

Neuron-specific isoforms of Endophilin-B1, also known as Bax-interacting factor-1 (Bif-1), are neuroprotective. Wang et al. reveal reduced expression of these variants in Alzheimer’s disease, and propose the existence of a feed-forward mechanism whereby beta-amyloid suppresses neuron-specific Bif-1, which in turn enhances beta-amyloid accumulation and neuronal vulnerability to stress.
Keywords: Alzheimer’s disease, cellular mechanisms, apoptosis, beta-amyloid, neuroprotection
Neuron-specific isoforms of Endophilin-B1, also known as Bax-interacting factor-1 (Bif-1), are neuroprotective. Wang et al. reveal reduced expression of these variants in Alzheimer’s disease, and propose the existence of a feed-forward mechanism whereby beta-amyloid suppresses neuron-specific Bif-1, which in turn enhances beta-amyloid accumulation and neuronal vulnerability to stress.
Abstract
Endophilin-B1, also known as Bax-interacting factor 1 (Bif-1, and encoded by SH3GLB1), is a multifunctional protein involved in apoptosis, autophagy and mitochondrial function. We recently described a unique neuroprotective role for neuron-specific alternatively spliced isoforms of endophilin-B1. To examine whether endophilin-B1-mediated neuroprotection could be a novel therapeutic target for Alzheimer’s disease we used a double mutant amyloid precursor protein and presenilin 1 (APPswe/PSEN1dE9) mouse model of Alzheimer’s disease and observed that expression of neuron-specific endophilin-B1 isoforms declined with disease progression. To determine if this reduction in endophilin-B1 has a functional role in Alzheimer’s disease pathogenesis, we crossed endophilin-B1−/− mice with APPswe/PSEN1dE9 mice. Deletion of endophilin-B1 accelerated disease onset and progression in 6-month-old APPswe/PSEN1dE9/endophilin-B1−/− mice, which showed more plaques, astrogliosis, synaptic degeneration, cognitive impairment and mortality than APPswe/PSEN1dE9 mice. In mouse primary cortical neuron cultures, overexpression of neuron-specific endophilin-B1 isoforms protected against amyloid-β-induced apoptosis and mitochondrial dysfunction. Additionally, protein and mRNA levels of neuron-specific endophilin-B1 isoforms were also selectively decreased in the cerebral cortex and in the synaptic compartment of patients with Alzheimer’s disease. Flow sorting of synaptosomes from patients with Alzheimer’s disease demonstrated a negative correlation between amyloid-β and endophilin-B1 levels. The importance of endophilin-B1 in neuronal function was further underscored by the development of synaptic degeneration and cognitive and motor impairment in endophilin-B1−/− mice by 12 months. Our findings suggest that endophilin-B1 is a key mediator of a feed-forward mechanism of Alzheimer’s disease pathogenesis where amyloid-β reduces neuron-specific endophilin-B1, which in turn enhances amyloid-β accumulation and neuronal vulnerability to stress.
Introduction
Endophilin-B1, also known as Bax-interacting factor-1 (Bif-1) or by its gene name SH3GLB1, is a multi-functional protein involved in autophagy, apoptosis, and mitochondrial function. A member of the endophilin family of proteins, endophilin-B1 was initially characterized as a protein that could bind and influence the shape of lipid membranes, including mitochondria (Cuddeback et al., 2001; Modregger et al., 2003; Karbowski et al., 2004). Endophilin-B1 was identified as a tumor suppressor gene that interacts with beclin (encoded by BECN1) and BAX to promote autophagy and apoptosis, respectively (Takahashi et al., 2005, 2007). Expression of endophilin-B1 is decreased in a number of different cancer types (Lee et al., 2006; Kim et al., 2008), and endophilin-B1 knockout in mice causes increased tumour formation (Takahashi et al., 2005, 2013).
Endophilin-B1 has primarily been studied in tumour cells, and the functional role of endophilin-B1 in neurons is not well understood. We recently reported that in contrast to what has been observed in non-neuronal cells (Karbowski et al., 2004; Takahashi et al., 2005), endophilin-B1 knockdown in neurons resulted in sensitization to apoptotic stimuli and mitochondrial fragmentation (Wang et al., 2014). These specific and unique functions of endophilin-B1 in neurons were attributable to novel splice isoforms endophilin-B1b (NP_001193581) and endophilin-B1c (XP_006710735) expressed specifically by neurons. Overexpression of endophilin-B1b or endophilin-B1c, but not the ubiquitously expressed endophilin-B1a (NP_057093), prevented apoptosis and mitochondrial fragmentation induced by endophilin-B1 knockdown or knockout (Wang et al., 2014). Ischaemic injury in mice produced by middle cerebral artery occlusion led to reduced endophilin-B1 protein in the ischaemic penumbra and mice lacking endophilin-B1 displayed increased infarct sizes and astrogliosis compared to wild-type littermates (Wang et al., 2014).
To determine if endophilin-B1 participates in the pathophysiology of chronic neurodegenerative disease we used in vivo and in vitro models of Alzheimer’s disease. We observed that the neuroprotective isoforms endophilin-B1b and endophilin-B1c (collectively referred to as endophilin-B1b/c) are specifically reduced by exposure to amyloid-β in vitro and in mice harbouring mutant APP and PSEN1 (APPswe/PSEN1dE9, abbreviated APP/PSEN1 hereafter). Crossing APP/PSEN1 mice to endophilin-B1−/− mice (abbreviated as Endo-B1−/− hereafter) exacerbated the behavioural and pathological phenotype. More remarkably, loss of endophilin-B1 also potentiated the accumulation of amyloid-β, suggesting that reduction of endophilin-B1 may have dual action in Alzheimer’s disease pathogenesis, affecting both amyloid-β metabolism and neuronal survival. Additionally, we demonstrate that endophilin-B1 deficiency alone is sufficient to disrupt synaptic integrity and neuronal function in vivo. We hypothesized that loss of endophilin-B1b/c might contribute to amyloid-β-induced neuronal degeneration through its influence on mitochondrial function and demonstrate here that restoring expression of endophilin-B1b/c prevents amyloid-β-mediated loss of mitochondrial membrane potential and neuronal apoptosis. Furthermore, we observed reduced endophilin-B1b/c expression relative to endophilin-B1a expression in CNS tissue from human Alzheimer’s disease cases. Taken together, the findings reported here suggest that endophilin-B1 may operate in a feed-forward loop at the nexus of Alzheimer’s disease pathophysiology whereby amyloid-β accumulation leads to decreased endophilin-B1b/c which subsequently enables both further amyloid-β accumulation and neurodegeneration. Thus restoration of endophilin-B1b/c expression may serve as a potential therapeutic target for Alzheimer’s disease.
Materials and methods
Animals
C57BL/6 mice were obtained from the Jackson laboratory. Endophilin-B1-deficient mice (Takahashi et al., 2007) that have been backcrossed to C57BL/6 16 times were maintained and genotyped as previously described (Wang et al., 2014). APP/PSEN1 mice on the C57BL/6 background [B6.Cg-Tg(APPswe,PSEN1dE9)85Dbo/Mmjax; (Jankowsky et al., 2004)] were confirmed using primers for APP (5′-AGGACTGACCACTCGACCAG-3′ and 5′-CGGGGGTCTAGTTCTGCAT-3′) and PSEN1 (5′-AATAGAGAACGGCAGGAGCA-3′ and 5′-GCCATGAGGGCACTAATCAT-3′). Heterozygous APP/PSEN1 mice were mated with either Endo-B1+/+ mice, or with Endo-B1−/− mice and the progeny backcrossed with Endo-B1−/− mice again, to obtain wild-type, APP/PSEN1, Endo-B1−/−, and APP/PSEN1/Endo-B1−/− mice. Animals were housed with bedding in groups of five in a specific pathogen-free facility under a 12 h light/dark cycle at 22 °C. Only male animals were used for experiments to minimize behavioural variability. Six wild-type and APP/PSEN1 mice at 6 weeks, and seven wild-type and APP/PSEN1 mice at 10 months, were compared for changes in endophilin-B1 protein expression. For 6-month studies, 16 wild-type, 16 APP/PSEN1, 16 Endo-B1−/−, and 17 APP/PSEN1/Endo-B1−/− animals were used to accurately assess possible subtle changes in behaviour, and to compensate for anticipated mortality in certain groups. For 9-month studies, five APP/PSEN1 and five APP/PSEN1/Endo-B1−/− animals were used for western blot analysis. For 12-month studies, eight wild-type and six Endo-B1−/− animals were used. The experimenter was blinded to each animal’s genotypes during behavioural analysis. Experiments with these animals were approved by the University of Washington institutional animal care committee.
Immunoblotting
Protein extracts for western blot analysis from mouse brain and neuronal cultures were prepared as described previously (Wang et al., 2014). Primary antibodies used were mouse monoclonal Bif-1 (clone 30A882.1.1, 1:500; Imgenex), mouse monoclonal β-actin (clone AC-15, 1:10 000; Sigma), rabbit polyclonal activated caspase-3 (#9661, 1:1000; Cell Signaling), mouse monoclonal neuronal class III β-tubulin (clone TuJ1, 1: 5000; Covance), mouse monoclonal synaptophysin (clone SY38, 1:5000; Millipore), mouse monoclonal amyloid precursor protein (clone 2.F2.19B4, 1:5000; Millipore), mouse monoclonal phosphorylated tau (clone AT8, 1:1000; Pierce), rabbit polyclonal tau (#A0024, 1:20 000; Dako), rabbit polyclonal presenilin-1 (#ARP58941_P050, 1:1000; Aviva Systems Biology), rabbit monoclonal presenilin-2 (clone EP1515Y, 1:5000; Epitomics). Horseradish peroxidase-conjugated secondary antibodies (1:2000) were from GE Healthcare. SuperSignal WestPico (Thermo Scientific) was used to expose X-ray films. For quantification, films were scanned and the protein bands of interest were measured for pixel intensity using NIH ImageJ software and normalized against β-actin values. When samples to be compared were run on multiple membranes, each membrane included a common reference sample to normalize against.
Morris water maze
Learning and memory were tested in mice using the Morris water maze (Morris, 1984). The maze consisted of a 165-gallon, circular galvanized stock tank, 122-cm in diameter and 61-cm tall, filled with water at room temperature made opaque by the addition of white tempera paint. A 10-cm square plexiglass stand was submerged just below the water level to serve as the escape platform in the middle of the northwest quadrant of the maze. Footage was captured using Noldus Media Recorder software (Noldus Information Technology). Animal location was traced and analysed using the Ethovision XT (Noldus) videotracking software. Stationary black and white shapes positioned on the walls surrounding the tank were used as spatial cues. Mice were trained to acquire the task in the morning for 4 days, with four trials per day with a 90 min intertrial interval. Mice were dropped randomly at one of the eight drop locations, facing outward towards the edge of the tank, and allowed to swim for 60 s. If a mouse failed to find the platform, it was guided to the platform, where it was allowed to sit for 10 s before being removed, dried with a paper towel, and placed under a heat lamp until dry before returning to its home cage. Acquisition data are presented as the average of the four trials for a particular day. Four days after the last acquisition day, memory retention was tested by removing the platform and monitoring mouse behaviour over 60 s. Time in the correct quadrant and cumulative distance to platform were measured.
Motor tests
One-year-old Endo-B1+/+ and Endo-B1−/− animals were subjected to further motor testing following the conclusion of the Morris water maze test. To test grip strength, mice were allowed to use either forelimbs or hindlimbs to grasp a metal grid attached to a digital grip strength meter (Columbus Instruments), and were slowly pulled away from the grid until they released their grasp. Maximum force (kg) applied prior to release was measured.
To test motor coordination, a four-lane rotarod (6-cm diameter; Ugo Basile) was accelerated 1 rpm every 5 s, from 4 rpm to 40 rpm. Latency to fall was recorded for three trials, with an intertrial interval of 2 h.
Gait and footfall patterns were measured in a dark room using a Catwalk apparatus (Noldus Information Technology), which provides detailed quantitative assessment of gait in mice as they walk along a walkway that illuminates as the paws make contact with the glass surface (Baiguera et al., 2012). Footage was recorded by a camera below the glass surface and analysed with Catwalk XT software. Testing consisted of placing the mouse at the end of the runway and allowing the mouse to traverse the runway before returning the mouse to their home cage. Three compliant runs were obtained for each mouse. A run was considered compliant for analysis if the mouse traversed the runway in ≤8 s and did not stop or turn around in the middle of the run.
Serial extraction of mouse brain tissue for amyloid-β Luminex assays
Mice were sacrificed by cervical dislocation and the cortex and the hippocampus were quickly dissected out on ice and snap frozen on dry ice to preserve protein integrity. The brain tissue was sonicated for ∼20 s in Tris-buffered saline (TBS; 50 mM Tris-Cl pH 7.6, 150 mM NaCl) containing protease inhibitor cocktail (Thermo Scientific). The lysate was centrifuged at 100 000 g for 15 min and the supernatant (TBS-soluble fraction) was collected and stored at −80 °C. The pellet was then washed once with TBS, dissolved in 6 M guanidine hydrochloride (GuHCl; Invitrogen) at 4 °C overnight and then centrifuged at 16 000 g for 30 min. The supernatant (GuHCl-soluble fraction) was collected and stored at −80 °C. All procedures were performed on ice to minimize protein degradation. Quantification of soluble and insoluble forms of human amyloid-β1-40 and amyloid-β1-42, expressed from the human mutant APP transgene, was done as previously described (Yang et al., 2013) using amyloid-β1-40 and amyloid-β1-42 Human Singleplex Bead Kits (Life Technologies) according to the manufacturer’s protocol.
Animal perfusion, tissue sectioning and immunofluorescence
Animals were rapidly anaesthetized with 2.5% avertin (Sigma) in saline via intraperitoneal injection (125 mg/kg) and perfused with phosphate-buffered saline (PBS) followed by 4% paraformaldehyde in PBS (Sigma). Brains were post-fixed in 4% paraformaldehyde overnight and cryoprotected in 30% sucrose (Sigma). Frozen sections were cut coronally at a thickness of 40 µm on a sliding microtome (Leica Microsystems). Free-floating sections were permeabilized in 0.2% Triton™ X-100, blocked in 5% goat serum and 1% bovine serum albumin, and labelled with antibodies before mounting onto coverslips with Aqua Poly/Mount (Polysciences). Primary antibodies used were mouse monoclonal amyloid-β (clone 6E10, 1:200; Covance), rabbit polyclonal GFAP (#Z0334, 1:400; Dako), mouse monoclonal pan-amyloid-β (MOAB-2 clone 6C3, 1:200; Millipore), rabbit polyclonal IBA-1 (#019-19741, 1:400; Wako Chemicals), mouse monoclonal synaptophysin (clone SY38, 1:200; Millipore). Nuclei were labelled with Hoechst 33258 (2.5 µg/ml). Alexa Fluor® dye-conjugated secondary antibodies (1:400) were from Invitrogen. Fluorescent microscopic images (five sections evenly spaced per brain region) were captured on an Axiovert 200 inverted microscope (Carl Zeiss Microimaging) equipped with a cooled CCD camera (SensiCam, Cooke Corp). For dentate gyrus and CA3 region, the field of view was sufficient to encompass the entire anatomical structure. For cortex, random regions dorsal to the hippocampus were taken. All samples were processed in parallel to control for variability in staining or image processing. Fluorescence was quantified using Slidebook 5.0 (Intelligent Imaging Innovations) by measuring the area with fluorescence intensity above a set threshold that was the same for every sample. The length of hippocampal CA3 pyramidal cell synaptic input was measured using ImageJ by drawing a radial line from the top of the cell layer (labelled with Hoechst) to the edge of synaptophysin-positive structures.
Primary neuronal culture
Postnatal cortical neurons from newborn C57BL/6 mice (within 24 h of birth) were prepared as previously described (Xiang et al., 1996) and maintained in Neurobasal®-A (Gibco) supplemented with B-27 (Gibco) and GlutaMAX-I (Gibco). One day after plating, cultures were infected with lentivirus at 10 MOI (multiplicity of infection) for 3 days before treatment with 10 µM amyloid-β25-35 peptide or inactive control amyloid-β35-25 peptide (Bachem; trifluoroacetate salt solubilized in water) for 24 h. Production of DNA constructs and lentivirus for enhanced green fluorescent protein (EGFP) and human endophilin-B1 isoforms has been described (Uo et al., 2009; Wang et al., 2014). Overall neuronal viability was determined by phase-contrast microscopy, based on specific morphological criteria, as previously described (Xiang et al., 1996).
To measure mitochondrial membrane potential, the JC-1 dye was used as previously described (Wang et al., 2014). Five fluorescent images per condition were captured at ×32 magnification, and the total pixel intensity of red (polarized mitochondria) and green (depolarized mitochondria) fluorescence above a set threshold was obtained.
To measure γ-secretase activity in primary mouse cortical neurons, a three-construct system was used as previously described (Jayadev et al., 2010). Briefly, neurons were plated in 96-well plates at 5 × 104 cells/well and infected with lentivirus carrying APP-Gal4-VP16, Gal4-Luc-GFP, and renilla luciferase (infection control). Four days later, cells were lysed and subject to luciferase assay using the manufacturer’s protocol (Promega). γ-Secretase activity was measured as the ratio of firefly to renilla luciferase. Assay specificity was confirmed using the γ-secretase inhibitor DAPT (Sigma), which decreased firefly luciferase activity by > 80% when treated at 100 µM for 24 h prior to lysis.
Western blot and reverse transcriptase-PCR with human patient samples
Brain autopsy samples of inferior parietal lobule cerebral cortex were obtained from the University of Washington Alzheimer’s Disease Research Centre (ADRC). A total of 37 samples (19 males and 18 females) were used, each with an assigned Braak stage based on neurofibrillary tangle staining. The mean age was 84.1 years, and the mean post-mortem interval was 4.6 h. Frozen samples were powderized on dry ice, and transferred into and sonicated in ice-chilled SDS (sodium dodecyl sulphate) sample buffer (50 mM Tris-Cl pH 6.8, 2% SDS, 10% glycerol).
RNA was isolated using an RNeasy® isolation kit (Qiagen), from an aliquot of the powderized human parietal cortex samples used for western blot analysis and reverse transcribed using SuperScript™ II Reverse Transcriptase according to the manufacturer’s instructions (Invitrogen). The cDNA was PCR amplified with Taq DNA polymerase (New England Biolabs) using primers that amplified both the b (NM_001206652) and a (NM_016009) transcripts of SH3GLB1 (5′-AAAAAAGGCAAAAGCTGCAGAAACTAGAAA-3′ and 5′-CATTCAGACAGCGAAGGTGATG-3′), the gene encoding endophilin-B1 protein. Expected PCR products for SH3GLB1a and SH3GLB1b were 147 and 210 bp, respectively. Data are presented as a ratio of SH3GLB1b to SH3GLB1a mRNA levels.
Human patient synaptosome samples and flow cytometry
Brain autopsy samples of parietal (A39 and A40) and superior parietal (A7) cortex were obtained from the UCLA, USC and UCI ADRCs. A total of 35 samples (18 females and 16 males) were used; control cases include three samples from cognitively normal elderly controls, two samples from spinocerebellar ataxia type II patients, and two from patients with Pick’s disease. One control case had hippocampal sclerosis but did not have other histopathological signs of Alzheimer’s disease. All samples were assigned a Braak stage based on neurofibrillary pathology. The mean age was 81.6 years for females, and 83.2 years for males. The mean post-mortem interval was 6.4 h. Intact synaptosomes in the P-2 fractions were prepared from cryopreserved brain tissue as described and characterized previously (Gylys et al., 2004), cryopreserved in 0.32 M buffered sucrose solution and stored at −80 °C as aliquots. On the day of the experiment the aliquots were quickly thawed at 37 °C and P-2 pellets were collected by centrifugation. P-2 fractions were then analysed by western blot or flow cytometry.
For flow cytometry, P-2 fractions were fixed in 0.25% paraformaldehyde/PBS (1 h at 4 °C), permeabilized in 0.2% Tween-20/PBS solution (15 min at 37 °C), and incubated with amyloid-β (10G4, which has been extensively characterized in synaptosome preparations; Sokolow et al., 2012) and endophilin-B1 specific antibodies, directly labelled with Alexa Fluor® 488 or Alexa Fluor® 647 fluorochromes using Zenon isotype-specific labelling kits (Molecular Probes). Immunolabeled P-2 pellets were washed with 0.2% Tween-20/PBS and resuspended in 500 µl of PBS for flow cytometry analysis. Data were acquired using a BD FACSCalibur analytical flow cytometer (Becton-Dickinson) equipped with argon 488 nm, helium-neon 635 nm, and helium-cadmium 325 nm lasers. Debris was excluded by establishing a size threshold set on forward light scatter. A total of 15 000 particles were collected and analysed for each sample. Fluorescence was detected by FL-1 (FITC) and FL-4 (APC) channel photomultiplier tubes, respectively. Analysis was performed using FCS Express software (DeNovo Software).
Statistics
All results were presented as mean ± standard error of the mean (SEM) unless otherwise stated. Student’s t-test, Log-Rank test, Pearson’s correlation, one-way, two-way, and repeated measures ANOVA with Tukey post hoc tests were applied where applicable using Prism Software (GraphPad). Results were considered statistically significant when P < 0.05.
Results
Neuron-specific endophilin-B1 expression is decreased in symptomatic Alzheimer’s disease mice
To determine whether endophilin-B1 (Endo-B1) protein is altered with disease onset and progression in a mouse model of Alzheimer’s disease, presymptomatic (6-week-old) and symptomatic (10-month-old) APP/PSEN1 mice were examined. Two major endophilin-B1 protein bands were detected for mouse brain (Fig. 1A), as previously described (Wang et al., 2014), with the lower band representing the ubiquitously expressed endophilin-B1a isoform and the upper band corresponding to neuron-specific isoforms, endophilin-B1b and endophilin-B1c (designated as endophilin-B1b/c because the endophilin-B1b and c isoforms cannot be individually resolved by SDS-PAGE; Wang et al., 2014). In 10-month-old APP/PSEN1 mice that display cognitive decline but no neuronal loss (West et al., 2009; Volianskis et al., 2010), there was a marked reduction in endophilin-B1b/c in the hippocampus and cortex, brain regions containing plaque pathology (Jankowsky et al., 2004), relative to wild-type littermates (Fig. 1A and B), but no change in endophilin-B1a (Fig. 1A and C). There was no change in endophilin-B1a or endophilin-B1b/c in presymptomatic 6-week-old APP/PSEN1 mice (Fig. 1D and E) or in the cerebellum of 10-month-old APP/PSEN1 mice (not shown), an area where plaque pathology has not yet developed (Thal et al., 2002). Thus, reduced endophilin-B1b/c expression in APP/PSEN1 mice is associated with disease progression in relevant brain regions.
Figure 1.

Endophilin-B1b/c is decreased in hippocampus and cortex of symptomatic APP/PSEN1 mice. (A) Hippocampus and cortex of 10-month-old APP/PSEN1 mice and wild-type littermates were analysed for endophilin-B1 (Endo-B1) by western blot. Actin was used as a loading control. (B and C) Densitometric quantification of western blots reveals that endophilin-B1b/c (B), but not endophilin-B1a (C) is decreased in hippocampus and cortex of 10-month-old APP/PSEN1 mice. *P < 0.05 versus control, Student’s t-test (n = 7 mice per group). Endophilin-B1b/c (D) and endophilin-B1a (E) in 6-week-old presymptomatic APP/PSEN1 mice are not different from wild-type controls (n = 6).
Loss of endophilin-B1 potentiates disease progression in APP/PSEN1 mice
To address whether loss of endophilin-B1 contributes to Alzheimer’s disease pathology, heterozygous APP/PSEN1 mice were crossed with Endo-B1+/+ (wild-type) or Endo-B1−/− mice to derive four genotypes (wild-type, APP/PSEN1, Endo-B1−/−, APP/PSEN1/Endo-B1−/−) and were then compared for development of Alzheimer’s disease-related pathology at 6 months of age, a time-point when plaques begin to form but before significant cognitive decline (Jankowsky et al., 2004; Volianskis et al., 2010). Early mortality in APP/PSEN1 mice (Fig. 2A) was observed within the reported range (Gimbel et al., 2010). Endo-B1−/− mice did not have enhanced mortality relative to wild-type controls. However, APP/PSEN1/Endo-B1−/− mice demonstrated a statistically significant increase in early mortality compared to APP/PSEN1 mice (Fig. 2A). Like APP/PSEN1 mice, deaths were sudden with no overt loss of motor function (data not shown) or changes in body weight in those that remained alive (Fig. 2B).
Figure 2.

Endophilin-B1 deficiency exacerbates mortality and cognitive decline in APP/PSEN1 mice. (A) Endophilin-B1 knockout (Endo-B1−/−) and Endo-B1+/+ (wild-type, WT) mice were crossed with heterozygous APP/PSEN1 mice to generate four genotypes. Mortality was monitored over 6 months. *P < 0.05 versus wild-type; #P < 0.05 versus APP/PSENn1, log-rank test (n = 16–17 mice per group). (B) There was no difference in animal weights at 6 months. (C) Mice at 6 months were trained in the Morris water maze over a period of 4 days, with acquisition curves shown for time to platform. *P < 0.05 versus Endo-B1 − /−; #P < 0.05 versus APP/PSEN1, two-way ANOVA (n = 7–16; symbols to the right of the curve represent significant differences in acquisition curves, while symbols on top represent significant differences in acquisition time for that specific trial day). (D and E) Four days after the last training day, memory retention was measured with the platform removed. Time in correct quadrant (D) and total distance to platform (E) are shown. *P < 0.05 versus Endo-B1−/−, two-way ANOVA (n = 7–16). (F) Behavioural impairment in APP/PSEN1/Endo-B1−/− animals was not due to differences in swimming velocity.
To assess the impact of endophilin-B1 knockout on spatial learning and memory in APP/PSEN1 mice all four genotypes were evaluated using the Morris water maze. At 6 months, APP/PSEN1 mice and Endo-B1−/− mice showed no detectable impairment in memory acquisition relative to wild-type mice (Fig. 2C). However, APP/PSEN1/Endo-B1−/− mice showed significant memory deficits relative to all other groups (Fig. 2C). Only APP/PSEN1/Endo-B1−/− mice demonstrated impaired memory retention, when tested without the platform 4 days after the last training session, spending significantly less time in the correct quadrant (Fig. 2D) and remaining at a significantly farther distance from the previous platform location (Fig. 2E). The memory impairment observed was not due to diminished motor function, as all four genotypes showed similar swimming velocities (Fig. 2F).
To determine if endophilin-B1 influences amyloid-β accumulation in APP/PSEN1 mice we assessed the relative levels of soluble and insoluble amyloid-β using the serial extraction technique (Yang et al., 2013). Hippocampal and cortical protein from 6-month-old APP/PSEN1 or APP/PSEN1/Endo-B1−/− mice was extracted using TBS to isolate soluble proteins followed by 6 M GuHCl to isolate insoluble proteins. Human amyloid-β peptide (1-40 and 1-42) was quantified using a Luminex-based assay (Yang et al., 2013). Endophilin-B1 deficiency led to significant elevations in soluble amyloid-β1-42 peptide in the cortex with a similar trend seen in the hippocampus, but did not alter the levels of soluble amyloid-β1-40 (Fig. 3A). The absence of endophilin-B1 increased both insoluble amyloid-β1-40 and amyloid-β1-42 in the cortex and hippocampus, although the difference in cortical amyloid-β1-42 did not reach statistical significance (Fig. 3B). Thus, endophilin-B1 knockout increased the amount of both soluble and insoluble amyloid-β in APP/PSEN1 mice.
Figure 3.

Endophilin-B1 deficiency exacerbates amyloid-β peptide burden in APP/PSEN1 mice. (A) TBS-soluble fraction, (B) GuHCl-soluble fraction. Levels of amyloid-β (Aβ) peptides 1–40 and 1-42 were determined for the two fractions obtained from cortex and hippocampus of APP/PSEN1/Endo-B1+/+ and APP/PSEN1/Endo-B1−/− mice at 6 months using standard Luminex assays. *P < 0.05 versus Endo-B1+/+ counterparts, Student’s t-test (n = 5–8 mice per group).
To determine if loss of endophilin-B1 alters amyloid plaque formation and astrogliosis, two pathological hallmarks of Alzheimer’s disease (Rodriguez et al., 2008; Ballard et al., 2011), we labelled mouse brain tissue with antibodies against amyloid-β and GFAP. In APP/PSEN1 mice endophilin-B1 deficiency substantially exacerbated Alzheimer’s disease-related pathology. Plaque burden was increased in the absence of endophilin-B1 in the cortex and the dentate gyrus and the CA3 region of hippocampus (Fig. 4A, D and G), consistent with the increased levels of insoluble amyloid-β detected (Fig. 3). Both the number of plaques and plaque area were significantly increased in the cortex and dentate gyrus of APP/PSEN1/Endo-B1−/− mice compared to APP/PSEN1 mice (Fig. 4B and E) although not significantly increased in the CA3 region due to large variations (Fig. 4H). Similar results were found with a second amyloid-β antibody, MOAB-2 (Supplementary Fig. 1), as well as with Thioflavin-S staining of mature plaques (not shown).
Figure 4.

Endophilin-B1 deficiency increases amyloid-β plaque accumulation, astrogliosis, synaptic degeneration, and tau hyperphosphorylation in APP/PSEN1 mice. (A–I) Immunoreactivity for amyloid-β (6E10 antibody, green) and astrocytes (GFAP antibody, red) was quantitated for cortex (A–C), and hippocampal dentate gyrus (D–F) and CA3 region (G–I) at 6 months. For amyloid-β immunoreactivity, the number of plaques per field and plaque area were measured only in APP/PSEN1/Endo-B1+/+ and APP/PSEN1/Endo-B1−/− animals, as no plaques were detectable in wild-type or Endo-B1−/− animals. *P < 0.05 versus APP/PSEN1/Endo-B1+/+, #P < 0.05 versus Endo-B1−/−, Student’s t-test or two-way ANOVA (n = 4 mice per group). (J) Immunoreactivity of presynaptic terminals (synaptophysin antibody, green) in the CA3 region of the hippocampus. Dotted white line marks the edge of the pyramidal cell layer, while dotted red line demarcates the extent of CA3 apical dendrites innervated by mossy fibres. (K and L) Length of CA3 synaptic input (K; distance from white to red dotted lines) and synaptophysin area (L; area of synaptophysin-positive structures within the two dotted lines) were quantified. *P < 0.05 versus APP/PSEN1/Endo-B1+/+, two-way ANOVA (n = 4). Nuclei were labelled with Hoechst 33258. Scale bars in A, D and G = 100 µm; J = 50 µm. (M) Cortex and hippocampus of 9-month-old APP/PSEN1/Endo-B1+/+ and APP/PSEN1/Endo-B1−/− mice were analysed for phosphorylated tau (clone AT8) and total tau by western blot. (N) The ratio of phosphorylated tau to total tau was elevated in the cortices of mice lacking endophilin-B1. *P < 0.05, Student’s t-test (n = 5 mice per condition).
Endophilin-B1 knockout did not induce astrogliosis at 6 months in any of the three brain regions examined (Fig. 4C, F and I). In the cortex of APP/PSEN1 mice, GFAP immunoreactivity was primarily restricted to the immediate vicinity of plaques. In the hippocampus (dentate gyrus and CA3), GFAP staining was diffuse and not strongly associated with plaques (Fig. 4D and G). Using GFAP immunoreactive area as a surrogate for the extent of the astroglial response, endophilin-B1 knockout increased astrogliosis in APP/PSEN1 mice across the cortex and the hippocampus (Fig. 4C, F and I). In contrast, no significant change in microglial activation was observed at this time point in any of the brain regions examined (Supplementary Fig. 1).
To determine if the increased plaque and astrogliosis correlated with neurodegenerative changes in APP/PSEN1/Endo-B1−/− mice, synaptic input to CA3 pyramidal neurons was assessed using immunoreactivity for synaptophysin, a presynaptic marker commonly lost in human Alzheimer’s disease and mouse models (Sze et al., 1997; Gimbel et al., 2010). At 6 months, neither Endo-B1−/− nor APP/PSEN1 mice exhibited any alteration in the synaptophysin expression pattern compared to wild-type mice (Fig. 4J–L). However, APP/PSEN1/Endo-B1−/− mice showed both a decreased number of synaptic endings (Fig. 4K) and a reduced area of innervation (Fig. 4L) in the dendritic compartment of CA3 pyramidal neurons compared to APP/PSEN1 mice, suggesting that loss of endophilin-B1 contributes to Alzheimer’s disease-related synaptic degeneration in this model.
Tau hyperphosphorylation is a key component of the amyloid cascade hypothesis missing in most rodent amyloid models (Duyckaerts et al., 2008). To determine if the increased amyloid-β and plaque accumulation in APP/PSEN1/Endo-B1−/− mice leads to increased hyperphosphorylated tau, we evaluated the level of tau phosphorylation by immunoblot (clone AT8) relative to total tau in the cortex and hippocampus. At 9 months, we observed increased phospho-tau in the cortex of APP/PSEN1/Endo-B1−/− mice compared to APP/PSEN1 mice (Fig. 4M and N), suggesting that loss of endophilin-B1 may contribute to the generation of hyperphosphorylated tau and subsequent neurodegeneration.
Endophilin-B1 knockout mice develop age-related synaptic degeneration and behavioural impairment
Because loss of endophilin-B1 hastened the onset of neurodegeneration in APP/PSEN1 mice, we examined whether endophilin-B1 knockout alone was sufficient to cause neurodegeneration. By 12 months, Endo-B1−/− mice demonstrated impaired memory acquisition (Fig. 5A) and retention (Fig. 5B) in the Morris water maze compared to wild-type littermates. Endo-B1−/− mice were not impaired in swim speed (Fig. 5C), grip strength (Supplementary Table 1), or rotarod performance (Supplementary Table 1) as measures of motor function. However, gait analysis using the more sensitive and specific CatWalk assay (Vandeputte et al., 2010) demonstrated that Endo-B1−/− mice took more numerous, shorter, and slower steps, resulting in slower walk speeds (Supplementary Table 1). Regularity index, a measure of normal step sequence patterns, and phase dispersions, a measure of inter-paw coordination, were also reduced (Supplementary Table 1). Front paws were more impaired than hind paws in Endo-B1−/− mice, with several parameters, such as step cycle and print area, affected only in the front paws (Supplementary Table 1). Synaptophysin immunoreactivity in the CA3 region of the hippocampus was also decreased in Endo-B1−/− mice (Fig. 5D and E). These data collectively suggest that loss of endophilin-B1 alone is sufficient to induce impairment in neuronal function and synaptic degeneration.
Figure 5.

Endo-B1−/− mice demonstrate cognitive deficits and synaptic degeneration at 12 months. (A) Mice at 12 months were trained in the Morris water maze over a period of 4 days, with acquisition curves shown for time to platform. *P < 0.05 versus Endo-B1+/+ for that day, two-way ANOVA (n = 6–8 mice per group). (B) Endo-B1−/− mice were impaired in the retention test measured by cumulative distance from platform. *P < 0.05 versus Endo-B1+/+, Student’s t-test (n = 6–8). (C) No differences in swimming velocity in Endo-B1−/− mice. (D and E) Endo-B1−/− mice showed synaptic degeneration in CA3 with a reduction in both the length of synaptic input (D) and the area of synaptophysin immunoreactivity (E) in the CA3 region of hippocampus. *P < 0.05 versus Endo-B1+/+, Student’s t-test (n = 4–5).
Western blots were used to assess whether endophilin-B1 deficiency is sufficient to alter proteins involved in Alzheimer’s disease pathogenesis. At 12 months, Endo-B1−/− mouse cortices had increased amyloid precursor protein compared to Endo-B1+/+ mice (Supplementary Fig. 2). Expression of PSEN1 and PSEN2, components of γ-secretase involved in cleavage of APP into amyloid-β, were not altered in Endo-B1−/− mice (Supplementary Fig. 2). Similarly, modulation of endophilin-B1 expression in cultured cortical neurons had no effect on γ-secretase activity using an in vitro reporter assay (Supplementary Fig. 3). The finding that endophilin-B1 deficiency alone leads to elevated amyloid precursor protein further supports a key role for the observed loss of endophilin-B1b/c in Alzheimer’s disease pathogenesis.
Neuron-specific endophilin-B1 isoforms protect against amyloid-β neurotoxicity in primary cultured neurons
To assess potential mechanisms by which endophilin-B1 modulates amyloid-β-mediated neurodegeneration, we employed an in vitro model of amyloid toxicity. Amyloid-β25-35, a potent neurotoxic form of the amyloid-β peptide (Yankner et al., 1990), was added to cultured postnatal mouse cortical neurons. Expression of endophilin-B1b/c, but not endophilin-B1a, was selectively reduced by amyloid-β25-35 exposure (Fig. 6A and B). Treatment with neurotoxic amyloid-β also resulted in activation of the pro-apoptotic enzyme caspase-3 (Fig. 6B) and reduction in mitochondrial membrane potential (Fig. 6E and F). Overexpression of the neuron-specific isoforms endophilin-B1b and endophilin-B1c, but not the ubiquitously expressed endophilin-B1a, attenuated amyloid-β-induced caspase-3 activation (Fig. 6A and C) and restored mitochondrial membrane potential (Fig. 6E and F), although only endophilin-B1c was able to preserve neuronal viability (Fig. 6D). Taken together, these findings demonstrate that amyloid-β toxicity reduces endophilin-B1b/c expression while restoration of endophilin-B1b/c protects neurons from amyloid-β toxicity, suggesting that endophilin-B1 may participate in a feed-forward loop in the Alzheimer’s disease pathogenesis cascade.
Figure 6.

Neuron-specific endophilin-B1 isoforms protect against amyloid-β (Aβ) neurotoxicity in vitro. (A) Postnatal cortical neurons were infected with lentivirus expressing either EGFP or endophilin-B1 isoforms for 3 days, then treated with 10 µM amyloid-β25-35 or reverse peptide amyloid-β35-25 for 24 h. Western blot analysis of endophilin-B1, activated caspase-3 (apoptosis marker), and actin (loading control) are shown. (B) Quantification of amyloid-β25-35-induced changes in the expression of endophilin-B1a, endophilin-B1b/c and activated caspase-3. Values are normalized to actin and expressed as per cent change relative to amyloid-β35-25 control. Amyloid-β25-35 induced an increase in activated caspase-3 and a decline in endophilin-B1b/c, but no change in endophilin-B1a (n = 4 separate experiments). *P < 0.05 versus amyloid-β35-25, Student’s t-test (n = 4). (C) Overexpression of neuron-specific endophilin-B1 isoforms endophilin-B1b and endophilin-B1c significantly decreased caspase-3 activation induced by amyloid-β25-35 (normalized to actin and relative to amyloid-β25-35/EGFP). *P < 0.05 versus amyloid-β25-35/EGFP, one-way ANOVA with Tukey post hoc test (n = 4). (D) Overexpression of endophilin-B1c, but not endophilin-B1b, reduced neuronal death induced by amyloid-β25-35. *P < 0.05 versus amyloid-β25-35/EGFP, one-way ANOVA with Tukey post hoc test (n = 3). (E) Cultured cortical neurons labelled with the JC-1 dye, an indicator of mitochondrial membrane potential, are shown. Healthy, polarized mitochondria emit red fluorescence, while damaged depolarized mitochondria show green fluorescence. Scale bar = 20 µm. (F) Quantification of JC-1 fluorescence intensity. *P < 0.05 versus amyloid-β35-25; #P < 0.05 versus amyloid-β25-35, one-way ANOVA with Tukey post hoc test (n = 4).
Neuron-specific endophilin-B1 isoforms are selectively decreased in patients with Alzheimer’s disease
To determine if reduced endophilin-B1 is observed in patients with Alzheimer’s disease, cortical tissue (inferior parietal lobule) from autopsies of neuropathologically confirmed Alzheimer’s disease and control patients was obtained from the University of Washington ADRC brain tissue repository. Post-mortem interval was <11 h in all cases and <7 h in all but four cases; each case underwent comprehensive neuropathological examination according to NIA-Reagan Criteria (Hyman and Trojanowski, 1997), which incorporate Braak staging for neurofibrillary tangle distribution (Braak and Braak, 1991) and Consortium to Establish a Registry for Alzheimer's Disease (CERAD) criteria for neuritic plaque density (Mirra et al., 1991). According to these criteria, there is a high likelihood that a patient's dementia is due to Alzheimer’s disease when neuropathological examination reveals high (cortical) Braak stage (V–VI) combined with CERAD frequent neuritic plaques. In our study, all Alzheimer’s disease cases (Braak V–VI) also had CERAD frequent neuritic plaques while controls (Braak I–II) had CERAD absent to sparse scores. Tissue lysates were assayed for endophilin-B1 protein using western blot. Endophilin-B1b/c was selectively decreased relative to endophilin-B1a in patients with Alzheimer’s disease (Fig. 7A). Quantitative results with all samples combined showed a progressive drop in endophilin-B1b/c expression, reaching a statistically significant level in the Braak V–VI group relative to controls (Fig. 7B), while endophilin-B1a levels were unaltered throughout disease stages (Fig. 7C). Messenger RNA was examined in a subset of samples using primers that amplified both SH3GLB1b, which encodes for the major neuron-specific isoform endophilin-B1b (Wang et al., 2014), and SH3GLB1a, which encodes for the ubiquitously expressed endophilin-B1a. Similar to what was seen at the protein level (Fig. 7A–C), SH3GLB1b mRNA significantly decreased with disease progression while SH3GLB1a mRNA was unchanged (not shown), resulting in a decreased SH3GLB1b to SH3GLB1a mRNA ratio (Fig. 7D). As a result, a moderate correlation (Fig. 7E) was observed between endophilin-B1b/c protein levels versus SH3GLB1b mRNA levels when normalized against SH3GLB1a levels. Because SH3GLB1b mRNA was selectively reduced in Alzheimer’s disease samples while SH3GLB1a mRNA was unchanged, it is likely that decreased expression of endophilin-B1b/c protein is secondary to altered splicing or mRNA stability rather than altered gene expression or reduced protein stability.
Figure 7.

Expression of neuron-specific endophilin-B1 isoforms is decreased in Alzheimer’s disease. (A) Representative western blot analysing endophilin-B1 protein expression in the inferior parietal lobule cerebral cortex of patients at various Braak stages. Actin was used as a loading control. (B and C) Scatter plot with median and interquartile range of endophilin-B1b/c (B) and endophilin-B1a (C) protein expression. One-way ANOVA showed significant difference in endophilin-B1b/c due to disease progression. *P < 0.05 versus Braak I-II; #P < 0.05 versus Braak III–IV, one-way ANOVA with Tukey post hoc test (n = 10–14 patients per group). (D) The ratio of SH3GLB1b to SH3GLB1a mRNA is decreased in patients with Alzheimer’s disease. *P < 0.05 versus Braak I–II, one-way ANOVA with Tukey post hoc test (n = 7–9). (E) Endophilin-B1b/c to endophilin-B1a protein ratio correlates with SH3GLB1b to SH3GLB1a mRNA ratio. *P < 0.05, Pearson correlation (n = 25). (F) Scatter plot with median and interquartile range of endophilin-B1b/c protein expression normalized to the neuronal cytoskeletal marker βIII-tubulin. One-way ANOVA showed significant difference in endophilin-B1b/c due to disease progression. *P < 0.05 versus Braak I–II. (G) Enriched synaptosome preparations from cortices of control, Braak II, Braak III–IV (not shown), and Braak V–VI patients were analysed for endophilin-B1 expression by western blot. (H and I) Endophilin-B1b/c (H), but not endophilin-B1a (I) expression was decreased in synaptosomes from Braak V–VI patients. One-way ANOVA showed significant difference in endophilin-B1b/c due to disease progression. *P < 0.05 versus control, one-way ANOVA with Tukey post hoc test (n = 6–8). (J) Synaptophysin was not altered in Alzheimer’s disease patient synaptosomes. (K) Endophilin-B1b/c expression was decreased relative to synaptophysin expression in Braak V–VI patients. *P < 0.05 versus control, one-way ANOVA with Tukey post hoc test (n = 6–8). (L) Synaptosomes from controls and Alzheimer’s disease (Braak V–VI) patients were sorted using flow cytometry with antibodies against amyloid-β and endophilin-B1. There was a negative correlation between the number of endophilin-B1 (+) synapses and amyloid-β (+) synapses in the Alzheimer’s disease group only. *P < 0.05, Pearson correlation (n = 7).
The observed reduction in endophilin-B1b/c was unlikely to be attributable to neuronal cell loss in the Alzheimer’s disease samples, as there was a significant decline in endophilin-B1b/c in Braak V–VI samples when normalized to levels of βIII-tubulin, a neuronal cytoskeletal marker (Fig. 7F). Further, we examined the inferior parietal lobule from autopsies of subjects with dementia with Lewy bodies, a form of dementia characterized by neocortical neuronal Lewy bodies in the absence of neuritic plaques and neurofibrillary tangles (Hyman et al., 2012; Montine et al., 2012). There was no detectable loss of endophilin-B1b/c in dementia with Lewy bodies samples (not shown) even though this disorder is associated with neuron loss, further supporting the hypothesis that reduced endophilin-B1b/c in Alzheimer’s disease cases did not result from generalized neurodegeneration.
To further support the specific reduction of endophilin-B1b/c in human Alzheimer’s disease pathogenesis, we examined intact synaptosomes purified from Alzheimer’s disease cortical autopsy samples and age-matched controls (Fig. 7G). We observed bands corresponding to both the endophilin-B1a and endophilin-B1b/c isoforms in synaptosomes, with an additional larger band that may reflect a post-translational modification of endophilin-B1 exclusive to synapses (Fig. 7G). With advanced disease progression, we saw a specific decrease in endophilin-B1b/c (Fig. 7H) relative to endophilin-B1a (Fig. 7I). Synaptophysin expression was not impacted by disease stage (Fig. 7J) in the enriched synaptosome preparation and the ratio of endophilin-B1b/c to synaptophysin was significantly reduced in Alzheimer’s disease patients (Fig. 7K), supporting the interpretation that the observed loss of endophilin-B1b/c is not secondary to a general reduction in neurons or synapses. Fluorescence-activated sorting of synaptosomes double-immunolabelled for amyloid-β and endophilin-B1 revealed a strong negative correlation between the percentage of synaptosomes containing amyloid-β and those containing endophilin-B1 among Alzheimer’s disease samples, but not in controls (Fig. 7L). Although the antibody used for sorting is not specific for endophilin-B1b/c, it likely represents changes seen in endophilin-B1b/c as endophilin-B1a was not altered in the synaptosomal fractions of Alzheimer’s disease samples (Fig. 7I). This suggests that amyloid-β accumulation within a synapse may negatively impact the trafficking of neuroprotective endophilin-B1b/c to that synapse and/or loss of endophilin-B1b/c from a synapse may promote the accumulation of amyloid-β within that synapse.
Discussion
Previously, we described a novel pro-survival function for endophilin-B1 that was unique to neurons and identified neuron-specific alternatively spliced endophilin-B1 isoforms that mediate this novel function (Wang et al., 2014). Here, we report that expression of neuroprotective endophilin-B1 isoforms (endophilin-B1b/c) was selectively decreased in a mouse model of Alzheimer’s disease, and that loss of endophilin-B1 exacerbated amyloid pathology, tau hyperphosphorylation, astrogliosis, cognitive decline, mortality, and synaptic degeneration. Reduced endophilin-B1b/c was also seen in human Alzheimer’s disease patient brains, but not in patients with dementia with Lewy bodies. Overexpression of endophilin-B1b/c protected primary cortical neuron cultures against amyloid-β toxicity, supporting a functional role for reduced endophilin-B1b/c in Alzheimer’s disease pathogenesis. Finally, loss of endophilin-B1 alone was also sufficient to induce behavioural deficits and synaptic degeneration, underscoring endophilin-B1’s critical role in maintaining neuronal viability and function.
The role of endophilin-B1 in plaque accumulation and neuronal viability
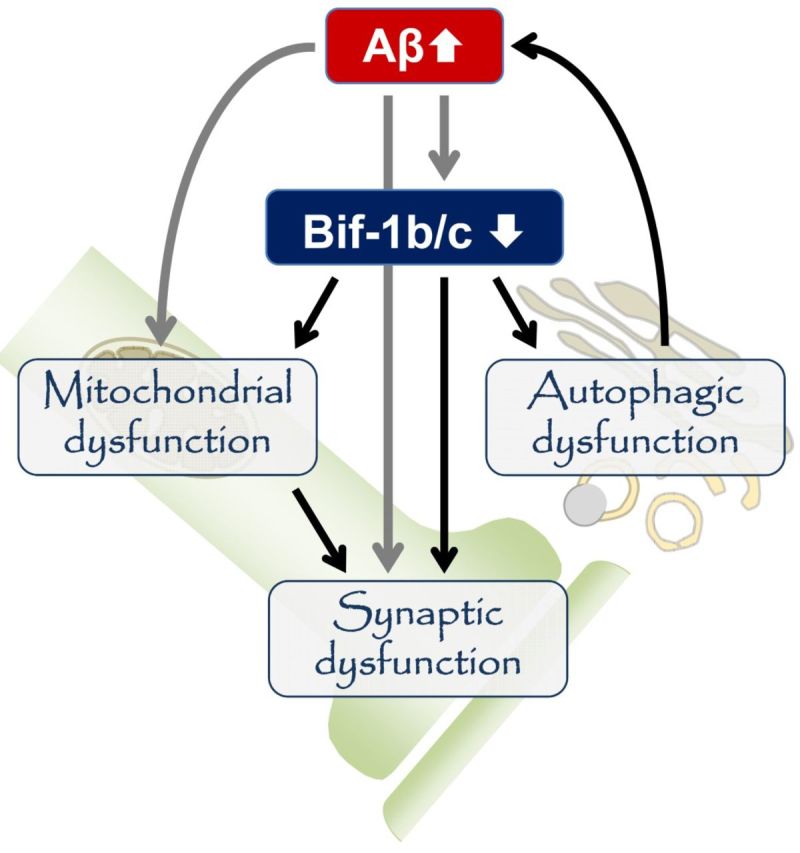
Endophilin-B1 is a multi-functional protein whose absence may affect Alzheimer’s disease pathology in a number of ways (Fig. 8). One mechanism by which loss of endophilin-B1 exacerbated the disease phenotype may involve increased levels of both soluble and insoluble amyloid-β (Fig. 3). The increase in insoluble amyloid-β levels correlated well with increased plaque burden (Fig. 4). The increase in soluble amyloid-β observed in APP/PSEN1/Endo-B1−/− mice could contribute to the observed early synaptic degeneration, as soluble oligomeric amyloid-β has been shown to be the most toxic amyloid-β species (Larson and Lesné, 2012). The increased amyloid-β load might be secondary to loss of endophilin-B1’s function in the process of autophagy (Takahashi et al., 2007, 2008) and resultant decreased clearance or increased generation of amyloid-β (Nixon, 2007). Small molecule autophagic activators have been shown to decrease amyloid-β levels and protect neurons from toxicity (Caccamo et al., 2010; Spilman et al., 2010; Tian et al., 2011; Chu et al., 2013), whereas mice deficient in Atg7, a gene critical to autophagy, were more vulnerable to Alzheimer’s disease pathology (Nilsson et al., 2013). Additionally, BECN1, a protein that interacts with endophilin-B1 to promote autophagy (Takahashi et al., 2007), is also downregulated in human Alzheimer’s disease (Pickford et al., 2008). Alternatively, it is also possible that endophilin-B1 deficiency leads to increased amyloid-β production, as disruption of autophagy has been implicated in γ-secretase activation and amyloid-β secretion (Ohta et al., 2010). While endophilin-B1 deficiency did not alter β- (data not shown) or γ-secretase expression levels (Supplementary Fig. 2) or γ-secretase enzymatic activity (Supplementary Fig. 3), Endo-B1−/− mice did show elevated levels of amyloid precursor protein (Supplementary Fig. 2), the substrate for amyloid-β production. Interestingly, and contrary to the observations reported here, inhibition of endophilin-B1-mediated autophagy was neuroprotective in Parkinson’s disease models (Wong et al., 2011). This discrepancy may reflect the different contribution of autophagy to the pathogenesis of these two diseases (Nixon, 2013).
Figure 8.

The multi-functional role of endophilin-B1 in Alzheimer’s disease pathogenesis. Amyloid-β accumulation leads to decreased endophilin-B1b/c expression in neurons, which in turn elevates amyloid-β and plaques, possibly through impairment of autophagy. Loss of endophilin-B1b/c may concomitantly increase neuronal vulnerability to amyloid-β toxicity through its potential function in synapses and mitochondria.
Aside from its potential role in amyloid-β homeostasis, endophilin-B1 deficiency may concomitantly sensitize neurons to amyloid-β stress as we previously demonstrated for ischaemia or DNA damage (Wang et al., 2014). Endophilin-B1 is found in synapses (Fig. 7G and L), and Endo-B1−/− mice develop synapse loss and behavioural deficits even in the absence of amyloid-β pathology (Fig. 5), underscoring the critical role of endophilin-B1 in synaptic maintenance. Endophilin-B1 is also reported to be involved in the turnover and trafficking of neurotransmitter receptors (Wan et al., 2008; Khan et al., 2014). While data presented here only evaluated synaptic architecture in the hippocampus, it is likely that endophilin-B1 is required for synaptic maintenance in a broader range of CNS circuitry as motor function was also compromised in Endo-B1−/− mice (Supplementary Table 1).
Loss of endophilin-B1 may also sensitize neurons to stress through its effects on mitochondrial maintenance. Previously, we reported that endophilin-B1 knockdown in neurons leads to fragmented, depolarized mitochondria (Wang et al., 2014), similar to what is seen when neurons are exposed to amyloid-β (Wang et al., 2008). Here, overexpression of endophilin-B1b or endophilin-B1c attenuated mitochondrial depolarization and caspase-3 activation induced by amyloid-β (Fig. 6). Dysfunctional mitochondria, particularly at the synapse where mitochondrial function is critical (Li et al., 2004), may result in heightened vulnerability to stress through mechanisms involving decreased respiration and increased reactive oxygen species generation (Galloway and Yoon, 2012). Interestingly, while overexpression of both endophilin-B1b and endophilin-B1c decreased caspase-3 activation and preserved mitochondrial function, only endophilin-B1c enhanced neuronal viability (Fig. 6D). Endophilin-B1c was more efficacious than endophilin-B1b in mitigating caspase-3 activation (Fig. 6C), possibly maintaining caspase-3 activity below the threshold necessary for promoting neuronal loss.
Although endophilin-B1a was not affected in Alzheimer’s disease models or in human brain samples, and its overexpression did not attenuate amyloid-β neurotoxicity in vitro, this ubiquitously expressed isoform may still play a role in Alzheimer’s disease pathogenesis in non-neuronal cells. Endophilin-B1a is expressed in astrocytes (Wang et al., 2014) and microglia (unpublished observations) and there was increased astrogliosis in response to Alzheimer’s disease-related pathology in endophilin-B1-deficient brains. The lack of endophilin-B1a expression in these glial cells may potentially affect their inflammatory response to injured neurons and/or their contribution to amyloid-β homeostasis.
Endophilin-B1 expression in human Alzheimer’s disease
Accurately measuring the neuronal biochemical changes in autopsy specimens from patients with neurodegenerative disease is complicated by the varied extent of neuronal loss in affected brain regions. We considered the possibility that reduced expression of neuron-specific endophilin-B1b/c reflects loss of neurons rather than specific loss of endophilin-B1b/c. However, several lines of evidence support the interpretation that endophilin-B1b/c reduction in human Alzheimer’s disease samples are not a result of neuronal loss. First, there was a significant reduction in endophilin-B1b/c normalized to both βIII-tubulin, a neuron-specific cytoskeletal protein (Fig. 7F), and the synaptic protein synaptophysin (Fig. 7K). Second, we observed a strong negative correlation between synapses positive for endophilin-B1 and those positive for amyloid-β only in Alzheimer’s disease samples (Fig. 7L), suggesting that endophilin-B1 is lost as amyloid-β accumulates in synapses. Finally, endophilin-B1b/c expression was unaltered in patient cases of dementia with Lewy bodies (data not shown), suggesting that loss of endophilin-B1b/c seen in Alzheimer’s disease is due to a specific interaction with Alzheimer’s disease-related pathology. This interpretation is further supported by loss of endophilin-B1b/c in symptomatic APP/PSEN1 mice (Fig. 1), as this mouse model does not exhibit neuronal loss (West et al., 2009).
Conclusions
Here, we present a pernicious cycle involved in Alzheimer’s disease pathology where the presence of amyloid-β can decrease expression of endophilin-B1b/c, and the subsequent loss of endophilin-B1b/c leads to increased amyloid-β and plaque formation, synaptic degeneration, and tau hyperphosphorylation. Conversely, overexpression of neuron-specific endophilin-B1 isoforms attenuates amyloid-β-induced caspase-3 activation and loss of mitochondrial membrane potential, while endophilin-B1c overexpression also sustains neuronal survival in the face of amyloid-β toxicity. The mechanism(s) by which endophilin-B1 influences amyloid-β homeostasis and promotes neuronal survival need to be further elucidated. Collectively, these results suggest that reduced endophilin-B1b/c contributes to Alzheimer’s disease pathogenesis. The identification of neuron-specific splice variants with neuroprotective action in both ischaemia (Wang et al., 2014) and Alzheimer’s disease models makes endophilin-B1 an intriguing therapeutic candidate for neurodegenerative disorders, especially for those, such as Alzheimer’s disease, where the neuron specific endophilin-B1 isoforms are selectively lost.
Supplementary Material
Acknowledgements
We thank Jeffrey Moore and Yeun Lee for immunostaining and quantification. Animal behaviour was assessed at the Center on Human Development and Disability Animal Behavior Core at the University of Washington (NICHD U54 HD083091). Brain samples were obtained from the ADRC Neuropathology Cores of University of Washington (NIA P50 AG05136), USC (NIA P50 AG05142), UCLA (NIA P50 AG16570), and UC Irvine (NIA P50 AG16573). The authors declare no competing financial interests.
Glossary
Abbreviations
- Bif-1
Bax-interacting factor-1
- EGFP
enhanced green fluorescent protein, Endo-B1 = endophilin-B1
Funding
This work was supported by grants from the National Institutes of Health NINDS R01 NS35533, NINDS R01 NS056031 and NINDS R21 NS084217 to R.S.M. and NIA R01 AG27465 to K.H.G, and by an NINDS Institutional Center Core Grant to support the viral core facility in the Neuroproteomics Center at the University of Washington (NINDS P30 NS055088).
Supplementary material
Supplementary material is available at Brain online.
References
- Baiguera C, Alghisi M, Pinna A, Bellucci A, De Luca MA, Frau L, et al. Late-onset Parkinsonism in NFκB/c-Rel-deficient mice. Brain 2012; 135: 2750–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballard C, Gauthier S, Corbett A, Brayne C, Aarsland D, Jones E. Alzheimer's disease. Lancet 2011; 377: 1019–31. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 1991; 82: 239–59. [DOI] [PubMed] [Google Scholar]
- Caccamo A, Majumder S, Richardson A, Strong R, Oddo S. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-β, and Tau: effects on cognitive impairments. J Biol Chem 2010; 285: 13107–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu C, Zhang X, Ma W, Li L, Wang W, Shang L, et al. Induction of autophagy by a novel small molecule improves aβ pathology and ameliorates cognitive deficits. PLoS One 2013; 8: e65367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuddeback SM, Yamaguchi H, Komatsu K, Miyashita T, Yamada M, Wu C, et al. Molecular cloning and characterization of Bif-1: a novel Src homology 3 domain-containing protein that associates with bax. J Biol Chem 2001; 276: 20559–65. [DOI] [PubMed] [Google Scholar]
- Duyckaerts C, Potier MC, Delatour B. Alzheimer's disease models and human neuropathology: similarities and differences. Acta Neuropathol 2008; 115: 5–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galloway CA, Yoon Y. What comes first, misshape or dysfunction? The view from metabolic excess. Jo Gen Physiol 2012; 139: 455–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimbel DA, Nygaard HB, Coffey EE, Gunther EC, Laurén J, Gimbel ZA, et al. Memory impairment in transgenic alzheimer mice requires cellular prion protein. J Neurosci 2010; 30: 6367–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gylys KH, Fein JA, Yang F, Wiley DJ, Miller CA, Cole GM. Synaptic changes in Alzheimer's disease: increased amyloid-β and gliosis in surviving terminals is accompanied by decreased PSD-95 fluorescence. Am J Pathol 2004; 165: 1809–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC, et al. National Institute on Aging–Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimer's Dement 2012; 8: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman BT, Trojanowski JQ. Editorial on consensus recommendations for the postmortem diagnosis of Alzheimer disease from the National Institute on Aging and the Reagan Institute Working Group on Diagnostic Criteria for the neuropathological assessment of Alzheimer disease. J Neuropathol Exp Neurol 1997; 56: 1095–7. [DOI] [PubMed] [Google Scholar]
- Jankowsky JL, Fadale DJ, Anderson J, Xu GM, Gonzales V, Jenkins NA, et al. Mutant presenilins specifically elevate the levels of the 42 residue β-amyloid peptide in vivo: evidence for augmentation of a 42-specific γ secretase. Hum Mol Genet 2004; 13: 159–70. [DOI] [PubMed] [Google Scholar]
- Jayadev S, Case A, Eastman AJ, Nguyen H, Pollak J, Wiley JC, et al. Presenilin 2 is the predominant γ-secretase in microglia and modulates cytokine release. PLoS One 2010; 5: e15743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karbowski M, Jeong S-Y, Youle RJ. Endophilin B1 is required for the maintenance of mitochondrial morphology. J Cell Biol 2004; 166: 1027–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan MM, Strack S, Wild F, Hanashima A, Gasch A, Brohm K, et al. Role of autophagy, SQSTM1, SH3GLB1, and TRIM63 in the turnover of nicotinic acetylcholine receptors. Autophagy 2014; 10: 123–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SY, Oh YL, Kim KM, Jeong EG, Kim MS, Yoo NJ, et al. Decreased expression of Bax-interacting factor-1 (Bif-1) in invasive urinary bladder and gallbladder cancers. Pathology 2008; 40: 553–37. [DOI] [PubMed] [Google Scholar]
- Larson ME, Lesné SE. Soluble Aβ oligomer production and toxicity. J Neurochem 2012; 120(Suppl 1): 125–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JW, Jeong EG, Soung YH, Nam SW, Lee JY, Yoo NJ, et al. Decreased expression of tumour suppressor Bax-interacting factor-1 (Bif-1), a Bax activator, in gastric carcinomas. Pathology 2006; 38: 312–5. [DOI] [PubMed] [Google Scholar]
- Li Z, Okamoto K-I, Hayashi Y, Sheng M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell 2004; 119: 873–87. [DOI] [PubMed] [Google Scholar]
- Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, et al. The Consortium to establish a registry for Alzheimer's disease (CERAD): Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology 1991; 41: 479. [DOI] [PubMed] [Google Scholar]
- Modregger J, Schmidt AA, Ritter B, Huttner WB, Plomann M. Characterization of endophilin B1b, a brain-specific membrane-associated lysophosphatidic acid Acyl transferase with properties distinct from endophilin A1. J Biol Chem 2003; 278: 4160–7. [DOI] [PubMed] [Google Scholar]
- Montine T, Phelps C, Beach T, Bigio E, Cairns N, Dickson D, et al. National Institute on Aging–Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol 2012; 123: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris R. Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods 1984; 11: 47–60. [DOI] [PubMed] [Google Scholar]
- Nilsson P, Loganathan K, Sekiguchi M, Matsuba Y, Hui K, Tsubuki S, et al. Aβ Secretion and plaque formation depend on autophagy. Cell Reports 2013; 5: 61–9. [DOI] [PubMed] [Google Scholar]
- Nixon RA. Autophagy, amyloidogenesis and Alzheimer disease. J Cell Sci 2007; 120: 4081–91. [DOI] [PubMed] [Google Scholar]
- Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med 2013; 19: 983–97. [DOI] [PubMed] [Google Scholar]
- Ohta K, Mizuno A, Ueda M, Li S, Suzuki Y, Hida Y, et al. Autophagy impairment stimulates PS1 expression and γ-secretase activity. Autophagy 2010; 6: 345–52. [DOI] [PubMed] [Google Scholar]
- Pickford F, Masliah E, Britschgi M, Lucin K, Narasimhan R, Jaeger PA, et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid β accumulation in mice. J Clin Invest 2008; 118: 2190–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez JJ, Olabarria M, Chvatal A, Verkhratsky A. Astroglia in dementia and Alzheimer's disease. Cell Death Differ 2008; 16: 378–85. [DOI] [PubMed] [Google Scholar]
- Sokolow S, Henkins KM, Bilousova T, Miller CA, Vinters HV, Poon W, et al. AD synapses contain abundant Aβ monomer and multiple soluble oligomers, including a 56-kDa assembly. Neurobiol Aging 2012; 33: 1545–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spilman P, Podlutskaya N, Hart MJ, Debnath J, Gorostiza O, Bredesen D, et al. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-β levels in a mouse model of alzheimer's disease. PLoS One 2010; 5: e9979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sze C, Troncoso J, Kawas C, Moution P, Price D, Martin L. Loss of the presynaptic vesicle protein synaptophysin in hippocampus correlates with cognitive decline in Alzheimer disease. J Neuropathol Exp Neurol 1997; 56: 933–44. [DOI] [PubMed] [Google Scholar]
- Takahashi Y, Coppola D, Matsushita N, Cualing HD, Sun M, Sato Y, et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat Cell Biol 2007; 9: 1142–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y, Hori T, Cooper TK, Liao J, Desai N, Serfass JM, et al. Bif-1 haploinsufficiency promotes chromosomal instability and accelerates Myc-driven lymphomagenesis via suppression of mitophagy. Blood 2013; 121: 1622–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y, Karbowski M, Yamaguchi H, Kazi A, Wu J, Sebti SM, et al. Loss of Bif-1 suppresses Bax/Bak conformational change and mitochondrial apoptosis. Mol Cell Biol 2005; 25: 9369–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y, Meyerkord CL, Wang H-G. BARgaining membranes for autophagosome formation: regulation of autophagy and tumorigenesis by Bif-1/Endophilin B1. Autophagy 2008; 4: 121–4. [DOI] [PubMed] [Google Scholar]
- Thal DR, Rüb U, Orantes M, Braak H. Phases of Aβ-deposition in the human brain and its relevance for the development of AD. Neurology 2002; 58: 1791–800. [DOI] [PubMed] [Google Scholar]
- Tian Y, Bustos V, Flajolet M, Greengard P. A small-molecule enhancer of autophagy decreases levels of Aβ and APP-CTF via Atg5-dependent autophagy pathway. FASEB J 2011; 25: 1934–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uo T, Dworzak J, Kinoshita C, Inman DM, Kinoshita Y, Horner PJ, et al. Drp1 levels constitutively regulate mitochondrial dynamics and cell survival in cortical neurons. Exp Neurol 2009; 218: 274–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandeputte C, Taymans JM, Casteels C, Coun F, Ni Y, Van Laere K, et al. Automated quantitative gait analysis in animal models of movement disorders. BMC Neurosci 2010; 11: 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volianskis A, Køstner R, Mølgaard M, Hass S, Jensen MS. Episodic memory deficits are not related to altered glutamatergic synaptic transmission and plasticity in the CA1 hippocampus of the APPswe/PS1ΔE9-deleted transgenic mice model of β-amyloidosis. Neurobiol Aging 2010; 31: 1173–87. [DOI] [PubMed] [Google Scholar]
- Wan J, Cheung AY, Fu W-Y, Wu C, Zhang M, Mobley WC, et al. Endophilin B1 as a Novel Regulator of Nerve Growth Factor/TrkA Trafficking and Neurite Outgrowth. The Journal of Neuroscience 2008; 28: 9002–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang DB, Uo T, Kinoshita C, Sopher BL, Lee RJ, Murphy SP, et al. Bax interacting factor-1 promotes survival and mitochondrial elongation in neurons. J Neurosci 2014; 34: 2674–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Su B, Siedlak SL, Moreira PI, Fujioka H, Wang Y, et al. Amyloid-β overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc Natl Acad Sci USA 2008; 105: 19318–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West MJ, Bach G, Søderman A, Jensen JL. Synaptic contact number and size in stratum radiatum CA1 of APP/PS1ΔE9 transgenic mice. Neurobiol Aging 2009; 30: 1756–76. [DOI] [PubMed] [Google Scholar]
- Wong ASL, Lee RHK, Cheung AY, Yeung PK, Chung SK, Cheung ZH, et al. Cdk5-mediated phosphorylation of endophilin B1 is required for induced autophagy in models of Parkinson's disease. Nat Cell Biol 2011; 13: 568–79. [DOI] [PubMed] [Google Scholar]
- Xiang H, Hochman DW, Saya H, Fujiwara T, Schwartzkroin PA, Morrison RS. Evidence for p53-mediated modulation of neuronal viability. J Neurosci 1996; 16: 6753–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Cudaback E, Jorstad NL, Hemingway JF, Hagan CE, Melief EJ, et al. APOE3, but not APOE4, bone marrow transplantation mitigates behavioral and pathological changes in a mouse model of Alzheimer disease. Am J Pathol 2013; 183: 905–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yankner B, Duffy L, Kirschner D. Neurotrophic and neurotoxic effects of amyloid beta protein: reversal by tachykinin neuropeptides. Science 1990; 250: 279–82. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.