

ABSTRACT
Phage therapy is a promising treatment of multi-drug resistant (MDR) bacterial infections but is limited by the narrow host range of phage. To overcome this limitation, we developed a host range expansion (HRE) protocol that expands the host range of Pseudomonas aeruginosa-specific phage by cycles of co-incubation of phage with multiple P. aeruginosa strains. Application of the HRE protocol to a mixture of 4 phages, using 16 P. aeruginosa strains for development, resulted in undefined phage mixtures with greatly expanded host range. Individual phage clones derived from the undefined mixture had expanded host ranges but no individual clone could lyse all of the strains covered by the undefined mixture from which it was isolated. Reconstituting host range-characterized clones into cocktails produced defined cocktails with predictable and broad host ranges. The undefined mixture from the 30th cycle of the mixed-phage HRE (4ϕC30) showed a dose-dependent ability to prevent biofilm formation by, and to reduce a pre-existing biofilm of, 3 P. aeruginosa clinical isolates that produced high amounts of biofilm. A defined cocktail reconstituted from 3 host range-characterized clones had activity on high biofilm-formers susceptible to the phage. Phage therapy was superior to antibiotic therapy (levofloxacin) in a strain of P. aeruginosa that was resistant to levofloxacin. The HRE protocol establishes a rapid approach to create libraries of phage clones and phage cocktails with broad host range, defined composition and anti-biofilm activity.
Keywords: Bacteriophage, biofilm, host range, Pseudomonas aeruginosa
Introduction
Nearly a decade ago, the Infectious Diseases Society of America released a series of policy statements calling attention to the paucity of new antimicrobial drugs for resistant bacterial pathogens.1,2 In 2013, the Centers for Disease Control issued a white paper which indicated that multidrug-resistant (MDR) organisms are a significant threat to the population. They divided organisms into 3 threat levels, classifying the agent examined here (Pseudomonas aeruginosa) as a serious threat.3 The lack of new drugs in the pipeline is especially acute for Gram-negative organisms,1 and has led to increased interest in alternative therapeutics such as bacteriophage (phage) therapy.
Since the work of d'Herelle in 1917,4 phage therapy has been considered a possible therapy for treatment of bacterial infections,5 but was largely ignored in the West after antibiotics became available. With the rise of antimicrobial resistance and paucity of new and effective antibiotics, phage therapy was rediscovered.6,7
A major limitation to using phage as therapy is their narrow host range; a specific phage is often capable of lysing only one or a small number of strains within a bacterial species. The resistance can be intrinsic to the host cell or acquired after exposure of the host cell to a phage to which it was previously sensitive. Four approaches can be used to overcome the narrow host range of phage for therapy: (i) broad host range phage can be isolated from the environment, 8,9 (ii) phage can be genetically engineered to recognize new hosts,10,11 (iii) the host range of individual phage or mixtures of phage can be expanded experimentally, 12 or (iv) cocktails can be made in which the combination of included phage have broad host range. The first approach results in phage selected only on the basis of their broad host range; more detailed information would be necessary for such phage to be used in clinical trials. The second approach requires extensive knowledge of the phage and its life cycle plus the ability to rescue manipulated genomes into infectious phage. While the manipulation of phage is possible, the rescue of manipulated genomes into infectious phage may be difficult and requires the prior determination of the sequence of the parental genome, a feasible but laborious approach. The third approach, while requiring cloning of individual phages from an expanded host range mixture and sequencing of the expanded host range derivative phages, has the advantage of allowing one to begin with fully characterized and sequenced parental phage, providing a scaffold for sequencing and identification of mutations in the expanded host range derivatives. The fourth approach is the creation of broad host range phage cocktails from individual broad host range phage clones. Here we discuss a combination of approaches 3 and 4.
Theoretically, both antibiotic resistant cells and persister cells are subject to killing by phage to which they are sensitive.10,13-15 However, host cells may acquire resistance to a specific phage, a second limitation of phage therapy. Phage resistance often arises from mutation of the host receptor for the phage.16,17 Phage resistance may be overcome with a cocktail (mixture) of phage targeting different receptors on host cells because multiple host mutations would be required for resistance to the cocktail.
Here, we seek to develop a phage therapy that will eradicate the P. aeruginosa biofilms thought to be the source of resurgent infection after treatment of MDR infections with antibiotics. We describe a host range expansion (HRE) method for phage specific to P. aeruginosa that results in undefined phage mixtures containing phages with the ability to lyse a greater number of P. aeruginosa strains than the input (parental) phage. Individual clones isolated from the mixture are characterized and reconstituted into defined cocktails with expanded host range in which each component can be thoroughly characterized (host range, sequence, etc.). Expanded-host range mixtures and cocktails generated from the HRE are demonstrated to be effective in reducing biofilm formation and reducing pre-existing biofilms of susceptible P. aeruginosa strains in vitro, including MDR strains. Finally, the relative effectiveness of phage cocktail and antibiotic are compared in biofilm prevention and treatment.
Results
Pseudomonas aeruginosa strains and their phenotypes
Characterization of the strains used in this study is shown in Figure 1. The ability to form biofilms varied greatly between strains, although the variation in metabolically active cells detected in the MTT assay was much less than variation in the total biomass detected in the crystal violet assay. All strains had some degree of antibiotic resistance; 14 of the strains were resistant to at least 2 of the anti-pseudomonal antibiotic classes tested, 11 strains were MDR, and 1 strain was XDR.18 The strains were also variable for twitching and swarming phenotypes which are associated with biofilm formation. Strains BWT111 and DS38 formed large amounts of alginates when incubated stationary for 24 hours (data not shown). When biofilm production, antibiotic resistance, and motility were taken into consideration, every strain had a distinct phenotype.
Figure 1.
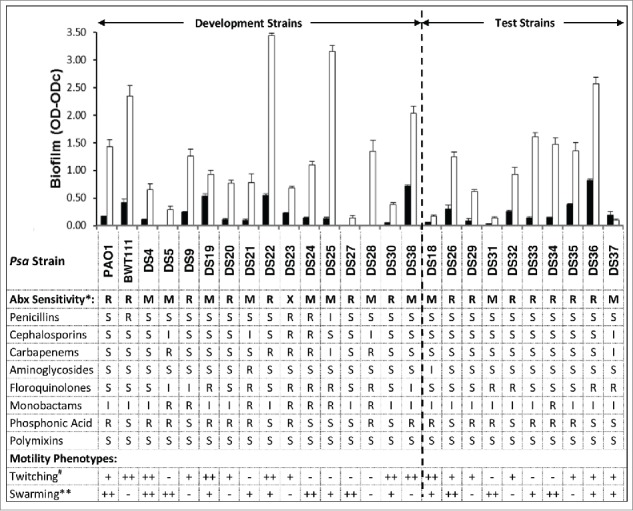
Phenotypes of the Pseudomonas aeruginosa strains used in this study. Top Panel: Twenty-4 hour biofilm formation was determined as described in materials and methods and the amount of biofilm was quantified by MTT assay (living cells, black bars) and CV assay (total biomass, white bars). Shown are means ± SEM for 3 separate assays. Middle Panel: Antibiotic (Abx) sensitivity (*) was determined for the 8 classes of anti-pseudomonal antibiotics 18 as described. Sensitivity was classified as: R = Resistant, non-sensitive to ≤2 antimicrobial agents ; M = MDR = non-sensitive to ≥ 1 agent in ≥ 3 classes; X = XDR = non-sensitive to ≥ 1 agent in all but ≤ 2 classes. CLSI cut-off values were used to classify strains as sensitive (S), intermediate (I), or resistant (R) to each antibiotic. Bottom Panel: Twitching and swarming phenotypes were determined. 19,20 Twitching activity (#) was defined as − = ≤ 25 mm2; + = ≥ 25 mm2 and ≤ 100 mm2; ++ = ≥ 100 mm2. Swarming activity (**) was defined as − = None; + = moderate, to within 2-4 cm of dish edge; and ++ = Strong, to within <1 cm of dish edge.
Host Range Expansion with a Mixture of Four P. aeruginosa-specific Phage
Prior to the HRE protocol, 6 of the 16 development strains (38%) were sensitive to the 1:1:1:1 mixture of parental Pseudomonas aeruginosa-specific phage (ϕKMV, ϕPA2, ϕPaer4, ϕE2005) (Fig. 2). By 5 cycles of the HRE protocol, the sensitivity of the development strains to the cocktail approached 75% and plateaued at that level in the subsequent cycles. In contrast, the sensitivity of the naïve test strains to the parental phage mixture was initially 20% and increased more slowly, reaching 100% by cycle 25 (Fig. 2). The specific development and test strains sensitive to the expanded host range cocktail at 30 cycles of expansion are shown in Figure 3 (group B).
Figure 2.
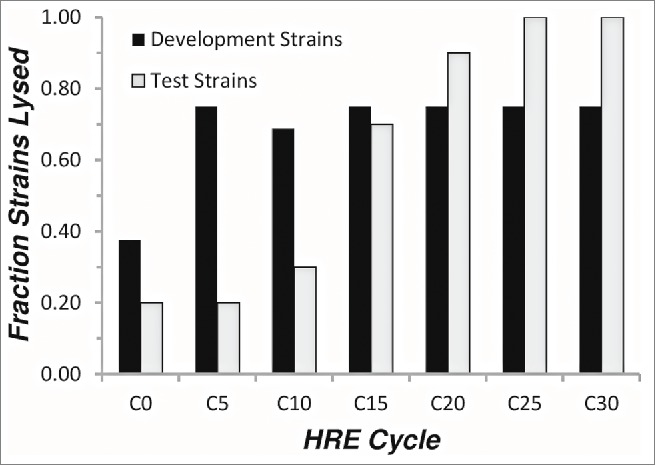
Host range expansion of a cocktail of 4 P. aeruginosa phages. Equal titers of 4 P.aeruginosa-specifc phage (ϕKMV, ϕPA2, ϕPaer4, ϕE2005) were mixed and subjected to the host-range expansion protocol as described in Materials and Methods. Every fifth cycle, spot tests were performed on lawns of each of the development and test strains to determine the sensitivity of each strain to the phage cocktail. The fraction of strains lysed is plotted versus the number of cycles of the host-range expansion protocol.
Figure 3.

Host Range of Parental Phage, HRE Mixtures, Isolated Clones, and Reconstituted Cocktails.
Isolation of phage clones with expanded host range from undefined mixtures
Isolation of plaques from cycle 30 of the HRE was performed using different strains as host. Prior to picking plaques, due to the small volume of the 4ϕC30 mixture, it was passaged on strain DS38 a high biofilm former. Passage on DS38 resulted in the loss of phage lysis on 4 strains (DS18, 21, 29, and 36) that were lysed by the undefined 4ϕC30 mixture (4ϕC30 mixture to 4ϕC30-DS38-P; Fig. 3). 108 plaques were picked from limited? dilutions of 4ϕC30-DS38-P plated on DS38 and plate stocks were made (clones). The clones were tested for host range by spotting a 10−2 dilution on the development and test strains. Overall, the 108 clones analyzed could be divided into 30 different host range spectra (Fig. 4, group D). Forty four of the clones shared a single host range spectrum that lysed only 2 strains not lysed by the parental phages. Clones with other host range spectra were found in the following numbers: 17 spectra (spectrum nos. 14-30), with each clone lysing a single spectrum; 7 spectra (7-13), each represented by 2 clones; 2 spectra (5-6), each represented by 3 clones; 1 spectrum (4), represented by 4 clones; 1 spectrum (3), represented by 10 clones, and 1 spectrum (2) represented by 13 clones. Similar results were obtained when clones were isolated from the 4ϕC30 mixture propagated on strain DS22. The 76 clones represented 23 different host range spectra and a single spectrum dominated the pool with 26 representatives (data not shown). Overall the clones isolated on DS22 showed narrower spectra when compared to the clones isolated on DS38, indicating that the host from which the clones are isolated affects the spectra of clones isolated. Defined cocktails of reconstituted mixtures of isolated clones could be made that recapitulated the host range of the original cocktail passaged in the isolation strain (DS38) (Fig. 3, group E). No clones were isolated on either strain DS22 or DS38 that lysed strains DS23 or DS24. It is possible that phage clones lysing DS23 and/or DS24 could be isolated by examining more clones or performing additional cycles of the HRE protocol. Alternatively, strains DS23 and DS24 may lack the receptors used by the phage or have an internal resistance mechanism.
Figure 4.

Stability of expanded host range with passage.
Stability of the expanded host range phenotype
Three individual phage plaques were picked from the HRE at cycles 2, 5, and 10 and passaged plate stock to plate stock 10 times. The P0, P5, and P10 stocks from each original plaques were then tested for host range on the development and test strains (Fig. 4). The host range of the clones changed when they were passaged on a single strain. Surprisingly, of the 16 changes in host range noted in the 3 passaged clones, only 2 were loss of host range changes and 14 were gain of host range changes. The gain of host range was unexpected because the phages were passaged on a single P. aeruginosa strain, which removed the selection for activity on the other strains.
Expanded host range phage cocktails reduce the formation of P. aeruginosa biofilms in vitro
Three clinical isolates (DS22, DS36 and DS38) that formed high amounts of biofilm (Fig. 1), none of which were lysed by any of the parental phage, were chosen for examination in reduction of biofilm formation assays using the unfractionated 4ϕC30 mixture. With 2 of the 3 strains (DS22 and DS36), phage significantly reduced biofilm formation in a dose-dependent manner in 24 hour assays (Fig. 5). In the other strain (DS38) statistically significant reduction was not observed but there was a trend toward reduction. In all 3 strains the phage titer at 24 hours was higher than the phage titer at inoculation. In all cases, planktonic cells were easily visible as haziness in the wells at 24 hours, suggesting ongoing multiplication of non-susceptible planktonic cells; the non-susceptible cells have had intrinsic resistance or have developed reduced phage susceptibility, such as growth phase changes, or mutation of the receptor for the phage.
Figure 5.
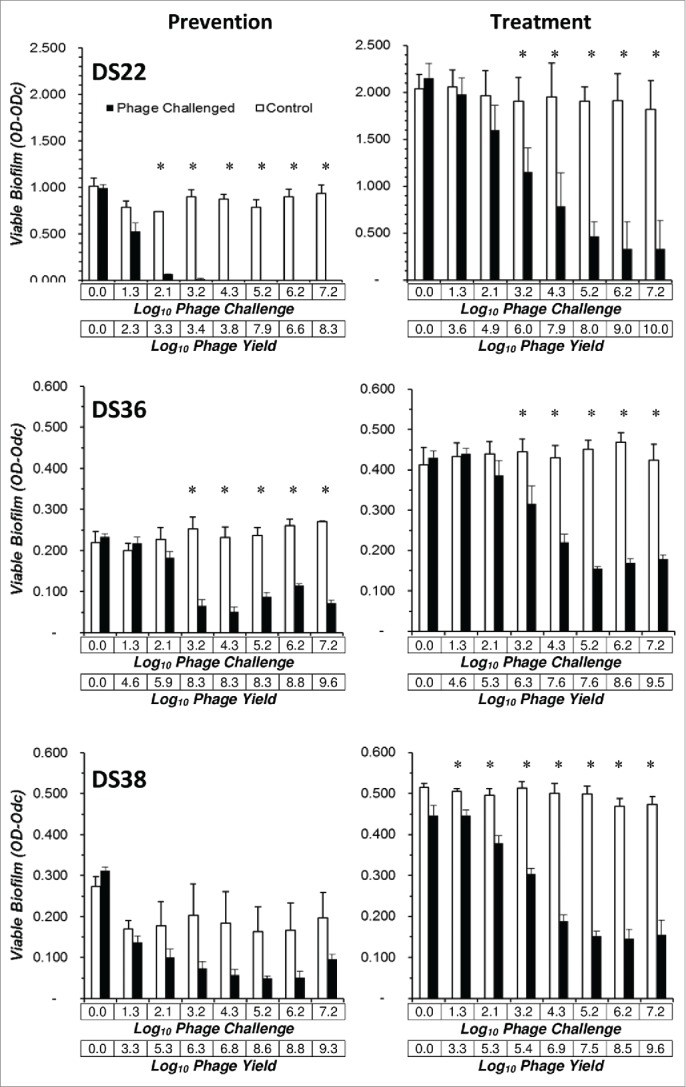
A 4ϕC30 mixture is active in prevention and treatment of Pseudomonas aeruginosa biofilms. Prevention of biofilm formation and treatment of pre-existing biofilm assays were performed using P. aeruginosa clinical isolates DS22 and DS36 and DS38, the strains forming the greatest amount of biofilm as detected by MTT assay (see Fig. 1). Phage challenge doses are shown on the upper X-axis and the phage yield is shown on the lower X-axis. Biofilm prevention (left column): 24 hour biofilm formation assays were performed in the presence of serial 10-fold dilutions of the 4ϕC30 mixture, washed, and biofilm determined with the MTT assay Treatment of pre-existing biofilm assays (right column): biofilms were formed for 24 hours, washed, and challenged with serial 10-fold dilutions of the 4ϕC30 mixture. After 6 hours in the presence of phage, the wells were washed and biofilm determined by MTT assay. All data are the mean SEM of 3 independent determinations. Student's t-test, (P =<0.05) was used to compare the control and phage treated OD-ODc values for each phage dose.
In another set of assays, strains with lower biofilm-forming abilities (DS19, DS20, and BWT111) were examined in biofilm formation reduction assays using a mixture from an earlier cycle of the HRE (4ϕC20). Strain DS19 was lysed by none of the parental phage, DS20 was lysed by one (ϕPaer4) and BWT111 was lysed by 2 (ϕPaer4 and ϕPA2). Strains DS19 and DS20 exhibited a statistically significant phage dose-dependent reduction in biofilm formation. No reduction in biofilm formation was observed for strain BWT111 (data not shown).
Expanded host range phage cocktails have activity against pre-existing biofilms
The three strains (DS22, DS36 and DS38) were also tested in reduction of pre-existing biofilm assays using the 4ϕC30 mixture. Statistically significant reduction of pre-existing biofilms of all 3 strains was observed in phage dose-dependent fashion (Fig. 5). Reduction of pre-existing biofilm was accompanied by an increase in phage titer. In no case was the biofilm totally eradicated.
In other experiments strains DS19, DS20 and BWT111 were also tested in reduction of pre-existing biofilm (treatment) assays using the earlier cycle 4ϕC20 mixture. Statistically significant dose-dependent reductions in pre-existing biofilm were noted for all 3 strains (data not shown).
Efficacy of a defined phage cocktail relative to antibiotic in biofilm prevention and treatment
Three high biofilm forming P. aeruginosa strains (DS22, DS36, and DS38) were used to compare the efficacy of a defined phage cocktail to that of antibiotic (levofloxacin) in biofilm prevention and treatment assays. A defined phage cocktail was reconstituted by mixing equal volumes of expanded host range clones derived from cycle 30 of the 4ϕC30 HRE. The cocktail contained spectrum 8 (clone 8), spectrum 23 (clone 67), and spectrum 30 (clone 99) (see Fig. 3, group E). With respect to levofloxacin, the P. aeruginosa strains used varied in their susceptibility to levofloxacin, being susceptible (strain DS22), intermediate (strain DS38) or resistant (strain DS36). As shown in Fig. 6, the levofloxacin-sensitive strain, DS22, had a similar response to phage cocktail or levofloxacin being significantly reduced by both agents. The effect of the phage cocktail on the highly resistant strain, DS38, was significant, while levofloxacin failed to prevent or treat biofilm formed by this strain. The strain showing intermediate levofloxacin susceptibility, DS36, was not susceptible to any of the phage clones in the cocktail used and was unaffected by phage or antibiotic treatment in either assay, although substantial phage replication occurred.
Figure 6.
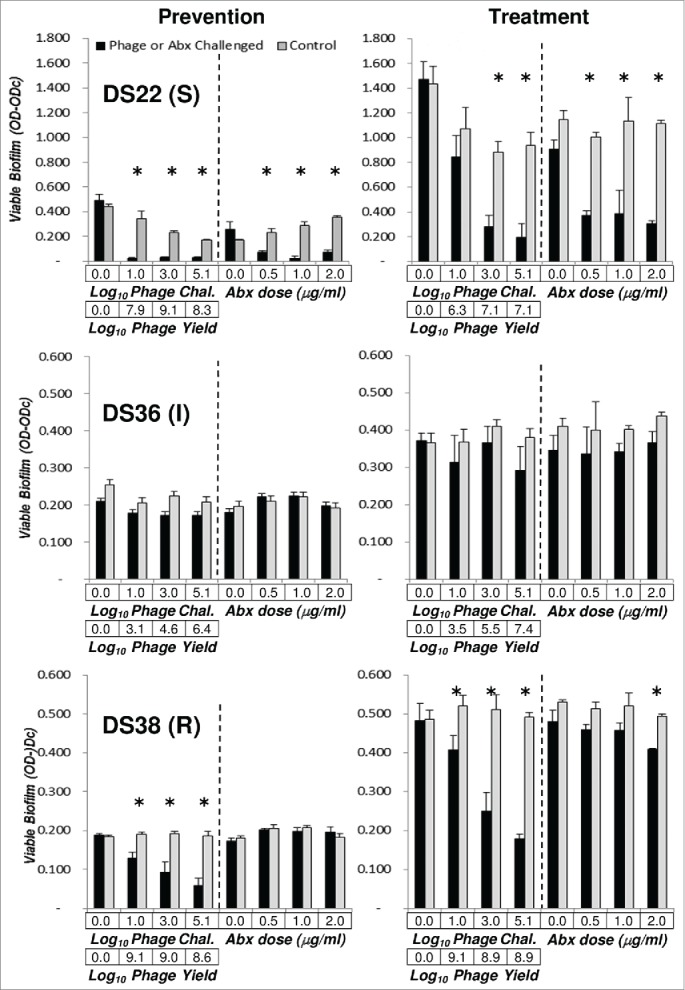
Comparison of a defined cocktail and antibiotic in prevention and treatment of Pseudomonas aeruginosa biofilms. Prevention of biofilm formation and treatment of pre-existing biofilm assays were performed as before, using clinical isolates DS22, DS36 and DS38, the strains forming the greatest amount of biofilm as detected by MTT assay (see Fig. 1). The defined cocktail used contained the following phages (Fig. 3): clone 88 (spectrum 8), clone 67 (spectrum 23) and clone 99 (spectrum 30). The prevention and treatment panels are as in Figure 4, except that 4 doses of phage cocktail are compared to 4 doses of levofloxacin (0.0, 0.5, 1.0, and 2.0 μg/ml). All data are the mean ± SEM of 3 independent determinations. Student's t-test, ( = P < 0.05) was used to compare the control and phage treated OD-ODc values for each phage and antibiotic dose.
Discussion
The results presented here indicate that (i) the host range of a mixture of phages can be expanded by the host range expansion protocol (HRE) described. (ii) Individual phage clones isolated from the HRE-generated mixtures have expanded host range and (iii) those expanded host ranges generally are stable or continue to increase upon passage in a single P. aeruginosa strain. (iv) Defined cocktails can be reconstituted from multiple individual expanded host range clones and have predictable host range. (v) Expanded host range mixtures and defined cocktails are active in reduction of biofilm formation or reduction of pre-existing biofilm assays against P. aeruginosa strains sensitive to the mixture or defined cocktail. (vi) Defined, expanded host range cocktails are more effective than antibiotic when the MIC of the target strain is high. (vii) Phage replication occurred in all cases where significant reductions in biofilm formation or reduction of pre-existing biofilms occurred, indicating that phage kill the target cells. In the case of the DS38 biofilm prevention assay (Fig. 5), substantial phage replication occurred, and a. We note non-significant trend toward biofilm reduction was observed. These results indicate that the HRE protocol described here can be used to generate a bank of expanded host range phage clones active against the P. aeruginosa strains present in a given clinical facility
The HRE protocol described here utilizes multiple P. aeruginosa strains for development of the expanded host range phage. Mixing the output phage from all the bacterial strains, tests all possible host range expansion events on each of the strains at subsequent cycles of the HRE. At each cycle the parental bacterial strains are exposed to the phage mixture, so that phages are tested against an unchanging baseline of development strains. The mixing of output phage resulted in rapid expansion of host range on the development strains; maximal levels of expansion were attained within 5 cycles (Fig. 2). The expansion of host range to the test strains occurred more slowly, requiring 25 cycles for maximal expansion. The slower expansion on the test strains most likely results from lack of selection, so that spontaneous, unselected changes extending the host range to the test strains accumulate more slowly. We do not currently understand the mechanism by which the changes resulting in host range expansion occur. The use of a mixture of 4 different phages in the HRE suggests that the expansion of host range could result from mutation, recombination, or both.31,32 This hypothesis is currently under test by sequencing parental and expanded host range phages.
Individual phage clones with expanded host range could be isolated from the output mixture of the HRE protocol. It is important to note that no individual clone completely recapitulated the host range spectrum of the phage mixture from which it was isolated (Fig. 3). This indicates that minor components of the mixture contribute significantly to its host range. When >100 clones were generated from a mixture, they represented 30 different spectra of host range on the development and test strains (Fig. 3, group D). One host range spectrum dominated among the clones, and it was expanded by only 2 strains when compared to the parental 4-phage mixture. Numerous clones were isolated that had a host range spectrum of 14-16 strains compared to 7 strains for the parental phage mixture. Two or 3 clones with unique spectra could be mixed to reconstitute a cocktail that recapitulated the host range spectrum of the phage mixture from which the clones were isolated; 19 of 26 strains were lysed by the defined, reconstituted cocktail (Fig. 3, group D). The spectra of clones isolated depended on the P. aeruginosa isolation strain (Fig. 3 and data not shown). Expanded host range phage mixtures or clones could not be isolated that targeted 2 of the P. aeruginosa strains (DS23, DS25; Fig. 3, groups B–E). It is possible that phage active on these strains could be isolated if more clones were screened, a different isolation strain was used, or if more cycles of the HRE were performed. However, it also possible that these strains are insensitive to the phages used because they lack the appropriate receptor or, alternatively, they possess an intracellular mechanism that renders them insensitive.33-35 Surprisingly, serial passage of individual clones on the laboratory P. aeruginosa strain PAO1 tended to maintain the expanded host range of the clones; of 16 host range changes observed on passage, 2 represented loss of host range and 14 were gain of host range (Fig. 4). This observation suggests that the expanded host range is stable enough to allow generation of large amounts of a phage clone.
Undefined phage mixtures directly from the HRE protocol (Fig. 5), or cocktails reconstituted from multiple individual clones (Fig. 6), had significant activity in reduction of biofilm formation and reduction of pre-existing biofilm assays. The assays utilized the 3 highest biofilm forming strains from our collection (DS22. DS36, DS38; Figs. 5 and 6) none of which were sensitive to the parental phages, and 3 moderate biofilm forming strains (DS19, DS20, BWT111; data not shown). A dose-response was observed for the phage mixtures or cocktails, with statistically significant reduction occurring at higher phage inocula. In no case was reduction of biofilm formation or reduction of pre-existing biofilms complete. However the significant reductions observed may be great enough that the host immune system could complete clearance of the bacteria. The undefined phage mixtures were not active against strains DS38 and BWT111 in the reduction of biofilm formation assay (Fig. 5 and data not shown). These two strains produce the greatest amounts of alginate of any strains in our collection and we speculate that the accumulation of alginate over the 24 hour course of the assay prevented activity. In contrast in the reduction of pre-existing biofilm assays, the phage mixture was active against both DS38 and BWT111, under conditions where alginates were washed away immediately before phage infection. Strain DS36 (Fig. 6) is not sensitive to the defined cocktail used so the lack of activity is not surprising. However, the results did indicate that active phage were necessary for reduction of biofilm formation or reduction of pre-existing biofilm. Finally, P. aeruginosa non-susceptible to phage were isolated from the reduction of biofilm formation and reduction of pre-existing biofilm assays, suggesting that phage therapy may be of limited utility in protecting against biofilm resurgence. However, non-susceptibility may carry a fitness cost36 or reduce virulence of the non-susceptible bacteria.37”
Comparison of defined cocktails and antibiotic in reduction of biofilm formation and reduction of pre-existing biofilm assays (Fig. 6) revealed that the cocktail was significantly active against a highly levofloxacin-resistant strain (DS38). In strain DS38 (MIC≥8μg/mL) the antibiotic was significantly active in treatment of pre-existing biofilm at 2μg/mL. The reason for this is unknown, but we note that MICs as determined by the micro-titer dilution method are notoriously difficult to align with biofilm MICs.32 No difference was noted between cocktail and antibiotic for the levofloxacin-sensitive strain DS22 (MIC=1μg/mL). This result suggests that cocktails reconstituted from individual clones will be effective against MDR P. aeruginosa strains, or strains highly resistant to single antibiotics. As noted above, the intermediate MIC strain DS36 (MIC=4μg/mL) was not sensitive to the cocktail used nor to levofloxacin. Although DS36 is not sensitive to any of the component phage of the cocktail used, the phage replicated. The basis of this replication is not understood but we speculate that replication occurred on planktonic Pseudomonas cells that may have gained sensitivity to the cocktail.
The goal of these studies is to generate broad host range phage that can be used to prevent formation of biofilms or reduce pre-existing biofilms in P. aeruginosa biofilm infections. Numerous studies examined the activity of phage on planktonic P. aeruginosa but relatively few papers have focused on P. aeruginosa biofilms in vitro or in vivo infections associated with biofilms.25,39,40 In the future we plan to utilize defined cocktails generated as demonstrated here for the treatment of catheter-associated urinary tract infections (CAUTI), where in vivo attempts at phage therapy are nonexistent. Phage therapy for P. aeruginosa has been studied in a number of infection models such as sepsis,41 ocular keratitis,42 cystic fibrosis and other lung infections,43,44 and burn wound infections23,45 with some success. In addition, a clinical trial to treat chronic otitis caused by MDR P. aeruginosa with phage therapy was successfully concluded. 46 These results suggest that similar approaches may be applicable to CAUTI.
We propose that libraries of expanded host range phage can be developed against the contemporary strains of P. aeruginosa or other MDR pathogens found in a given clinical setting. Such cocktails used in combination with traditional antimicrobial agents might reduce the load of pathogens to a level where the host immune response could clear the infection, while the anti-biofilm properties of the phage might prevent resurgence of the infection after cessation of traditional antimicrobial agents. The HRE protocol represents an advance for overcoming the limitation of phage's highly specific host range.
Materials and methods
Bacterial strains and phage
Pseudomonas aeruginosa clinical isolates (DS series) were obtained from the clinical microbiology laboratory at the Veterans Affairs Medical Center, Houston, TX. We designated 14 isolates, all resistant to at least one antibiotic class including 8 MDR, and 1 extremely drug resistant (XDR),18 from our collection as “development strains” to be used for generation of phage mixtures with expanded host range (Fig. 1). Additionally, 2 laboratory strains (PAO1 (BWT121) and BWT111) were used as development strains known to support growth of the 4 parental phage.15 Ten additional clinical isolates (3 MDR) from our collection were used as naive “test strains” to test the host range of phage on strains that were not used for host range expansion. Bacterial stocks were stored in Luria Broth with 15% glycerol at −80°C. Overnight bacterial cultures were inoculated from glycerol stocks and propagated in Luria Broth at 225 rpm, 37°C.
All P. aeruginosa strains used were characterized for the ability to form biofilms in vitro, antibiotic resistance, twitching motility and swarming motility (Fig. 1). Twenty-four hour biofilms were formed by diluting an overnight culture 1:100 in tryptic soy broth (TSB), inoculating the wells of a 96-well plate with 100 μl, and incubating at 37°C as static cultures. Twenty-four hours later the wells were washed 3 times with PBS and the biofilm content of each well determined using the MTT assay (below) or the standard crystal violet staining assay. Antibiotic resistance (Clinical and Laboratory Standards Institute standards) was determined using VITEK2 (bioMerieux) for Penicillins (ampicillin, ampicillin/sulbactam, piperacillin/tazobactam), Cephalosporins (cefazolin, ceftazidime, ceftriaxone, cefepime), Carbapenems (imipenem), Aminoglycosides (amikacin, gentamicin, tobramycin) and Fluoroquinolones (ciprofloxacin, levofloxacin). Etest strips (bioMerieux) were used to determine resistance to Monobactams (aztreonam), Phosphonic Acids (fosfomycin) and Polymixins (colistin). Resistance phenotypes were classified according to Magiorakos et al.18 Twitching motility was determined by stabbing colonies into 1% agar as described by Kazmierczak et al19 and swarming motility by spot testing liquid culture onto 0.5% agar swarming plates as described by Tremblay.20
Four phages were used in this work. ϕKMV, is fully sequenced21 and utilizes Type IV pili as its host receptor.22 ϕPA2 (ATCC 14203-B1) has not been sequenced, was previously tested in a mouse burn model of phage therapy23 and is genetically similar to ϕLIT1 which uses Type IV pili as the host receptor,24 suggesting that ϕPA2 may also use Type IV pili. ϕPaer425 was obtained from R. Donlan (CDC), and has not been sequenced or characterized for host receptor usage. ϕE2005-24-39 (hereafter called ϕE2005), obtained from R. Donlan (CDC), has not been sequenced or characterized for receptor usage but was used in prior biofilm eradication studies on urinary catheters.15 All 4 phages are members of Podoviridae and were chosen for their expected small genomes and robust physical structure.
The host range expansion (HRE) protocol
The HRE protocol is based on the work of Burrowes. 12 To initiate the HRE, serial dilutions of a mixture of parental phage were incubated with each of the bacterial development strains in a 96-well format. Progeny phage from wells at the lowest phage concentration in which lysis occurred were pooled, and this mixture of progeny phage was used as the inoculum for the next cycle of HRE (Fig. 7). The phage inoculum for the first cycle of the HRE was a 1:1:1:1 mixture of the 4 parental phages (ϕKMV, ϕPA2, ϕPaer4, and ϕE2005). The phage inoculum (100 μL) was added to the first well of each row of the culture plate then serially diluted 1:10 across 10 columns, leaving columns 11 and 12 as bacterial and media controls, respectively. Bacterial inocula for each cycle of the HRE were the 16 P. aeruginosa development strains (Fig. 1). Overnight cultures were normalized to an OD600=0.2-0.3 and then diluted 1:100 in LB broth. Then, wells 1-11 of each row were inoculated with 90 μL bacterial inoculum, one development strain per row. The plate was incubated overnight at 225 rpm, 37°C. After incubation, the wells were inspected for lysis. The highest phage dilution well showing complete lysis and the next dilution at which lysis was incomplete were pooled (from all bacterial strains; boxes, Fig. 7). If no lysis was seen for a specific strain, the well with undiluted phage and 10−1dilution was added to the pool. The pooled lysate was treated with 10 μL chloroform, vortexed, aliquoted into microcentrifuge tubes, and centrifuged at 14,000 rpm for 2 minutes. The aqueous phase was passed through a 0.22 μm filter and stored at 4°C until it was used as inoculum for the next cycle of the HRE. Thirty cycles of the HRE were performed. The host range of the pooled cocktail was tested every 5 cycles by spot testing 26 on lawns of the development strains and the test strains. Spot tests were performed with several dilutions of each phage mixture to avoid complications from lysis from without on the various strains.27,28
Figure 7.
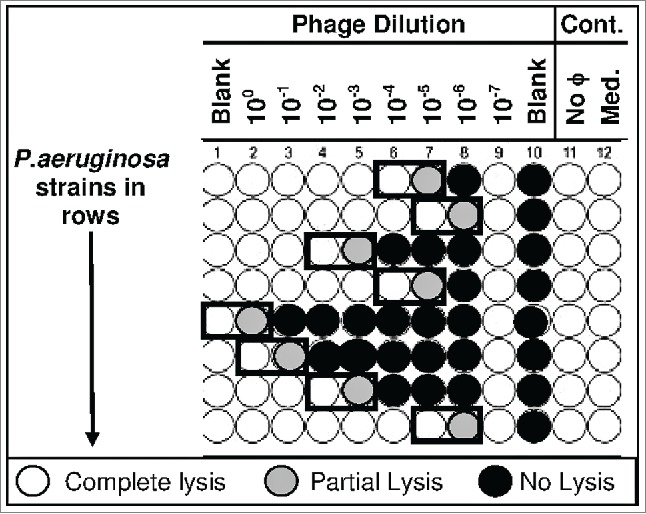
Host Range Expansion () Protocol. A 96-well plate is inoculated with the appropriate P. aeruginosa strains (one strain per row) and dilutions of single phages or phage cocktails (columns). The plate is incubated at 37°C for 18 hours. The contents of the well with the highest phage dilution showing complete lysis and the next well showing partial or no lysis (black boxes) are pooled across the plate. In this way the phage from the highest phage dilution showing complete lysis and the next well showing partial or no lysis are pooled for all the strains into a single tube. The contents of that tube constitute the mixture of phages used to inoculate the next plate (cycle) with the same phage dilutions and P. aeruginosa strains. This process is reiterated for the desired number of cycles. At various cycle numbers the host-range of the phage mixture is determined by spotting on lawns of a number of P. aeruginosa strains (development strains and test strains).
Isolation of phage clones from undefined mixtures and preparation of stocks
Phage mixtures obtained directly from the HRE protocol were considered undefined because they contained a mixture of phage with many unique host ranges. To enable the creation of defined cocktails, we isolated clones and tested their host ranges. We use “defined” to indicate that the cocktail is composed of individual, isolated clones that we have characterized for host range. To isolate clones, we first expanded the undefined cocktails by making plate stocks on each of 2 P. aeruginosa clinical isolates (DS22 and DS38). Plate stocks were generated by plating 100 μL bacterial host with 100 μL phage mixture in 1% top agar and incubating overnight at 37°C. Phage was collected from plates with confluent or near-confluent lysis by scraping the top agar into 4.0 mL LB broth, pelleting the agar, and collecting the supernatant as the stock. The stock was treated with chloroform to lyse any remaining cells. The lysate was then terminally diluted on the same bacterial strain. One hundred well-isolated plaques were picked into 1.0 mL phage storage buffer (100 mM NaCl, 6.7 mM Tris-HCl, 3.2 mM Tris-Base, 10 mM MgSO4•7H2O). Each picked plaque represented a clone derived from a single phage in the original undefined mixture. One half mL of each plaque solution was used to generate plate stocks of the plaque-isolated clone on the same strain from which it was originally isolated. Serial dilutions of the plate stocks were used to test host range of the individual phage clones by spot tests on the development and test strains.
Stability of the host range phenotype
Clones from the HRE cycle and clone number indicated were subjected to limit dilution and plated on strain PAO1. An individual plaque was picked into 1.0 mL phage storage buffer and 0.5 mL of the picked plaque suspension was plated on PAO1 and a plate stock was generated. This process was repeated 10 times for each original plaque. The host ranges of the resulting stocks were tested by spot test on the development and test strains at passages 1, 5 and 10.
Biofilm prevention assays
These assays were performed with the undefined 4 phage cycle 30 mixture (4ϕC30) and P. aeruginosa clinical isolates DS22, DS36, and DS38, the highest biofilm formers among our strains as determined by the MTT assay (Fig. 1). The biofilm prevention assay was performed in 96-well flat-bottomed polystyrene microtiter plates (Costar 3585). Plate columns 2-8 were inoculated with 100 μl of the desired P. aeruginosa strain (1:100 dilution of overnight culture in TSB); plate columns 1 and 10 were left blank, and plate columns 11 and 12 received 100 μl sterile TSB. Immediately after placing the bacterial host cells in the wells, rows A-D of columns 2-8 were inoculated with 10 μl of serial dilutions of the 4ϕC30 mixture (ranging from 10−1 in column 2 to 10−7 in column 8). Rows E-H of columns 2-8 were not inoculated and served as controls for biofilm formation. The plates were incubated for 24 hours at 37°C as stationary cultures. Twenty-four hours after inoculation, the media from the phage infected wells were pooled (column by column) for determination of phage titer by spot test. 24 The remaining media was flicked from the wells, and the wells were washed 3 times with 150 μL PBS and then refilled with 100 μL of PBS. To each well 10 μl of a 5 mg/ml solution of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT; Sigma) was added and the plate was incubated 4 hours at 37°C. In this assay, only metabolically active cells able to reduce MTT to the formazan product are detected, unlike crystal violet staining, which detects total biomass. 29,30 The MTT assay was used because we were interested in determining the metabolically active cells that survived phage treatment and were capable of resurgence from biofilm. After incubation with MTT, 100 μl of extraction solution (20% SDS, 50% N,N,-Dimethylformamide) was added to each well to solubilize the formazan, and the plates were incubated overnight at 37°C. The plates were read at 560 nm in an automated plate reader. The wells from columns 11 and 12 which originally received only sterile TSB served as the negative control. The cutoff of the assay (ODc) was calculated as the mean optical density (OD) for the negative control wells plus 3x the standard deviation of the mean of the negative control wells. The OD-ODc (optical density minus cutoff) values of the wells from the columns receiving phage were averaged. These values represented the metabolically active biofilm formed in the presence of phage. The OD-ODc of the wells from the columns not receiving phage represented biofilm formation in the absence of phage. Assays were replicated 3 times for each P. aeruginosa strain tested. The OD-ODc values for the 3 plates were averaged, and the data presented as the mean ± the standard error of the mean. Student's t-test was performed to compare biofilm optical density for wells treated with phage and untreated wells for each phage dilution.
Treatment of pre-existing biofilm assays
The treatment of pre-existing biofilm assays were performed as for the biofilm prevention assays with the following changes. (i) Following inoculation of cells into columns 2-8 and sterile TSB into columns 11 and 12, the plates were incubated for 24 hours at 37°C as stationary cultures to allow biofilm formation. (ii) Twenty-4 hours after plate inoculation, the media was flicked from the wells, the wells were washed once with PBS, and re-filled with 100 μL TSB. At this time rows A-D of columns 2-8 of the plate were inoculated with 10 μL of a 10-fold dilution series of phage cocktail as above. The plates were incubated at 37°C for 6 hours to allow phage to act on the pre-formed biofilm. In some cases, removal of the medium present during biofilm formation also removed alginates formed by certain P. aeruginosa strains. Thus, the biofilm treatment assays were initiated in the absence of alginates. The experiments were repeated 3 times each with P. aeruginosa strains DS22, DS36, and DS38, and the data were analyzed in identical fashion to that described for the biofilm reduction assay.
Comparison of phage and antibiotic therapies
Assays to compare the efficacy of phage therapy and antibiotic therapy on biofilm formation (prevention) and on pre-existing biofilms (treatment) were performed as described above except that only 4 doses of phage (defined cocktail consisting of 3 characterized phage clones) were used so that the other half of the plate could be used for 4 doses of antibiotic. The experiments were repeated 3 times each with P. aeruginosa strains DS22, DS36, and DS38, and the data were analyzed in identical fashion to that described for the biofilm reduction assay. The antibiotic used was levofloxacin. With respect to levofloxacin, the P. aeruginosa strains were susceptible (strain DS22, MIC = 1 μg/ml), intermediate (strain DS38, MIC = 4 μg/ml) and resistant (strain DS36, MIC ≥ 8 μg/ml) as determined by VITEK.
Abbreviations
- CAUTI
Catheter-Associated Urinary Tract Infection
- EOP
Efficiency of Plating
- HRE
Host Range Expansion
- LB
Luria-Bertani Broth
- MDR
Multi-Drug Resistant
- MIC
Minimum Inhibitory Concentration
- MTT
3-(4,5,dimethylthiazole-2-yl)-2,5-diphenyl tetrazolium bromide
- OD
Optical Density
- ODc
Control Optical Density
- PBS
Phosphate Buffered Saline Phage
- PSB
Phage Storage Buffer
- TSB
Trypticase Soy Broth
- VITEK
Machine for microbial identification and antibiotic susceptibility testing
- XDR
Extremely Drug Resistant
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by grant DK092293 from the National Institutes of Health. Dr. Trautner's work is also supported by resources and use of facilities at the Houston VA Center for Innovations in Quality, Effectiveness and Safety [CIN13-413] at the Michael E. DeBakey VA Medical Center, Houston, TX.
References
- [1].Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, Scheld M, Spellberg B, Bartlett J. Bad Bugs, No Drugs: No ESKAPE! An Update from the Infectious Diseases Society of America. Clin Infect Dis, 2009; 48(1):1–12; http://dx.doi.org/ 10.1086/595011 [DOI] [PubMed] [Google Scholar]
- [2].Infectious Diseases Society of America . Combating antimicrobial resistance: Policy recommendations to save lives. Clin Infect Dis 2011; 52(Suppl 5):S397–S428; PMID:21474585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].CDC Antibiotic Resistance Threats in the United States, 2013. http://www.cdc.gov/drugresistance/threat-report-2013/pdf/ar-threats-2013-508.pdf [Google Scholar]
- [4].D'Herelle F. On an invisible microbe antagonistic to dysentery bacilli. Compt Rend Acad Sci 1917; 165:373–5 [Google Scholar]
- [5].Abedon ST, Kuhl SJ, Blasdel BG, Kutter EM. Phage treatment of human infections. Bacteriophage 2011; 1:66–85; PMID:22334863; http://dx.doi.org/ 10.4161/bact.1.2.15845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Smith HW, Huggins MB, Shaw KM. The control of experimental Escherichia coli diarrhoea in calves by means of bacteriophages. J Gen Microbiol 1987; 133:1111–26; PMID:3309177 [DOI] [PubMed] [Google Scholar]
- [7].Smith HW, Huggins MB. Successful treatment of experimental Escherichia coli infections in mice using phage: Its general superiority over antibiotics. J Gen Microbiol 1982; 128:307–18; PMID:7042903 [DOI] [PubMed] [Google Scholar]
- [8].Santos SB, Fernandes E, Carvalho CM. Selection and characterization of a multivalent Salmonella phage and its production in a nonpathogenic Escherichia coli strain. Appl Environ Microbiol 2010; 76:7338–42; PMID:20817806; http://dx.doi.org/ 10.1128/AEM.00922-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Knezevic P, Kostanjsek R, Obreht D, Petrovic O. Isolation of Pseudomonas aeruginosa specific phages with broad activity spectra. Curr Microbiol 2009; 59:173–80; PMID:19472004; http://dx.doi.org/ 10.1007/s00284-009-9417-8 [DOI] [PubMed] [Google Scholar]
- [10].Lu TK, Collins JJ. Dispersing biofilms with engineered enzymatic bacteriophage. Proc Natl Acad Sci USA 2007; 104:11197–202;PMID:17592147; http://dx.doi.org/ 10.1073/pnas.0704624104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Lu TK, Koeris MS. The next generation of bacteriophage therapy. Curr Opin Microbiol 2011; 14:524–31; PMID:21868281; http://dx.doi.org/ 10.1016/j.mib.2011.07.028 [DOI] [PubMed] [Google Scholar]
- [12].Burrowes B. 2011. Analysis of the Appelman protocol for the generation of therapeutic bacteriophages. Ph.D. thesis. Texas Tech Univeristy Health Sciences Center, Lubbock, TX. [Google Scholar]
- [13].Khawaldeh A, Morales S, Dillon B, Alavidze A, Ginn AN, Thomas L, Chapman SJ, Dublanchet A, Smithyman A, Iredell JR. Bacteriophage therapy for refractory Pseudomonas aeruginosa urinary tract infection. J Med Microbiol 2011; 60:1697–1700; PMID:21737541; http://dx.doi.org/ 10.1099/jmm.0.029744-0 [DOI] [PubMed] [Google Scholar]
- [14].Phee A, Bondy-Denomy J, Kishen A, Basrani B, Azarpazhooh A, Maxwell K. Efficacy of bacteriophage treatment on Pseudomonas aeruginosa biofilms. J Endond 2013; 39:364–9; http://dx.doi.org/ 10.1016/j.joen.2012.10.023 [DOI] [PubMed] [Google Scholar]
- [15].Liao KS, Lehman SM, Tweardy DJ, Donlan RM, Trautner BW. Bacteriophages are synergistic with bacterial interference for the prevention of Pseudomonas aeruginosa biofilm formation on urinary catheters. J Appl Microbiol 2012; 113:1530–9; PMID:22985454; http://dx.doi.org/ 10.1111/j.1365-2672.2012.05432.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Paterson S, Vogwill T, Buckling A, Benmayor R, Spiers AJ, Thomson NR, Quail M, Smith F, Walker D, Libberton B, Fenton A, Hall N, Brockhurst MA. Antagonistic coevolution accelerates molecular evolution. Nature 2010; 464:275–8; PMID:20182425; http://dx.doi.org/ 10.1038/nature08798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Poullain V, Gandon S, Brockhurst MA, Buckling A, Hochberg ME. The evolution of specificity in evolving and coevolving antagonistic interactions between a bacteria and its phage. Evolution 2008; 62:1–11; PMID:18005153 [DOI] [PubMed] [Google Scholar]
- [18].Magiorakos A-P, Srinivasan A, Carey RB, Carmeli Y, Falagas ME, Giske CG, Harbarth S, Hindler JF, Kahlmeter G, Olsson-Liljequist B, et al.. Multidrug-resistant, extensively drug-resistant and pan drug-resistant bacteria: an international expert proposal for interim stndard definitions for acquired resistance. Clin Microbiol Infect 2012; 18:268–81; PMID:21793988; http://dx.doi.org/ 10.1111/j.1469-0691.2011.03570.x [DOI] [PubMed] [Google Scholar]
- [19].Kazmierczak BI, Lebron MB, Murray TS. Analysis of FimX, a phosphodiesterase that governs twitching motility in Pseudomonas aeruginosa. Mol Microbiol 2006; 64:1026–43; http://dx.doi.org/ 10.1111/j.1365-2958.2006.05156.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Tremblay J, Deziel E. Improving the reproducibility of Pseudomonas aeruginosa swarming motility assays. J Basic Microbiol 2008; 48:509–15,; PMID:18785657; http://dx.doi.org/ 10.1002/jobm.200800030 [DOI] [PubMed] [Google Scholar]
- [21].Lavigne R, Burkal'tseva MV, Robben J, Sykilinda NN, Kurochkina LP, Grymonprez B, Jonckx B, Krylov VN, Mesyanzhinov VV, Vlockaert G. The genome of bacteriophage phiKMV, a T7-like virus infecting Pseudomonas aeruginosa. Virology 2003; 312:49–59; PMID:12890620; http://dx.doi.org/ 10.1016/S0042-6822(03)00123-5 [DOI] [PubMed] [Google Scholar]
- [22].Chibeu A, Ceyssens PJ, Hertveldt K, Volckaert G, Lavigne R. The adsorption of Pseudomonas aeruginosa bacteriophage phiKMV is dependent on expression regulation of type IV pili genes. FEMS Microbiol Lett 2009; 296(2):210–8; PMID:19459952; http://dx.doi.org/ 10.1111/j.1574-6968.2009.01640.x [DOI] [PubMed] [Google Scholar]
- [23].McVay CS, Velasquez M, Fralick JA. Phage therapy of Pseudomonas aeruginosa infection in a mouse burn wound model. Antimicrob Agents Ch 2007; 51:1934–8; http://dx.doi.org/ 10.1128/AAC.01028-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Ceyssens PJ, Noben JP, Ackermann HW, Verhaegen J, De Vos D, Pirnay JP, Merabishvili M, Vaneechoutte M, Chibeu A, Voldkaert G, Lavigne R. Survey of Pseudomonas aeruginosa and its phages: de novo peptide sequencing as a novel tool to assess the diversity of worldwide collected viruses. Environ Microbiol 2009; 11(5):1303–13; PMID:19207572; http://dx.doi.org/ 10.1111/j.1462-2920.2008.01862.x [DOI] [PubMed] [Google Scholar]
- [25].Fu W, Forster T, Mayer O, Curtin JJ, Lehman SM, Donlan RM. Bacteriophage cocktail for the prevention of biofilm formation by Pseudomonas aeruginosa on catheters in an in vitro model system. Antimicrob Agents Ch 2010; 54(1):397–404; http://dx.doi.org/ 10.1128/AAC.00669-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kutter E, Sulakvelidze A (eds). 2005. Bacteriophages: biology and applications. CRC Press, Boca Raton, FL. [Google Scholar]
- [27].Abedon ST. Lysis from without. Bacteriophage 2011; 1(2):46–9; PMID:21687534; http://dx.doi.org/ 10.4161/bact.1.1.13980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Mirzaei ML, Nilsson AS. Isolation of phages for phage therapy: a comparison of spot tests and efficiency of plating analyses for determination of host range and efficacy. PLoS ONE 2015; 10(3): e0118557; PMID:25761060; http://dx.doi.org/ 10.1371/journal.pone.0118557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wang H, Cheng H, Wang F, Wei D, Wang X. An improved 3-(4,5-dimethylthiazol-2-yl)-2,5,-diphenyl tetrazolium bromide (MTT) reduction assay for evaluating the viability of Escherichia coli cells. J Microbiol Meth 2010; 82:330–3; http://dx.doi.org/ 10.1016/j.mimet.2010.06.014 [DOI] [PubMed] [Google Scholar]
- [30].Mshana RN, Tadesse G, Abate G, Miorner H. Use of 3-(4,5-dimethylthiazol-2-yl)-2,5,-diphenyl tetrazolium bromide for rapid detection of rifampin-resistant Mycobacterium tuberculosis. J Clin Microbiol 1998; 36:1214–9; PMID:9574679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Lin T-Y, Lo Y-H, Tseng P-W, Chang S-F, Lin Y-T, Chen T-S. A T3 and T7 recombinant phage acquires efficient adsorption and a broader host range. PLoS ONE 2012; 7(2): e30954; PMID:22347414; http://dx.doi.org/ 10.1371/journal.pone.0030954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Le S, He X, Tan Y, Huang G, Zhang L, Lux R, Shi W, Hu F. Mapping the tail fiber as the receptor binding protein responsible for differential host specificity of Pseudomonas aeruginosa bacteriophages PaP1 and JG004. PLoS ONE 2013; 8(7):e68562; PMID:23874674; http://dx.doi.org/ 10.1371/journal.pone.0068562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Labrie SJ, Samson JE, Moineau S. Bacteriophage resistance mechanisms. Nat Rev Microbiol 2010; 8:317–27; PMID:20348932; http://dx.doi.org/ 10.1038/nrmicro2315 [DOI] [PubMed] [Google Scholar]
- [34].Samson JE, Magadan AH, Sabri M, Moineau S. Revenge of the phages: defeating bacterial defences. Nat Rev Microbiol 2013; 11:675–86; PMID:23979432; http://dx.doi.org/ 10.1038/nrmicro3096 [DOI] [PubMed] [Google Scholar]
- [35].Goldfarb T, Sberro H, Weinstock E, Cohen O, Doron S, Charpak-Amikam Y, Afik S, Ofir G, Sorek R. BREX is a novel phage resistance system widespread in microbial genomes. EMBO J 2015; 34:169–83; PMID:25452498; http://dx.doi.org/ 10.15252/embj.201489455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Mai-Prochnow A, Hui JG, Kjelleberg S, Rakonjac J, McDougald D, Rice SA. Big things in small packages: the genetics of filamentous phage and effects on fitness of their host. FEMS Microbiol Revs 2015; 39(4):465–87; PMID:25670735; http://dx.doi.org/ 10.1093/femsre/fuu007; Epub ahead of print [DOI] [PubMed] [Google Scholar]
- [37].Leon M, Bastias R. Virulence reduction in bacteriophage resistant bacteria. Front Microbiol 2015; 6:343; PMID:25954266; Published online 29 April 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Dosler S, Karaaslan E. Inhibition and destruction of Pseudomonas aeruginosa biofilms by antibiotics and antimicrobial peptides. Peptides 2014; 62:32–7; PMID:25285879; http://dx.doi.org/ 10.1016/j.peptides.2014.09.021 [DOI] [PubMed] [Google Scholar]
- [39].Ahiwale S, Tamboli N, Thorat K, Kulkarni R, Ackermann H, Kapadnis B. 2011. In vitro management of hospital Pseudomonas aeruginosa biofilm using indigenous T7-like lytic phage. Curr Microbiol 2011; 62:335–40; PMID:20711581; http://dx.doi.org/ 10.1007/s00284-010-9710-6 [DOI] [PubMed] [Google Scholar]
- [40].Kim S, Rahman M, Seol SY, Yoon SS, Kim J. Pseudomonas aeruginosa bacteriophage PA10 requires type IV pili for infection and shows broad bactericidal and biofilm removal activities. Appl Environ Microbiol 2012; 78:6380–5; PMID:22752161; http://dx.doi.org/ 10.1128/AEM.00648-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Watanabe R, Matsumoto T, Sano G, Ishii Y, Tateda K, Sumiyana Y, Ushiuama J, Sakurai S, Matsuzaki S, Imai S, Yamaguchi K. Efficancy of bacteriophage therapy against gut-derived sepsis caused by Pseudomonas aeruginosa in mice. Antimicrob Agents Ch 2007; 51:446–52; http://dx.doi.org/ 10.1128/AAC.00635-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Fukuda K, Ishida W, Uchiyama J, Rashel M, Kato S, Morita T, Muraoka A, Sumi T, Matsuzaki S, Daibata M, Fukushima A. Pseudomonas aeruginosa keratitis in mice: effects of typical bacteriophage KPP12 administration. PLoS ONE 2012; 7(10); e47742; PMID:23082205; http://dx.doi.org/ 10.1371/journal.pone.0047742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Cao Z, Zhang J, Niu YD, Cui N, Ma Y, Cap F, Jin L, Li Z, Xu Y. Isolation and characterization of a “phiKMV-like” bacteriophage and its therapeutic effect on mink hemorrhagic pneumonia. PLoS ONE 2015; 10(1):e0116571; PMID:25615639; http://dx.doi.org/ 10.1371/journal.pone.0116571 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [44].Debarbuieux L, Leduc D, Maura D, Morello E, Criscuolo A, Grossi O, Balloy V, Touqui L. Bacteriophages can treat and prevent Pseudomonas aeruginosa lung infections. J Infect Dis 2010; 201:1096–104; PMID:20196657; http://dx.doi.org/ 10.1086/651135 [DOI] [PubMed] [Google Scholar]
- [45].Rose T, Berbeken G, De Vos D, Merabishvili M, Vaneechoutte M, Lavigne R, Jennes S, Zizi M, Pirnay J-P. Experimental phage therapy of burn wound infection; difficult first steps. Int J Burn Trauma 2014; 4:66–73 [PMC free article] [PubMed] [Google Scholar]
- [46].Wright A, Hawkins CH, Anggard EE, Harper DR. A controlled clinical trial of a therapeutic bacteriophage preparation in chronic otitis due to antibiotic-resistant Pseudomonas aeruginosa: a preliminary report of efficacy. Clin Otolaryngol 2009; 34:349–57; PMID:19673983; http://dx.doi.org/ 10.1111/j.1749-4486.2009.01973.x [DOI] [PubMed] [Google Scholar]