

Abstract
HIV integrase (IN) strand transfer inhibitors (INSTIs) are among the newest anti-AIDS drugs; however, mutant forms of IN can confer resistance. We developed noncytotoxic naphthyridine-containing INSTIs that retain low nanomolar IC50 values against HIV-1 variants harboring all of the major INSTI-resistant mutations. We found by analyzing crystal structures of inhibitors bound to the IN from the prototype foamy virus (PFV) that the most successful inhibitors show striking mimicry of the bound viral DNA prior to 3′-processing and the bound host DNA prior to strand transfer. Using this concept of “bi-substrate mimicry,” we developed a new broadly effective inhibitor that not only mimics aspects of both the bound target and viral DNA but also more completely fills the space they would normally occupy. Maximizing shape complementarity and recapitulating structural components encompassing both of the IN DNA substrates could serve as a guiding principle for the development of new INSTIs.
Inhibiting enzymes that modify DNA or RNA substrates has the potential to be used in the treatment of a wide range of diseases. Polynucleotide transferases are important targets for drug development; however, the catalytic centers of these enzymes are often extended to accommodate their large substrates. It has been difficult to design potent, specific, and selective small molecule inhibitors. Fortunately, this has not been an intractable problem, as exemplified by the development of drugs that act as interfacial inhibitors of HIV-1 integrase (IN),1 one of three viral enzymes that are essential for viral replication. IN inserts viral DNA into the host genome through two sequential reactions. The first reaction, termed “3′-processing” (3′-P), involves the cleavage of the 3′-dinucleotides from viral DNA; the second reaction “strand transfer” (ST)2 involves the subsequent insertion of the processed ends of the viral DNA into host DNA. These reactions take place in the context of a stable nucleoprotein complex containing a multimer of IN assembled on viral DNA ends, referred to as the intasome. Divalent metal cofactors are essential for IN-mediated catalysis, and chelation of the active site Mg2+ ions is a key component of the successful integrase strand transfer inhibitors (INSTIs), which represent the newest class of anti-AIDS drugs.3 However, in infected patients, the HIV-1 genome diversifies rapidly due to an accumulation of mutations that arise during viral replication.4,5 Consequently, the emergence of resistance is a limitation for all anti-HIV therapeutics, including INSTIs, and resistant forms of the virus have been isolated from patients who received raltegravir (RAL, 1) or elvitegravir (EVG, 2), the first two INSTIs approved by the FDA for the treatment of HIV/AIDS.4,6,7 More recently, dolutegravir (DTG, 3) has come to market as a second generation INSTI. DTG appears to be considerably less prone to virological failure than RAL and EVG.8−12 However, viruses that carry IN resistance mutations can be selected by growing the virus in the presence of DTG, and mutations that confer cross-resistance to all three INSTIs have been identified.13−17
A primary objective of continued INSTI development is to discover compounds with minimal toxicity that retain good effectiveness against existing resistant mutants.13 Until now, efforts to create new drugs that retain broad efficacy against the resistant forms of IN have largely been empirical. This is in spite of the availability of “cocrystal” structures of the leading first and second-generation INSTIs bound to the intasome formed by the othologous enzyme from prototype foamy virus (PFV) in a complex with metal cofactors and the cognate viral DNA substrate (referred to as the “intasome”).18−21 Although these advances elucidated the structural basis for INSTI function, they have not yet led to principles that can guide the design of the next generation INSTIs, which would retain effectiveness against the known resistant IN mutants.16,22
In our current work, we empirically varied the structure of our recently disclosed 1-hydroxy-2-oxo-1,8-naphthyridine-containing INSTIs (4) to improve their ability to potently block in cell culture, the replication of a panel of HIV-1 based vectors that carry all of the major INSTI-resistant IN mutants.23,24 We also obtained “cocrystal” structures of the PFV intasome with the best inhibitors and found that when they bind to IN, they show striking mimicry of the binding of viral DNA in its initial state, prior to the cleavage of the dinucleotide in the 3′-P reaction. The inhibitors also mimic features of target DNA in their binding to the intasome active site.25 Our findings suggest that employing aspects of molecular mimicry of viral and host DNA may be useful in the design of improved INSTIs. Importantly, these principles may also be applicable to the broader class of enzymes that have DNA or RNA substrates.
Results and Discussion
Development of INSTIs That Retain Efficacy against RAL-resistant Mutant Forms of IN
INSTIs selectively bind at the interface of IN and the viral DNA end following its 3′-P.18,19,25 INSTIs contain a triad of heteroatoms (shown in red in Figure 1) that chelate the essential pair of Mg2+ ions in the IN active site. INSTIs also typically include a halobenzyl ring that stacks against the base of the penultimate deoxycytidine near the processed 3′-end of the viral DNA (shown in blue in Figure 1).3,26 Our long-term goal is to develop small molecules that are active against IN mutants that are resistant to current INSTIs. Initially, our efforts were directed at the retention of efficacy against the Y143R and N155H mutants and the double mutant, Q148H/G140S.13−16 While these are associated with clinical HIV-1 resistance to RAL, there is a considerable overlap in the resistance profiles of RAL and EVG.27,28 Prior to the current work, we had performed extensive investigations on a core 1-hydroxy-2-oxo-1,8-naphthyridine platform.23,24 These efforts yielded compound 4a, with a ST inhibitory value of IC50 = 19 nM in an in vitro IN assay (compound 4a, Table 1) and an antiviral potency equivalent to RAL (EC50 ≃ 1 nM) against HIV vectors carrying wild-type (WT) IN in single-round replication assays (Table 2). In similar assays, RAL shows a significant loss of antiviral efficacy when the HIV-1 vectors employed carried the drug resistance mutations Y143R, N155H, or Q148H/G140S mutants (EC50 ≃ 160 nM, 150 nM, and 1900 nM, respectively). In contrast, 4a shows effective retention of antiviral potency against both the Y143R and N155H variants (EC50 ≃ 2 nM and 5 nM, respectively, Table 2).24 These values are similar to those displayed by DTG; however, DTG is more potent than 4a against the Q148H/G140S double mutant (4a EC50 ≃ 35 nM; DTG EC50 ≃ 6 nM).24
Figure 1.
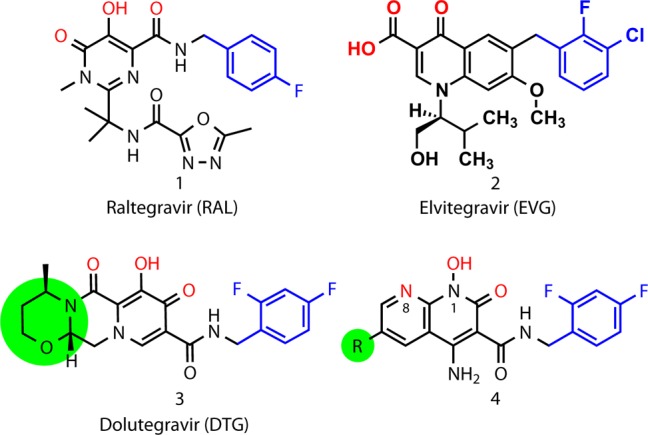
Structures of the FDA-approved INSTIs (RAL, 1; EVG, 2; and DTG, 3) and heterobicyclic compounds of the current series (4). Colors highlight key functional features: metal-chelating triad of heteroatoms (red), halobenzyl rings (blue). The terminal ring of DTG and the corresponding 6-substituents described in the present work shown in green shading.
Table 1. Inhibitory Potencies of Compounds 4a–4f Determined Using an in Vitro IN Assaya.
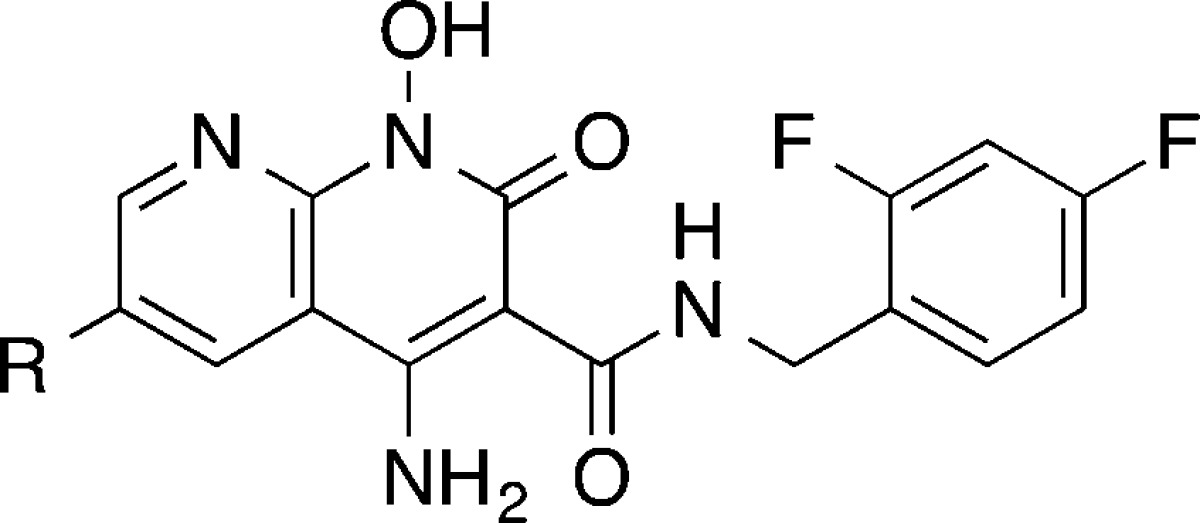
| IC50 (μM) |
|||
|---|---|---|---|
| no. | R | 3′-processing | strand transfer |
| 4ab | H | 2.5 ± 0.3 | 0.019 ± 0.002 |
| 4b | (CH2)3OH | 4.3 ± 0.6 | 0.0113 ± 0.0027 |
| 4c | (CH2)5OH | 8.2 ± 1.2 | 0.0027 ± 0.0004 |
| 4d | (CH2)6OH | 19 ± 1 | 0.00059 ± 0.00011 |
| 4e | (CH2)8OH | 32 ± 6 | 0.0054 ± 0.0008 |
| 4f | (CH2)2 SO2Ph | 0.0037 ± 0.0013 | |
Table 2. Antiviral Potencies in Cells Infected with HIV-1 Vectors That Carry WT or Resistant IN Mutants.
| EC50 (nM)c,d |
||||||
|---|---|---|---|---|---|---|
| no. | CC50 (μM)a | EC50 (nM)b | Y143R | N155H | G140S/Q148H | G118R |
| 1e | > 250 | 4 ± 2 | 162 ± 16 (41) | 154 ± 33 (39) | 1900 ± 300 (475) | 36 ± 5 (9)e |
| 2 | > 250 | 6.4 ± 0.8 | 7.9 ± 2.3 (1.2) | 90 ± 18 (14) | 5700 ± 1100 (891) | 21 ± 10 (3.3) |
| 3e | > 250e | 1.6 ± 0.9e | 4.3 ± 1.2 (2.7)e | 3.6 ± 1.3 (2.3)e | 5.8 ± 0.5 (3.6)e | 13 ± 5 (8.1)e |
| 4ae | > 250e | 1.1 ± 0.7e | 2.5 ± 0.6 (2.3)e | 5.3 ± 2.3 (4.8)e | 35 ± 9 (32)e | 16 ± 5 (15)e |
| 4b | > 250 | 3.1 ± 2.0 | 2.8 ± 0.9 (0.90) | 8.5 ± 3.1 (2.7) | 13.4 ± 6.5 (4.3) | 10 ± 1.6 (3.2) |
| 4c | > 250 | 1.3 ± 0.2 | 3.0 ± 0.5 (2.3) | 2.4 ± 0.8 (1.8) | 6.9 ± 2.3 (5.3) | 5.3 ± 1.6 (4.1) |
| 4d | > 250 | 2.3 ± 0.6 | 2.1 ± 1.4 (0.91) | 2.7 ± 1.0 (1.2) | 9.4 ± 3.6 (4.1) | 6.4 ± 2.5 (2.8) |
| 4e | > 250 | 3.7 ± 0.4 | 3.2 ± 0.8 (0.86) | 6.5 ± 2.7 (1.8) | 7.7 ± 1.9 (2.1) | 11 ± 1 (3.0) |
| 4f | > 250 | 2 ± 0.1 | 0.6 ± 0.1 (0.3) | 2.1 ± 1.2 (1) | 5.2 ± 0.3 (3) | 11.4 ± 3.5 (6) |
| EC50 (nM)c,d |
|||||
|---|---|---|---|---|---|
| no. | R263K | T66I | E92Q | H51Y | H51Y/R263K |
| 1e | 5.7 ± 2.3 (1.4)e | 2.8 ± 0.4 (0.7) | 30 ± 10 (7.5) | 3.4 ± 0.2 (0.2) | 6 ± 2.3 (1.5) |
| 2 | 10 ± 6 (1.6) | 66 ± 1 (10) | 154 ± 34 (24) | 4.5 ± 2.1 (0.7) | 53 ± 18 (8) |
| 3e | 11 ± 3 (6.9)e | 0.9 ± 0.8 (0.56) | 2.3 ± 0.4 (1.4) | 3.2 ± 0.2 (2) | 16 ± 2 (10) |
| 4ae | 6.4 ± 2.3 (5.8)e | 0.6 ± 0.3 (0.54) | 3.0 ± 1.8 (2.7) | 1.0 ± 0.6 (0.91) | 3.2 ± 0.9 (2.9) |
| 4b | 2.5 ± 0.8 (0.81) | 1.1 ± 0.1 (0.35) | 4.8 ± 2.0 (1.5) | 2.6 ± 0.5 (0.84) | 15 ± 2 (4.8) |
| 4c | 2.6 ± 0.1 (2.0) | 0.93 ± 0.24 (0.72) | 3.8 ± 2.3 (2.3) | 3.8 ± 0.6 (2.9) | 2.6 ± 1.4 (2.0) |
| 4d | 8.4 ± 2.3 (3.7) | 0.50 ± 0.35 (0.22) | 0.74 ± 0.13 (0.32) | 1.6 ± 0.1 (0.70) | 5.2 ± 1.6 (2.3) |
| 4e | 3.8 ± 1.1 (1.0) | 1.7 ± 0.5 (0.46) | 2.6 ± 0.2 (0.70) | 4.8 ± 1.1 (1.3) | 22 ± 8 (5.9) |
| 4f | 5.3 ± 0.8 (2) | 0.7 ± 0.1 (0.3) | 1.2 ± 0.6 (0.6) | 0.8 ± 0.5 (0.4) | 5.3 ± 0.4 (2) |
Cytotoxic concentration resulting in 50% reduction in the level of ATP in human osteosarcoma (HOS) cells.
Values obtained from cells infected with a lentiviral vector carrying WT IN.
Cells were infected with viral vectors carrying IN mutations. The table shows the EC50 values.
Fold-change (FC) in EC50 relative to WT.
Previously reported values.24
Overcoming Loss of Potency against the Integrase Double Mutant Q148H/G140S
The extended tricyclic ring system of DTG permits its third ring (highlighted in green in Figure 1) to contact the β4-α2 loop in the active site of IN (highlighted in orange, Figure 2). Maintenance of interactions in this region has been suggested to be important for the binding of DTG, as well as for the binding of other second generation INSTIs.20,29−31 In order to determine the importance of these interactions, we modified 4a by adding substituents at its 6-position (highlighted in green in Figure 1). Our intent was to create hydrogen bonds with the protein or with bound water molecules proximal to the 6-position. Although there are a variety of constructs that could be used, the work reported herein is focused on simple linear methylene chains of increasing lengths with terminal hydroxyl groups (compounds 4b–4e, Table 1 and Supporting Information Scheme S1). The in vitro ST inhibitory potencies were relatively independent of chain length in going from three methylenes (4b, IC50 value = 11 nM) to eight methylenes (4e, IC50 value = 5 nM). The most potent analogue (by a small margin) was 4c with IC50 = 2.7 nM; Table 1), which also displayed an antiviral inhibitory potency against the Q148H/G140S mutant (4c, EC50 ≃ 7 nM), which was similar to that of DTG (Table 2).
Figure 2.
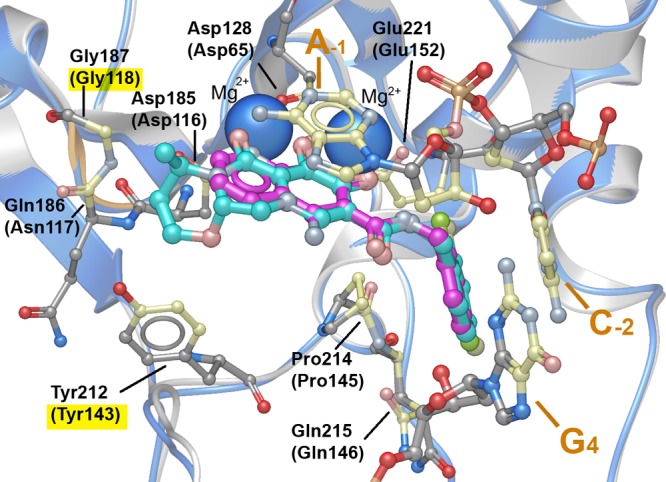
DTG and 4a binding in the active site of the PFV intasome. PFV-bound DTG (cyan with protein ribbon in gray) is shown with Mg2+ ions (solid blue spheres) along with the A-1 and C-2 nucleotides of 3′-processed viral DNA and the G4 nucleotide of the uncleaved complementary strand (PDB code 3S3M). Interacting residues are shown with contacting carbons shown in cream (corresponding IN residues are indicated in parentheses). The protein β4−α2 loop is highlighted in orange. Contact residues associated with RAL resistance (Y143R and G118R) are highlighted in yellow. PFV-bound 4a is superimposed onto the DTG-bound structure with the ligand carbons in magenta and the protein ribbon in blue.
Compounds with 6-Substituents Potently Inhibit DTG-resistant IN Mutants
We expanded the panel of HIV vectors used in the antiviral assays to include additional IN mutants that have a reduced susceptibility to DTG, including the G118R, R263K, T66I, E92Q, H51Y single mutants and the H51Y/R263K double mutant.13−16 We were particularly interested in whether 4c retained potency against these resistant mutants, because 4c had the best profile against the original panel of mutants. This compound inhibited the replication of the T66I, E92Q, and H51Y mutants as well as DTG and, encouragingly, also exhibited improved antiviral potencies relative to DTG against the G118R and R263K single mutants and the H51Y/R263K double mutant (Table 2). Importantly, none of the analogues of 4 showed appreciable cytotoxicity.
X-ray Crystal Structures of 4a and 4c Bound to the PFV Intasome
We refined a structure of the PFV intasome in complex with 4a to a resolution of 2.58 Å and compared it to the previously reported structure with DTG (PDB accession code: 3S3M).20 The 8-naphthyridine nitrogen and N-hydroxyl of 4a correspond to the 6-oxo and 7-hydroxyl heteroatoms of DTG, and the naphthyridine 2-oxo carbonyl of 4a corresponds to the ring 8-oxo carbonyl of DTG (Figure 2). As we had anticipated in our original design strategy,23 the halobenzyl carboxamido group of 4a is not involved in metal chelation. We also examined the contact residues for DTG (shown in Figure 2 and listed in the Supporting Information Table S2) and noted (as has previously been reported20) that DTG makes contact with PFV integrase Gly187 and Tyr212, which correspond to HIV-1 IN residues Gly118 and Tyr143, which are associated with RAL-resistant mutations (Table 2). In contrast, 4a does not make contact with either the Gly187 or Tyr212 residues (see Supporting Information Table S2). The 15-fold loss of potency of the G118R virus to 4a is likely due to steric exclusion of the compound by the bulky Arg side chain. Neither DTG nor 4a directly contacts other IN residues associated with INSTI resistance. This is consistent with prior observations that for inhibitors binding to the IN active site, resistance mutations do not usually arise due to a loss of IN–drug interactions.19,20
Next, we determined the structure of the PFV intasome crystallized with the 6-substituted compound 4c (Figure 3). The structure revealed that the hydroxyl group of the pentan-5-ol side chain of 4c forms a hydrogen bond with the backbone amide of PFV IN Tyr212, a residue that corresponds to HIV-1 IN Tyr143 (Figure 3A). Of particular interest, this same amide group interacts with the phosphoryl groups of both the viral and target DNAs during the 3′-P and ST reactions, respectively.25 Until now, RAL was the only INSTI known to make interactions with the backbone amide of Tyr143. However, in case of RAL, the interaction depends on the stacking of its 1,3,4-oxadiazole ring with the side chain of Tyr143, making the drug highly susceptible to the Y143R mutation.18,19,32
Figure 3.
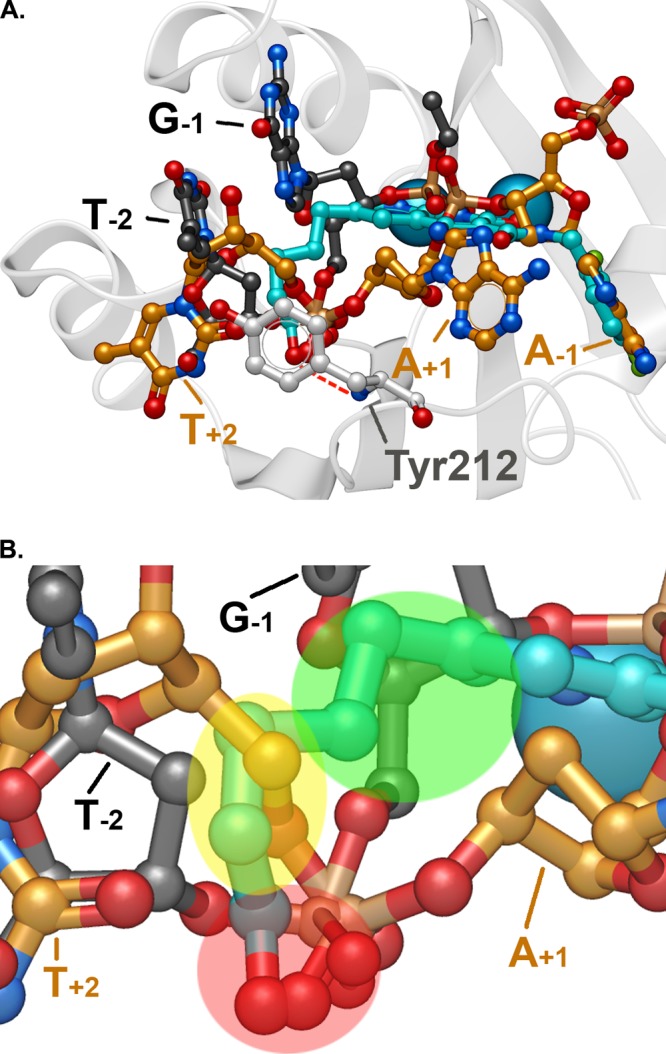
Compound 4c in the active site of the PFV intasome. (A) Superposition of the binding of 4c (blue) with pre-3′-processed DNA (carbons in gold; PDB code: 4E7I(25)) and target DNA (carbons in black; PDB code: 4E7K(25)). The bases of proximal nucleotides of the viral and host DNAs are annotated. The hydrogen bond between the 6- substituent terminal hydroxyl of 4c with the backbone amide nitrogen of Tyr212 is shown. (B) Closeup of panel A highlighting regions of correspondence between the 6-side chain of 4c and the nucleotides of the substrate DNAs: red, phosphoryl oxygens; yellow, T+2 deoxysugar; and green, G–1 deoxysugar. The IN residue Tyr212 is not shown for clarity.
By comparing the crystal structure of the PFV intasome with 4c bound to the structure of the PFV intasome with viral DNA bound prior to 3′-P (PDB code 4E7I), we discovered that the position of the terminal portion of the 6-side chain of 4c coincides with parts of the T+2 deoxyribose sugar of the unprocessed viral DNA. We also compared the 4c bound structure with the target capture complex, which has both the 3′-P viral DNA and the host DNA held in orientations suitable for insertion of the viral DNA into the host DNA (PDB code 4E7K). The naphthyridine-proximal region of the 6-side chain of 4c overlaps with a portion of the deoxyribose sugar of the G–1 nucleotide in the target DNA structure (Figure 3B). Thus, there is overlap of the 6-side chain of 4c with both unprocessed viral DNA in a pre-3′-P complex and the target DNA in a ST complex. It is noteworthy that the entire series of compounds 4b–4e had similar antiviral potencies against the panel of resistant IN mutants shown in Table 2. This is consistent with several of the linear polymethylene 6-substituents maintaining key interactions through conformational flexibility.
Interactions of the Intasome with a Sulfonyl-containing 6-Substituent
On the basis of the structural results discussed above, we introduced a sulfonyl functionality within the 6-substituent to yield compound 4f. This was done in an attempt to make 4f mimic the interactions IN shows with its DNA substrate phosphatediesters. Accordingly, 4f showed potencies equal to or slightly better than DTG in cell-based antiviral assays against a panel of viral vectors that included the major RAL and DTG-resistant IN mutants (Table 2).
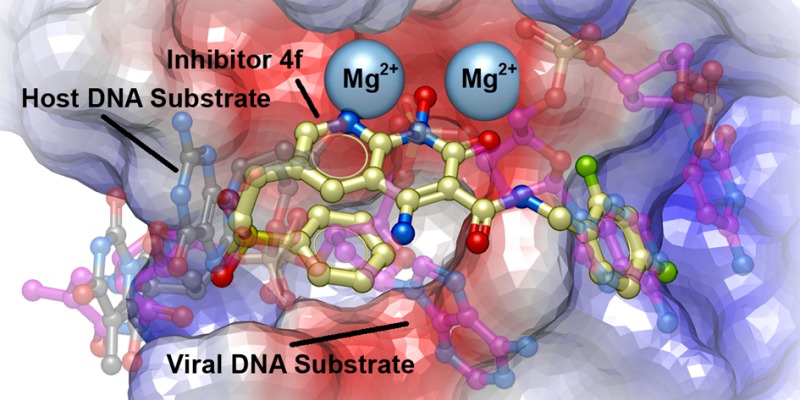
To visualize the interactions 4f makes with the active site of IN, we determined a crystal structure of 4f bound in the active site of the PFV intasome (Figure 4). The structure revealed that one of the sulfonyl oxygens of 4f forms hydrogen bonds with the main chain amide and the phenolic side chain of Tyr212 via bridging water molecules, while the other makes close van der Waals contacts with backbone atoms of Gln186 and Gly187 in the β4-α2 loop (Figure 4A). Strikingly, the sulfonylphenyl ring is bent back under the plane of the metal-chelating naphthyridinone ring system, where it makes an intramolecular π–π stacking interaction (3.3 Å separation) (Figure 4A and B). By superimposing this structure with the structures of both the uncleaved (pre-3′-P) viral DNA and the target DNA (ST) substrates (PBD codes 4E7I and 4E7K, respectively),25 it is apparent that the sulfonyl group fills the envelope formed by the overlap in the positions of both the bound uncleaved viral and the bound target DNAs (Figure 4B). In addition, the position of the sulfonylphenyl ring is similar to the position of the deoxyribose sugar of the adenosine A–1 in the scissile A-T dinucleotide of the uncleaved viral DNA, while other portions of the 6-side chain are similar to the position of the deoxyribose sugar of the G–1 guanosine in the target DNA (Figure 4B).
Figure 4.

PFV Intasome crystal structure complexed with 4f. (A) Crystal structure oriented to show the complexed Mg2+ ions (blue spheres) and the semitransparent surface of 4f (orange surface with carbons in cream), 3′P DNA (purple surface), and protein (gray surface) with key protein residues shown (carbons in light gray). Important binding interactions are indicated. (B) Bound 4f with superimposed pre-3′-processed DNA (carbons in purple; PDB code: 4E7I(25)) and target DNA (carbons in black; PDB code: 4E7K(25)). Regions where the side chain of 4f overlaps with DNA are indicated by black dashed ovals.
The face-on-face stacking of the sulfonylphenyl ring of 4f onto its own heterobicyclic core allows it to almost completely fill the catalytic pocket. When 4f is bound, its sulfonylphenyl ring packs against the side chain β-methylene of the metal-chelating residue D185 (≃ 3.8 Å) and against the α-methylene of the Tyr212 backbone (≃ 3.7 Å). The sulfonylphenyl ring also nestles against the back face of the Pro214 pyrrolidine ring, a key residue that helps form the hydrophobic binding pocket where the halobenzyl moiety is bound (Figure 4A). These results demonstrate improved overlap in the bound form of 4f and portions of both target and viral DNA substrates as compared to the bound form of 4c, which was our goal in designing 4f.
INSTIs are among the most effective antiretroviral drugs for the treatment of HIV/AIDS. However, as has been true for all anti-HIV drugs, the development of resistant IN mutants has been a limitation for RAL and EVG, the first two of the three current FDA-approved members of this family. Although the most recently introduced INSTI, DTG, appears to be considerably more resilient to viral resistance, it is not infallible. Therefore, continued research is needed to obtain INSTIs that are active against all of the known resistant mutants.
In the current work, we introduced functionality at the 6-position of our previously reported INSTI 4a to take advantage of potential binding interactions afforded by the third [(R)-4-methyl-1,3-oxazinane] ring of DTG. Although the approach was initially empirical, it yielded analogues, such as 4c, that retain excellent inhibitory potencies against the major drug-resistant IN mutants. By analyzing crystal structures of the PFV intasome in complex with some of the new compounds, we realized that the 6-substitutent of 4c recapitulates functionalities present in both an uncleaved viral DNA substrate in the pre-3′-P complex and the target DNA prior to ST. This represents an unusual form of “bi-substrate” mimicry, which we attempted to enhance by introducing a sulfone group at the 6-position. Among the resulting compounds is the phenylsulfone 4f, which shows potent activity against recombinant HIV IN in biochemical assays and good efficacy in assays done in cell culture against a broad panel of viral mutants carrying the major RAL and DTG IN resistance mutations. Importantly, a crystal structure of 4f within the PFV intasome shows that 4f occupies the sites where both the target and viral DNA are bound and largely fills the space that is available at the catalytic center.
The molecular interactions of 4c and, to a greater extent, 4f exemplify a more general concept of “substrate envelope” originally used to explain why some HIV protease inhibitors are able to retain efficacy against multiple resistance mutations.33 The rationale underlying this principle is that any mutant form of an enzyme that retains good enzymatic activity must be able to bind its cognate substrate(s). Thus, an inhibitor that binds to an enzyme in a fashion such that the inhibitor remains entirely within the envelope defined by the structure of the normal substrate(s) will be able to bind efficiently to both WT and (enzymatically active) mutant forms of the enzyme. The fact that our 6-substituted analogues have structural components that bind in ways that are similar to the target and viral DNAs points the way to developing new and more broadly effective INSTIs.25
Shape complementarity also plays an important role in protein–ligand recognition.34 While simultaneously remaining with an envelope defined by the cognate substrates, the 6- substituted analogues, 4c and 4f, occupy more of the catalytic-site volume (as defined by the structure of the protein and the 3′-processed DNA) than the unsubstituted parent 4a. Thus, the compounds have good shape complementarity without protruding beyond the substrate envelope. This has resulted in potent inhibitors that retain high efficacy against the entire panel of resistant mutant forms of IN.
Our findings provide a unifying theme for understanding why there are common structural features in some of the most successful second generation INTSIs. For example, although it is known that the terminal (R)-4-methyl-1,3-oxazinane ring of DTG is important for its potency against resistant mutant forms of IN, this portion of the molecule was developed empirically.30 In the PFV cocrystal structures of DTG (PDB code: 3S3M),20 the position where the third ring is bound overlaps with portions of a bound MK-2048 molecule (PDB code: 3OYB).19 MK-2048 is another second generation tricyclic INSTI that retains good inhibitory potency against RAL-resistant mutants. For both DTG and MK-2048, the overlapping regions project toward the β4-α2 loop in the respective PFV intasome cocrystal structures (see Figure 2). Examination of structures of the PFV intasome with other second generation INSTIs bound, including “Compound G” (PDB code: 3OYG)19 and MK0536 (PDB code: 3OYH;19 these structures are shown in the Supporting Information, Figure S2) and its more recent analogues,31 shows that these molecules bind within a volume that is defined by the bound substrates. We previously proposed that the contacts made between the compounds and HIV-1 IN may be an important feature that helps some INSTIs retain efficacy against RAL-resistant mutants.19
Our current work shows that portions of the most successful INSTIs can be viewed as occupying regions defined by the binding of the precleaved viral DNA and target DNA, which helps to explain why the compounds are broadly effective against the known drug resistant IN mutants. The principle of maximizing shape complementarity and recapitulating the structural components (structural complementarity) of two of the substrates, the target and viral DNAs, could serve as a guiding principle for the development of new INSTIs.
Methods
Synthetic
Detailed synthetic procedures and analytical data for the synthesis of final products 4b–4f are provided in the Supporting Information.
Recombinant Proteins, Biochemical Assays, and Crystallography
IN proteins were produced in E. coli; HIV-1 IN biochemical assays and PFV crystallography were carried out as previously described.18,19,35 Detailed experimental protocols are provided in the Supporting Information.
Single-round HIV-1 Infectivity Assay
Assays were performed using the human embryonic kidney cell culture cell line 293 acquired from the American type culture collection (ATCC) and the human osteosarcoma cell line, HOS, obtained from Dr. Richard Schwartz (Michigan State University, East Lansing, MI) using methods that have been previously reported36,37 and are described in detail in the Supporting Information.
X-ray Crystallography
PFV intasome crystals grown as previously described19,20 were soaked in the presence of 0.5–1 mM INSTIs in cryoprotection solution prior to snap freezing in liquid nitrogen. X-ray diffraction data were collected on beamline I04 of the Diamond Light Source (Oxfordshire, UK) and processed using XDS38 and Scala.39 Structures were solved using rigid-body refinement of 4BDZ (protein and DNA components), and small molecules were fitted into difference maps. The structures were built in Coot,40 refined in Phenix,41 and validated using MolProbity.42 Data collection and refinement statistics are given in Supporting Information Table S1.
Acknowledgments
We thank the staff of the Diamond Light Source beamline I04 for assistance with X-ray data collection. Our studies are supported by the NIH Intramural Program, Center for Cancer Research (ZIA BC 007363, Z01 BC 007333), National Cancer Institute and by grants from the NIH Intramural AIDS Targeted Antiviral Program (IATAP).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschembio.5b00948.
Detailed chemical synthesis and compound characterization, details of HIV-1 IN biochemical assays, HIV-1 vector constructs, single-round HIV-1 infectivity assay, and X-ray crystallography data collection and refinement statistics (PDF)
Accession Codes
Coordinates and structure factors were deposited with the Protein Data Bank under accession codes 5FRM, 5FRN, and 5FRO.
The authors declare the following competing financial interest(s): Compounds described in the paper are included in pending or filed patent applications.
Supplementary Material
References
- Pommier Y.; Marchand C. (2012) Interfacial inhibitors: Targeting macromolecular complexes. Nat. Rev. Drug Discovery 11, 25–36. 10.1038/nrd3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherepanov P.; Maertens G. N.; Hare S. (2011) Structural insights into the retroviral DNA integration apparatus. Curr. Opin. Struct. Biol. 21, 249–256. 10.1016/j.sbi.2010.12.005. [DOI] [PubMed] [Google Scholar]
- Di Santo R. (2014) Inhibiting the HIV integration process: Past, present, and the future. J. Med. Chem. 57, 539–566. 10.1021/jm400674a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abram M. E.; Ferris A. L.; Shao W.; Alvord W. G.; Hughes S. H. (2010) Nature, position, and frequency of mutations made in a single cycle of HIV-1 replication. J. Virol. 84, 9864–9878. 10.1128/JVI.00915-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuevas J. M.; Geller R.; Garijo R.; Lopez-Aldeguer J.; Sanjuan R. (2015) Extremely high mutation rate of HIV-1 in vivo. PLoS Biol. 13, e1002251. 10.1371/journal.pbio.1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wainberg M. A.; Zaharatos G. J.; Brenner B. G. (2011) Development of antiretroviral drug resistance. N. Engl. J. Med. 365, 637–646. 10.1056/NEJMra1004180. [DOI] [PubMed] [Google Scholar]
- Garrido C.; Villacian J.; Zahonero N.; Pattery T.; Garcia F.; Gutierrez F.; Caballero E.; Van Houtte M.; Soriano V.; de Mendoza C. (2012) Broad phenotypic cross-resistance to elvitegravir in HIV-infected patients failing on raltegravir-containing regimens. Antimicrob. Agents Chemother. 56, 2873–2878. 10.1128/AAC.06170-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geretti A. M.; Armenia D.; Ceccherini-Silberstein F. (2012) Emerging patterns and implications of HIV-1 integrase inhibitor resistance. Curr. Opin. Infect. Dis. 25, 677–686. 10.1097/QCO.0b013e32835a1de7. [DOI] [PubMed] [Google Scholar]
- Quashie P. K.; Mesplede T.; Wainberg M. A. (2013) Evolution of HIV integrase resistance mutations. Curr. Opin. Infect. Dis. 26, 43–49. 10.1097/QCO.0b013e32835ba81c. [DOI] [PubMed] [Google Scholar]
- Ballantyne A. D.; Perry C. M. (2013) Dolutegravir: First global approval. Drugs 73, 1627–1637. 10.1007/s40265-013-0121-4. [DOI] [PubMed] [Google Scholar]
- Hurt C. B.; Sebastian J.; Hicks C. B.; Eron J. J. (2014) Resistance to HIV integrase strand transfer inhibitors among clinical specimens in the United States, 2009–2012. Clin. Infect. Dis. 58, 423–431. 10.1093/cid/cit697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack P. L. (2014) Dolutegravir: A review of its use in the management of HIV-1 infection in adolescents and adults. Drugs 74, 1241–1254. 10.1007/s40265-014-0256-y. [DOI] [PubMed] [Google Scholar]
- Mesplede T.; Quashie P. K.; Wainberg M. A. (2012) Resistance to HIV integrase inhibitors. Curr. Opin. HIV AIDS 7, 401–408. 10.1097/COH.0b013e328356db89. [DOI] [PubMed] [Google Scholar]
- Wainberg M. A.; Mesplede T.; Raffi F. (2013) What if HIV were unable to develop resistance against a new therapeutic agent?. BMC Med. 11, 249. 10.1186/1741-7015-11-249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah B. M.; Schafer J. J.; DeSimone J. A. Jr. (2014) Dolutegravir: A new integrase strand transfer inhibitor for the treatment of HIV. Pharmacotherapy 34, 506–520. 10.1002/phar.1386. [DOI] [PubMed] [Google Scholar]
- Malet I.; Gimferrer Arriaga L.; Artese A.; Costa G.; Parrotta L.; Alcaro S.; Delelis O.; Tmeizeh A.; Katlama C.; Valantin M.-A.; Ceccherini-Silberstein F.; Calvez V.; Marcelin A.-G. (2014) New raltegravir resistance pathways induce broad cross-resistance to all currently used integrase inhibitors. J. Antimicrob. Chemother. 69, 2118–2122. 10.1093/jac/dku095. [DOI] [PubMed] [Google Scholar]
- de Souza Cavalcanti J.; de Paula Ferreira J. L.; Morenade Souza Guimaraes P.; Vidal J. E.; de Macedo Brigido L. F. (2015) High frequency of dolutegravir resistance in patients failing a raltegravir-containing salvage regimen. J. Antimicrob. Chemother. 70, 926–929. 10.1093/jac/dku439. [DOI] [PubMed] [Google Scholar]
- Hare S.; Gupta S.; Valkov E.; Engelman A.; Cherepanov P. (2010) Retroviral intasome assembly and inhibition of DNA strand transfer. Nature 464, 232–236. 10.1038/nature08784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hare S.; Vos A. M.; Clayton R. F.; Thuring J. W.; Cummings M. D.; Cherepanov P. (2010) Molecular mechanisms of retroviral integrase inhibition and the evolution of viral resistance. Proc. Natl. Acad. Sci. U. S. A. 107, 20057–20062. 10.1073/pnas.1010246107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hare S.; Smith S. J.; Metifiot M.; Jaxa-Chamiec A.; Pommier Y.; Hughes S. H.; Cherepanov P. (2011) Structural and functional analyses of the second-generation integrase strand transfer inhibitor dolutegravir (S/GSK1349572). Mol. Pharmacol. 80, 565–572. 10.1124/mol.111.073189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menendez-Arias L. (2013) Molecular basis of human immunodeficiency virus type 1 drug resistance: Overview and recent developments. Antiviral Res. 98, 93–120. 10.1016/j.antiviral.2013.01.007. [DOI] [PubMed] [Google Scholar]
- Serrao E.; Odde S.; Ramkumar K.; Neamati N. (2009) Raltegravir, elvitegravir, and metoogravir: The birth of ″me-too″ HIV-1 integrase inhibitors. Retrovirology 6, 25. 10.1186/1742-4690-6-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X. Z.; Smith S. J.; Metifiot M.; Johnson B. C.; Marchand C.; Pommier Y.; Hughes S. H.; Burke T. R. Jr. (2014) Bicyclic 1-hydroxy-2-oxo-1,2-dihydropyridine-3-carboxamide-containing HIV-1 integrase inhibitors having high antiviral potency against cells harboring raltegravir-resistant integrase mutants. J. Med. Chem. 57, 1573–1582. 10.1021/jm401902n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X. Z.; Smith S. J.; Metifiot M.; Marchand C.; Boyer P. L.; Pommier Y.; Hughes S. H.; Burke T. R. Jr. (2014) 4-Amino-1-hydroxy-2-oxo-1,8-naphthyridine-containing compounds having high potency against raltegravir-resistant integrase mutants of HIV-1. J. Med. Chem. 57, 5190–5202. 10.1021/jm5001908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hare S.; Maertens G. N.; Cherepanov P. (2012) 3′-Processing and strand transfer catalysed by retroviral integrase in crystallo. EMBO J. 31, 3020–3028. 10.1038/emboj.2012.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johns B. A.; Kawasuji T.; Velthuisen E. J. (2013) HIV integrase inhibitors. RSC Drug Discovery Ser. 32, 149–188. 10.1039/9781849737814-00149. [DOI] [Google Scholar]
- Charpentier C.; Karmochkine M.; Laureillard D.; Tisserand P.; Belec L.; Weiss L.; Si-Mohamed A.; Piketty C. (2008) Drug resistance profiles for the HIV integrase gene in patients failing raltegravir salvage therapy. HIV Med. 9, 765–770. 10.1111/j.1468-1293.2008.00628.x. [DOI] [PubMed] [Google Scholar]
- Cooper D. A.; Steigbigel R. T.; Gatell J. M.; Rockstroh J. K.; Katlama C.; Yeni P.; Lazzarin A.; Clotet B.; Kumar P. N.; Eron J. E.; Schechter M.; Markowitz M.; Loutfy M. R.; Lennox J. L.; Zhao J.; Chen J.; Ryan D. M.; Rhodes R. R.; Killar J. A.; Gilde L. R.; Strohmaier K. M.; Meibohm A. R.; Miller M. D.; Hazuda D. J.; Nessly M. L.; DiNubile M. J.; Isaacs R. D.; Teppler H.; Nguyen B.-Y. (2008) Subgroup and resistance analyses of raltegravir for resistant HIV-1 infection. N. Engl. J. Med. 359, 355–365. 10.1056/NEJMoa0708978. [DOI] [PubMed] [Google Scholar]
- Kawasuji T.; Johns B. A.; Yoshida H.; Weatherhead J. G.; Akiyama T.; Taishi T.; Taoda Y.; Mikamiyama-Iwata M.; Murai H.; Kiyama R.; Fuji M.; Tanimoto N.; Yoshinaga T.; Seki T.; Kobayashi M.; Sato A.; Garvey E. P.; Fujiwara T. (2013) Carbamoyl pyridone HIV-1 integrase inhibitors. 2. Bi- and tricyclic derivatives result in superior antiviral and pharmacokinetic profiles. J. Med. Chem. 56, 1124–1135. 10.1021/jm301550c. [DOI] [PubMed] [Google Scholar]
- Johns B. A.; Kawasuji T.; Weatherhead J. G.; Taishi T.; Temelkoff D. P.; Yoshida H.; Akiyama T.; Taoda Y.; Murai H.; Kiyama R.; Fuji M.; Tanimoto N.; Jeffrey J.; Foster S. A.; Yoshinaga T.; Seki T.; Kobayashi M.; Sato A.; Johnson M. N.; Garvey E. P.; Fujiwara T. (2013) Carbamoyl pyridone HIV-1 integrase Inhibitors 3. A diastereomeric approach to chiral nonracemic tricyclic ring systems and the discovery of dolutegravir (S/GSKi349572) and (S/GSK1265744). J. Med. Chem. 56, 5901–5916. 10.1021/jm400645w. [DOI] [PubMed] [Google Scholar]
- Raheem I. T.; Walji A. M.; Klein D.; Sanders J. M.; Powell D. A.; Abeywickrema P.; Barbe G.; Bennet A.; Clas S. D.; Dubost D.; Embrey M.; Grobler J.; Hafey M. J.; Hartingh T. J.; Hazuda D. J.; Miller M. D.; Moore K. P.; Pajkovic N.; Patel S.; Rada V.; Rearden P.; Schreier J. D.; Sisko J.; Steele T. G.; Truchon J. F.; Wai J.; Xu M.; Coleman P. J. (2015) Discovery of 2-pyridinone aminals: A prodrug strategy to advance a second generation of HIV-1 Integrase strand transfer inhibitors. J. Med. Chem. 58, 8154–8165. 10.1021/acs.jmedchem.5b01037. [DOI] [PubMed] [Google Scholar]
- Metifiot M.; Vandegraaff N.; Maddali K.; Naumova A.; Zhang X.; Rhodes D.; Marchand C.; Pommier Y. (2011) Elvitegravir overcomes resistance to raltegravir induced by integrase mutation Y143. AIDS 25, 1175–1178. 10.1097/QAD.0b013e3283473599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y.; Altman M. D.; Ali A.; Nalam M. N. L.; Cao H.; Rana T. M.; Schiffer C. A.; Tidor B. (2013) Testing the substrate-envelope hypothesis with designed pairs of compounds. ACS Chem. Biol. 8, 2433–2441. 10.1021/cb400468c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls A.; McGaughey G. B.; Sheridan R. P.; Good A. C.; Warren G.; Mathieu M.; Muchmore S. W.; Brown S. P.; Grant J. A.; Haigh J. A.; Nevins N.; Jain A. N.; Kelley B. (2010) Molecular shape and medicinal chemistry: A perspective. J. Med. Chem. 53, 3862–3886. 10.1021/jm900818s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metifiot M.; Maddali K.; Johnson B. C.; Hare S.; Smith S. J.; Zhao X. Z.; Marchand C.; Burke T. R. Jr.; Hughes S. H.; Cherepanov P.; Pommier Y. (2013) Activities, crystal structures and molecular dynamics of dihydro-1H-isoindole derivatives, inhibitors of HIV-1 integrase. ACS Chem. Biol. 8, 209–217. 10.1021/cb300471n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X. Z.; Maddali K.; Metifiot M.; Smith S. J.; Vu B. C.; Marchand C.; Hughes S. H.; Pommier Y.; Burke T. R. Jr. (2012) Bicyclic hydroxy-1H-pyrrolopyridine-trione containing HIV-1 integrase inhibitors. Chem. Biol. Drug Des. 79, 157–165. 10.1111/j.1747-0285.2011.01270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brachmann A.; Koenig J.; Julius C.; Feldbruegge M. (2004) A reverse genetic approach for generating gene replacement mutants in Ustilago maydis. Mol. Genet. Genomics 272, 216–226. 10.1007/s00438-004-1047-z. [DOI] [PubMed] [Google Scholar]
- Kabsch W. (2010) Xds. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66, 125–132. 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. (2011) An introduction to data reduction: space-group determination, scaling and intensity statistics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 67, 282–292. 10.1107/S090744491003982X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 60, 2126–2132. 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkóczi G.; Chen V. B.; Echols N.; Headd J. J.; Hung L.-W.; Jain S.; Kapral G. J.; Grosse Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R. D.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. (2011) The Phenix software for automated determination of macromolecular structures. Methods 55, 94–106. 10.1016/j.ymeth.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen V. B.; Arendall W. B. III; Headd J. J.; Keedy D. A.; Immormino R. M.; Kapral G. J.; Murray L. W.; Richardson J. S.; Richardson D. C. (2010) MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66, 12–21. 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X. Z.; Semenova E. A.; Vu B. C.; Maddali K.; Marchand C.; Hughes S. H.; Pommier Y.; Burke T. R. (2008) 2,3-Dihydro-6,7-dihydroxy-1H-isoindol-1-one-Based HIV-1 Integrase Inhibitors. J. Med. Chem. 51, 251–259. 10.1021/jm070715d. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.