

abstract
Previous studies have elucidated a neuroendocrine mechanism consisting of the hypothalamus (growth hormone releasing hormone, GHRH) – pituitary (growth hormone, GH) – STAT5a/b axis that underlies sex-biased gene expression in the liver. It is now established that male vs female patterned secretion of GHRH, and thus of circulating GH levels (“pulsatile” vs “more continuous” respectively), leading to differently patterned activation of PY-STAT5a/b in hepatocytes results in sex-biased gene expression of cohorts of hundreds of downstream genes. This review outlines new data in support of a STAT5a/b-based mechanism of sex bias in the vascular disease pulmonary hypertension (PH). Puzzling observations in PH include its 2-4-fold higher prevalence in women but a male-dominance in many rodent models, and, paradoxically, inhibition of PH development by estrogens in such models. We observed that conditional deletion of STAT5a/b in vascular smooth muscle cells (SMC) in mice converted the male-dominant model of chronic hypoxia-induced PH into a female-dominant phenotype. In human idiopathic PH, there was reduced STAT5a/b and PY-STAT5 in cells in late-stage obliterative pulmonary arterial lesions in both men and women. A juxtaposition of the prior liver data with the newer PH-related data drew attention to the hypothalamus-GH-STAT5 axis, which is the major target of estrogens at the level of the hypothalamus. This hypothesis explains many of the puzzling aspects of sex bias in PH in humans and rodent models. The extension of STAT5-anchored mechanisms of sex bias to vascular disease emphasizes the contribution of central neuroendocrine processes in generating sexual dimorphism in different tissues and cell types.
KEYWORDS: arcuate nucleus; BCL6; growth hormone; hypothalamus; neuroendocrine mechanisms; pulmonary hypertension, sex and gender bias; STAT5a and STAT5b; vascular disease; women's health
ABBREVIATIONS
- 2-ME
2-methoxyestradiol
- 5-HT
5-hydroxytrptamine
- BCL6
B-cell lymphoma protein 6
- BMP
bone morphogenetic protein
- BMPR2
BMP receptor 2
- CYP
cytochrome P450 enzymes
- E2
estradiol-17β
- EC
endothelial cells
- eNOS
endothelial nitric oxide synthase
- GH
growth hormone
- IPAH
idiopathic pulmonary arterial hypertension
- MCT
monocrotaline
- MCTP
monocrotaline pyrrole
- PASMC
pulmonary artery smooth muscle cells
- PH
pulmonary hypertension
- RVH
right ventricular hypertrophy
- RVSP
right ventricular systolic pressure
- RV/S+LV
ratio of wet weights of right ventricle to that of septum plus left ventricle
- SERT
serotonin transporter
- SMC
smooth muscle cells
INTRODUCTION
Men and women and men have clear differences in prevalence of vascular disease.1,2 As examples, the prevalence of hypertension, atherosclerosis and coronary artery disease is higher in men than in women.1,2 However, as a converse example, the prevalence of pulmonary hypertension (PH)a is 2-4-fold higher in women than in men, even though after diagnosis, women have a better outcome than men.3-6 In many rodent models of arterial remodeling after vascular injury (femoral artery cuffing or common carotid artery ligation) or chronic hypoxia-induced PH males show more extensive vascular remodeling than females.6-19 In these models the administration of estrogens [estradiol-17β (E2) or 2-methoxyestradiol (2-ME)] is typically protective.6,16-19 This produces the apparently paradoxical situation that in a disease like PH, while women have a higher prevalence than men, the typical rodent models of PH (chronic hypoxia or monocrotaline administration) show a male dominance and E2 administration inhibits the latter.6,16-19 This dichotomy between humans and rodents, and, more generally, the varied differences in sex biasb between men and women in vascular disease prevalence and models of vascular injury remain incompletely understood.6,16-20 Indeed, it has been especially perplexing to understand the different sex-bias observations in similar vascular disease situations in different species. As examples, understanding the differences between sex bias in PH in humans compared to PH in rodent models, or even in understanding the differences between models of PH in the rat compared to the mouse (e.g. monocrotaline is efficient in producing PH in the male rat, but not in the mouse) has been a challenge.6,16-20 Thus far, the main mechanistic focus of various studies to understand these situations has been to focus almost exclusively on the direct effects of E2 or other sex hormones on vascular cells. As examples, there is an extensive literature on the effects of E2 on increasing the function of eNOS in endothelial cells, the ability of E2 to stimulate vascular smooth muscle cell proliferation, and to inhibit the trafficking of vasorelevant receptors such as BMPR2 (bone morphogenetic receptor 2) to the cell surface.6,21-28 However, these studies of direct effects of sex hormones on vascular cells, taken together, have been unable to explain the many disparate sex bias observations in a vascular disease such as PH in humans and different rodent species.
A Neuroendocrine Mechanism (Hypothalamus-GH-STAT5) Underlying Sex-Biased Gene Expression in the Liver
There are a series of relevant and critical insights pertaining to mechanisms determining sex-biased gene expression in the liver that have not quite percolated into the vascular disease literature.29 The pharmacologic issue driving extensive studies of sex bias in liver gene expression was the difference between men and women (and male vs female animals) to metabolize administered xenobiotics. Thus a major focus of these studies was the sex-biased expression of individual members of the P450 cytochrome (CYP) enzyme family that metabolize medications. It was observed that, in the rat, CYP2A2, 2C11 and 3A2 were male-biased, but 2C12 female-biased, with 2C6 expressed in both sexes.29-35 There were species-specific differences: CYP3A subfamily was male-biased in rats, but female-biased in humans and mice.29-35 Administration of estrogens or testosterone into rodents, respectively “feminized” or “masculinized” CYP enzyme expression.29 As in most of the vascular biology literature today, investigators in liver-gene expression in the early 1970s considered this sex-biased expression to be mainly the result of direct effects of sex hormones (estrogens and testosterone) on the hepatocyte.36
A critical discovery in 1973 by Colby et al.37 was the observation that the feminizing effect of an injection of E2 into a rat or a mouse on sex-biased liver gene expression was indirect and had an absolute dependence on the pituitary. This has since been extensively confirmed - the feminizing effects of exogenously administered E2 or the masculinizing effects of testosterone on sex-biased gene expression in the liver are blocked by hypophysectomy.29,38-48 The major target of injected E2 (and other sex hormones) is the hypothalamus, in particular the arcuate nucleus.49-54 [Note that there is no blood-brain barrier at the arcuate nucleus and additional “circumventricular” regions in the brain.] As subsequently elucidated in detail by Waxman et al and other investigators in the last 2 decades a neuroendocrine mechanism of sex-bias, operating through an axis consisting of the arcuate nucleus-growth hormone releasing hormone (GHRH)-growth hormone (GH)-signal transducer and activator of transcription 5 (STAT5) accounts for sex-biased expression of >1000 genes in the liver, and other tissues of the body.29,46-59 The generation of sexual dimorphism at the level of the hypothalamus results from the male vs female patterned secretion of GHRH and corresponding patterns of GH secretion (Fig. 1).29,30,60-66 This has been extensively confirmed in mice, rats and humans (Fig. 2 is an example of human data).29,30,60-66 Circulating GH levels in the male (M) are referred to as “pulsatile” (2-4 peaks per day) with very low interpulse levels; in the female (F) there is a higher frequency of pulses (>7 peaks/day) with significant interpulse levels, thus this is called “more continuous” (Figs. 1 and 2). This results in M vs F patterned activation of PY-STAT5a/b in the distal tissues (Fig. 3) and downstream of that a major cascade of sex-specific gene expression of >1,000 genes.29,67 Even when assayed at a single time-point in humans (in the morning after fasting) the median value of serum GH levels was 80-120 fold higher in women than in men.66 This is a higher sexual dimorphism ratio than that for E2 (ratio: 2.2 female bias) or testosterone (ratio: 14 male bias) observed in the same sera.66
FIGURE 1.
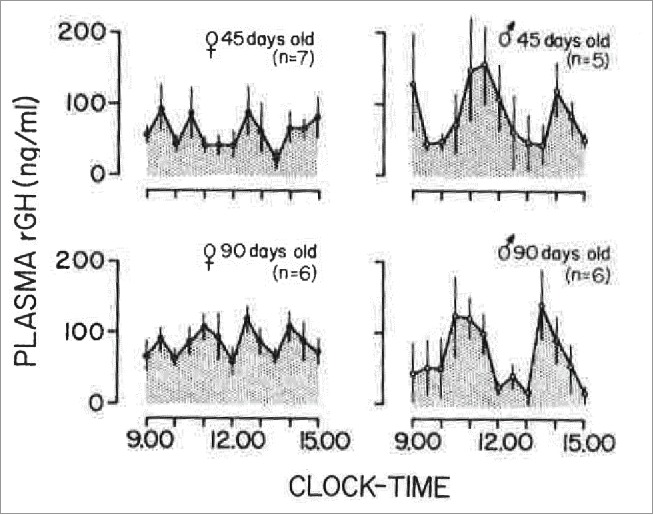
Mean plasma GH levels in female and male rats, 45 and 90 days old, sampled at 30-min intervals for a 6-h period. The number of animals in each group is shown in parenthesis. Vertical lines represent the SEM. Adapted from Edén (1979) with permission of The Endocrine Society.60
FIGURE 2.
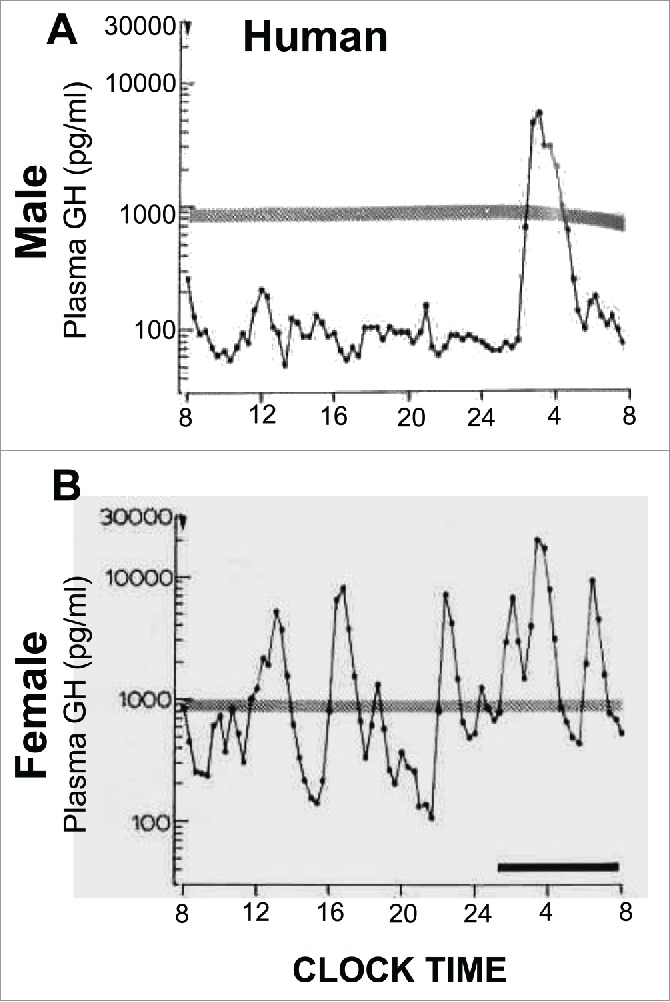
Sex-bias in circulating GH levels in male and female humans. Panels A and B: representative profiles of plasma GH levels showing that both men and women have pulsatile GH levels, but there is a higher overall level of GH in women. Adapted from Winer et al (1990) with permission of The Endocrine Society.61
FIGURE 3.
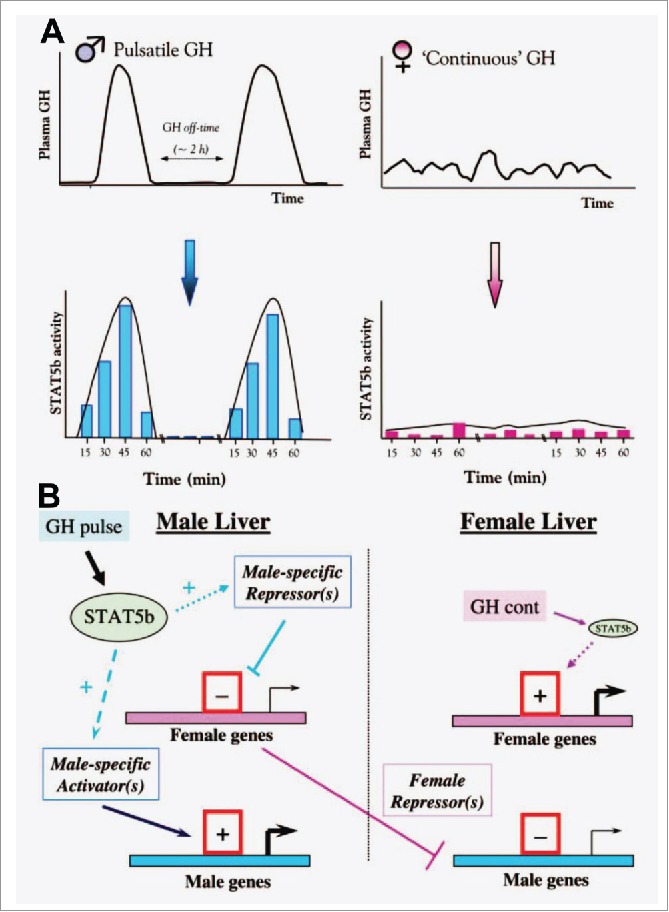
Schematics illustrating concepts of how male (pulsatile) vs female (more continuous) patterns of circulating GH elicit patterned activation of PY-STAT5 in the rat liver (Panel A), and thus sex-biased gene expression (Panel B). Schematics adapted from Waxman and O'Connor (2006) with permission of The Endocrine Society.67
Mice, rats and humans have quantitative differences in the respective male vs female patterns of circulating GH.27 (Figs. 1 and 2). Human males show GH pulses of high magnitude, with very low interpulse levels; however, women show a more continuous level of GH 100-fold higher than in the male (Fig. 2).30,60-66 In contrast, male rats show discrete pulses approximately every 3-4 hr with little or no circulating GH detectable during the interpulse interval.30 (Fig. 1). In female rats, the pulses are more frequent, the pulse heights are lower and the interpulse levels are higher.30 (Fig. 1). Male mice also have a pulsatile pattern; female mice have a pattern that tends to have a higher frequency but with low interpulse levels.62 Thus a difference between female humans and female rodents (rat or mouse) is in the much higher continuous GH levels in women compared to female rodents.(Figs. 1 and 2).
It needs emphasis that a major signaling mechanism activated by GH in target tissues is the Jak2-STAT5a/b pathway (Fig. 3A).29,67 Thus the regulation of gene expression by GH (activation or repression) is mediated by low-level activation of PY-STAT5 in M vs F patterns.29,67 (Fig. 3A). This patterned PY-STAT5 activation, in combination with other transcription factors (HNF4, HNF6, C/EBP), transcriptionally activated or repressed cohorts of hundreds of genes in hepatocytes. Cascades of downstream gene expression included regulation by STAT5-mediated patterned activation of master transcription repressors or activators such as BCL6 and Cux2.29,57-59,67 This generates a cascade of sex-biased gene expression but without the direct effect of any steroid sex hormone (Fig. 3B). Parenthetically, STAT5a and STAT5b are ubiquitous 90 kDa cytoplasmic proteins that are 96% related to each other and are derived from adjacent genes at the STAT5a/b locus in both mouse and man.29,67 STAT5a and STAT5b have overlapping as well as some discrete functions.29,67 Of the 7 STAT transcription factors, STAT5a and STAT5b are the only ones implicated in mediating sex-biased gene expression.29,67
Waxman and colleagues, and others, have shown in detailed studies (29,67 and citations therein) that sex-biased expression of genes by GH-STAT5 activation in the hepatocyte depends on a “dynamical” signaling process that involves multiple activation and inactivation cycles (frequency as in “pulsatile” or “continuous”), differences in magnitude of signal strength (“level” of GH), the rates of these changes (different slopes) resulting in different rates of association of different transcription regulatory proteins (including PY-STAT5a/b) at the level of the chromatin encompassing different genes (Fig. 3). It is these differences in signal strength, frequency, slopes of the activation and inactivation reactions, and in co-associated proteins that lead to different chromatin conformations (active or inactive for RNA transcription) in the DNA context of different genes. The net result is a cellular phenotype driven by the GH-STAT5 axis consisting of the sex-biased expression of hundreds of genes differently in different species (Fig. 3). This patterned activation of cells includes longer-lived chromatin remodeling at the respective male and female-specific genes in that respective male-derived hepatocytes were more responsive to the male pulsatile pattern of GH in culture, and female-derived less so.29,33
Different aspects of the specific GH patterns affect sex-biased expression of the same genes differently in different species.31-35 As an example the CYP3A subfamily is male-biased in rats, but female-biased in mice and humans.29,31-35 We note that CYP3A has been shown to be induced by GH through STAT5,68 and in an issue relevant to pulmonary hypertension, it is the CYP3A subfamily of enzymes that convert the injected inactive monocrotaline (MCT) to the bioactive monocrotaline pyrrole (MCTP) in male rats,69,70 Thus it is no surprise that MCT administration into a male rat produces PH more efficiently than in the female.6,17
Sex Bias and STAT5a/b in Vascular Disease
We mentioned earlier that in rodents aortic remodeling after a balloon injury or pulmonary arterial remodeling after exposure to chronic hypoxia was more pronounced in males, although the mechanisms have not been elucidated.6-15 In vascular smooth muscle cells (SMCs), PY-STAT5b has been identified as a transcription factor which facilitates growth and motility, and neointima formation in response to thrombin, PDGF and arterial injury.21-25,71-74 Suppression of activation of PY-STAT5b signaling in the vessel wall reduced balloon injury-induced neointima formation.73 Remarkably, that hypophysectomy in the male rat markedly impaired arterial remodeling after aortic balloon injury due to reduced vascular SMC proliferation and myointima formation, was reported in 1978,75 and confirmed,76 but the mechanisms remained to be elucidated. It is now known that GH promotes vascular SMC proliferation and migration, and is required for normal vascular reactivity and remodeling.77-80 Indeed, the prevalence of systemic hypertension is 20-50% in patients with acromegaly (in whom the plasma GH levels are high) due to “stiffer arteries.”77-80 Nevertheless, the issue of the GH-STAT5 axis contributing to sex bias in vascular remodeling has received little attention.
Our interest in the functions of STAT5 species (both unphosphorylated U-STAT5 and Tyr-phosphorylated PY-STAT5) in vascular biology and sex bias arose several years ago from the discovery that U-STAT5a/b and STAT5a-GFP associated with the endoplasmic reticulum (ER) and Golgi apparatus in pulmonary arterial endothelial and smooth muscle cells.81,82 siRNA mediated knockdown of U-STAT5a/b in vascular cells (a) produced a remarkable cystic ER/lunate nucleus phenotype, and (b) inhibited trafficking of the tsO45 VSV-G-GFP glycoprotein, and vasorelevant receptors such as BMPR2 to the cell surface.81,82 Moreover, the inhibition of intracellular trafficking of BMPR2 to the cell surface by siRNA-mediated knockdown of STAT5a/b was combinatorially exacerbated by estradiol-17β (E2).82 These observations, and the data of Waxman and colleagues implicating STAT5 in mediating sex bias in liver gene expression,29,67 led us to ask whether STAT5 might underlie the sex bias seen in a vascular disease such as PH.
Our approach83 to test the hypothesis relating sex and STAT5a/b in vascular remodeling has been to develop novel mice lines which have a conditional vascular smooth muscle-specific deletion of the floxed STAT5a/b locus using the SM22α-Cre method [heterozygous SM22α-Cre, STAT5a/b fl/wt (designated “+/−”) and homozygous SM22α-Cre, STAT5a/b fl/fl (designated “−/−”)]. In such mice both the adjacent STAT5a and STAT5b were deleted in SMCs, especially vascular SMCs.83 In contrast to endothelial-cell-specific STAT5a/b knockout mice (produced using the Tie2-Cre approach) which have microcytic anemia and are difficult to maintain postnatally,54 the SMC-specific STAT5a/b+/− and −/− knockout mice were fertile and viable.83
For our initial studies in these conditional SMC:STAT5a/b knockout mice, we focused on the sex-biased response to chronic hypoxia. It is well known that in the wt mouse chronic hypoxia typically triggers a male-dominant pulmonary arterial remodeling with female mice showing less extensive changes.6,16-19 Hypoxia has been shown to activate PY-STAT5 in cancer cells in culture.84 We observed that although male and female mice had equal levels of STAT5a/b expression in pulmonary arterial walls in lung sections and in isolated PASMCs,83 mice subjected to chronic hypoxia showed a sex-bias in the level of PY-STAT5 activation in the pulmonary arterial walls - greater level of activation in the female than in the male (Fig. 4). An investigation of the effect of conditional STAT5a/b knockout on PH induced by chronic hypoxia revealed that the male dominance was abrogated in the hypoxic STAT5a/b +/− and −/− mice (Figs. 5 and 6).83 Overall, female STAT5a/b +/− and −/− developed the greatest increase in right ventricular systolic pressure (RVSP), in right ventricular hypertrophy (RVH) and in pulmonary arterial remodeling (Figs. 5 and 6).83 Additionally, knockout males also had more severe PH than wt males (Fig. 6). These data provided the first evidence implicating STAT5a/b in the sexual dimorphism observed in a vascular disease process. At the cellular level this increased severity of PH in hypoxic STAT5a/b knockout mice involved marked hypertrophy of SMCs in the pulmonary arterial tunica media.83
FIGURE 4.
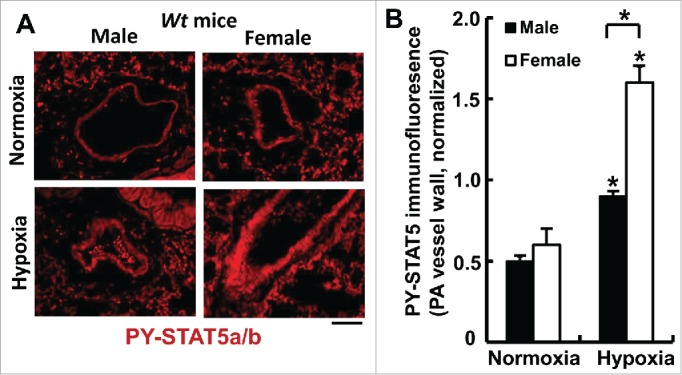
Sex-biased activation of PY-STAT5a/b in pulmonary arterial walls in wild-type mice after 7 weeks of chronic hypoxia (expt. as in Fig. 6). At the conclusion of the experiment in Fig. 6, quantitative immunofluorescence was used to evaluate levels of PY-STAT5a/b in the arterial walls of pulmonary arterial segments in sections of lungs using methods previously described (83). *P <0.05 in comparisons between hypoxia and normoxia groups of the 2 sexes; and also in the male vs female hypoxia comparison; scale bar = 50 µm.
FIGURE 5.
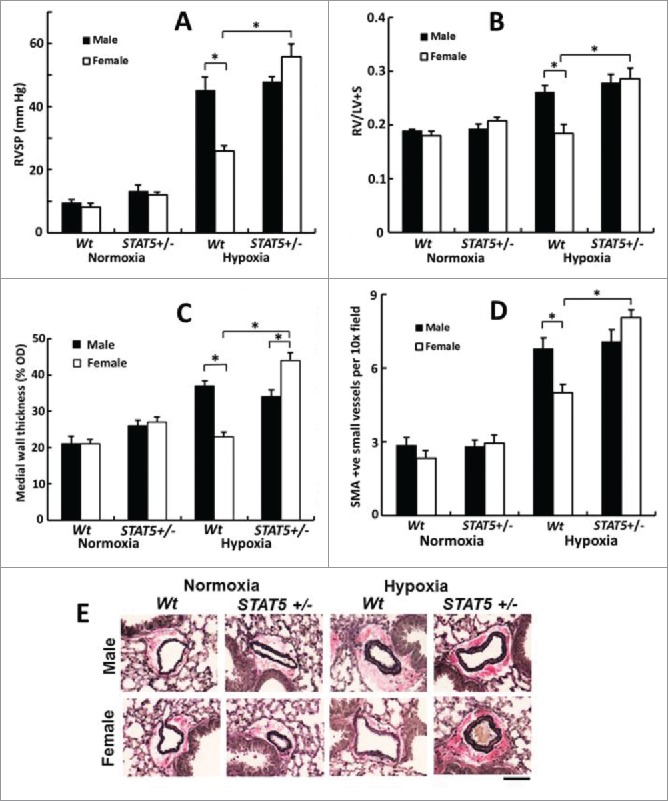
Abrogation of the male dominance of PH in the chronic hypoxia model in mice with heterozygous SM22-Cre, STAT5a/b+/− deletion (7 weeks' of hypoxia; n= 5 per group). Panel A, RVSP; Panel B, RVH; Panels C, PA remodeling in terms of wall thickness; Panel D: PA remodeling in terms of SMA-positive vessels; Panel E, Van Gieson's elastin staining. Scale bar = 45 µm). *P <0.05. Adapted from Sehgal et al (2014).83
FIGURE 6.
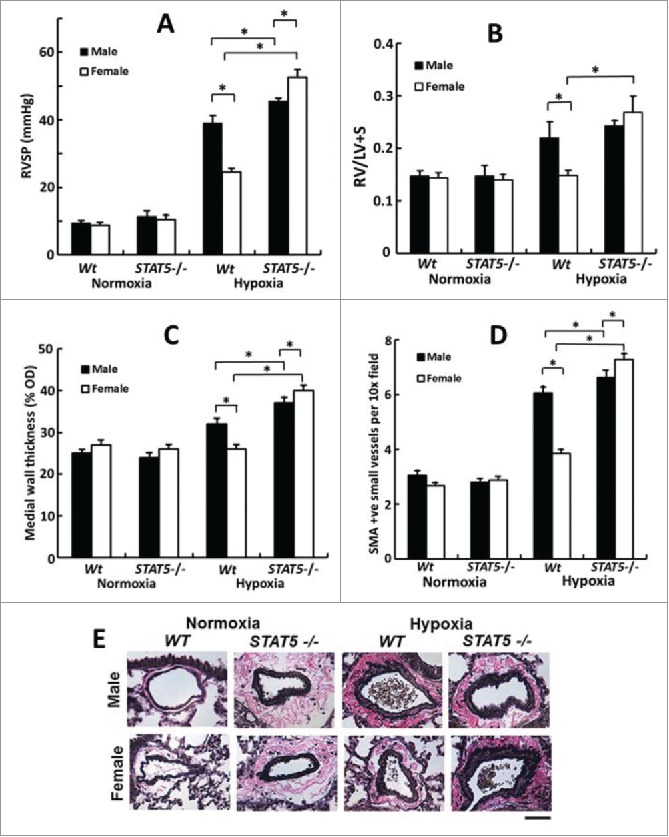
Female mice develop the severest PH in response to chronic hypoxia after homozygous SM22-Cre, STAT5a/b−/− deletion (7 weeks' of hypoxia; n= 5 per group). Panel A, RVSP; Panel B, RVH; Panels C, PA remodeling in terms of wall thickness; Panel D: PA remodeling in terms of SMA-positive vessels; Panel E, Van Gieson's elastin staining. Scale bar = 45 µm). *P <0.05. Adapted from Sehgal et al (2014).83
The effects of a knockout of STAT5a/b in vascular SMCs likely extend beyond the pulmonary circulation. Although, the male and female SMC:STAT5a/b−/− mice had similar systolic, diastolic and mean blood pressures as respective wt mice,83 “resistance” arteries such as second-order mesenteric arteries isolated from the knockout mice were 10-100-fold less responsive to vasodilation by acetylcholine (Fig. 7). Whether there is a STAT5-dependent sex bias in the mesenteric artery vasodilation response to acetylcholine is as yet unclear.
FIGURE 7.
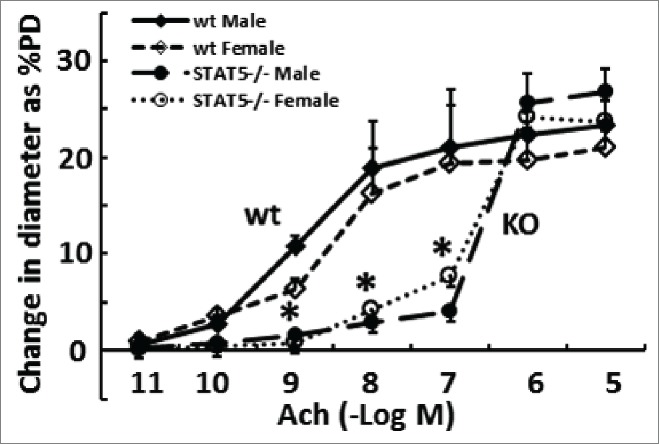
Resistance arteries (second-order mesenteric arteries) are “stiffer” in mice with homozygous SM22-Cre, STAT5a/b−/− deletion. The phenotypes of isolated pressurized (80 mm Hg) resistance arteries derived from groups of male and female wt and mutant mice were evaluated in terms of the vasorelaxation response to acetylcholine (Ach) using methods as in ref.112 and expressed as % change in peripheral diameter (% PD)(pooled data from n = 4 per group; mean ±SE). *P < 0.05 comparing respective wt and knockout groups (pooling both sexes) by ANOVA.
Reduced of STAT5a/b in SMCs in Vascular Lesions in Human Idiopathic Arterial Pulmonary Hypertension (IPAH)
Quantitative immunofluorescence analyses showed a marked reduction of STAT5a/b and of PY-STAT5 in SMCs in obliterative vascular lesions in lung sections derived from both male and female patients with late-stage IPAH, albeit with some variations between patients and in lesions in the same patient (Fig. 8).20,83 We have previously reported changes in the organization of the endoplasmic reticulum (ER)-associated proteins atlastin-3 (ATL3) and reticulon-4 (RTN4) (also called NogoB) in vascular ECs and SMCs following siRNA-mediated knockdown of STAT5a/b.81 Thus we evaluated expression of ATL3 and RTN4 in cells in IPAH lesions. Expression of ATL3 was markedly reduced but that of RTN4 was increased.83 These data, taken together with our previous observations of an increase in Golgi apparatus-associated tether giantin and SNARE Vti1a in such cells,85 point to the occurrence of ER stress and of changes in intracellular trafficking in cells in such obliterative arterial lesions.
FIGURE 8.
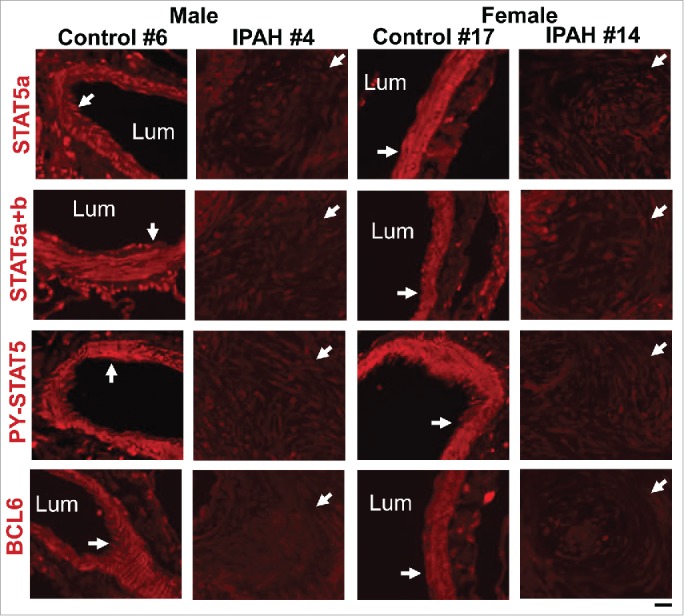
Representative immunofluorescence images showing coordinate reductions in STAT5a/b, PY-STAT5 and BCL6 in obliterative pulmonary arterial lesions in male and female patients with late-stage IPAH compared to control arterial walls (white arrows). The patient numbers correspond to the listing in Supplemental Table 2 in ref. 83. Scale bar = 50 µm.
We then investigated the expression of a known STAT5-dependent gene target in such lesions – the master transcription repressor BCL6. Waxman and colleagues have shown that BCL6 expression is driven by the GH-STAT5 axis in hepatocytes (male-biased in the liver), that the DNA binding motifs of STAT5 and BCL6 are very similar, and that BCL6 can repress STAT5 expression.57-59 The data obtained showed a marked reduction of BCL6 in cells in obliterative IPAH lesions (Fig. 8).20,83 This loss of BCL6 is consistent with the overall thinking about IPAH pathogenesis today in that (a) it is known that genetic deletion of BCL6 in mice results in a hypercytokine production state which includes pulmonary vasculitis,86-88 and (b) that several investigators have proposed that the PH disease process involves localized pulmonary vascular inflammation.89-92 Thus, a reduction in BCL6 in cells in obliterative lesions of IPAH (Fig. 8) is consistent with development of a localized proinflammatory state. However, lung sections from both men and women with IPAH showed reduced BCL6 expression, perhaps because the sections available were from patients with late-stage disease (Fig. 8).20,83
In considering sex bias in the pathogenesis of PH in humans (and in rodent models) we need to keep in mind 2 separate issues. First is the contribution of intact levels of STAT5a/b and PY-STAT5 as part of the GH-STAT5 axis in the early stages of the disease to mediating sex bias in humans (the 2-4-fold higher prevalence in women than in men) and in rodent models (typically male-dominance with, paradoxically, inhibition of PH by E2). Second is loss of STAT5a/b, PY-STAT5 and BCL6 in the late stages of IPAH in both men and women. These two issues get combined in the hypoxic SMC:STAT5a/b−/− mouse model in which (a) the sex bias is reversed, and (b) females get more severe disease than males. What downregulates STAT5a/b levels in IPAH lesions in late stage disease in both men and women remains to be understood.
Reduced STAT5a/b in Hypertrophic PASMC Lines Isolated From IPAH Patients
The above immunofluorescence data derived from lung sections of IPAH patients showing reductions in STAT5a/b obliterative lesion raised the question whether cells derived from lung vessels of IPAH patients might also show such reductions. A corollary question was whether vascular cells with reduced STAT5 derived from IPAH patients might display cell hypertrophy and organellar changes. Cell imaging studies of the primary PASMC lines derived from female controls and IPAH patients showed that 9 out of 9 control SMC lines consisted of small cells, while 7 out of 11 IPAH SMC lines consisted of enlarged hypertrophic cells, with markedly enlarged Golgi apparatus (displayed using either anti-giantin or ant-Vti1a antibodies).83 Western blot analyses showed a correlation between the cluster of these hypertrophic SMC lines and reduced STAT5a expression (using the k-means cluster statistic).83 Thus, low STAT5 expression clustered with cell hypertrophy, and enlarged Golgi apparatus in female IPAH-derived SMC lines in a multi-parameter pattern (not enough numbers of male-derived lines were available for study). These data provided further evidence for the involvement of a loss of STAT5 in the pathogenesis of PH.
A Neuroendocrine Perspective (Hypothalamus-GH-STAT5) of Sex Bias in PH
The previous literature about how the hypothalamus-GH-STAT5 axis underlies sex bias in liver gene expression, juxtaposed with the new data on the role of STAT5 in determining sex bias in hypoxic PH in a mouse model, and the puzzling sex-bias questions in the PH literature provides us with an opportunity to suggest a synthesis of these different lines of research.(Fig. 9; a more detailed synthesis is in ref. 20) .
FIGURE 9.
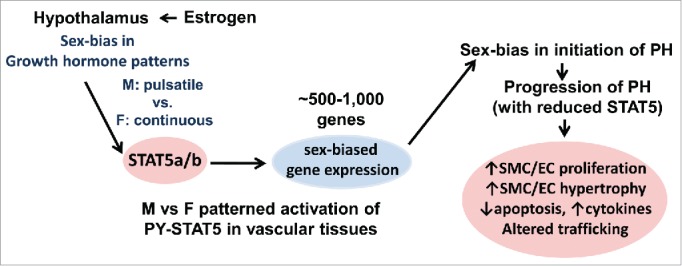
The GH-STAT5-BCL6 neuroendocrine axis as it relates to sex-bias in the initiation and progression of the pathogenesis of pulmonary hypertension.
Why is prevalence of IPAH higher in women than in men? On average, women have 80-120-fold higher levels of circulating GH than men.66 Moreover this is in a more continuous pattern than in men (Fig. 2).20,29,61 It is known that GH promotes vascular smooth muscle cell proliferation and migration, and is required for normal vascular reactivity and modeling.77-80 We propose that these 80-120-fold higher levels and a more continuous pattern of GH, and thus activation of different sets of cell-cycle, cell-proliferation and cell-migration regulatory genes,29 is why IPAH is more prevalent in women than men.(Fig. 9).
What is the relevant difference between a female human and a female rodent underlying differences in sex bias in PH? There are quantitative differences in patterns of circulating GH in males and females in mice, rats and humans resulting in the same gene being regulated differently in terms of sex-specificity in the 3 species (Figs. 1, 2 and 3)(discussed in detail in refs).20,29,67 The major relevant difference between female humans and female rodents is in the much higher continuous GH levels in women compared to female rodents (Fig. 2). Thus expression of P450 CYP 3A subfamily genes is male-biased in the rat,30,31 but female-biased in humans.34
Is there really an estrogen paradox? We have suggested above that the basis for why women get more IPAH than men may be related to the 80-120-fold higher GH levels and not so much E2.66 In animal models, typically in male rodents, E2 would be protective by targeting the hypothalamus directly.49-54 Thus, as has been shown in the liver literature, E2 would "feminize" GH patterns changing the male pattern of expression in rodents to a female pattern (Fig. 1). Therefore the apparent paradox can be explained by involving GH in human IPAH, and by the direct targeting of the hypothalamus by E2 (Fig. 9).20,83
Why is the MCT model male-biased in the rat?6,16,17 Downstream of the GH/STAT5 axis is the sex-specific expression of P450 CYP3A subfamily members.29,30,31,34 STAT5 is the transcription factor that upregulates CYP3A.29,68 and it is the CYP3A subfamily enzymes that metabolize MCT to its active MCTP.69,70 In the rat this is male-biased but is female-biased in mouse and humans.29-31,34 Thus, a single injection of MCT efficiently induces PH in the male rat. However, MCT does not readily produce PH in the female mouse either suggesting the occurrence of additional inter-species differences.
Why do female mice overexpressing the serotonin transporter (SERT) or the S100 calcium-binding protein S100A4/mts1 or the dexfenfluramine-administered mice, but not male mice, develop modest PH after 5 months?93-97 Although increased serotonin (5-HT) has been implicated in the pathogenesis of PH,93-97 the mechanistic focus has largely been on direct effects of 5-HT on pulmonary vascular tissues. We note that it is already known that the PH-causing anorexigens fenfluramine, aminorex, phentermine and fluoxetine increase 5-HT in the hypothalamus.98-101 and that fenfluramine blunted GH responsiveness to GHRH.102 Additionally, it is already known that PY-STAT5 signaling in the hypothalamus is involved in regulating appetite and sex-biased changes in body weight.103 It has been shown already that 5-HT suppresses STAT5 expression and PY-STAT5 activation,104 and 5-HT-receptor and dopaminergic D1, D2 receptor antagonists also inhibit PY-STAT5 activation (e.g., pimozide).105,106 We suggest that consideration of mechanisms in such models also include the central effects of 5-HT at the level of the hypothalamus and the arcuate nucleus, and sex-specific changes in the patterns of GH secretion and STAT5 activation. The female-biased PH in these circumstances is likely generated at the level of the hypothalamus.
Why is there no sex bias in the hypoxia-SU5416 model?6,18 SU5416 (Sugen) has been described in the PH literature as an inhibitor of receptor 2 for vascular endothelial growth factor (VEGF R2)(6 and citations therein). However, it has been shown already that SU5416 inhibits activation of PY-STAT5.107 The inhibition by SU5416 of PY-STAT5 activation [and of additional receptor tyrosine kinases108,109] suggests why this model does not show a sex bias.
We emphasize that from our perspective, outlined in Figure 9, we specifically combine both central neuroendocrine mechanisms with peripheral tissue-level mechanisms in the pathogenesis of a vascular disease such as PH.
Discovering Sex-Biased and STAT5-Dependent Gene Expression Patterns in Vascular Cells
Investigations of sex bias and its GH-STAT5-dependence in liver gene expression using global unbiased microarray analyses of expressed RNA have established an important paradigm – the different expression patterns include cohorts of hundreds of genes that are male- or female-biased, that are up- or down-regulated after hypophysectomy, that are up- or downregulated upon GH administration into hypohysectomized mice, and that are up- or down-regulated in hepatocytes from mice with a liver-specific STAT5a/b−/− knockout, or those that are sex-biased but unaffected by any of these manipulations (Fig. 3).29,67 Moreover, in each of these instances, while there are some genes that are coordinately affected similarly in males and females (e.g. reduction of SOCS2 expression after STAT5a/b−/− knockout in males and females), the cohorts of the affected genes in males and females are largely different. The important point in this paradigm is that sex-bias determinism through the GH-STAT5 axis at the level of peripheral tissues resides in changes in patterns of gene expression of cohorts of hundreds of genes (comprising transcription factors, growth factors and cytokines, intracellular trafficking mediators, cell adhesion molecules, and proteins that regulate cell proliferation, the cell cycle and apoptosis) and not just one or a few “mediators.” Thus this paradigm is different from the reductionist approach of most investigators who ask for identification of changes in one or a few genes, and expect that to account for the development of sex-biased phenotypes in different tissues. Moreover, such patterns of sex-biased GH-STAT5-driven gene expression patterns are likely to be different in different tissue types in a particular species, and also different in the same tissue type in different species.
The availability of mice with SMC-specific STAT5a/b−/− deletion allows for the derivation of primary PASMC and aortic SMC lines from males and females and an investigation of sex- and STAT5-specific patterns of gene expression using global unbiased RNA microarray or RNA sequencing approaches. Similarly, the availability of primary human PASMC lines derived from IPAH patients (females in this instance) with low STAT5 expression and corresponding control lines with high STAT5 allows for a similar investigation of STAT5-biased patterns of gene expression. The identification of BCL6 as a master transcription factor downstream of STAT5 in sex determinism hepatocytes by Waxman and colleagues,57-59 suggests the possibility that derivation of mice with SMC-specific deletion of BCL6, singly or in combination with STAT5a/b, may also lead to mice that have lost the male-dominant PH phenotype after exposure to chronic hypoxia. Such studies are likely to be informative with respect to the pathogenesis of the human disease in that we observed a marked decrease in BCL6 in SMCs in obliterative arterial lesions in IPAH (Fig. 8). The nestin-Cre-STAT5a/b−/− mice already developed by Lee et al.103 which have deletion of STAT5a/b in the central nervous system including the hypothalamus provide a substrate to directly test the role of the hypothalamus in sex bias in the chronic hypoxia model as proposed in Figure 9 (STAT5 and PY-STAT5 are the relevant transcription factors in the hypothalamus that regulate appetite, sexual dimorphism of body weight, and insulin resistance).103
We note that GH and STAT5 are involved in vascular SMC proliferation, motility and remodeling after injury,71-75,77-80 and that transgenic male mice overexpressing bovine GH developed hypertension between 5 and 6 months of age, independent of their body-weight.110 This hypertension then persisted long-term.110 Such observations, together with our data in Figure 7, implicate the GH-STAT5 axis in systemic vascular biology. The extent to which the GH-STAT5 axis contributes to sex bias in systemic vascular remodeling remains an open question, but one that can now be addressed using the conditional SMC-specific STAT5a/b−/− mice.
Conclusions
Fifty years ago Frantz and Rabkin reported observing markedly higher plasma GH levels in fasting ambulatory women than in men.111 They also observed that administration of an estrogen (diethylstilbestrol) to men “feminized” the pattern of circulating GH, and postulated that this was due to an effect of estrogen on the pituitary or higher centers. Over the next several decades these seminal observations led to the development of the neuroendocrine perspective of sex-biased liver gene expression anchored in hypothalamus-GH-STAT5 mediated mechanisms. These mechanisms, elucidated in great detail by numerous investigators, have now proven useful in understanding puzzling sex-bias issues in the pathogenesis of the vascular disease pulmonary hypertension in humans and rodent models. A critical insight transposed into vascular biology from the prior liver literature is that exogenously administered estrogens (and other sex hormones) affect sex-biased gene expression in peripheral tissues by affecting neuronal cells in the hypothalamus, and thus “feminizing” (or “masculinizing”) the pattern of GH secretion. This is an insight missing from the vascular biology literature. The focus of sex-bias studies in vascular biology thus far has almost exclusively been on the direct effects of sex hormones on vascular cells. The inclusion of the neuroendocrine GH-STAT5 pathways in considering sexual dimorphism in human disease and in rodent models broadens our mechanistic perspective of how sex and gender bias comes about. Part of this broadening of perspective includes the appreciation that the generation of a net sex-biased phenotype in a particular cell type in a particular species involves changes in the expression and function of cohorts of hundreds of genes through STAT5-anchored mechanisms, but also inclusive of STAT5-independent mechanisms. The paradigm at hand is one in which STAT5 transcription factors, in association with other cell-type-specific transcription factors, are transient activators or repressors of the expression of large cohorts of genes in different patterns depending upon the dynamical properties of GH-STAT5 activation such that the same pathways can contribute to both inhibiting or enhancing a particular disease process depending upon sex and species, and the particular pulmonary hypertension circumstance. The novel neuroendocrine concept, for the moment, is that the GH-STAT5 axis connects sexual dimorphism phenotype at the level of peripheral vascular tissues to mechanisms at the level of the hypothalamus.
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST
No potential conflicts of interest were disclosed.
NOTES
Some investigators use the phrase “pulmonary arterial hypertension (PAH)” to generically encompass the human disease and rodent models. Others use the phrase “pulmonary hypertension (PH)” as the generic term and reserve “pulmonary arterial hypertension (PAH)” for different forms of the human disease [such as idiopathic pulmonary arterial hypertension (IPAH) or hereditary pulmonary arterial hypertension (HPAH)]. In this essay we follow the second approach, and use PH as the generic term.
The phrase “sex bias” refers to biological differences, while the phrase “gender bias” refers to behavioral and congnitive differences. Thus “sex bias” is appropriate throughout this article.
Funding
Research in the authors' was supported, in part, by National Heart, Lung, and Blood Institute Grants HL-087176 (PBS), HL-114509 (PBS) and HL-111469 (EJM). We also thank the Pulmonary Hypertension Breakthrough Initiative (PHBI) for providing sections of human lung tissue studied in ref. 28 (adapted data illustrated, in part, in the present Fig. 8); funding for PHBI is provided by the Cardiovascular Medical Research and Education Fund (CMREF).
REFERENCES
- 1.Xing D, Nozell S, Chen YF, Hage F, Oparil S. Estrogen and mechanisms of vascular protection. Anterioscler Thromb Vasc Biol 2009; 29:289-95; http://dx.doi.org/ 10.1161/ATVBAHA.108.182279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pepine CJ, Nichols WW, Pauly DF. Estrogen and different aspects of vascular disease in women and men. Cir Res 2006; 99:459-61; PMID: 26252185; 17338926 10.2119/molmed.2015.00122 [DOI] [PubMed] [Google Scholar]
- 3.Tuder RM, Marecki JC, Richter A, Fijlkowska I, Flores S. Pathology of pulmonary hypertension. Clin Chest Med 2007; 28:23-42; PMID:17338926; http://dx.doi.org/ 10.1016/j.ccm.2006.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rabinovitch M. Molecular pathogenesis of pulmonary hypertension. J Clin Invest 2008; 118:2372-9; PMID:18596905; http://dx.doi.org/ 10.1172/JCI33452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Austin ED, Cogan JD, West JD, Hedges LK, Hamid R, Dawson EP, Wheeler LA, Parl FF, Loyd JE, Phillips JA 3rd. Alterations in oestrogen metabolism: implications for higher penetrance of familial pulmonary arterial hypertension in females. Eur Respir J 2009; 34:1093-9; PMID:19357154; http://dx.doi.org/ 10.1183/09031936.00010409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lahm T, Tuder R, Petrache I. Progress in solving the sex hormone paradox in pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 2014; 307:L7-L26; PMID:24816487; http://dx.doi.org/ 10.1152/ajplung.00337.2013 [DOI] [PubMed] [Google Scholar]
- 7.Kumar A, Lindner V. Remodeling with neointima formation in the mouse carotid artery after cessation of blood flow. Arterio Thromb Vasc Biol 1997; 17:2238-44; http://dx.doi.org/ 10.1161/01.ATV.17.10.2238 [DOI] [PubMed] [Google Scholar]
- 8.Boucher JM, Harrington A, Rostama B, Lindner V, Liaw L. A receptor-specific function for Notch2 in mediating vascular smooth muscle cell growth arrest through p27kip1. Circ Res 2013; 113:975-85; PMID:23965337; http://dx.doi.org/ 10.1161/CIRCRESAHA.113.301272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kumar A, Hoover JL, Simmons CA, Lindner V, Shebuski RJ. Remodeling and neointimal formation in the carotid artery of normal and P-selectin-deficient mice. Circulation 1997; 96:4333-42; PMID:9416901; http://dx.doi.org/ 10.1161/01.CIR.96.12.4333 [DOI] [PubMed] [Google Scholar]
- 10.Vermeersch P, Buys Em, Sips P, Pokreisz P, Marsboom G, Gillijns H, Pellens M, Dewerchin M, Bloch KD, Brouckaert P, et al.. Gender-specific modulation of the response to arterial injury by soluble guanylate cyclase α1. Open Cardio Med J 2009; 3:98-104; http://dx.doi.org/ 10.2174/1874192400903010098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sullivan TR Jr, Karas RH, Aronovitz M, Faller GT, Ziar JP, Smith JJ, O'Donnell TF Jr, Mendelsohn ME. Estrogen inhibits the response-to-injury in a mouse carotid artery model. J Clin Invest 1995; 96:2482-8; PMID:7593638; http://dx.doi.org/ 10.1172/JCI118307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Savolainen-Peltonen H, Luoto NM, Kangas L, Häyry P. Selective estrogen receptor modulators prevent neointima formation after vascular injury. Mol Cell Endocrinol 2004; 227:9-20; PMID:15501580; http://dx.doi.org/ 10.1016/j.mce.2004.08.004 [DOI] [PubMed] [Google Scholar]
- 13.Akishita M, Ouchi Y, Miyoshi H, Kozaki K, Inoue S, Ishikawa M, Eto M, Toba K, Orimo H. Estrogen inhibits cuff-induced initimal thickening of rat femoral artery: effects on migration and proliferation of vascular smooth muscle cells. Altherosclerosis 1997; 130:1-10; http://dx.doi.org/ 10.1016/S0021-9150(96)06023-6 [DOI] [PubMed] [Google Scholar]
- 14.Malorni W, Straface E, Matarrese P, Ascione B, Coinu R, Canu S, Galluzzo P, Marino M, Franconi F. Redox state and gender differences in vascular smooth muscle cells. FEBS Letters 2008; 582:635-42; PMID:18242172; http://dx.doi.org/ 10.1016/j.febslet.2008.01.034 [DOI] [PubMed] [Google Scholar]
- 15.Hogg ME, Vavra AK, Banerjee MN, Martinez J, Jiang Q, Keefer LK, Chambon P, Kibbe MR. The role of estrogen receptor α and β in regulating vascular smooth muscle cell proliferation is based on sex. J Surg Res 2012; 173:1-10; PMID:21658718; http://dx.doi.org/ 10.1016/j.jss.2011.09.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mair KM, Johansen AKZ, Wright AF, Wallace E, MacLean MR. Pulmonary arterial hypertension: basis of sex differences in incidence and treatment response. Brit J Pharmacol 2014; 171:567-79; http://dx.doi.org/ 10.1111/bph.12281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Umar S, Rabinovitch M, Eghbali M. Estrogen paradox in pulmonary hypertension – current controversies and future perspectives. Am J Respir Crit Care Med 2012; 186:125-31; PMID:22561960; http://dx.doi.org/ 10.1164/rccm.201201-0058PP [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu A, Schreier D, Tian L, Eickhoff JC, Wang Z, Hacker TA, Chesler NC. Direct and indirect protection of right ventricular function by estrogen in an experimental model of pulmonary arterial hypertension. Am J Physiol Heart Circ Physiol 2014; 307:H273-83; PMID:24906919; http://dx.doi.org/ 10.1152/ajpheart.00758.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frump AL, Goss KN, Vayl A, Albrecht M, Fisher A, Tursunova R, Fierst J, Whitson J, Cucci AR, Brown MB, Lahm T. Estradiol improves right ventricular function in rats with severe angioproliferative pulmonary hypertension: effects of endogenous and exogenous sex hormones. Am J Physiol Lung Cell Mol Physiol 2015; 308:L873-90; PMID:25713318; http://dx.doi.org/ 10.1152/ajplung.00006.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sehgal PB, Yang YM, Miller EJ. Hypothesis: neuroendocrine mechanisms (hypothalamus-growth hormone-STAT5 axis) contribute to sex nias in pulmonary hypertension. Mol Med 2015; Epub July 30; http://dx.doi.org/ 10.2119/molmed.2015.00122; PMID: 2625218522997253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Montague CR, Hunter MG, Cavrilin MA, Phillips GS, Goldschmidt-Clermont PJ, Marsh CB. Activation of estrogen receptor-α-reduces aortic smooth muscle differentiation. Cir Res 2006; 99:477-84; http://dx.doi.org/ 10.1161/01.RES.0000238376.72592.a2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ueda K, Lu Q, Baur W, Aronovitz MJ, Karas RH. Rapid estrogen receptor signaling mediates estrogen-induced inhibition of vascular smooth muscle cell proliferation. Anterioscler Thromb Vasc Biol 2013; 33:1837-43; http://dx.doi.org/ 10.1161/ATVBAHA.112.300752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bernelot Meons SJ, Schnitzler GR, Nickerson M, Guo H, Ueda K, Lu Q, Aronovitz MJ, Nickerson H, Bauer WE, Hansen U, et al.. Rapid estrogen receptor signaling is essential for the protective effects of estrogen against vascular injury. Circulation 2012; 126:1993-2004; PMID:22997253; http://dx.doi.org/ 10.1161/CIRCULATIONAHA.112.124529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kappert K, Caglayan E, Hunteburth M, Bäumer AT, Sparwel J, Uebel M, Rosenkranz S. 17beta-estradiol attenuates PDGF signaling in vascular smooth muscle cells at the postreceptor level. Am J Physiol Heart Circ Physiol 2006; 290:H538-46; PMID:16227346; http://dx.doi.org/ 10.1152/ajpheart.00240.2005 [DOI] [PubMed] [Google Scholar]
- 25.Austin ED, Hamid R, Hemnes AR, Loyd JE, Blackwell T, Yu C, Phillips JA III, Gaddipati R, Gladson S, Gu E, et al.. BMPR2 expression is suppressed by signaling through the estrogen receptor. Biol Sex Differences 2012; 3:6; http://dx.doi.org/ 10.1186/2042-6410-3-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fessel JP, Chen X, Frump A, Gladson S, Blackwell T, Kang C, Johnson J, Loyd JE, Hemnes A, Austin E, et al.. Interaction between bone morphogenetic protein receptor type 2 and estrogeneic compounds in pulmonary arterial hypertension. Pulm Circ 2013; 3:564-77; PMID:24618541; http://dx.doi.org/ 10.1086/674312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mair KM, Yang XD, Long L, White K, Wallace E, Ewart MA, Docherty CK, Morrell NW, MacLean MR. Sex affects BMPR-II signaling in pulmonary artery smooth muscle cells. Am J Respir Crit Care Med 2015; 191:693-703; PMID:25608111; http://dx.doi.org/ 10.1164/rccm.201410-1802OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wright AF, Ewart MA, Mair K, Nilsen M, Dempsie Y, Loughlin L, Maclean MR. Oestrogen receptor α in pulmonary hypertension. Cardiovasc Res 2015; 106:206-16; PMID:25765937; http://dx.doi.org/ 10.1093/cvr/cvv106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Waxman DJ, Holloway MG. Sex differences in the expression of hepatic drug metabolizing enzymes. Mol Pharmacol 2009; 76:215-28; PMID:19483103; http://dx.doi.org/ 10.1124/mol.109.056705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dhir RN, Shapiro BH. Interpulse growth hormone secretion in the episodic plasma profile causes the sex reversal of cytochrome P450s in senescent mate rats. Proc Natl Acad Sci USA 2003; 100:15224-8; PMID:14638941; http://dx.doi.org/ 10.1073/pnas.2434273100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Waxman DJ, Ram PA, Pampori NA, Shapiro BH. Growth hormone regulation of male-specific rat liver P450s 2A2 and 3A2: induction by intermittent growth hormone pulses in male but not in female rats rendered growth hormone deficient by neonatal monosodium glutamate. Mol Pharmacol 1995; 48:790-7; PMID:7476908 [PubMed] [Google Scholar]
- 32.Thangavel C, Garcia MC, Shapiro BH. Intrinsic sex differences determine expression of growth hormone-regulated female cytochrome P450s. Mol Cell Endocrinol 2004; 220:31-9; PMID:15196697; http://dx.doi.org/ 10.1016/j.mce.2004.04.002 [DOI] [PubMed] [Google Scholar]
- 33.Thangavel C, Dworakowski W, Shapiro BH. Inducibility of male-specific isoforms of cytochrome P450 by sex-dependent growth hormone profiles in hepatocyte cultures from male but not female rats. Drug Metab Dispos 2006; 34:410-9; PMID:16339352 [DOI] [PubMed] [Google Scholar]
- 34.Martignoni M, Groothuis GMM, Kanter R. Species differences between mouse, rat, dog, monkey and human CYP-mediated drug metabolism, inhibition and induction. Expert Opin Drug Metab Toxicol 2006; 2:875-94; PMID:17125407; http://dx.doi.org/ 10.1517/17425255.2.6.875 [DOI] [PubMed] [Google Scholar]
- 35.Cheung C, Yu AM, Chen CS, Krausz KW, Byrd LG, Feigenbaum L, Edwards RJ, Waxman DJ, Gonzalez FJ. Growth hormone determines sexual dimorphism of hepatic cytochrome P450 3A4 expression in transgenic mice. J Pharmacol Exptl Ther 2006; 316:1328-34; http://dx.doi.org/ 10.1124/jpet.105.094367 [DOI] [PubMed] [Google Scholar]
- 36.Kato R, Onoda K. Studies on the regulation of the activity of drug oxidation in rat liver microsomes by androgen and estrogen. Biochem Pharmacol 1970; 19:1649-60; PMID:4398016; http://dx.doi.org/ 10.1016/0006-2952(70)90328-X [DOI] [PubMed] [Google Scholar]
- 37.Colby HD, Gaskin JH, Kitay JI. Requirement of the pituitary gland for gonadal hormone effects on hepatic corticosteroid metabolism in rats and hamsters. Endocrinol 1973; 92:769-74; http://dx.doi.org/ 10.1210/endo-92-3-769 [DOI] [PubMed] [Google Scholar]
- 38.Kramer RE, Greiner JW, Rumbaugh RC, Sweeney TD, Colby HD. Requirement of the pituitary gland for gonadal hormone effects on hepatic drug metabolism in rats. J Pharmacol Exptl Ther 1979; 208:19-23 [PubMed] [Google Scholar]
- 39.Rumbaugh RC, Colby HD. Is growth hormone the pituitary feminizing factor mediating the actions of estradiol on hepatic drug and steroid metabolism? Endocrinol 1980; 107:719-24; http://dx.doi.org/ 10.1210/endo-107-3-719 [DOI] [PubMed] [Google Scholar]
- 40.Sakuma T, Endo Y, Mashino M, Kuroiwa M, Ohara A, Jarukamjorn K, Nemoto N. Regulation of the expression of two female-predominant CYP3A mRNAs (CYP3A1 and CYP3A44) in mouse liver by sex and growth hormone. Arch Biochem Biophys 2002; 404:234-42; PMID:12147261; http://dx.doi.org/ 10.1016/S0003-9861(02)00329-6 [DOI] [PubMed] [Google Scholar]
- 41.Muller EE, Locatelli V, Cocchi D. Neuroendocrine control of growth hormone secretion. Physiol Rev 1999; 79:511-607; PMID:10221989 [DOI] [PubMed] [Google Scholar]
- 42.Nishida Y, Yoshioka M St. Amand J. Sexually dimorphic gene expression in the hypothalamus, pituitary gland, and cortex. Genomics 2005; 85:679-87; PMID:15885495; http://dx.doi.org/ 10.1016/j.ygeno.2005.02.013 [DOI] [PubMed] [Google Scholar]
- 43.MacLeod JN, Pampori NA, Shapiro BH. Sex differences in the ultradian pattern of plasma growth hormone concentrations in mice. J Endocrinol 1991; 131:395-9; PMID:1783886; http://dx.doi.org/ 10.1677/joe.0.1310395 [DOI] [PubMed] [Google Scholar]
- 44.Low MJ, Otero-Corchon V, Parlow AF, Ramirez JL, Kumar U, Patel YC, Rubinstein M. Somatostatin is required for masculinization of growth hormone-regulated hepatic gene expression but not of somatic growth. J Clin Invest 2001; 107:1571-80; PMID:11413165; http://dx.doi.org/ 10.1172/JCI11941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Coutant R, Lahlou N, Bouvattier C, Bougneres P. Circulating leptin level and growth hormone response to stimulation in obese and normal children. Eur J Endocrinol 1998; 139:591-7; PMID:9916863; http://dx.doi.org/ 10.1530/eje.0.1390591 [DOI] [PubMed] [Google Scholar]
- 46.Waxman DJ, Ram PA, Park SH, Choi HK. Intermittant plasma growth hormone triggers tyrosine phosphorylation and nuclear translocation of a liver-expressed, Stat 5-related DNA binding protein: proposed role as an intracellular regulator of male-specific liver gene transcription. J Biol Chem 1995; 27:13262-70; http://dx.doi.org/ 10.1074/jbc.270.22.13262 [DOI] [PubMed] [Google Scholar]
- 47.Gebert CA, Park SH, Waxman DJ. Regulation of signal transducer and activator of transcription (STAT) 5b activation by the temporal pattern of growth hormone stimulation. Mol Endocrinol 1997; 11:400-14; PMID:9092792; http://dx.doi.org/ 10.1210/mend.11.4.9904 [DOI] [PubMed] [Google Scholar]
- 48.Tannenbaum GS, Choi HK, Gurd W, Waxman DJ. Temporal relationship between the sexually dimorphic spontaneous GH secretory profiles and hepatic STAT5 activity. Endocrinol 2001; 142:4599-606; http://dx.doi.org/ 10.1210/endo.142.11.8480 [DOI] [PubMed] [Google Scholar]
- 49.Garcia-Segura LM, Baetens D, Naftolin F. Synaptic remodeling in arcuate nucleus after injection of estradiol valerate in adult female rats. Brain Res 1986; 366:131-6; PMID:3697673; http://dx.doi.org/ 10.1016/0006-8993(86)91287-4 [DOI] [PubMed] [Google Scholar]
- 50.Shirasu K, Stumpf WE, Sar M. Evidence for direct action of estradiol on growth hormone-releasing factor (GRF) in rat hypothalamus: localization of [3H]estradiol in GRF neurons. Endocrinol 1990; 127:344-9; http://dx.doi.org/ 10.1210/endo-127-1-344 [DOI] [PubMed] [Google Scholar]
- 51.Senaris RM, Lago F, Lewis MD, Dominguez F, Scanlon MF, Dieguez C. Differential effects of in vivo estrogen administration on hypothalamic growth hormone releasing hormone and somatostatin gene expression. Neurosci Lett 1992; 141:123-6; PMID:1354846; http://dx.doi.org/ 10.1016/0304-3940(92)90349-C [DOI] [PubMed] [Google Scholar]
- 52.Desjardins GC, Brawer JR, Beaudet A. Estradiol is selectively neurotoxic to hypothalamic β-endorphin neurons. Endocrinol 1993; 132:86-93 [DOI] [PubMed] [Google Scholar]
- 53.Brawer JR, Beaudet A, Desjardins GC, Schiffer HM. Pathologic effect of estradiol on the hypothalamus. Biol Reprod 1993; 49:647-52; PMID:8218628; http://dx.doi.org/ 10.1095/biolreprod49.4.647 [DOI] [PubMed] [Google Scholar]
- 54.Kelly MJ, Ronnekleiv OK. Neural signaling of estradiol in the hypothalamus. Mol Endocrinol 2015; 29(5):645-57; PMID:25751314; http://dx.doi.org/ 10.1210/me.2014-1397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Udy GB, Towers RP, Snell RG, Wilkins RJ, Park SH, Ram PA, Waxman DJ, Davey HW. Requirement of STAT5b for sexual dimorphism of body growth rates and liver gene expression. Proc Natl Acad Sci USA 1997; 94:7239-44; PMID:9207075; http://dx.doi.org/ 10.1073/pnas.94.14.7239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Holloway MG, Cui Y, Laz EV, Hosui A, Hennighausen L, Waxman DJ. Loss of sexually dimorphic liver gene expression upon hepatocyte-specific deletion of Stat5a-Stat5b locus. Endocrinol 2007; 148:1977-86; http://dx.doi.org/ 10.1210/en.2006-1419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang Y, Laz EV, Waxman DJ. Dynamic, sex-differential STAT5 and BCL6 binding to sex-biased, growth hormone-regulated genes in adult mouse liver. Mol Cell Biol 2012; 32:880-96; PMID:22158971; http://dx.doi.org/ 10.1128/MCB.06312-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Meyer RD, Laz EV, Su T, Waxman DJ. Male-specific hepatic Bcl6: Growth hormone-induced block of transcription elongation in females and binding to target genes inversely coordinated with STAT5. Mol Endocrinol 2009; 23:1914-26; PMID:19797429; http://dx.doi.org/ 10.1210/me.2009-0242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sugathan A, Waxman DJ. Genome-wide analysis of chromatin states reveals distinct mechanisms of sex-dependent gene regulation in male and female mouse liver. Mol Cell Biol 2013; 33:3594-610; PMID:23836885; http://dx.doi.org/ 10.1128/MCB.00280-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Edén S. Age- and sex-related differences in episodic growth hormone secretion in the rat. Endocrinol 1979; 105:555-60; http://dx.doi.org/ 10.1210/endo-105-2-555 [DOI] [PubMed] [Google Scholar]
- 61.Winer LM, Shaw MA, Baumann G. Basal plasma growth hormone levels in man: new evidence for rhythmicity of growth hormone secretion. J Clin Endocrinol Metab 1990; 70:1678-86; PMID:2347901; http://dx.doi.org/ 10.1210/jcem-70-6-1678 [DOI] [PubMed] [Google Scholar]
- 62.MacLeod JN, Pampori NA, Shapiro BH. Sex differences in the ultradian pattern of plasma growth hormone concentrations in mice. J Endocrinol 1991; 131:395-9; PMID:1783886; http://dx.doi.org/ 10.1677/joe.0.1310395 [DOI] [PubMed] [Google Scholar]
- 63.Painson JC, Tannenbaum GS. Sexual dimorphism of stomatostatin and growth hormone-releasing factor signaling in the control of pulsatile growth hormone secretion in the rat. Endocrinol 1991; 128:2858-66; http://dx.doi.org/ 10.1210/endo-128-6-2858 [DOI] [PubMed] [Google Scholar]
- 64.van den Berg G, Veldhuis JD, Frolich M, Roelfsema F. An amplitude-specific divergence in the pulsatile mode of growth hormone (GH) secretion underlies the gender differences in mean GH concentrations in men and premenopausal women. J Clin Endocrinol Metab 1996; 81:2460-7; PMID:8675561 [DOI] [PubMed] [Google Scholar]
- 65.Pincus SM, Gevers EF, Robinson IC, van den Berg G, Roelfsema F, Hartman ML, Veldhuis JD. Females secrete growth hormone with more process irregularity than males in both humans and rats. Am J Physiol 1996; 270:E107-15; PMID:8772482 [DOI] [PubMed] [Google Scholar]
- 66.Engström BE, Karlsson FA, Wide L. Marked gender differences in ambulatory morning growth hormone values in young adults. Clin Chem 1998; 44:1289-95; PMID:9625055 [PubMed] [Google Scholar]
- 67.Waxman DJ, O'Connor C. Growth hormone regulation of sex-dependent liver gene expression. Molec Endocrinol 2006; 20:2613-29; http://dx.doi.org/ 10.1210/me.2006-0007 [DOI] [PubMed] [Google Scholar]
- 68.Subramanian A, Teixeira J, Wang J, Gil G. A STAT factor mediates the sexually dimorphic regulation of hepatic cytochrome P450 3A10/lithocholic acid 6 β-hydroxylase gene expression by growth hormone. Mol Cell Biol 1995; 15:4672-82; PMID:7651384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kasahara Y, Kiyatake K, Tatsumi K, Sugito K, Kakusaka I, Yamagata S, Ohmori S, Kitada M, Kuriyama T. Bioactivation of monocrotaline by P-450 3A in rat liver. J Cardiovasc Pharmacol 1997; 30:124-9; PMID:9268231; http://dx.doi.org/ 10.1097/00005344-199707000-00018 [DOI] [PubMed] [Google Scholar]
- 70.Reid MJ, Lame MW, Morin D, Wilson DW, Segall HJ. Involvement of cytochrome P450 3A in the metabolism and covalent binding of 14C-monocrotaline in rat liver microsomes. J Biochem Mol Toxicol 1998; 12:157-66; PMID:9522275; http://dx.doi.org/ 10.1002/(SICI)1099-0461(1998)12:3%3c157::AID-JBT4%3e3.0.CO;2-K [DOI] [PubMed] [Google Scholar]
- 71.Paukku K, Valgeirsdóttir S, Saharinen P, Bergman M, Heldin CH, Silvennoinen O. Platelet-derived growth factor (PDGF)-induced activation of signal transducer and activator of transcription (Stat) 5 is mediated by PDGF β-receptor and is not dependent on c-src, fyn, jak1 or jak2 kinases. Biochem J 2000; 345:759-66; PMID:10642538; http://dx.doi.org/ 10.1042/bj3450759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cao H, Dronadula N, Rizvi F, Li Q, Srivastava K, Gerthoffer WT, Rao GN. Novel role for STAT-5B in the regulation of Hsp27-FGF-2 axis facilitating thrombin-induced vascular smooth muscle cell growth and motility. Cir. Res 2006; 98:913-22. [DOI] [PubMed] [Google Scholar]
- 73.Kundumani-Shridharan V, Wang D, Karpurapu M, Liu Z, Zhang C, Dronadula N, Rao GN. Suppression of activation of signal transducer and activator of transcription-5B signaling in the vessel wall reduces balloon injury-induced neointima formation. Am J Pathol 2007; 171:1381-94; PMID:17823285; http://dx.doi.org/ 10.2353/ajpath.2007.061258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Han M, Li AY, Meng F, Dong LH, Zheng B, Hu HJ, Nie L, Wen JK. Synergistic co-operation of signal transducer and activator of transcription 5B with activator protein 1 in angiotensin IIK-induced angiotensinogen gene activation in vascular smooth muscle cells. FEBS J 2009; 276:1720-8; PMID:19220857; http://dx.doi.org/ 10.1111/j.1742-4658.2009.06902.x [DOI] [PubMed] [Google Scholar]
- 75.Tiell ML, Stemerman MB, Spaet TH. The influence of the pituitary on arterial intimal proliferation in the rat. Circ Res 1978; 42:644-9; PMID:639188; http://dx.doi.org/ 10.1161/01.RES.42.5.644 [DOI] [PubMed] [Google Scholar]
- 76.Khorsandi M, Fagin JA, Fishbein MC, Forrester JS, Cercek B. Effects of hypophysectomy on vascular insulin-like growth factor-I gene expression after balloon denudation in rats. Atherosclerosis 1992; 93:115-22; PMID:1596294; http://dx.doi.org/ 10.1016/0021-9150(92)90205-U [DOI] [PubMed] [Google Scholar]
- 77.Isgaard J, Arcopinto M, Karason K, Cittadini A. GH and the cardiovascular system: an update on a topic at heart. Endocrine 2015; 48:25-35; PMID:24972804; http://dx.doi.org/ 10.1007/s12020-014-0327-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jin MH, Yokoyama U, Sato Y, Shioda A, Jiao Q, Ishikawa Y, Minamisawa S. DNA microarray profiling identified a new role of growth hormone in vascular remodeling of rat ductus arteriosus. J Physiol Sci 2011; 61:167-79; PMID:21287305; http://dx.doi.org/ 10.1007/s12576-011-0133-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Capaldo B, Guardasole V, Pardo F, Matarazzo M, Di , Rella F, Numis F, Merola B, Longobardi S, Saccà L. Abnormal vascular reactivity in growth hormone deficiency. Circulation 2001; 103:520-4; PMID:11157716; http://dx.doi.org/ 10.1161/01.CIR.103.4.520 [DOI] [PubMed] [Google Scholar]
- 80.Borson-Chazot F, Serusclat A, Kalfallah Y, Ducottet X, Sassolas G, Bernard S, Labrousse F, Pastene J, Sassolas A, Roux Y, Berthezène F. Decrease of carotid intima-media thickness after one year growth hormone (GH) treatment in adults with GH deficiency. J Clin Endocrin Metabol 1999; 84:1329-33 [DOI] [PubMed] [Google Scholar]
- 81.Lee JE, Yang YM, Liang FX, Gough DJ, Levy DE, Sehgal PB. Nongenomic STAT5-dependent effects on Golgi apparatus and endoplasmic reticulum structure and function. Am J Physiol Cell Physiol 2012; 302:C804-20; PMID:22159083; http://dx.doi.org/ 10.1152/ajpcell.00379.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yang YM, Lane KB, Sehgal PB. Subcellular mechanisms in pulmonary arterial hypertension: combinatorial modalities that inhibit anterograde trafficking and cause BMPR2 mislocalization. Pulm Circ 2013; 3:533-50; PMID:24618539; http://dx.doi.org/ 10.1086/674336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yang YM, Yuan H, Edwards JG, Skayian Y, Ochani K, Miller EJ, Sehgal PB. Deletion of STAT5a/b in vascular smooth muscle abrogates the male bias in hypoxic pulmonary hypertension in mice: implications in the human disease. Mol Med 2014; 20:625-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Joung YH, Park JH, Park T, Lee CS, Kim OH, Ye SK, Yang UM, Lee KJ, Yang YM. Hypoxia activates signal transducers and activators of transcription 5 (STAT5) and increases its binding activity to the GAS element in mammary epithelial cells. Exp Mol Med 2003; 35:350-7; PMID:14646587; http://dx.doi.org/ 10.1038/emm.2003.46 [DOI] [PubMed] [Google Scholar]
- 85.Sehgal PB, Mukhopadhyay S, Patel K, Xu F, Almodóvar S, Tuder RM, Flores SC. Golgi dysfunction is a common feature in idiopathic human pulmonary hypertension and vascular lesions in SHIV-nef-infected macaques. Am J Physiol Lung Cell Mol Physiol 2009; 297:L729-37; PMID:19648286; http://dx.doi.org/ 10.1152/ajplung.00087.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dent AL, Shaffer AL, Yu X, Allman D, Staudt LM. Control of inflammation, cytokine expression, and germinal center formation by BCL-6. Science 1997; 276:589-92; PMID:9110977; http://dx.doi.org/ 10.1126/science.276.5312.589 [DOI] [PubMed] [Google Scholar]
- 87.Toney LM, Cattoretti G, Graf JA, Merghoub T, Pandolfi PP, Dalla-Favera R, Ye BH, Dent AL. BCL-6 regulates chemokine gene transcription in macrophages. Nat Immunol 2000; 1:214-20; PMID:10973278 http://dx.doi.org/ 10.1038/79749 [DOI] [PubMed] [Google Scholar]
- 88.Liao W, Spolski R, Li P, Du N, West EE, Ren M, Mitra S, Leonard WJ. Opposing actions of IL-2 and IL-21 on Th9 differentiation correlates with their differential regulation of BCL6 expression. Proc Natl Acad Sci USA 2014; 111:3508-13; PMID:24550509; http://dx.doi.org/ 10.1073/pnas.1301138111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Burke DL, Frid MG, Kunrath CL, Karoor V, Anwar A, Wagner BD, Strassheim D, Stenmark KR. Sustained hypoxia promotes the development of a pulmonary artery-specific inflammatory microenvironment. Am J Physiol Lung Cell Mol Physiol 2009; 297:L238-50; PMID:19465514; http://dx.doi.org/ 10.1152/ajplung.90591.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Soon E, Holmes AM, Treacy CM, Doughty NJ, Southgate L, Machado RD, Trembath RC, Jennings S, Barker L, Nicklin P, et al.. Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation 2010; 122:920-7; PMID:20713898; http://dx.doi.org/ 10.1161/CIRCULATIONAHA.109.933762 [DOI] [PubMed] [Google Scholar]
- 91.Daley E, Emson C, Guignabert C, de Waal Malefyt R, Louten J, Kurup VP, Hogaboam C, Taraseviciene-Stewart L, Voelkel NF, Rabinovitch M, et al.. Pulmonary arterial remodeling induced by a Th2 immune response. J Exp Med 2008; 205:361-72; PMID:18227220; http://dx.doi.org/ 10.1084/jem.20071008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Savai R, Al-Tamari HM, Sedding D, Kojonazarov B, Muecke C, Teske R, Capecchi MR, Weissmann N, Grimminger F, Seeger W, et al.. Pro-proliferative and inflammatory signaling converge on FoxO1 transcription factor in pulmonary hypertension. Nat Med 2014; 20:1289-300; PMID:25344740; http://dx.doi.org/ 10.1038/nm.3695 [DOI] [PubMed] [Google Scholar]
- 93.MacLean MR, Deuchar GA, Hicks MN, Morecroft I, Shen S, Sheward J, Colston J, Loughlin L, Nilsen M, Dempsie Y, et al.. Overexpression of the 5-hydroxytryptamine transporter gene: effect on pulmonary hemodynamics and hypoxia-induced pulmonary hypertension. Circulation 2004; 109:2150-5; PMID:15078799; http://dx.doi.org/ 10.1161/01.CIR.0000127375.56172.92 [DOI] [PubMed] [Google Scholar]
- 94.White K, Dempsie Y, Nilsen M, Wright AF, Loughlin L, MacLean MR. The serotonin transporter, gender, and 17β estradiol in the development of pulmonary arterial hypertension. Cardiovasc Res 2011; 90:373-82; PMID:21177701; http://dx.doi.org/ 10.1093/cvr/cvq408 [DOI] [PubMed] [Google Scholar]
- 95.Dempsie Y, Nilsen M, White K, Mair KM, Loughlin L, Ambartsumian N, Rabinovitch M, Maclean MR. Development of pulmonary arterial hypertension in mice over-expresssing S100A4/Mts1 is specific to females. Respir Res 2011; 12:159; PMID:22185646; http://dx.doi.org/ 10.1186/1465-9921-12-159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dempsie Y, Morecroft I, Welsh DJ, MacRitchie NA, Herold N, Loughlin L, Nilsen M, Peacock AJ, Harmar A, Bader M, et al.. Converging evidence in support of the serotonin hypothesis of dexfenfluramine-induced pulmonary hypertension with novel transgenic mice. Circulation 2008; 117:2928-37; PMID:18506000; http://dx.doi.org/ 10.1161/CIRCULATIONAHA.108.767558 [DOI] [PubMed] [Google Scholar]
- 97.Launay J-M, Hervé P, Peoc'h K, Tournois C, Callebert J, Nebigil CG, Etienne N, Drouet L, Humbert M, Simonneau G, et al.. Function of the serotonin 5-hydroxytryptamine 2B receptor in pulmonary hypertension. Nature Medicine 2002; 8:1129-35; PMID:12244304; http://dx.doi.org/ 10.1038/nm764 [DOI] [PubMed] [Google Scholar]
- 98.Tao R, Fray A, Aspley S, Brammer R, Heal D, Averbach S. Effects on serotonin in rat hypothalamus of D-fenfluramine, aminorex, phentermine and fluoxetine. Eur J Pharmacol 2002; 445:69-81; PMID:12065196; http://dx.doi.org/ 10.1016/S0014-2999(02)01751-X [DOI] [PubMed] [Google Scholar]
- 99.Prow MR, Lancashire B, Aspley S, Heal DJ, Kilpatrick IC. Additive effects on rat brain 5HT release of combining phentermine with dexfenfluramine. Int J Obes Relat Metab Disord 2001; 25:145-1453; http://dx.doi.org/ 10.1038/sj.ijo.0801717 [DOI] [PubMed] [Google Scholar]
- 100.Jia Y, El-Haddad M, Gendy A, Nguyen T, Ross MG. Serotonin-induced region-specific responses of the arcuate nucleus and ventromedial hypothalamic nuclei. Int J Neurosci 2010; 120:386-95; PMID:20402579; http://dx.doi.org/ 10.3109/00207450802336683 [DOI] [PubMed] [Google Scholar]
- 101.Voigt JP, Fink H. Serotonin controlling feeding and satiety. Behave Brain Res 2015; 277:14-31; http://dx.doi.org/ 10.1016/j.bbr.2014.08.065 [DOI] [PubMed] [Google Scholar]
- 102.Argenio GF, Bernini GP, Sgró M, Vivaldi MS, Del , Corso C, Santoni R, Franchi F. Blunted growth hormone (GH) responsiveness to GH-releasing hormone in obese patients: influence of prolonged administration of serotoninergic drug fenfluramine. Metabolism 1991; 40:724-7; PMID:1870427; http://dx.doi.org/ 10.1016/0026-0495(91)90091-A [DOI] [PubMed] [Google Scholar]
- 103.Lee JY, Muenzberg H, Gavrilova O, Reed JA, Berryman D, Villanueva EC, Louis GW, Leinninger GM, Bertuzzi S, Seeley RJ, et al.. Loss of cytokine-STAT5 signaling in the CNS and pituitary gland alters energy balance and leads to obesity. PLoS One 2008; 3:e1639; PMID:18286195; http://dx.doi.org/ 10.1371/journal.pone.0001639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chiba T, Kimura S, Takahashi K, Morimoto Y, Sanbe A, Ueda H, Kudo K. Serotonin suppresses β-casein expression via inhibition of the signal transducer and activator of transcription 5 (STAT5) protein phosphorylation in human mammary epithelial cells MCF-12A. Biol Pharm Bull 2014; 37:1336-40; PMID:25087955; http://dx.doi.org/ 10.1248/bpb.b14-00273 [DOI] [PubMed] [Google Scholar]
- 105.Lieberman LA, Higgins DE. A small-molecule screen identifies the antipsychotic drug pimozide as an inhibitor of Listeria monocytogenes infection. Antimicrob Agents Chemother 2009; 53:756-64; PMID:19015342; http://dx.doi.org/ 10.1128/AAC.00607-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Nelson EA, Walker SR, Weisberg E, Bar-Natan M, Barrett R, Gashin LB, Terrell S, Klitgaard JL, Santo L, Addorio MR, et al.. The STAT5 inhibitor pimozide decreases survival of chronic myelogenous leukemia cells resistant to kinase inhibitors. Blood 2011; 117:3421-09; PMID:21233313; http://dx.doi.org/ 10.1182/blood-2009-11-255232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Yee KW, O'Farrell AM, Smolich BD, Cherrington JM, McMahon G, Wait CL, McGreevey LS, Griffith DJ, Heinrich MC. SU5416 and SU5614 inhibit kinase activity of wild-type and mutant FLT3 receptor tyrosine kinase. Blood 2002; 100:2941-9; PMID:12351406; http://dx.doi.org/ 10.1182/blood-2002-02-0531 [DOI] [PubMed] [Google Scholar]
- 108.Roskovski R, Jr. Sunitinib: a VEGF and PDGF receptor protein kinase and angiogenesis inhibitor. Biochem Biophys Res Commun 2007; 356:323-8; PMID:17367763; http://dx.doi.org/ 10.1016/j.bbrc.2007.02.156 [DOI] [PubMed] [Google Scholar]
- 109.Mezrich JD, Nguyen LP, Kennedy G, Nukaya M, Fechner JH, Zhang X, Xing Y, Bradfield CA. SU5416, a VEGF receptor inhibitor and ligand of the AHR represents a new alternative for immunomodulation. PLoS One 2012; 7(9):e44547; PMID:22970246; http://dx.doi.org/ 10.1371/journal.pone.0044547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Jara A, Benner CM, Sim D, Liu X, List EO, Householder LA, Berryman DE, Kopchik JJ. Elevated systolic blood pressure in male GH transgenic mice is age dependent. Endocrinology 2014; 155:975-86; PMID:24424040; http://dx.doi.org/ 10.1210/en.2013-1899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Frantz AG, Rabkin MT. Effects of estrogen and sex difference on secretion of human growth hormone. J Clin Endocr 1965; 25:1470-80; PMID:5843702; http://dx.doi.org/ 10.1210/jcem-25-11-1470 [DOI] [PubMed] [Google Scholar]
- 112.Yang YM, Huang A, Kaley G, Sun D. eNOS uncoupling and endothelial dysfunction in aged vessels. Am J Physiol Heart Circ Physiol 2009; 297:H1829-1836; PMID:19767531; http://dx.doi.org/ 10.1152/ajpheart.00230.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]