles of WT and Nrf2−/− mice was evaluated at different times by histology (Fig. 8). Necrotic degeneration of muscle fibers with prominent edema was observed in WT and Nrf2−/− mice on d 2 after CTX injury (Fig. 8C, D). The inflammatory infiltrate was greater in the post-CTX injury group. At d 4 postinjury, significant regeneration had occurred in WT TA muscle with numerous newly generated myofibers containing centrally located nuclei (Fig. 8E). In contrast, the damage with edema and inflammation still persisted in the Nrf2−/− TA muscle (Fig. 8F). On d 8, regenerating multinucleated myofibers were found with increased cross section of myofibers in WT mice when compared with d 4 post-CTX injury. Although regeneration and formation of myogenic fibers of TA muscle is detected in Nrf2−/− mice on d 8, the size of the myogenic fibers remained smaller than those of WT mice (Fig. 8H). Notably, the improvement of TA muscle morphology 8 d postinjury of Nrf2−/− was comparable only to that of WT mice 4 d postinjury (Fig. 8). On d 15 postinjury, the size and structural orientation of WT muscle fibers were almost identical among WT and Nrf2−/− PBS-injected mice. Smaller TA muscle myofibers were seen in Nrf2−/− mice than the WT (Fig. 8I–J), suggesting a still incomplete regeneration process.
Figure 8.

Regeneration of CTX-induced TA muscle injury was delayed in Nrf2−/− mice. Bright field images of hematoxylin and eosin-stained cross sections of TA muscle injected with PBS (A, B) and post-CTX injury TA muscles collected on d 2 (C, D), d 4 (E, F), d 8 (G, H) and d 15 (I, J) of WT (A, C, E, G, I) and Nrf2−/− (B, D, F, H, J) mice (n = 4–5 mice/group). On d 2 postinjury, TA muscles of WT and Nrf2−/− mice show formation of edema and inflammatory infiltration (C, D) (black arrow). On d 4 (E, G) and d 8 postinjury (H), muscles show the presence of multinucleated myofibers (white arrow). On d 15 postinjury (I, J) larger cross-sectional and multinucleated myofibers can be seen in WT compared with Nrf2−/− mice (white arrow).
Myogenesis is impaired in Nrf2−/− mice following CTX injury
To determine the effect of genetic loss of Nrf2 in muscle repair, we performed immunostaining for MHC-emb, a marker of newly formed muscle during regenerating process (39). As shown in Fig. 9A, the triple immunostaining for DAPI (blue nucleus, centrally located nuclei is an indicator of newly formed muscle), laminin (Oregon green muscle membrane, indicating individual muscle fiber), and MHC-emb (red, newly formed fibers) showed a greater MHC-emb + fibers with overdeveloped nuclei in representative WT and Nrf2−/− mice at 2 d post-CTX treatment compared with the appropriate vehicle controls (Fig. 9Aa, b vs. c, d). The total number of MHC-emb positive fibers was significantly greater by 2-fold in WT mice as early as 2 d post-CTX injury when compared with Nrf2−/− mice (P < 0.05, Fig. 9Ac vs. d; B, CTX-d 2). In contrast, the MHC-emb staining began to intensify only on d 4 in Nrf2−/− and was maximal with a 2-fold increase in mean fluorescence intensity on d 8 postinjury when compared with WT (Fig. 9Ag vs. h, P < 0.05; B, CTX d 8). By d 8 post-CTX injury, the muscle regeneration appear to be almost complete in WT animals that is evident from the basal normal intensity of MHC-emb fluorescence (Fig. 9Aa vs. g). However, the MHC-emb staining remained elevated in Nrf2−/− animals even after d 15 post-CTX injury, when the WT showed almost negligible staining of MHC-emb (Fig. 9Ai vs. j). Similarly, the area of MHC-emb expression was also found to be significantly differed between regenerating TA muscles of WT and Nrf2−/− mice (Fig. 9B). On d 2 and 4 postinjury, the area of MHC-emb expression was significantly higher in WT compared with Nrf2−/− mice, whereas on d 8 the area of MHC-emb expression was decreased in WT and significantly increased in Nrf2−/− mice. Furthermore, as can be seen from the mean diameter, based on laminin immunostaining, the size of regenerating myofibers appeared to be smaller in Nrf2−/− animals compared with WT (Fig. 9Ai vs. j; C). Taken together, this result suggests that when the process of muscle fiber regeneration was complete in WT, it was still continued in the Nrf2−/− animals (compare Fig. 9Ag, i vs. h, j) indicating a slower rate of myogenesis along with a dystrophic pattern of regeneration.
Figure 9.

Delayed expression of MHC-emb in TA muscle of Nrf2−/− mice injured with CTX. A) Cross sections of regenerating TA muscle of WT (a, c, e, g, i) and Nrf2−/− (b, d, f, h, j) mice showing anti-MHC-emb (red fluorescence). Muscle cell plasma membrane and nucleus were counter stained with anti-laminin (green) and DAPI (blue), respectively. Cross section of PBS-injected TA muscles of WT and Nrf2−/− mice (a, b), and cross-sections at 2 d (c, d), 4 d (e, f), 8 d (g, h), and 15 d (i, j) following CTX injury of TA muscle of WT and Nrf2−/− mice. B) Average area of TA muscle expressing MHC-emb of n = 3 section of each group was determined using ImageJ software. C) Average diameter of ∼100 green positive myotubes per field (Ai, j) of n = 3 sections of each group was determined using ImageJ software. n = 3/d per group. Statistical significance was calculated by 1-way ANOVA. *P < 0.05 CTX-WT vs. Nrf2−/−.
CTX treatment impairs C2C12 myoblast proliferation and differentiation
Using an in vitro cell culture system, we tested the effect of CTX on C2C12 satellite cell proliferation and differentiation. A dose-dependent effect of CTX on cell survival was determined (Fig. 10A). We observed no significant effect on the rate of survival up to 0.5 µM CTX. A slight to severe decrease in proliferation of C2C12 cells in the concentrations ranging from 1 to 4 µM CTX was observed (Fig. 10A). In the subsequent experiments, myoblasts treated with nontoxic dose of CTX (0.5 µM) displayed little to no myotubes formation on d 5 of differentiation (Fig. 10B). The vehicle (PBS) treated C2C12 cells exhibited long myotube formation indicating effective differentiation of myoblasts (Fig. 10B). Next, we determined the effect of 0.5 µM CTX on myoblast proliferation and cell migration in Nrf2-silenced C2C12 cells in the wound-healing assay. Figure 10C, D shows that the control shRNA-transfected cells showed almost complete closure 24 h postwounding. In the Nrf2-silenced group receiving PBS showed only 80% wound closure (P < 0.05). Although 24 h postwounding of control shRNA transfected cells receiving CTX showed ∼40% migration of cells into and across the denuded area of the monolayer, it was further reduced to ∼15% in Nrf2-silenced cells (P < 0.05; Fig. 10C, D). These results suggest that loss of Nrf2 greatly inhibits CTX-induced satellite cell motility into the wounded area. On the other hand, the rate of proliferation of C2C12 myoblast was not affected by either CTX treatment or silencing Nrf2. However, the proliferation was significantly decreased by ∼10% under CTX treatment to Nrf2 knockdown C2C12 myoblast.
Figure 10.

CTX treatment hampers C2C12 myoblast differentiation and wound healing. A) Graph illustrates the percentage C2C12 cells viability measured by MTT assay in PBS and CTX treatment. B) Bright field light microscopy images showing the effect of 0.5 µM CTX treatment on C2C12 myoblast differentiation (×10 magnification). C) Artificial scratch wounds were created in Nrf2-silenced C2C12 monolayers, and the wound closure was captured by light microscope (×10 magnification). D) Graph represents the percentage of wound healing calculated by measuring the area of wound using ImageJ software. E) MTT cell viability assay of C2C12 myoblast transfected with control and Nrf2 shRNA for 48 h and treated with CTX (0.5 µM) for 24 h. Statistical significance was calculated by Student’s t test. *P < 0.05 between percent of wound healing of PBS and CTX-treated myoblast. ***P < 0.05 between control-shRNA+CTX vs. Nrf2-shRNA+CTX.
DISCUSSION
Molecular mechanisms to slow down the progression of muscular pathologies and enhance the regenerative process are currently being investigated. The results of the current study reveal that genetic ablation of Nrf2: 1) exacerbates CTX-induced TA muscle injury, 2) compromises the antioxidant genes and protein expression and promotes ROS generation and sustained the oxidative stress in TA muscle injured with CTX, 3) delays the activation and proliferation of Pax7-positive (stem cells) cells and impairs the muscle differentiation process, 4) interferes with the muscle repair process by increasing the mean diameter of MHC-emb-positive neomyotubes, and 5) induced strong inflammatory response 2, 4, and 8 d post-CTX injury prior to returning back to almost normal morphometry as demonstrated by histologic analysis. Because Nrf2 is highly dysregulated and decreased during aging, chronic and/or severe oxidative setting, toxin/pollutant exposures, ischemic condition, and viral infections (40–42), and because these conditions are known to exert profound influence on skeletal muscle injury, loss of Nrf2 related mechanisms may represent a significant underlying factor to the heightened risk for skeletal muscle pathologies (43, 44).
In the Nrf2 knockouts, CTX severely strains the Nrf2-dependent antioxidant response (Figs. 2 and 3) and sustains the increased levels of superoxide and other ROS (Fig. 4). Although there was not an immense variation in TA muscle damage among WT and Nrf2 knockouts basally, CTX injury severely affected the gain of TA muscle mass in Nrf2−/− mice. This indicates that the dynamic regenerative/growth process is impacted in Nrf2 knockouts similar to those observed in an acute transient mouse model of muscle degeneration using CTX and dystrophin-deficient X-linked muscular dystrophy mouse model (32, 45). Although we and others have demonstrated a critical role for Nrf2 in skeletal muscle, mice deficient for Nrf2 develop into phenotypically normal adults (Fig. 1A), suggesting that nonenzymatic ascorbate glutaredoxin pathways can preserve the basal antioxidant defense mechanisms in muscle cells. However, a variety of insults including treatment with toxic chemicals, oxidants, endurance exercise stress, and accelerated aging in the absence or deficiency of Nrf2 is highly challenging because they become extremely sensitive to these assaults (46). Our results here show that the TA muscles possessing remarkable adaptability are also not an exception if Nrf2 is absent. Recent findings have shown that Nrf2 is indispensable to protect the skeletal muscle from TWEAK and ischemic-mediated cell death and injury (47, 48). Moreover, Nrf2 is widely expressed in all types of skeletal muscle, especially slow (i.e., soleus muscle) and fast muscles (i.e., TA muscle). Of note, the fast muscles (TA muscle) are hypersensitive to oxidative stress than the slow type muscle, which ultimately promote the oxidative stress-induced damage in adult myofibers (49). In this context, our study has illustrated a critical role for Nrf2 antioxidant signaling during the regenerative process of TA muscle post-CTX injury.
Regenerative response to skeletal muscle injury is a complex, multistep process that involves satellite cell activation, proliferation, migration, and differentiation (50). Satellite cells play a central role in retaining the muscle mass during the entire life span and these cells are normally quiescent but can be stimulated to enter the cell cycle, proliferate, undergo self-renewal, and differentiate into mature myofibers in response to injured or inflamed muscle (8–13). Herein, we observed a robust expression of Pax7 gene and protein in WT mice following CTX injury on d 2 and 4 that declines thereafter (8 d post-CTX injury) and returned to basal level. This indicates that the prompt increase of Pax7 early after the injury and until d 4 is to promote the activation and proliferation of satellite cell population with a later decrease to facilitate regeneration and transitioning to myogenic lineage. Such a typical biphasic pattern for Pax7 expression is common during myogenesis (51, 52). Notably, a persisting increase of Pax7 was still found on d 8 post-CTX injury in Nrf2−/− mice, with a faint trace of Pax7 protein (Fig. 6) still observed on d 15. In addition, the extent of Pax7 increases in Nrf2−/− was relatively less when compared with WT on d 2 and 4 after CTX injury (Fig. 6A, B). This suggests that there could be an interference of satellite cells at 2 levels in Nrf2−/− mice, one at the activation stage (corresponding to 2 and 4 d) and the other at the differentiation stage (corresponding to 8 and 15 d) (i.e., delayed onset of differentiation process that is reflected by the failure of timely down-regulation of Pax7). Our results are supported by 2 independent genetic knockout studies that suggest a decrease in Pax7 expression is essential to initiate the differentiation kinetics and associated muscle regeneration (53) that is connected to impaired oxidative metabolism (54).
The early differentiation step during the regenerative process is a highly orchestrated process with a decrease in Pax7 that is combined with increase in the expression of other intrinsic muscle-specific transcriptional factors such as MyoD and myogenin (55). Along these lines, we illustrate here that genetic ablation of Nrf2 resulted in weak activation of MyoD when compared with WT in a CTX injury model during the early phase of regeneration (Figs. 5B and 6A, B). The reason for decreased MyoD transcript and protein expression in Nrf2−/− mice during the early stage of differentiation could be attributed to the decreased Pax3/7 levels that coordinately recruit RNA pol-II and form a preinitiation complex at the MyoD promoter to induce its transcription (56). In addition, it has been shown that depletion of GSH and the resultant oxidative intracellular environment impairs the differentiation process of C2C12 myoblasts by reducing MyoD and myogenin expression (57–60). Because loss of Nrf2 is known to disturb the GSH-redox homeostasis, it could impact the MyoD and myogenin expression, thus delaying the differentiation process during CTX injury in TA muscles. Histologic analysis demonstrated that the improvement of TA muscle morphology was more rapid in WT with d 4 of recovery similar to d 8 in Nrf2−/− (Fig. 8), indicating a requirement for Nrf2 in controlling the rate of recovery. Although the WT and Nrf2−/− animals exhibited comparable characteristics of regenerative fibers in response to CTX at the end of 15 d, the size of TA muscle myofibers from the Nrf2−/− was smaller and also consistent with a likelihood of myofibrillar misalignment and disarray. We also observed that Nrf2−/− mice displayed smaller MHC-emb-positive neomyotubes and increased muscle fiber heterogeneity (Fig. 9Ai, j; C), indicating a poor regenerative outcome. Previous reports have shown that besides developmental expression, MHC-emb is also expressed in adult skeletal muscle under a numerous experimental conditions and is provided as an evidence for the occurrence of ongoing muscle regeneration (61, 62). Notably, this result is consistent with the increase of dystrophied regenerating fibers observed in CTX injury in senescence and oxidative stress induced redox disturbances, wherein Nrf2 dysregulation is common (27, 63, 64). Furthermore, the in vitro wound study confirmed the Nrf2-silenced C2C12 cells showed impaired migration in response to CTX treatment (Fig. 10). It is noteworthy that efficient muscle regeneration is marked by activation of satellite cells that is accompanied by extensive cell mobility/migration. Overall, our findings reveal the connection between Nrf2-based redox sensitivity and muscle differentiation process during regeneration and identified a plausible candidate that can be targeted to enhance muscle regeneration (Fig. 11). This may be particularly relevant in the context of injury, aging and pathologies such as AIDS or cancer wherein skeletal muscle molecular abnormalities and muscle wasting is common. Currently, studies are underway in our laboratory to address if amplifying Nrf2 can preserve and sustains the TA muscle regenerative function in response to chronic and/or severe injury in the background of altered redox homeostasis.
Figure 11.

Schematic representation of the effect of Nrf2 deficiency on skeletal muscle regeneration after injury. Under normal physiologic conditions, when redox homeostasis is being maintained by the transcription factor Nrf2; activation, proliferation, and differentiation of satellite cells occurs to regenerate skeletal muscle after injury. However, when the redox imbalance occurs due to loss and/or inactivation of Nrf2, skeletal muscle regeneration could be delayed.
Supplementary Material
Acknowledgments
These studies were supported by awards/research funds from the U.S. National Institutes of Health (NIH) National Heart, Lung, and Blood Institute (R01HL118067), NIH National Institute on Aging (1R03AG042860-01), the American Heart Association (BGIA-0865015F), University of Utah Center for Aging (Pilot Grant-2009), the University of Utah, and the University of Alabama at Birmingham startup funds (to N.S.R.). The authors thank Ms. Indira Guevera, Ms. Jennifer Hong, and Ms. Nancy Atieno (University of Utah School of Medicine, Salt Lake City, Utah, USA) for their technical assistance, including animal colony maintenance and genotyping. The authors also thank Dr. Christopher Davidson for his review and critiques to strengthen the manuscript.
Glossary
- CTX
cardiotoxin
- DHE
dihydroethidium
- G6pd
glucose 6-phosphate dehydrogenase
- Gclc
glutamate-cysteine ligase, catalytic subunit
- Gc1m
glutamate-cysteine ligase, modifier subunit
- Gsr
glutathione reductase
- Keap1
Kelch like-ECH-associated protein 1
- MHC-emb
embryonic myosin heavy chain
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide
- Myh2
myosin heavy chain 2
- MyoD
myogenic differentiation
- Nqo1
NAD(P)H dehydrogenase quinone 1
- Nrf2
nuclear factor (erythroid-derived-2)–like 2
- Pax
paired box
- PM
proliferation media
- ROS
reactive oxygen species
- sh
small hairpin
- Sod1
superoxide dismutase 1
- Sod2
superoxide dismutase 2
- TA
tibialis anterior
- WT
wild-type
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
REFERENCES
- 1.Blaisdell F. W. (2002) The pathophysiology of skeletal muscle ischemia and the reperfusion syndrome: a review. Cardiovasc. Surg. 10, 620–630 [DOI] [PubMed] [Google Scholar]
- 2.Di Stasi S. L., MacLeod T. D., Winters J. D., Binder-Macleod S. A. (2010) Effects of statins on skeletal muscle: a perspective for physical therapists. Phys. Ther. 90, 1530–1542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ribchester R. R., Thomson D., Wood N. I., Hinks T., Gillingwater T. H., Wishart T. M., Court F. A., Morton A. J. (2004) Progressive abnormalities in skeletal muscle and neuromuscular junctions of transgenic mice expressing the Huntington’s disease mutation. Eur. J. Neurosci. 20, 3092–3114 [DOI] [PubMed] [Google Scholar]
- 4.Shrier I. (2004) Muscle dysfunction versus wear and tear as a cause of exercise related osteoarthritis: an epidemiological update. Br. J. Sports Med. 38, 526–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chargé S. B., Rudnicki M. A. (2004) Cellular and molecular regulation of muscle regeneration. Physiol. Rev. 84, 209–238 [DOI] [PubMed] [Google Scholar]
- 6.Arrington E. D., Miller M. D. (1995) Skeletal muscle injuries. Orthop. Clin. North Am. 26, 411–422 [PubMed] [Google Scholar]
- 7.Tidball J. G. (2005) Inflammatory processes in muscle injury and repair. Am. J. Physiol. Regul. Integr. Comp. Physiol. 288, R345–R353 [DOI] [PubMed] [Google Scholar]
- 8.Collins C. A., Gnocchi V. F., White R. B., Boldrin L., Perez-Ruiz A., Relaix F., Morgan J. E., Zammit P. S. (2009) Integrated functions of Pax3 and Pax7 in the regulation of proliferation, cell size and myogenic differentiation. PLoS One 4, e4475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schultz E., Jaryszak D. L., Valliere C. R. (1985) Response of satellite cells to focal skeletal muscle injury. Muscle Nerve 8, 217–222 [DOI] [PubMed] [Google Scholar]
- 10.Snow M. H. (1983) A quantitative ultrastructural analysis of satellite cells in denervated fast and slow muscles of the mouse. Anat. Rec. 207, 593–604 [DOI] [PubMed] [Google Scholar]
- 11.Watt D. J., Morgan J. E., Clifford M. A., Partridge T. A. (1987) The movement of muscle precursor cells between adjacent regenerating muscles in the mouse. Anat. Embryol. (Berl.) 175, 527–536 [DOI] [PubMed] [Google Scholar]
- 12.Kuang S., Gillespie M. A., Rudnicki M. A. (2008) Niche regulation of muscle satellite cell self-renewal and differentiation. Cell Stem Cell 2, 22–31 [DOI] [PubMed] [Google Scholar]
- 13.Tedesco F. S., Dellavalle A., Diaz-Manera J., Messina G., Cossu G. (2010) Repairing skeletal muscle: regenerative potential of skeletal muscle stem cells. J. Clin. Invest. 120, 11–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Relaix F., Zammit P. S. (2012) Satellite cells are essential for skeletal muscle regeneration: the cell on the edge returns centre stage. Development 139, 2845–2856 [DOI] [PubMed] [Google Scholar]
- 15.Collins C. A., Olsen I., Zammit P. S., Heslop L., Petrie A., Partridge T. A., Morgan J. E. (2005) Stem cell function, self-renewal, and behavioral heterogeneity of cells from the adult muscle satellite cell niche. Cell 122, 289–301 [DOI] [PubMed] [Google Scholar]
- 16.Sherwood R. I., Christensen J. L., Conboy I. M., Conboy M. J., Rando T. A., Weissman I. L., Wagers A. J. (2004) Isolation of adult mouse myogenic progenitors: functional heterogeneity of cells within and engrafting skeletal muscle. Cell 119, 543–554 [DOI] [PubMed] [Google Scholar]
- 17.Zammit P. S., Heslop L., Hudon V., Rosenblatt J. D., Tajbakhsh S., Buckingham M. E., Beauchamp J. R., Partridge T. A. (2002) Kinetics of myoblast proliferation show that resident satellite cells are competent to fully regenerate skeletal muscle fibers. Exp. Cell Res. 281, 39–49 [DOI] [PubMed] [Google Scholar]
- 18.Lepper C., Partridge T. A., Fan C. M. (2011) An absolute requirement for Pax7-positive satellite cells in acute injury-induced skeletal muscle regeneration. Development 138, 3639–3646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sambasivan R., Yao R., Kissenpfennig A., Van Wittenberghe L., Paldi A., Gayraud-Morel B., Guenou H., Malissen B., Tajbakhsh S., Galy A. (2011) Pax7-expressing satellite cells are indispensable for adult skeletal muscle regeneration. Development 138, 3647–3656 [DOI] [PubMed] [Google Scholar]
- 20.Huard J., Li Y., Fu F. H. (2002) Muscle injuries and repair: current trends in research. J. Bone Joint Surg. Am. 84-A, 822–832 [PubMed] [Google Scholar]
- 21.Cohn R. D., van Erp C., Habashi J. P., Soleimani A. A., Klein E. C., Lisi M. T., Gamradt M., ap Rhys C. M., Holm T. M., Loeys B. L., Ramirez F., Judge D. P., Ward C. W., Dietz H. C. (2007) Angiotensin II type 1 receptor blockade attenuates TGF-beta-induced failure of muscle regeneration in multiple myopathic states. Nat. Med. 13, 204–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Conboy I. M., Conboy M. J., Smythe G. M., Rando T. A. (2003) Notch-mediated restoration of regenerative potential to aged muscle. Science 302, 1575–1577 [DOI] [PubMed] [Google Scholar]
- 23.Palacios D., Mozzetta C., Consalvi S., Caretti G., Saccone V., Proserpio V., Marquez V. E., Valente S., Mai A., Forcales S. V., Sartorelli V., Puri P. L. (2010) TNF/p38α/polycomb signaling to Pax7 locus in satellite cells links inflammation to the epigenetic control of muscle regeneration. Cell Stem Cell 7, 455–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hosokawa K., Arai F., Yoshihara H., Nakamura Y., Gomei Y., Iwasaki H., Miyamoto K., Shima H., Ito K., Suda T. (2007) Function of oxidative stress in the regulation of hematopoietic stem cell-niche interaction. Biochem. Biophys. Res. Commun. 363, 578–583 [DOI] [PubMed] [Google Scholar]
- 25.Pervaiz S., Taneja R., Ghaffari S. (2009) Oxidative stress regulation of stem and progenitor cells. Antioxid. Redox Signal. 11, 2777–2789 [DOI] [PubMed] [Google Scholar]
- 26.Capell B. C., Collins F. S., Nabel E. G. (2007) Mechanisms of cardiovascular disease in accelerated aging syndromes. Circ. Res. 101, 13–26 [DOI] [PubMed] [Google Scholar]
- 27.Narasimhan M., Hong J., Atieno N., Muthusamy V. R., Davidson C. J., Abu-Rmaileh N., Richardson R. S., Gomes A. V., Hoidal J. R., Rajasekaran N. S. (2014) Nrf2 deficiency promotes apoptosis and impairs PAX7/MyoD expression in aging skeletal muscle cells. Free Radic. Biol. Med. 71, 402–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McMahon M., Itoh K., Yamamoto M., Hayes J. D. (2003) Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression. J. Biol. Chem. 278, 21592–21600 [DOI] [PubMed] [Google Scholar]
- 29.Irintchev A., Wernig A. (1987) Muscle damage and repair in voluntarily running mice: strain and muscle differences. Cell Tissue Res. 249, 509–521 [DOI] [PubMed] [Google Scholar]
- 30.Lefaucheur J. P., Sébille A. (1995) The cellular events of injured muscle regeneration depend on the nature of the injury. Neuromuscul. Disord. 5, 501–509 [DOI] [PubMed] [Google Scholar]
- 31.Pavlath G. K., Thaloor D., Rando T. A., Cheong M., English A. W., Zheng B. (1998) Heterogeneity among muscle precursor cells in adult skeletal muscles with differing regenerative capacities. Dev. Dyn. 212, 495–508 [DOI] [PubMed] [Google Scholar]
- 32.Ramadasan-Nair R., Gayathri N., Mishra S., Sunitha B., Mythri R. B., Nalini A., Subbannayya Y., Harsha H. C., Kolthur-Seetharam U., Srinivas Bharath M. M. (2014) Mitochondrial alterations and oxidative stress in an acute transient mouse model of muscle degeneration: implications for muscular dystrophy and related muscle pathologies. J. Biol. Chem. 289, 485–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chien C. M., Chang S. Y., Lin K. L., Chiu C. C., Chang L. S., Lin S. R. (2010) Taiwan cobra cardiotoxin III inhibits Src kinase leading to apoptosis and cell cycle arrest of oral squamous cell carcinoma Ca9-22 cells. Toxicon 56, 508–520 [DOI] [PubMed] [Google Scholar]
- 34.Raynor R. L., Zheng B., Kuo J. F. (1991) Membrane interactions of amphiphilic polypeptides mastoparan, melittin, polymyxin B, and cardiotoxin. Differential inhibition of protein kinase C, Ca2+/calmodulin-dependent protein kinase II and synaptosomal membrane Na,K-ATPase, and Na+ pump and differentiation of HL60 cells. J. Biol. Chem. 266, 2753–2758 [PubMed] [Google Scholar]
- 35.Rajasekaran N. S., Varadharaj S., Khanderao G. D., Davidson C. J., Kannan S., Firpo M. A., Zweier J. L., Benjamin I. J. (2011) Sustained activation of nuclear erythroid 2-related factor 2/antioxidant response element signaling promotes reductive stress in the human mutant protein aggregation cardiomyopathy in mice. Antioxid. Redox Signal. 14, 957–971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bismuth K., Relaix F. (2010) Genetic regulation of skeletal muscle development. Exp. Cell Res. 316, 3081–3086 [DOI] [PubMed] [Google Scholar]
- 37.Bryson-Richardson R. J., Currie P. D. (2008) The genetics of vertebrate myogenesis. Nat. Rev. Genet. 9, 632–646 [DOI] [PubMed] [Google Scholar]
- 38.Pownall M. E., Gustafsson M. K., Emerson C. P. Jr (2002) Myogenic regulatory factors and the specification of muscle progenitors in vertebrate embryos. Annu. Rev. Cell Dev. Biol. 18, 747–783 [DOI] [PubMed] [Google Scholar]
- 39.D’Albis A., Couteaux R., Janmot C., Roulet A., Mira J. C. (1988) Regeneration after cardiotoxin injury of innervated and denervated slow and fast muscles of mammals. Myosin isoform analysis. Eur. J. Biochem. 174, 103–110 [DOI] [PubMed] [Google Scholar]
- 40.Islam K. N., Polhemus D. J., Donnarumma E., Brewster L. P., Lefer D. J. (2015) Hydrogen sulfide levels and nuclear factor-erythroid 2-related factor 2 (NRF2) activity are attenuated in the setting of critical limb ischemia (CLI). J. Am. Heart Assoc. 4, 4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Komaravelli N., Tian B., Ivanciuc T., Mautemps N., Brasier A. R., Garofalo R. P., Casola A. (2015) Respiratory syncytial virus infection down-regulates antioxidant enzyme expression by triggering deacetylation-proteasomal degradation of NRF2. Free Radic. Biol. Med. 88, 391–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Muthusamy V. R., Kannan S., Sadhaasivam K., Gounder S. S., Davidson C. J., Boeheme C., Hoidal J. R., Wang L., Rajasekaran N. S. (2012) Acute exercise stress activates Nrf2/ARE signaling and promotes antioxidant mechanisms in the myocardium. Free Radic. Biol. Med. 52, 366–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xiu J. H., Zhu H., Xu Y. F., Liu J. N., Xia X. Z., Zhang L. F. (2013) Necrotizing myositis causes restrictive hypoventilation in a mouse model for human enterovirus 71 infection. Virol. J. 10, 215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang J. C., Lin M. W., Rau C. S., Jeng S. F., Lu T. H., Wu Y. C., Chen Y. C., Tzeng S. L., Wu C. J., Hsieh C. H. (2015) Altered exosomal protein expression in the serum of NF-κB knockout mice following skeletal muscle ischemia-reperfusion injury. J. Biomed. Sci. 22, 40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen C. N., Brown-Borg H. M., Rakoczy S. G., Thompson L. V. (2008) Muscle disuse: adaptation of antioxidant systems is age dependent. J. Gerontol. A Biol. Sci. Med. Sci. 63, 461–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Aoi W., Sakuma K. (2011) Oxidative stress and skeletal muscle dysfunction with aging. Curr. Aging Sci. 4, 101–109 [DOI] [PubMed] [Google Scholar]
- 47.Al-Sawaf O., Fragoulis A., Rosen C., Kan Y. W., Sönmez T. T., Pufe T., Wruck C. J. (2014) Nrf2 protects against TWEAK-mediated skeletal muscle wasting. Sci. Rep. 4, 3625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Al-Sawaf O., Fragoulis A., Rosen C., Keimes N., Liehn E. A., Hölzle F., Kan Y. W., Pufe T., Sönmez T. T., Wruck C. J. (2014) Nrf2 augments skeletal muscle regeneration after ischaemia-reperfusion injury. J. Pathol. 234, 538–547 [DOI] [PubMed] [Google Scholar]
- 49.Armand A. S., Laziz I., Djeghloul D., Lécolle S., Bertrand A. T., Biondi O., De Windt L. J., Chanoine C. (2011) Apoptosis-inducing factor regulates skeletal muscle progenitor cell number and muscle phenotype. PLoS One 6, e27283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Karalaki M., Fili S., Philippou A., Koutsilieris M. (2009) Muscle regeneration: cellular and molecular events. In Vivo 23, 779–796 [PubMed] [Google Scholar]
- 51.Halevy O., Piestun Y., Allouh M. Z., Rosser B. W., Rinkevich Y., Reshef R., Rozenboim I., Wleklinski-Lee M., Yablonka-Reuveni Z. (2004) Pattern of Pax7 expression during myogenesis in the posthatch chicken establishes a model for satellite cell differentiation and renewal. Dev. Dyn. 231, 489–502 [DOI] [PubMed] [Google Scholar]
- 52.Schultz E., Chamberlain C., McCormick K. M., Mozdziak P. E. (2006) Satellite cells express distinct patterns of myogenic proteins in immature skeletal muscle. Dev. Dyn. 235, 3230–3239 [DOI] [PubMed] [Google Scholar]
- 53.Angione A. R., Jiang C., Pan D., Wang Y. X., Kuang S. (2011) PPARδ regulates satellite cell proliferation and skeletal muscle regeneration. Skelet. Muscle 1, 33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Luquet S., Lopez-Soriano J., Holst D., Fredenrich A., Melki J., Rassoulzadegan M., Grimaldi P. A. (2003) Peroxisome proliferator-activated receptor delta controls muscle development and oxidative capability. FASEB J. 17, 2299–2301 [DOI] [PubMed] [Google Scholar]
- 55.Füchtbauer E. M., Westphal H. (1992) MyoD and myogenin are coexpressed in regenerating skeletal muscle of the mouse. Dev. Dyn. 193, 34–39 [DOI] [PubMed] [Google Scholar]
- 56.Hu P., Geles K. G., Paik J. H., DePinho R. A., Tjian R. (2008) Codependent activators direct myoblast-specific MyoD transcription. Dev. Cell 15, 534–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ardite E., Barbera J. A., Roca J., Fernández-Checa J. C. (2004) Glutathione depletion impairs myogenic differentiation of murine skeletal muscle C2C12 cells through sustained NF-kappaB activation. Am. J. Pathol. 165, 719–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hansen J. M., Klass M., Harris C., Csete M. (2007) A reducing redox environment promotes C2C12 myogenesis: implications for regeneration in aged muscle. Cell Biol. Int. 31, 546–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sestili P., Barbieri E., Martinelli C., Battistelli M., Guescini M., Vallorani L., Casadei L., D’Emilio A., Falcieri E., Piccoli G., Agostini D., Annibalini G., Paolillo M., Gioacchini A. M., Stocchi V. (2009) Creatine supplementation prevents the inhibition of myogenic differentiation in oxidatively injured C2C12 murine myoblasts. Mol. Nutr. Food Res. 53, 1187–1204 [DOI] [PubMed] [Google Scholar]
- 60.Wu Y. J., Fang Y. H., Chi H. C., Chang L. C., Chung S. Y., Huang W. C., Wang X. W., Lee K. W., Chen S. L. (2014) Insulin and LiCl synergistically rescue myogenic differentiation of FoxO1 over-expressed myoblasts. PLoS One 9, e88450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ontell M., Ontell M. P., Sopper M. M., Mallonga R., Lyons G., Buckingham M. (1993) Contractile protein gene expression in primary myotubes of embryonic mouse hindlimb muscles. Development 117, 1435–1444 [DOI] [PubMed] [Google Scholar]
- 62.Judson R. N., Gray S. R., Walker C., Carroll A. M., Itzstein C., Lionikas A., Zammit P. S., De Bari C., Wackerhage H. (2013) Constitutive expression of Yes-associated protein (Yap) in adult skeletal muscle fibres induces muscle atrophy and myopathy. PLoS One 8, e59622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Putman C. T., Sultan K. R., Wassmer T., Bamford J. A., Skorjanc D., Pette D. (2001) Fiber-type transitions and satellite cell activation in low-frequency-stimulated muscles of young and aging rats. J. Gerontol. A Biol. Sci. Med. Sci. 56, B510–B519 [DOI] [PubMed] [Google Scholar]
- 64.Powers S. K., Kavazis A. N., DeRuisseau K. C. (2005) Mechanisms of disuse muscle atrophy: role of oxidative stress. Am. J. Physiol. Regul. Integr. Comp. Physiol. 288, R337–R344 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.