

ABSTRACT
American Foulbrood Disease (AFB) is an infection of honeybees caused by the bacterium Paenibacillus larvae. One potential remedy involves using biocontrol, such as bacteriophages (phages) to lyse P. larvae. Therefore, bacteriophages specific for P. larvae were isolated to determine their efficacy in lysing P. larvae cells. Samples from soil, beehive materials, cosmetics, and lysogenized P. larvae strains were screened; of 157 total samples, 28 were positive for at least one P. larvae bacteriophage, with a total of 30. Newly isolated bacteriophages were tested for the ability to lyse each of 11 P. larvae strains. Electron microscopy demonstrated that the phage isolates were from the family Siphoviridae. Seven phages with the broadest host ranges were combined into a cocktail for use in experimental treatments of infected bee larvae; both prophylactic and post-infection treatments were conducted. Results indicated that although both pre- and post-treatments were effective, prophylactic administration of the phages increased the survival of larvae more than post-treatment experiments. These preliminary experiments demonstrate the likelihood that phage therapy could be an effective method to control AFB.
KEYWORDS: bacteriophages, environmental sampling, genomes, Paenibacillus larvae, phage therapy
Introduction
One of many diseases affecting honeybees, Apis mellifera, is American Foulbrood (AFB), which is caused by the bacterium, Paenibacillus larvae.13,36 This Gram-positive, rod-shaped bacterium produces spores, which are inadvertently picked up by adult bees and transported back to hives.13 While adults are unaffected by the disease, they are vectors of transmission to the susceptible larvae.20,45 In general, spores are resistant to antibiotics and heat, and studies specific to P. larvae have shown that their spores are capable of persisting for several decades,13 making eradication of this disease difficult. Furthermore, following infection and death of one larva, millions of spores can be produced and released in the hive.38 Honeybee larvae are most susceptible to P. larvae infection during the first 36 h after the egg hatches.13 As the larvae of a hive succumb to the disease, resulting in fewer bees reaching adulthood, the hive is unable to maintain its population and eventually collapses. Some beekeepers have used powdered antibiotics to treat AFB, but due to the presence of antibiotic resistant spores and increasing antibiotic resistance in bacterial cells, it is not a permanent cure.12,41 showed that 8 antibiotic resistance genes to tetracycline, long used as a treatment for AFB, were found in high frequency in gut microbiota of honeybees in the U. S. where the practice is common; however, only 2 of these resistance genes were found in gut microbiota of honeybees in countries where the practice is forbidden, and those only at a low frequency. In addition to increased antibiotic resistance among bacterial strains, antibiotics are not a solution to treatment of AFB because they have the potential to disrupt the normal microbial ecology of the honeybee gut microorganisms.28 Furthermore, in areas where antibiotics are used, residual amounts are found in honey meant for human consumption.29,34 Currently, the only effective method for eliminating the disease is burning the diseased hives.13 This practice is costly to the beekeeping community, resulting in loss of hive materials, productive hives, and it produces significant economic loss.13 Therefore, the prevalence and seriousness of AFB, combined with the lack of effective and safe methods of treatment, has created a need for an alternative approach.
One potential method of treatment is phage therapy, which is the therapeutic use of bacteriophages (phages) to kill bacterial cells. Phages are capable of infecting and lysing bacteria,6 with specificity for targeting certain bacterial species. Previous studies have explored the characteristics of single phages isolated from lysogenic strains of P. larvae. Several have suggested using phages as a potential treatment strategy for AFB4,5,10,11,15,16,43; however, phage therapy specifically for AFB has not been reported. Phage therapy has been used to clinically treat a variety of diseases of both humans and other animals17-19,39; therefore, the potential use of phage therapy for AFB should be explored.
Bacteriophages are abundant in natural environments40 and are self-propagating only when host bacteria are present. Furthermore, they target specific bacterial species. Phages, if used against P. larvae, would decrease the need for destructive treatments of infected hives and would not be harmful to bees, humans, or other microbes because of their specificity. Therefore, researching the potential use of phage therapy in treating AFB is of interest for economic and environmental health reasons. Unfortunately, no ready repository of P. larvae phages is available for use. The purpose of this study was to screen various environmental samples for the presence of P. larvae-specific bacteriophages. Following isolation, phages were purified and characterized prior to use as an experimental treatment of honeybee larvae infected with P. larvae spores.
Results
A total of 157 samples were screened, 28 of which were positive for viral particles (Table 1). Two samples contained 2 separate viral entities each, resulting in 30 unique phage isolates. Samples were organized into categories, and the proportions of positive samples containing phages from each sample category are as follows: lysogenic phages obtained from bacterial strains of P. larvae, 54.5%; cosmetics, 22.7%; soil under beehives, 18.89%; hive samples, 15.9%; and other environmental samples (consisting of soil not in proximity to beehives, water sources, farm waste, and plant material); 8.0%. Approximately 20% of all the samples tested yielded phages capable of lysing Paenibacillus larvae NRRL B-2605. While the highest number of the total isolated phages were obtained from soil under and around beehives (10 phage isolates), the proportion of isolated phages was highest from lysogenic incorporation in strains of P. larvae (6 phage isolates from the 11 strains tested). It is not uncommon to find lysogenic phages of P. larvae.33
Table 1.
Sources and designations of phage isolates.
| Category | Source | Phage Designations |
|---|---|---|
| Cosmetics | Hand cream (with beeswax and honey) | Scottie |
| Body wash (with royal jelly) | Beta | |
| Lipbalm #1 | Valery | |
| Lipbalm #2 | Vadim | |
| Lipbalm #3 | Vegas | |
| Hive Samples | Scale from infected hive | Xenia |
| Hive sample from Iowa | Iowa | |
| Hive sample from Iowa (honey and wax) | Ivy | |
| Propilis – Gilcrease Orchards, Nevada | Halcyone | |
| Propilis – Gilcrease Orchards, Nevada | Holly | |
| Propilis – Gilcrease Orchards, Nevada | Heidi | |
| Propilis – Gilcrease Orchards, Nevada | Harvey | |
| Soil Under Hives | Gilcrease Orchards, Nevada | Harrison |
| Gilcrease Orchards, Nevada | Hermione | |
| Gilcrease Orchards, Nevada | Hayley | |
| Gilcrease Orchards, Nevada | Hope | |
| Gilcrease Orchards, Nevada | Heath | |
| Pennsylvania | Penny | |
| Pennsylvania | Charlie | |
| Pennsylvania | Paisley | |
| UNLV, Nevada | Tristan | |
| Washington | Willow | |
| Other | Garden soil, Summerlin, Las Vegas, Nevada | Summerlin |
| Garden soil, Summerlin, Las Vegas, Nevada | Sunny | |
| Lysogenic Phages | Phage from ATCC-49843 | Alexis |
| Phage from ATCC-25368 | Bella | |
| Phage from ATCC-25367 | Carly | |
| Phage from ATCC-25747 | Diane | |
| Phage from ATCC-49843 | Erin | |
| Phage from wild strain 2231 | Fern |
Plaque morphology
Individual phage filtrates produced plaques in soft agar overlays, which were observed and described based on size and morphology (Table 2). Although there was a distribution of sizes, in general, clear plaques were more prevalent than turbid plaques. Additionally, the turbid plaques tended to be smaller than 0.5 mm.
Table 2.
Plaque and phage morphologies of the 7 cocktail phages.
| Plaque Morphology |
EM Imaging Comparison |
||
|---|---|---|---|
| Phage Designation | Size | Clarity | Head Shape |
| Xenia | Large | Clear | No data |
| Halcyone | Pinpoint | Turbid | Prolate |
| Willow | Medium | Clear | Prolate |
| Fern | Large | Clear | Prolate |
| Vadim* | Large | Clear | Icosahedral |
| Harrison | Medium | Clear | Icosahedral |
| Hayley | Medium | Clear | Prolate |
Plaque diameters were described classified as follows: pinpoint (<0.1 mm), small (0.1 mm – 0.5 mm), medium (0.5 mm – 1.0 mm), and large (>1.0 mm).
= plaques with a turbid halo around a clear plaque center. TEM images were provided by the CAMCOR facilities at the University of Oregon. Measurements are based on the averages of 2–4 images. Family classifications are based on descriptions of morphology only (Ackermann, 1987).
Host range distribution
Specificity of the isolated phages on the genus Paenibacillus, and more precisely, to the species P. larvae, was determined by spot test experiments (Fig. 1). Bacterial strains were selected to test closely related organisms (such as P. alvei) and species not closely related (such as E. coli W3104). The phages showed high specificity to P. larvae strains with some isolates capable of a minor lysis of an unknown species of Paenibacillus isolated from an infected hive. This organism was not identified to the species level after 16sRNA gene sequencing failed to identify this species (data not shown).
Figure 1.

Host range of isolated P. larvae bacteriophages determined by soft agar overlay spot tests. Results range from no lysis (blank cell) to complete lysis (black cell). The bacterial species are represented across the top and are ranked from left to right in order of susceptibility to lysis. The isolated phages are listed on the left side of the table and are ranked from top to bottom in order of the percentage of P. larvae strains they are capable of lysing.
Isolated bacteriophages failed to lyse the following strains: Paenibacillus polymyxa, P. alvei, P. lentimorbus, P. popilliae, Escherichia coli C600, E. coli W3104, E. coli MC4100, Shigella flexneri AWY3, S. flexneri BS103, Bacillus subtilis, B. anthracis, B. circulans, and Chromohalobacter sp, indicating host specificity to the genus Paenibacillus. Three phages, Halcyone, Willow, and Harrison, lysed the tested P. larvae strains, and Fern lysed all strains with the exception of its host strain, 2231. The isolated lysogenic phages were generally not capable of lysing the host strain from which they were isolated, with the exception of Diane and Alexis, and these only produced minimal lysis. There is no apparent correlation between the source category or geographical location and the effectiveness of the phages against P. larvae strains. Although host ranges were determined for all 30 isolates, only 7 were characterized further, as they were selected for use in experimental treatments as a phage cocktail.
Comparison of phage morphology using TEM
Results for these 7 phages were imaged using TEM (Table 2). All seven phages had icosahedral or prolate heads with long, flexible tails, which places them in the family Siphoviridae. Even among phages in the same family, there are size variations of heads and tails.9 Images representing a prolate phage isolate and an icosahedral phage isolate are provided in Fig. 2.
Figure 2.

Representative TEM images of 2 phage isolates. The left image displays a phage isolated from a propolis sample with a prolate head and the right image displays a phage isolated from a cosmetic source with an icosahedral head. Scale bar = 100 nm.
Comparisons of phage genomes
Sequencing analysis of the phage isolates was completed (data not shown) to determine isolate relatedness. The results demonstrated that the majority of these isolates were genetically unique.42 GenBank accession numbers for 9 of the phages are as follows: KT361649, KT361650, KT361651, KT361652, KT361653, KT361654, KT361655, KT361656, KT361657.
Larval infections
The percent larval survival on the last day of pupation, Day 8, was compared using the student's t-test for p-value calculations. Significant differences were present in each of the following comparisons: negative controls versus ATTC 49843 spore infections (p = 3.99 × 10-8); negative controls versus NRRL B-3554 spore infections (p = 3.58 × 10-8); negative controls versus 2188 spore infections with one dose (p = 2.11 × 10-6); 2188 spore infection with one dose versus phage cocktail prophylaxis (p = 0.0193). All other comparisons yielded p-values that were not significantly different. Results showing comparisons of the actual survival rates between different groups during the experiments can be visualized in Figs. 3–5.
Figure 3.

Larvae survival rates of media controls. Survival of larvae treated with a negative control (food only) and with the addition of broth (200 µl per 1.0 ml food). Larvae were harvested on Day 0 and fed daily according to Table 3. Negative control data represents 10 control replicates with n = 12 or 13 each. Error bars = standard error.
Controls
No significant difference was found between the broth control (200 µl GmBHI broth : 1 ml larvae food) and the negative control with larvae food only (p = 0.204). This preliminary experiment is important because phages suspended in broth were added to the larvae food.
Spore infections
The difference in survival rates between each experimental group fed spores, regardless of the strain and regardless of the number of doses, was statistically significant when compared to the negative controls (Fig. 4). However, there was not a significant difference in survival between larvae fed one dose of 2188 spores and larvae fed a daily dose of 2188 spores for the duration of the experiment (p = 0.203).
Figure 4.
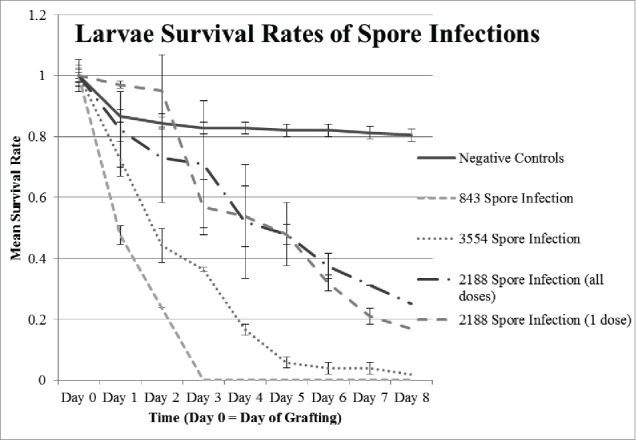
Larvae survival rates with spore infections. Survival rates after infection with P. larvae spores from strains ATCC 49843, NRRL B-3554, and newly isolated 2188. Spores were fed to as indicated (Table 3). Two different treatments with 2188 were conducted: spores given daily or in one dose on Day 0. Negative control data represents 10 control replicates with n = 12 or 13 each. Error bars = standard error.
Phage cocktail experiments
The phage cocktail control displays a slight decrease in survival compared to broth controls, but a student t-test indicated p = 0.153, suggesting the decrease was not statistically significant (Fig. 5). Regardless of whether the phages were administered before or after spores were introduced, the survival increased by approximately 59% when compared to spore-treated larvae. This indicates that the addition of phage cocktail decreased mortality of infected larvae and might potentially be used in prophylaxis or treatment of AFB.
Presence of P. larvae DNA and phages in deceased larvae
Deceased larvae were frozen and later tested using primers and PCR amplification to determine the presence of P. larvae DNA.31 Larvae obtained from negative control and phage cocktail control experiments (both of which had no bacteria added) showed no evidence of P. larvae DNA. Approximately 25% of the larvae taken from spore-only experiments were positive for P. larvae DNA after death. The average proportion of larvae positive for P. larvae DNA from phage cocktail treatments, regardless of whether phage was administered prior to or after spore infection, was slightly lower; 20% (data not shown). Additionally, there were no phages recovered from larvae not infected with phages (negative controls, broth controls, and spore infections). Phages were, however, recovered from larvae that underwent prophylactic and post-infection treatments. Larvae that were fed a phage cocktail control did not yield recoverable phages (data not shown).
Discussion
Soil around beehives is easily available and was a reliable source of lytic phages that infected all or nearly all of the 11 P. larvae strains tested. Soil samples from Nevada (2 sites), Pennsylvania and Washington yielded phages capable of lysing P. larvae. P. larvae phages were also easily isolated from soil around hives in Utah.25 A higher proportion of phages were isolated from cosmetic samples with bee-related ingredients; however, the use of phages isolated from cosmetic sources might entail legal ramifications.
Phages were found in every type of sample tested except water, flowers, and leaves not closely associated with bees. They appear to be present in hives, soil surrounding hives and other environmental samples (59–66% of the phages), begging the question of whether phages routinely keep P. larvae infections in check until either spore numbers overwhelm the hive or environmental stressors such as cold temperatures cause disease susceptibility in the bees. A careful analysis of hive infections, phage presence, and environmental stressors might provide insight on this question.
After isolation of the 30 phages, it was unclear whether the isolates represented 30 unique phage types or if the same phages had been isolated multiple times. Two potentially different lytic phages were isolated from the same soil sample (Charlie and Paisley). Their plaque morphologies were different, one being turbid and pinpoint in size and another clear and medium in size, respectively. The host range pattern of each isolate was also different with 5 differences in infectivity and intensity of lysis of the 11 P. larvae bacterial strains. Transmission electron microscopy revealed that Charlie and Paisley have the same phage morphology, i.e., Siphoviridae heads with flexible tails1; however, they are not the same size and proportionately, Charlie has a much longer tail. They are, therefore, unlikely to be the same phage despite isolation from the same source. Based on a genomic comparison of the 7 phage isolates used in the treatment cocktail, only 2 pairs displayed relatedness higher than 90% (data not shown). Willow and Fern are likely the same phage isolated from 2 different sources, having 99.99% similarity. Hayley and Vadim are likely very closely related, if not the same phage, having 96.83% similarity. Halcyone, isolated from propolis in a hive, and Harrison, isolated from soil under the same hive, have only 32.26% similarity, indicating that these 2 isolates are unrelated (data not shown).
Host range experiments revealed a lack of clearing on other genera or species besides P. larvae, indicating extreme host specificity (Fig. 1). As a potential treatment for AFB, such severe host specificity is encouraging because the complex interactions of microbes in the hive are not well understood. Lack of specificity would undoubtedly disrupt the ecology of microbes not intentionally targeted with P. larvae phages. A parallel study revealed that a P. larvae phage lysin did not disrupt bacteria typically associated with honeybee gut microbiota, indicating the phages themselves would also likely not lyse the natural microbiota.22
Phages displayed host preference for the ERIC group from which they were isolated. All 30 phages were isolated on strain NRRL-2605 from ERIC group I.14 Preference for using strains from ERIC group I over strains from ERIC group IV was significant (p = 0.0078) (data not shown). To further test this hypothesis, experiments were conducted to determine the host ranges of 3 phages isolated from environmental sources using a P. larvae strain from ERIC group IV (ATCC-25367). These phages showed a significant preference for strains from ERIC group IV over strains from ERIC group I (p = 0.016) (data not shown). This indicates the likelihood that an effective phage cocktail would include phages isolated from multiple ERIC groups to ensure treatment effectiveness in an AFB-infected hive.
Using the most effective phages; for instance, Halcyone and Willow, which are capable of lysing the 11 P. larvae strains tested, an effective phage cocktail could be created. However, a more robust cocktail could be designed by testing the lysing capabilities of these phages on additional strains of P. larvae. Because bacterial receptors are another factor that might affect the host range of a phage, the development of resistance should be considered. Bacteria are known to become resistant to phages by mutations in their receptors and/or becoming lysogenic,23 and therefore the use of a cocktail of multiple phages, rather than a single phage, reduces the potential for development of phage resistance. As such, determining selection criteria for the most suitable phages is important. For example, a P. larvae strain from an AFB-infected hive could be isolated and undergo phage testing, allowing for the design of an efficient treatment or prophylaxis; flexibility to adjust the phage components of a cocktail would ensure treatment efficacy.
The significant decrease in survival of larvae infected with spores (Fig. 4) is consistent with previous literature, where as few as 10 spores have been reported to cause the disease (Tarr, 1937; Toumanoff, 1929; Woodrow, 1941; 46). The results also demonstrate that there was not a significant difference between the proportion of larval survival when infected with one dose of spores on the first day compared to doses administered each day with the same strain (2188). A single exposure of 90 P. larvae spores in this study caused the same decrease in survival as multiple exposures, which supports the claim that low doses of spores can cause infection of AFB in honeybee larvae.46
The slightly decreased survival of the larvae given food diluted with phage cocktails may be due to the same reason larvae survival is slightly decreased when fed food diluted with broth; namely, death occurred as a result of nutrient dilution and not the phage cocktails themselves. Experiments indicate an overall improvement of approximately 59% survival when phage cocktails are administered either prior to or following infection with P. larvae spores. Prophylactic treatment with phage cocktail is slightly more effective than the post infection treatment, although not significantly.
PCR results demonstrate that there was no bacterial cross-contamination of samples during preparation nor incubation, as none of the samples showed evidence of bacterial DNA that had not received bacteria. PCR positive deceased larvae likely died from a P. larvae infection after spore germination in the larvae gut. There was a lower mortality rate among spore-infected but phage-treated larvae, which is supported by a lower proportion of specimens positive for P. larvae DNA in larvae for this experiment; however, a negative result for DNA would not be expected since P. larvae DNA would still be present due to lysis by phage. Results showing the larvae that received phage cocktail but yielded no recoverable phages might indicate that the bacterial host was necessary for phages to propagate.
Materials and methods
Growth of bacterial strains
The following strains of Paenibacillus larvae from ERIC group I were used: NRRL B-2605, NRRL B-3554, NRRL B-3650, ATCC-25748, ATCC-25747; the following strains of P. larvae from ERIC group IV were used: ATCC-49843, ATCC-25367, ATCC-25368; and the following strain from either ERIC group III or IV was used: ATCC-3688. In addition, 2 naturally occurring strains that were isolated from infected hives, both determined to be from ERIC group I, were included: 2188 and 2231. The latter numbers are for identification and differentiation, and do not relate to any depository numbers. P. larvae strains were routinely grown in Brain Heart Infusion broth (Criterion)8 modified with the addition of 1 mM CaCl2 (J. T. Baker) and 1 mM MgCl2 (Sigma Aldrich) to create mBHI, both of which were added to enhance viral attachment.21 Agar plates were made by the addition of 1.5% (w/v) agar to the mBHI broth. Either mBHI or R2A (Difco) plates were used to maintain stock cultures. Although the P. larvae strains took 3 d to grow on R2A at 37°C, cells remained viable longer on this medium as opposed to mBHI agar (data not shown). All agar plates were incubated at 37°C (NapCo E Series Model 303). P. larvae broth cultures were grown at 37°C and shaken at 100 rpm in an environmental shaker (Barnstead LabLine MaxQ 4000) for 24–48 h to obtain a maximum density (OD600 ≈0.7). Overnight cultures of P. larvae strains were preserved in 15% glycerol and maintained at −80°C (VWR) until use.8
Bacteriophage enrichment and purification
Bacteriophage enrichment was achieved using standard techniques as described by.21 To enrich for P. larvae-specific bacteriophage, P. larvae NRRL B-2605 was used because it lacks lysogenic phages and it was previously used as a universal host in phage research.46
Lysogenic phages
Lysogenic phages were obtained by screening 11 strains of P. larvae. A procedure adapted from 10 was used. No special methods were needed to induce prophage from P. larvae strains as suggested by 24; sufficient numbers of phage became lytic during the growth of their host bacteria. Cultures of P. larvae were grown in mBHI broth at 37°C and shaken at 100 rpm overnight. These were pelleted by centrifugation at 3,220 × g for 15 minutes (Eppendorf Model 5810) to remove bacterial cells. Supernatants were filtered through 0.45 µm sterile syringe filters with cellulose acetate membranes (VWR) to remove remaining cells. The filtrate served as the starting material for testing phage presence, which was determined by plaque formation on a bacterial lawn of P. larvae 2605 using a soft agar overlay method.21 Soft agar overlays of 3 ml mBHI with 0.95% (w/v) agar and 1% (w/v) yeast extract were used for phage screenings and titer determinations; yeast extract was added to enhance the clarity of plaques.16
Environmental samples and phage isolation
Samples were obtained using alcohol flame-sterilized metal spoons and placed into sterile WHIRL-PAK bags (Nasco). After collection, samples were stored at 4°C until transport to the lab. Environmental phages were obtained from screening various soil samples, cosmetics containing materials derived from beehives, and materials directly from beehives such as royal jelly, wax, propolis, and honey. These samples were obtained from the following geographic locations: Nevada, Washington, New Mexico, Oregon, Pennsylvania, New York, and Iowa. Cosmetic sample sources, obtained from traditional retail settings, included various brands of lip balms.
To isolate bacteriophage from environmental or commercial samples, portions ranging in mass from 1 g to 5 g were weighed and placed in 10 ml sterile phosphate buffered saline (PBS) pH 7.4. The mixtures were shaken in 37°C at 100 rpm overnight, then pelleted by centrifugation at 3,220 × g for 15 minutes and the supernatant was collected. Supernatants were filtered through 0.45 µm sterile syringe filters to remove remaining cells. Soil samples were re-filtered using 0.2 µm sterile syringe filters. The resulting filtrates, free from bacterial contamination, were used as the starting material for enrichment and isolation of lytic bacteriophages.
Phage purification
A soft agar overlay method similar to that described by21 was used to demonstrate individual phage plaques on the surface of mBHI agar plates. In this case, a melted 3.0 ml soft mBHI agar tube was used to suspend 1.0 ml overnight bacterial culture and 1.0 ml potential phage filtrate. This mixture was gently rocked back and forth by hand to mix the contents, then poured onto the surface of a solid mBHI agar plate and swirled to distribute the soft agar. Plates were not disturbed until solidified, at which point they were turned and incubated overnight at 37°C. After incubation, plaques were clearly visible from filtrates positive for bacteriophage capable of lysing P. larvae. Individual isolated plaques were sampled from the soft agar overlay plates with a sterile wooden applicator stick (VWR). Each plaque was used to inoculate 20 ml sterile mBHI broth, along with the addition of 1.0 ml of log phase P. larvae from an overnight culture. After overnight incubation at 100 rpm and 37°C, the cells were pelleted by centrifugation at 3,220 × g for 15 minutes, and the supernatant was filtered using 0.45 µm sterile syringe filters. This process was performed at least 3 times, which allowed for pure, independent bacteriophage isolates. This triple-pass method was similar to the double-pass procedure described by.4
Determination of phage titers
Phage titers were determined by following the soft agar overlay technique. Standard methods using 2 plates from each of multiple serial dilutions resulted in choosing the dilution that produced plaque numbers ranging from 30–200 to ensure statistical accuracy. Plaques were counted, counts were averaged, and titers of the filtrate were calculated based on the dilution.26
Soft agar overlay spot test
After amplification, each lysate was tested to determine its ability to form plaques, and therefore the efficacy of phage lysing bacterial hosts.37 Lysates were tested on each of 11 P. larvae strains and other bacterial species including: Paenibacillus polymyxa, Paenibacillus alvei, Paenibacillus lentimorbus, Paenibacillus popillae, Escherichia coli, Shigella flexneri, Bacillus cereus, Bacillus subtilis, Bacillus anthracis, Bacillus circulans, and Chromohalobacter sp. A 1.0 ml aliquot of sterile broth and 1.0 ml of an overnight culture of a single bacterial strain were added to a tube of melted mBHI agar (0.95%) with the addition of 4 g dextrose (Sigma). thereafter designated as GmBHI broth or agar. This mixture was then poured over a GmBHI agar (1.5%) plate to create a bacterial lawn. Plates were divided into quadrants (marked on the bottom of the plate), and 10 μl of a single lysate was dropped onto the surface of each quadrant, creating quadruplicate testing. Each lysate had a sufficiently high titer to contain ˜100 phage particles in 10 μl. The ability of a phage to lyse a P. larvae bacterial strain was measured by clearing. Each phage isolate was tested against each bacterial strain using a scale from no evidence of lysis (−) to complete clearing (+++) (Fig. 1). The scoring method was similar to that used by 30 and is a typical subjective test for biological activity.
Phage concentration
To prepare a highly concentrated phage lysate, 20 identical soft agar overlay plates were prepared by mixing P. larvae NRRL B-2605 and the highest titer phage lysate to result in complete cell lysis. Plates were prepared with GmBHI containing 1.5% agarose (not agar) and overlays were made of GmBHI with 0.95% agarose. Plates were incubated overnight at 37°C. Five ml of PBS containing 0.144 g/l KH2PO4 phosphate, 0.795 g/l K2HPO4, and 9.0 g/l NaCl and adjusted to pH 7.4 was added to the surface of each plate and remained for 20 min. The agarose overlay was then scraped off using a sterile pipette tip, making sure the underlying medium was not disturbed. The agarose plus PBS was collected and transferred to a funnel lined with 4 layers of cheesecloth to remove particles. The resulting liquid was then filtered through a sterile 0.2 µm filter (Sartorius) using vacuum filtration to remove bacterial cells. The filtrate was distributed into sterile 50 ml polysulfone centrifuge tubes (VWR) and phages were pelleted by centrifugation for 15 h at 4°C and 18,000 × g (Beckman J2-HS). The supernatant was removed and the centrifuge tubes were briefly inverted, being careful to prevent complete drying. The phage pellet was gently resuspended using a 1.0 ml sterile, disposable pipette tip with 1–2 mm of the tip removed prior to sterilization. Phages were suspended in 1.0 ml of phage buffer consisting of 10 mM Tris-HCl, 10 mM MgSO4, and 68 mM NaCl at pH 7.5. The concentrated phages were transferred to a 1.5 ml microcentrifuge tube. The starting volume of approximately 100 ml was concentrated to a final volume of 3.0 ml.
EM grid preparation
To a carbon-coated copper grid (Ted Pella), 10 µl of each concentrated preparation was applied to the surface and allowed to sit for 10 min prior to wicking with Whatman 541 paper wedges. The grid was rinsed (2X) for 2 min with sterile filtered ddH2O, and the liquid was wicked away. The grid was stained for 2 min with 10 µl 2% uranyl acetate (pH 4.4), and the stain was wicked away before allowing the grid to air dry (approx. 15 min). Grids were sent to the CAMCOR facilities at the University of Oregon for imaging.
DNA isolation and sequencing
The concentrated filtrates described above for electron microscopy were also used as a source of phage DNA. A series of treatments resulted in pure phage DNA that was then analyzed by Illumina sequencing for genome size.
DNase treatment
One ml of each concentrated phage preparation was added to a 1.5 ml sterile microcentrifuge tube (4 − 1.5 ml tubes each). The tubes were incubated at room temperature for 15 min following the addition of 20 μl (40 units total) of DNase to each tube. During incubation, the tubes were gently inverted 2–3 times every 5 min. The DNase was heat inactivated by incubation at 75°C for 20 min before tubes were transferred to ice for 10 min.
Protein coat degradation
Proteinase K (20 μl of 20 mg/ml) was added per ml of original concentrated phage lysate, then tubes were incubated at 55°C for 2 h. The tubes were gently inverted several times every 15 min during incubation, then again after incubation prior to starting the DNA extraction procedure.
DNA extraction and purification
Each bacteriophage isolate was prepared using one of 2 DNA extraction kits. Both Qiagen (DNeasy Blood and Tissue Kit, Cat# 69581) and Norgen (Phage DNA Isolation Kit, Cat# 46700) spin column kits were used according to the manufacturer's instructions. DNA from the kit providing the highest concentration for that specific phage was maintained at – 20°C until further use.
DNA sequence analysis and comparison
DNA concentration was determined by PicoGreen (Life Technologies, Carlsbad, CA) fluorescence and all samples normalized to 0.2 ng/ul. One ng DNA per sample was used to produce random sequencing libraries using the Nextera XT DNA Sample Preparation Kit (Illumina Inc., San Diego, CA) that was sequenced on a MiSeq Desktop Sequencer (Illumina Inc.). The sequencing run yielded a cluster density of 541k/mm^2 (+/−24 k/mm^2) with 95.96% of clusters passing filter (+/−0.84%) for a total of DNA concentration was determined by PicoGreen (Life Technologies, Carlsbad, CA) fluorescence and all samples normalized to 0.2 ng/μl. One ng DNA per sample was used to produce random sequencing libraries using the Nextera XT DNA Sample Preparation Kit (Illumina Inc, San Diego, CA) that was sequenced on a MiSeq Desktop Sequencer (Illumina Inc). The sequencing run yielded a cluster density of 541k/mm2 (+/−24k/mm2) with 95.96% of clusters passing filter (+/−0.84%) for a total of 9.95 million paired end reads. Sample representation ranged from 1.87% to 6.86% of total sequences. Similarities were determined using complete sequences (Oliviera et al., 2013) and the program Geneious 7.0.3.
Experimental phage therapy treatments
Specific phages were selected for the creation of phage cocktails based on lysing efficacy of phages and DNA characterization to determine genetic similarity. The phage cocktails were used in experimental treatments of larvae infected with P. larvae spores.
Larvae food preparation
Larvae food consisted of the following: 14.4 ml sterile, distilled water, 4.2 g royal jelly powder (Glory Bee), 0.6 g glucose (Difco), 0.6 g fructose (Difco), and 0.2 g yeast extract (Difco) as described by.32 The sugars and yeast extract were added to the water, this mixture was filtered, and then UV treated for 1 h. The royal jelly powder (4.2 g) was aseptically added to the water mixture but was otherwise untreated. The mixture was made homogenous by vortexing to ensure complete dispersion of the royal jelly. Food was prepared and stored at −20°C until needed. Larvae were fed increasing amounts of food each day,7 as indicated in Table 3. As a negative control, larvae were fed food without amendments while all other larvae were fed a mixture of food with treatment additives. In each case, 200 µL of concentrated spores or phage cocktails were added to 1.0 ml of larvae food as described above. Larvae were given the following treatments: negative control = food with no additives, broth control = food with GmBHI broth added to the same dilution as other additives, food amended with either NRRL B-3554 spores, ATCC 49843 spores, or 2188 spores, and food amended with 200 µl phage cocktail suspended in GmBHI. All larvae in the experimental phage cocktail treatments were infected with spores from P. larvae 2188. The phage cocktail was tested as both a prophylactic and post-infection treatment. The broth control data represents 2 replicates with n = 48 for each. Negative control data (food only) represents 10 replicates with n = 12 or 13, as a negative control was conducted alongside each treatment experiment to account for anomalies in survival on different egg grafting days.
Table 3.
Daily doses of food, number of spores, and number of phages fed to larvae.
| Days after Grafting | Day 0 | Day 1 | Day 2 | Day 3 | Day 4 | Day 5 | Day 6 | Day 7 | Day 8 |
|---|---|---|---|---|---|---|---|---|---|
| Volume of Larvae Food (µl) | 10 | 10 | 20 | 30 | 40 | 50 | 50 | 60 | 0 |
| # ATCC 49843 Spores | 100 | 100 | 200 | 300 | 400 | 500 | 500 | 600 | 0 |
| # NRLL B-3554 Spores | 90 | 90 | 180 | 270 | 360 | 450 | 450 | 540 | 0 |
| # Isolated 2188 Spores | 90 | 90 | 180 | 270 | 360 | 450 | 450 | 540 | 0 |
| # of PFUs in Phage cocktail | 3.003 | 3.003 | 6.003 | 9.003 | 1.204 | 1.504 | 1.504 | 1.804 | 0 |
Spore preparation
Spores were prepared by first inducing sporulation, then harvesting spores as described by the methods in 3 and 2 with the exception of replacing the HistoDenz (Sigma) density gradient with d-Sorbitol of the same concentrations. All experiments where larvae were infected with spores were conducted in duplicate. Sample sizes were as follows: ATCC 49843 spores, n = 52 and 53; NRRL B-3554 spores, n = 52 and 50; 2188, n = 48 and 48; one dose of 2188, n = 51 and 49. Spore concentration was determined using OD readings compared to standard densities previously calculated with a Petroff Hausser counting chamber (3). Calculations of spore load fed to each larva per day are given in Table 3.
Larvae were fed ATCC 49843 (843) and NRRL B-3554 (3554) spores daily. Two different treatments with 2188 spores were conducted; one in which larvae were fed spores daily or just one dose on Day 1. Spore treatment sample sizes ranged from 48–53 with a mean size of 50 and all spore infection treatments were conducted in duplicate.
Phage cocktail preparation
Titers per ml of phage lysates used for the phage cocktail were determined as previously described and are as follows: Halcyone, 5 × 104; Willow, 3 × 106; Fern, 5 × 106; Vadim, 4 × 105; Hermione, 104; H3S, 4 × 105; Xenia, 4 × 106. The cocktail contained equal aliquots of the aforementioned 7 phages, resulting in a titer of 1.8 × 106 per ml. The final phage concentration was both calculated from initial titers and confirmed by soft agar overlays after combination. Phage cocktail experiments were conducted in duplicate using larvae which ranged from 48 – 54 with a mean value of 51. Sample sizes were as follows: 2188 (1 dose), n = 51 and 49; phage cocktail control, n = 48 and 49; treatment (cocktail administered after infection with spores), n = 54 and 53; and prophylaxis treatment (cocktail administered prior to infection), n = 52 and 53. A volume of 200 µl of each cocktail was added to 1 ml of prepared larvae food immediately prior to feeding. Calculated PFUs fed to each larva per day are listed in Table 3.
Larvae rearing
Larvae were reared by methods similar to those described by.7 Queens were caged each Monday using plastic or metal wire mesh and removed on Friday.44 Eggs, turned to a horizontal position shortly before hatching, were then closely observed and the hatched larvae were grafted from the frames within a day of hatching. Eggs were randomly grafted from 4 different beehives to reduce hive-specific effects. Each treatment included a corresponding negative control consisting of larvae taken from the same frame on the same day. Grafted larvae were placed into sterile petri dishes (VWR). Incubation microcosms were created by placing 1 l of 10% glycerol in the bottom of plastic containers followed by a layer of plastic support frames on which the Petri dishes were placed. The boxes were closed with loosely fitting plastic lids, allowing for maintenance of 90% humidity within the box. Metal trays filled with water were placed on the bottom of the incubator to maintain 80% humidity within the incubator. The temperature was kept at 34°C.
For controls, larvae were fed either unamended food (negative controls), food diluted with GmBHI, or food diluted with water. Each experimental treatment had a corresponding negative control prepared on the same day from the same frame and fed unamended food. Negative control data for Figs. 3–5 represent the average of 10 control replicates with n = 12 or 13 each. Sample sizes were dependent upon the number of eggs available. In the prophylactic experiment, larvae were given food with phage cocktail 4 hr before food with spores. In the post-infection treatment experiment, larvae were given food with spores 4 hr before food with phage cocktail. Calculated spore load and viral load per day are provided in Table 3.
Figure 5.
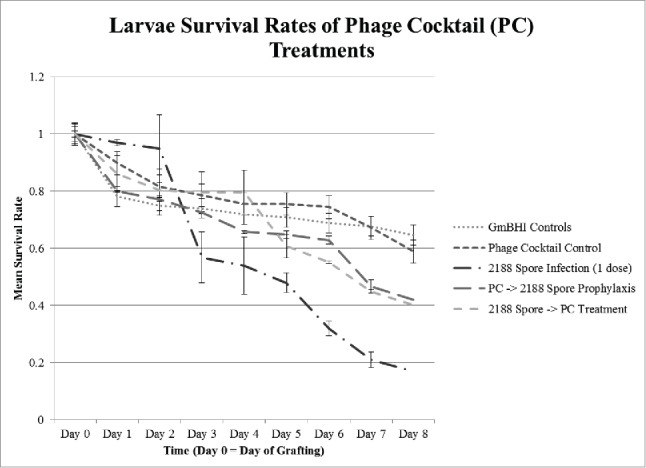
Larvae survival rates with spores and phage cocktail (PC) treatments. Larvae were fed spores, phage cocktail, spores and then phage cocktail 4 hours later, or phage cocktail and then spores 4 hours later. Sample sizes were as follows: 2188 (1 dose), n = 51 and 49; broth control, n = 48 and n = 48; phage cocktail control, n = 48 and 49; treatment (cocktail administered after infection with spores), n = 54 and 53; and prophylaxis treatment (cocktail administered prior to infection), n = 52 and 53. Error bars = standard error.
Daily observations
Larvae were viewed under a dissecting microscope (Nikon) daily and observed for signs of life: opening and closing spiracles, growth or food consumption in a chronic exposure scheme.7,14 In the event that no movement could be seen for the first 2 days, larvae were kept until day 3 in the event that they were alive but movement was not easily visualized. On the third day, if no growth or movement was observed, larvae were assumed dead and removed. The number of surviving larvae was recorded daily.
Statistical analyses
Each experimental treatment was conducted in duplicate, and attempted samples sizes were n = 50. Attempted sample sizes for the negative control larvae were n = 12 due to constraints on the number of eggs deposited by the queen at one time. Surviving larvae were counted and recorded daily. The number of surviving larvae at the end of the larval stage were counted, and then compared to the number of survivors from other treatments using a student t-test. The parameters of the t-test were set as 2-tailed and either homoscedastic if the variances between the 2 test groups were equal or as heteroscedastic if the variances between the 2 test groups were unequal. Variances were calculated from the average of the percentage of surviving larvae on the final day. Statistical significance was determined when p < a, and a = 0.05.
Disclosure of potential conflicts of interest
The authors declare filed patent US20140213144 A1.
Acknowledgments
The following individuals provided assistance with research: Lucy LeBlanc, Erin Davis, Carolyn Chang, Ian Lam, Syed Ahmed, and Anthony Harrington. The following individuals provided lab supplies or bacterial cultures: Drs. Helen J. Wing, Eduardo Robleto, Ernesto Abel-Santos, Brian Hedlund, Ms. Jasmin Khilnani and Mr. Israel Alvarado. Paula Jacoby Garrett provided assistance with report editing. The following people donated beehive or soil samples: K. Bean, C. Cohen, L. Hudson, M. Reed, M. Tonkinson, P. Crellin, S. Hendrickson, F. Chaves, H. Bard, and W. Haight. Dr. Vincent Fischetti from the Rockefeller University provided insight for compiling phage cocktails and Dr. Malcolm Zellars from the University of Georgia and Kurt Langworthy from the University of Oregon provided helpful suggestions on EM preparation procedures. Andrew Krohn provided significant assistance sequencing DNA. The following individuals obtained images from the CAMCOR facilities at the University of Oregon: Kurt Langworthy, Hayley Johanesen, A. Bruce Johnson, James Raasch, and Lincoln Conner. We acknowledge the use of CAMCOR, which has been purchased with a combination of federal and state funding in Oregon.
Funding
This research was supported by USDA Grant NEVR-2010-03755.
References
- [1].Ackermann HW. Bacteriophage taxonomy. Microbiol Sci 1987; 4:214-8; PMID:3153614 [PubMed] [Google Scholar]
- [2].Akoachere M, Squires RC, Nour AM, Angelov L, Brojatsch J, Abel-Santos E. Identification of an in vivo inhibitor of Bacillus anthracis spore germination. J Biol Chem 2007; 282:12112-8; PMID:17296608; http://dx.doi.org/ 10.1074/jbc.M611432200 [DOI] [PubMed] [Google Scholar]
- [3].Alvarado I, Phui A, Elekonich MM, Abel-Santos E. Requirements for in vitro germination of Paenibacillus larvae spores. J Bacteriol 2013: 195(5):1005-11; PMID:23264573; http://dx.doi.org/ 10.1128/JB.01958-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Bakheit N, Stahly DP. Properties of clear plaque mutants of the Bacillus larvae bacteriophage PBL0.5 and PBL2. J Invertebr Pathol 1988; 52:78-83; http://dx.doi.org/ 10.1016/0022-2011(88)90105-X [DOI] [Google Scholar]
- [5].Campana CF, Bakheit N, Stahly DP. Morphology of Bacillus larvae bacteriophage PBL3 and physical map of its DNA. J Invertebr Pathol 1991; 57:141-3; http://dx.doi.org/ 10.1016/0022-2011(91)90055-U [DOI] [Google Scholar]
- [6].Carlton RM. Phage therapy: past history and future prospects. Arch Immunol Ther Exp 1991; 47:267-74 [PubMed] [Google Scholar]
- [7].Crailsheim K, Brodschneider R, Aupinel P, Behrens D, Genersch E, Vollmann J, Riessberger-Galle U. Standard methods for artificial rearing of Apis mellifera larvae In The COLOSS BEEBOOK, Volume I: standard methods for Apis mellifera research. Dietemann V, Ellis JD, P Neumann (eds). J Api Res 2013; 52(1):http://dx.doi.org/ 10.3896/IBRA.1.52.1.05. [DOI] [Google Scholar]
- [8].De Graaf DC, Alippi AM, Antúnez K, Aronstein KA, Budge G, De Koker D, De Smet L, Dingman DW, Evans JD, Foster LJ, et al.. Standard methods for American foulbrood research In The COLOSS BEEBOOK, Volume II: standard methods for Apis mellifera pest and pathogen research. Dietemann V, Ellis JD, Neumann P (eds). J Api Res 2013; 52(1):http://dx.doi.org/ 10.3896/IBRA.1.52.1.11. [DOI] [Google Scholar]
- [9].Demuth J, Neve H, Witzel KP. Direct electron Microscopy study on the morphological diversity of bacteriophage populations in lake plussee. App Env Microbiol 1993; 59:3378-84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Dingman DW, Bakheit N, Field CC, Stahly DP. Isolation of two bacteriophage from Bacillus larvae, PBL1 and PBL0.5, and partial characterization of PBL1. J Gen Virol 1984; 65:1101-5; PMID:6726188; http://dx.doi.org/ 10.1099/0022-1317-65-6-1101 [DOI] [PubMed] [Google Scholar]
- [11].Drobnikova V, Ludvik J. Bacteriophage of Bacillus larvae. J Api Res 1982; 21:53-6 [Google Scholar]
- [12].Evans JD. Diverse origins of tetracycline resistance in the honey bee bacterial pathogen P. larvae. J Invertebr Pathol 2003; 83:46-50; PMID:12725811; http://dx.doi.org/ 10.1016/S0022-2011(03)00039-9 [DOI] [PubMed] [Google Scholar]
- [13].Genersch E. American foulbrood in honeybees and its causative agent, P. larvae. J Invertebr Pathol 2010; 103:510-9 [DOI] [PubMed] [Google Scholar]
- [14].Genersch E, Forsgren E, Pentikäinen J, Ashiralieva A, Rauch S, Kilwinski J, Fries I. Reclassification of Paeniacbillus larvae supsp. pulvifaciens and Paenibacillus larvae subsp. larvae as Paenibacillus larvae without supspecies differentiation. Intl J Syst Evol Microbiol 2006; 56:501-11; PMID:16514018; http://dx.doi.org/ 10.1099/ijs.0.63928-0 [DOI] [PubMed] [Google Scholar]
- [15].Gochnauer TA. The isolation of a bacteriophage (bacterial virus) from Bacillus larvae. Bee World 1955; 36:101-3; http://dx.doi.org/ 10.1080/0005772X.1955.11094880 [DOI] [Google Scholar]
- [16].Gochnauer TA. Some properties of a bacteriophage from Bacillus larvae. J Invertebr Pathol 1970; 15:149-56; PMID:5435792; http://dx.doi.org/ 10.1016/0022-2011(70)90228-4 [DOI] [PubMed] [Google Scholar]
- [17].Guenther S, Huwyler D, Richard S, Loessner MJ. Virulent bacteriophage for efficient biocontrol of Listeria monocytogenes in ready-to-eat foods. Appl Environ Microbiol 2009; 75:93-100; PMID:19011076; http://dx.doi.org/ 10.1128/AEM.01711-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hagens S, Loessner MJ. Application of bacteriophages for detection and control of foodborne pathogens. Appl Microbiol Biotechnol 2007; 76:513-9; PMID:17554535; http://dx.doi.org/ 10.1007/s00253-007-1031-8 [DOI] [PubMed] [Google Scholar]
- [19].Hawkins C, Harper D, Burch D, Anggard E, Soothill J. Topical treatment of Pseudomonas aeruginosa otitis of dogs with a bacteriophage mixture: A before/after clinical trial. Vet Microbiol 2010; 146(3–4):309-13; PMID:20627620; http://dx.doi.org/ 10.1016/j.vetmic.2010.05.014 [DOI] [PubMed] [Google Scholar]
- [20].Hitchcock JD, Stoner A, Wilson WT, Menapace DM. Pathogenicity of Bacillus pulvifaciens to honeybee larvae of various ages (Hymenoptera: Apidae). J Kans Entomol Soc 1979; 52:238-46 [Google Scholar]
- [21].Hurst CJ, Reynolds KA. Chapter 48: Sampling viruses from soil In Manual of environmental microbiology. Hurst CJ, Crawford RL, R KG, McInerney MJ, D SL (eds) Washington, D.C., USA: American Society for Microbiology Press, 2002. [Google Scholar]
- [22].LeBlanc L, Nezami S, Yost D, Tsourkas P, Amy PS. Isolation and characterization of a novel phage lysin active against Paenibacillus larvae, a honeybee pathogen. Bacteriophage J 2015; 5:e1080787-1-e1080787-12. http://dx.doi.org/ 10.1080/21597081.2015.1080787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Lindberg AA. Bacteriophage receptors. Annu Rev Microbiol 1973; 27:205-41; PMID:4584686; http://dx.doi.org/ 10.1146/annurev.mi.27.100173.001225 [DOI] [PubMed] [Google Scholar]
- [24].Mayer VW, Gabridge MG, Oswald EJ. Rapid plate test for evaluating phage induction capacity. Appl Microbiol 1969; 18:697-8; PMID:4905042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Merrill BD, Grose JH, Breakwell DP, Burnett SH. Characterization of Paenibacillus larvae bacteriophages and their genomic relationships to firmicute bacteriophages. BMC Genomics 2014; 15:745; PMID:25174730; http://dx.doi.org/ 10.1186/1471-2164-15-745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Miller RV. Methods for enumeration and characterization of bacteriophage from environmental samples In Techniques in Microbial Ecology. Burlage RS, Atlas R, Stahl D, Geesey G, Sayler G. (eds). Oxford, UK: Oxford University Press, 1998. [Google Scholar]
- [27].Oliveira A, Melo LDR, Kropinski AM, Azeredo J. Complete genome sequence of the broad-host-range Paenibacillus larvae phage philBB_P123. Genome Announc 2013; 1:e00438-13; PMID:24009112; http://dx.doi.org/ 10.1128/genomeA.00438-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Olofsson TC, Vásquez A. Detection and identification of a novel lactic acid bacterial flora within the honey stomach of the honeybee Apis mellifera. Curr Microbiol 2008; 57:356-63; PMID:18663527; http://dx.doi.org/ 10.1007/s00284-008-9202-0 [DOI] [PubMed] [Google Scholar]
- [29].Ortelli D, Edder P, Corvi C. Analysis of chloramphenicol residues in honey by liquid chromatography-tandem mass spectrometry. Chromatographia 2004; 59:61-4; http://dx.doi.org/ 10.1365/s10337-004-0244-6 [DOI] [Google Scholar]
- [30].Owens J, Barton MD, Heuzenroeder MW. The isolation and characterization of Campylobacter jejuni bacteriophages from free range and indoor poultry. Vet Microbiol 2013; 162:144-50; PMID:22980913; http://dx.doi.org/ 10.1016/j.vetmic.2012.08.017 [DOI] [PubMed] [Google Scholar]
- [31].Piccini C, D'Alessandro B, Antunez K, Zunino P. Detection of Paenibacillus larvae subspecies larvae spores in naturally infected bee larvae and artificially contaminated honey by PCR. World J Microbiol Biotechnol 2002; 18:761-5; http://dx.doi.org/ 10.1023/A:1020435703165 [DOI] [Google Scholar]
- [32].Peng YSC, Mussen E, Fong A, Montague MA, Tyler T. Effects of chlortetracycline of honey bee worker larvae reared in vitro. J Invertebr Pathol 1992; 60:127-33; http://dx.doi.org/ 10.1016/0022-2011(92)90085-I [DOI] [Google Scholar]
- [33].Qin X, Evans JD, Aronstein KA, Murray KD. Genome sequences of the honey bee pathogens Paenibacillus larvae and Ascosphaera apis. Insect Mol Biol 2006; 15:715-8; PMID:17069642; http://dx.doi.org/ 10.1111/j.1365-2583.2006.00694.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Saridaki-Papakonstadinou M, Andredakis S, Burriel A, Tsachev I. Determination of tetracycline residues in Greek honey. Trakia J Sci 2006; 4:33-6 [Google Scholar]
- [35].Sheflo MA, Gardner AV, Merrill BD, Fisher JNB, Lunt BL, Breakwell DP, Grose JH, Burnett SH. Complete genome sequences of five Paenibacillus larvae bacteriophages. Genome Announc 2013; 1:e00668-13; PMID:24233582; http://dx.doi.org/ 10.1128/genomeA.00668-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Shimanuki H, Knox D. Diagnosis of honey bee diseases. U.S. Department of Agriculture, Agriculture Handbook No. AH-690; 2000. URL http://www.ars.usda.gov/is/np/honeybeediseases/honeybeediseases.pdf. [Google Scholar]
- [37].Silva YJ, Costa L, Pereira C, Mateus L, Cunha A, Calado R , et al. Phage therapy as an alternative to prevent Vibrio anguillarum infections in fish larvae production. Mar Drugs 2013; 9:e114197. http://dx.doi.org/ 10.1371/journal.pone.0114197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Sturtevant AP. Relation of commercial honey to the spread of American foulbrood. J Agric Res 1932; 45:257-85 [Google Scholar]
- [39].Sulakvelidze A, Alavidze Z, Morris G Jr.. Bacteriophage therapy. Antimicrob Agents Chemother 2001; 45:649-59; PMID:11181338; http://dx.doi.org/ 10.1128/AAC.45.3.649-659.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Suttle CA, Fuhrman JA. Enumeration of virus particles in aquatic or sediment samples by epifluorescence microscopy In Manual of Aquatic Viral Ecology. Wilhelm SW, Weinbauer MG, Suttle CA (eds). Waco, TX, USA: American Society of Limnology and Oceanography, 2010. [Google Scholar]
- [41].Tian B, Fadhil NH, Powell JE, Kwong WK, Moran NA. Long-term exposure to antibiotics has caused accumulation of resistance determinants in the gut microbiota of honeybees. mBio 2012; 3:e00377-12; PMID:23111871; http://dx.doi.org/ 10.1128/mBio.00377-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Tsourkas P, Yost DG, Krohn A, LeBlanc L, Zhang A, Stamereilers C, Amy PS. Complete genome sequences of nine phages capable of infecting Paenibacillus larvae, the causative agent of American foulbrood disease in honeybees. Genome Announc 2015; 3:e01120-15; PMID:26472825; http://dx.doi.org/ 10.1128/genomeA.01120-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Valerianov T, Popova A, Toschkoff A. Isolation from soil of a bacteriophage lysing Bacillus larvae. Acta Microbiol Virol Immunol 1976; 4:81-5 [PubMed] [Google Scholar]
- [44].Williams GR, Alaux C, Costa C, Csaki T, Doublet V, Eisenhardt D , et al. Standard methods for maintaining adult Apis mellifera in cages under in vitro laboratory conditions. In The COLOSS BEEBOOK, Volume I: standard methods for Apis mellifera research. Dietemann V, Ellis JD, Neumann P. (eds). J Api Res 2013; 52(1):1-36. http://dx.doi.org/ 10.3896/IBRA.1.52.1.04. [DOI] [Google Scholar]
- [45].Wilson WT. Resistance to American foulbrood in honey bees XI. Fate of Bacillus larvae spores ingested by adults. J Invertebr Pathol 1971; 17:247-55; PMID:5575743; http://dx.doi.org/ 10.1016/0022-2011(71)90099-1 [DOI] [PubMed] [Google Scholar]
- [46].Woodrow AW. Susceptibility of honeybee larvae to individual inoculations with spores of Bacillus larvae. J Econ Entomol 1942; 35:892-5; http://dx.doi.org/ 10.1093/jee/35.6.892 [DOI] [Google Scholar]