

Abstract
In this study, we explore the mechanistic relationship between growth factor signaling and kinase activity that supports the protein synthesis-dependent phase of long-term memory (LTM) consolidation for sensitization of Aplysia. Specifically, we examine LTM for tail shock-induced sensitization of the tail-elicited siphon withdrawal (T-SW) reflex, a form of memory that requires both (i) extracellular signal-regulated kinase (ERK1/2; MAPK) activity within identified sensory neurons (SNs) that mediate the T-SW and (ii) the activation of transforming growth factor β (TGFβ) signaling. We now report that repeated tail shocks that induce intermediate-term (ITM) and LTM for sensitization, also induce a sustained post-training phase of MAPK activity in SNs (lasting at least 1 h). We identified two mechanistically distinct phases of post-training MAPK: (i) an immediate phase that does not require ongoing protein synthesis or TGFβ signaling, and (ii) a sustained phase that requires both protein synthesis and extracellular TGFβ signaling. We find that LTM consolidation requires sustained MAPK, and is disrupted by inhibitors of protein synthesis and TGFβ signaling during the consolidation window. These results provide strong evidence that TGFβ signaling sustains MAPK activity as an essential mechanistic step for LTM consolidation.
In the field of memory research, consolidation refers to the sensitive time period following a learning event during which the brain develops a stable memory representation (McGaugh 2000; Alberini and Kandel 2015). This process gives rise to memories that can last days, years, or even a lifetime. Therefore, it is perhaps not surprising that an extensive array of molecular mechanisms is called into play to achieve these lasting changes. The prevailing evidence suggests that, although posttranslational modifications are sufficient to support short-term memory, the induction of LTM requires additional signaling that includes new protein and RNA synthesis (Costa-Mattioli et al. 2009; Santini et al. 2014; Alberini and Kandel 2015). However, the temporal and mechanistic dynamics that govern this transition are poorly understood. One possibility is that a “transient” activation of upstream signaling pathways (e.g., kinases such as cAMP-dependent protein kinase A [PKA] and MAPK) induced by the learning event gives rise to the up-regulation of transcription- and translation-dependent effectors (e.g., Zif268, C/EBP, c-Fos) that lead to long-term synaptic strengthening and memory. Another possibility is that PKA and MAPK signaling is “sustained” in an active state for many hours following training to direct synaptic strengthening and memory formation. For instance we, and others, have shown that PKA activity is critical during the later phases of consolidation at times that are well beyond the initiation of gene transcription (for up to 6 h; Muller and Carew 1998; Chain et al. 1999). Stimuli sufficient to produce long-term synaptic facilitation (LTF) at Aplysia sensorimotor synapses (e.g., 4–5 spaced tail-nerve shocks or exogenous pulses of the neuromodulator, serotonin) and LTM for sensitization of Aplysia defensive reflexes (e.g., spaced tail shocks) are also capable of inducing MAPK activation that persists for up to 3 h post-training (Sharma et al. 2003b; Kopec et al. 2015). Both PKA and MAPK can then translocate to the nucleus to recruit the CREB-mediated transcriptional activity necessary to promote LTF and LTM (Bartsch et al. 1995; Abel et al. 1998).
We recently reported that signaling by the growth factor TGFβ “during learning” induces a post-training phase of MAPK and C/EBP gene expression, and is necessary for LTM for sensitization in Aplysia (Kopec et al. 2015). In the present paper, we report that TGFβ signaling is additionally required “beyond” the learning event, “during consolidation” of LTM, to sustain MAPK signaling through a protein synthesis-dependent mechanism. In addition, we show that this post-training TGFβ-MAPK signaling cascade is required for LTM consolidation. Taken together, our findings suggest that growth factor-dependent sustained kinase activity may represent a general mechanism that maintains persistent signaling during the consolidation of long-term memories.
Results
Dynamics of MAPK signaling within the LTM consolidation window
Previous work has indicated that the formation of long-term memory for sensitization, and the recruitment of its underlying molecular mechanisms, is sensitive to the number and pattern of training trials (Sutton et al. 2002; Wainwright et al. 2002; Philips et al. 2007; Farah et al. 2009). For example, Sutton et al. (2002) demonstrated that a pattern of at least four temporally spaced tail shocks (TSs) was necessary for the induction of LTM. Using the same paradigm, Sharma et al. (2003b) next showed that sensitization training with five spaced TSs induces robust, persistent MAPK activation. Taken together, these findings raise the question: what is the minimum number of training trials required for the induction of persistent MAPK activity? To examine this question, we delivered two, three, or four spaced TSs (120 mA for 1.5 sec; intershock interval = 10 min) to the intact animal and examined MAPK activation at 1 h post training in the sensory cluster. Consistent with our previous behavioral observations, semiquantitative Western blot analysis found that two or three shocks produced no persistent MAPK activation, whereas four spaced shocks produced robust activation (Fig. 1A; one-way ANOVA: F(2,16) = 6.67, P = 0.008; 2TS: n = 6, mean ± SEM, 102 ± 11% MAPK activation, P = 0.84, NS; 3TS: n = 6, 100 ± 8%, P = 0.96, NS; 4TS: n = 7, 154 ± 15% P < 0.05). These results suggested that a threshold number of training trials is required for the recruitment of sustained MAPK activation.
Figure 1.
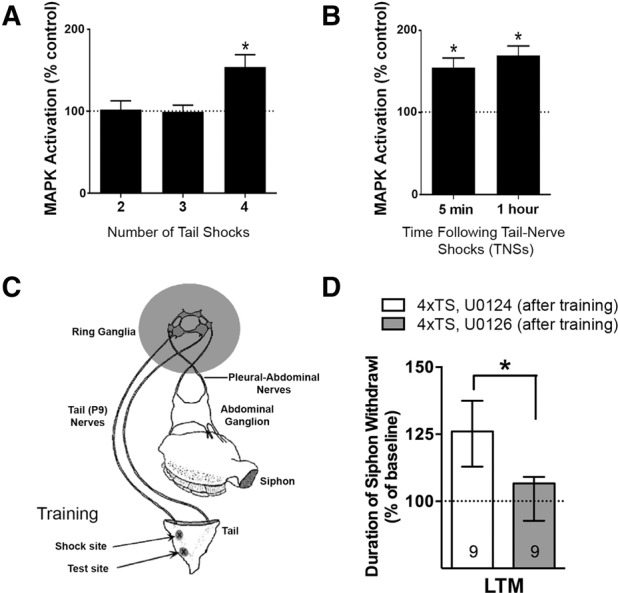
A sustained ERK/MAPK signaling recruited by multiple training trials is required for the consolidation of LTM in Aplysia. (A) A minimum of four spaced (ITI = 10 min) training tail shocks (trials) is required for the induction of post-training MAPK activity in sensory neuron (SN) somata. (B) MAPK activation is sustained at least 1 h following repeated tail-nerve shocks (4xTNSs, ITI = 10 min), an in vitro analog of repeated-trial sensitization training. Data in A, B are mean ± SEM. (C) Model of reduced behavioral preparation of the Aplysia tail-elicited siphon withdrawal (T-SW) reflex (Sutton et al. 2001). Unilateral testing and training sites are indicated (see arrows) and the ring ganglia of the central nervous system (CNS) is shaded to indicate that it can be further isolated for pharmacological manipulation of the tail SN component of the T-SW reflex circuitry. (D) Disruption of sustained MAPK activation by application of the active MEK inhibitor, U0126, delivered to the CNS after training (4xTS, U0126 [after training]), reveals that MAPK is required for consolidation of LTM for sensitization of the T-SW. Data are presented as median ± IQR.
In order to better study the dynamics of MAPK activation we developed a reduced preparation amenable to pharmacological manipulation that utilizes tail-nerve shock (TNS) as reliable training analog for TS (see Materials and Methods). We dissected out the CNS (ring ganglia) with the tail nerves still attached, so that we could readily shock one-tail nerve and quickly harvest both ipsilateral and contralateral sensory clusters (as described for the tail-shock studies above; see Materials and Methods). We delivered four tail-nerve shocks (ITI = 10 min) and assayed MAPK at 5 min as well as at 1 h. Consistent with our findings in intact animals (Fig. 1A), repeated tail-nerve shocks induced robust immediate MAPK activation (5 min, n = 19, 155 ± 12% MAPK activation, P < 0.05, paired t-test) that was sustained at 1 h following training (1 h, n = 29, 170 ± 11%; P < 0.05) over the unshocked paired control cluster (Fig. 1B).
LTM requires MAPK activity during consolidation
We previously demonstrated that blocking MAPK activity both during and after behavioral training (5xTS) impairs the induction of LTM (Sharma et al. 2003b). We also previously reported that MAPK activity is required during training for LTM induced by a Two-Trial training paradigm (Philips et al. 2013; Kopec et al. 2015). While these data clearly implicate MAPK as important for LTM, they do not inform the question of whether MAPK activity is required (i) selectively for acquisition (during training), (ii) for consolidation (after training), or (iii) for both. To examine the role of persistent MAPK activation during LTM consolidation, we restricted the application of the MAPK kinase (MEK) inhibitor, U0126 (20 µM), or its inactive isomer, U0124 (20 µM), to a period after the completion of training (thus preserving the early phase of MAPK activation that is recruited during acquisition). To monitor behavior, we used the reduced behavioral preparation in which the tail-elicited siphon withdrawal (T-SW) reflex is activated by stimulation to the tail, and memory is revealed by an enhancement of the siphon withdrawal response following training (Fig. 1C). Animals were trained with four TSs (120 mA for 1.5 sec, ITI = 10 min), and then the central nervous system chamber, which contains the tail sensory neurons, was exposed to U0126 or U0124 (20 μM) for 3 h. This duration was chosen because it represents the time period during which post-training MAPK activation is sustained in tail SNs (Sharma et al. 2003b). Importantly, previous work has shown that U0126 treatment does not disrupt baseline responding as assessed for up to 20 h after treatment (Sharma et al. 2003b; Menges et al. 2015). After training and drug incubation, the preparations were tested the next day (18–20 h) for LTM. As expected, the group receiving the inactive isomer, U0124, displayed significant LTM (Fig. 1D; n = 10, median ± IQR, 126 ± 25% baseline T-SW, P = 0.003 Wilcoxon signed rank test). In contrast, U0126 treatment significantly impaired the induction of LTM (n = 10, 107 ± 16%, P = 0.56, NS); moreover, there was a significant difference between groups (P < 0.05 Mann–Whitney U-test). These data add strong support to the hypothesis that the consolidation phase of LTM for sensitization requires the persistent activation of MAPK.
Sustained, but not immediate, post-training MAPK activation requires translation
We next examined the mechanisms responsible for the behaviorally relevant sustained MAPK activation in the LTM consolidation window. LTM consolidation requires both new protein synthesis and de novo gene transcription (Alberini and Kandel 2015). Thus, we examined, in turn, whether persistent MAPK activation required translation and transcription. Ring ganglia were preincubated with the general protein synthesis inhibitor emetine (100 µM; Sutton et al. 2001; Menges et al. 2015) or vehicle (ASW) for 1 h and then trained (four tail-nerve shocks, ITI = 10 min) in the presence of drug. We harvested sensory neuron clusters at either 5 min or 1 h post-training. Immediate (5 min) MAPK activation was not disrupted by emetine treatment (Fig. 2A; VEH: n = 8, 151 ± 23%; EME: n = 6, 197 ± 41%; VEH versus EME, P = 0.32, NS), whereas 1 h MAPK activation was significantly disrupted relative to vehicle levels (VEH: n = 13, 153 ± 12%; EME: n = 10, 115 ± 10%; VEH versus EME, P < 0.05). To address whether translation was specifically required during consolidation (after training) for 1 h MAPK activation, we introduced emetine immediately following training and found that 1 h MAPK activation levels were again significantly reduced (Fig. 2A; VEH: n = 11, 150 ± 13%; EME: n = 11, 122 ± 7%; VEH versus EME, P = 0.04, one-tail).
Figure 2.
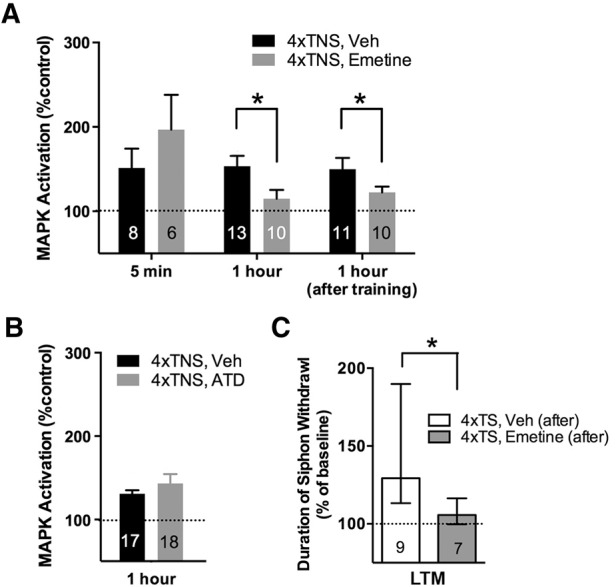
Protein synthesis is required after training for sustained MAPK activation and LTM consolidation. (A) Preexposure and continued incubation with the protein synthesis inhibitor, emetine, disrupts 1 h MAPK activated by 4xTNS training, but not immediate (5 min) MAPK activation. Applying emetine immediately after 4xTNS training also significantly reduces 1 h MAPK activation. (B) 1 h MAPK is not disrupted by the presence of an irreversible inhibitor of transcription, actinomycin D (ATD). Data in A, B are expressed as mean ± SEM. (C) The introduction of emetine in the consolidation window after repeated tail shock training (4xTS, Emetine [after]) significantly disrupts LTM consolidation. Data are expressed as median ± IQR.
We next asked whether the protein synthesis-dependent persistent phase of MAPK activation also required transcription. Interestingly, the RNA synthesis inhibitor, actinomycin D (50 μg/mL, ATD; Sutton et al. 2001), did not disrupt 1 h MAPK activation (Fig. 2B; VEH: n = 17, 131 ± 4%; ATD: n = 18, 143 ± 11%1; VEH versus ATD, P = 0.33, NS). Taken together these results support the view that MAPK activation within the consolidation phase exists in at least two mechanistically distinct phases: (i) a translation-independent immediate phase, and (ii) a sustained phase of MAPK activation that requires ongoing translation during the consolidation of LTM.
LTM requires translation during consolidation
Our data thus far suggest that a phase of translation-dependent MAPK activation is required during the consolidation of LTM. These results support the prediction that LTM should also require translation during consolidation. To directly test this hypothesis we used the semi-intact preparation (Fig. 1C) to apply emetine to the CNS for 3 h following training (to block the persistent, translation-dependent phase of MAPK activation). This treatment significantly disrupted the induction of LTM (Fig. 2C; VEH: n = 10, median ± IQR, 128 ± 77% baseline T-SW; EME: n = 7, 106 ± 17%; VEH versus EME, P < 0.05 Mann–Whitney U-test). In a separate experiment, untrained animals (no shock [NS]) received identical drug/vehicle treatment to control for the possibility that emetine treatment could disrupt baseline responding. However, long-term behavioral responses were stable following emetine treatment (data not shown; VEH: n = 6, 99 ± 45% and EME: n = 10, 108 ± 16%; VEH versus EME, P = 0.45), supporting the conclusion that LTM formation requires ongoing protein synthesis during the consolidation window.
Sustained MAPK activity requires TGFβ signaling
TGFβ signaling plays a key role in the induction of long-term synaptic strengthening and neuronal excitability (Chin et al. 1999, 2006). We recently reported that TGFβ signaling is upstream of the MAPK activity observed 1 h following a two-trial sensitization paradigm, and plays a significant role during training for two-trial LTM induction (Kopec et al. 2015). While informative, these experiments did not indicate whether TGFβ signaling is exclusively required for LTM induction (i.e., during learning), or whether TGFβ signaling may also be required for LTM consolidation. To explore this question, we examined the role of post-training TGFβ signaling in sustained MAPK activation by training (4xTNSs) in the presence of a TGFβ receptor chimera (TGFβsR-Fc). This chimera sequesters extracellular ligand to block endogenous TGFβ signaling (Chin et al. 1999; Kopec et al. 2015). The immediate phase of MAPK activation was not disrupted by the TGFβsR-Fc (Fig. 3A; VEH: n = 8, 133 ± 14%; TGFβsR-Fc: n = 8, 145 ± 16%; VEH versus TGFβsR-Fc, P = 0.59, NS). However, the persistent (1 h) phase of MAPK activation was significantly disrupted by the chimera (VEH: n = 10, 125 ± 7%; TGFβsR-Fc: n = 10, 103 ± 8%; VEH versus TGFβsR-Fc, P < 0.05). Additionally, we replicated the earlier observation that recombinant human TGFβ ligand can activate MAPK in Aplysia sensory neurons (Chin et al. 2002). Specifically, a 1.5 h treatment of 100 ng/mL TGFβ1 delivered to the CNS, a duration equivalent to our repeated TNSs training duration and 1 h collection time point, resulted in the significant activation of MAPK relative to the vehicle treated control ganglia from the same animal (Fig. 3B; VEH [0.1% BSA in ASW]: n = 21, 110 ± 9%; TGFβ: n = 21, 133 ± 8%; VEH versus TGFβ, P < 0.05).
Figure 3.
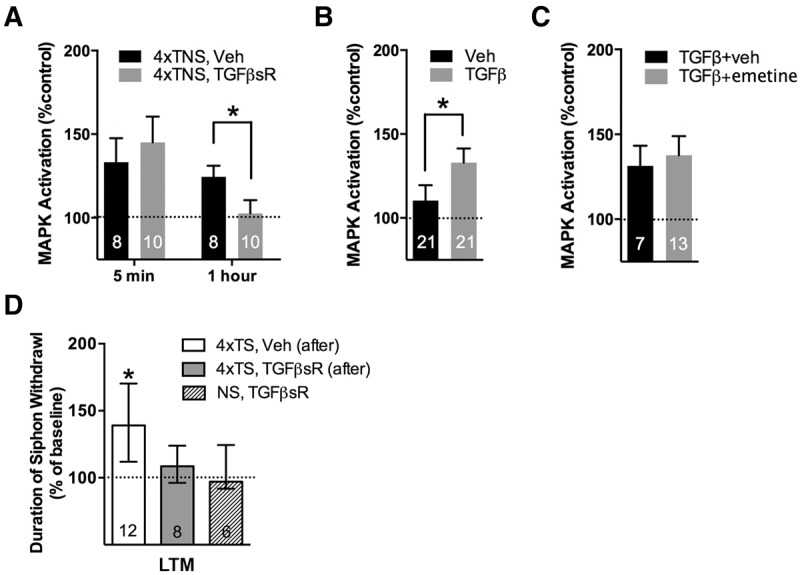
TGFβ signaling is required during the consolidation window after training for both sustained MAPK activation and LTM consolidation. (A) Disruption of extracellular TGFβ signaling using the soluble TGFβ receptor chimera (TGFβsR) blocks 1 h MAPK activation by 4xTNS, whereas 5-min MAPK activation is unaffected by the chimera. (B) Bath application of recombinant human TGFβ (100 ng/mL, 90 min) to the isolated pleural-pedal pair is sufficient to activate MAPK. (C) TGFβ-induced MAPK activation is not disrupted by the protein synthesis inhibitor, emetine. Data in A–C are expressed as mean ± SEM. (D) Exposure to TGFβsR after 4xTS training disrupts LTM consolidation (4xTS, TGFβsR [after]). Data are expressed as median ± IQR.
Given that 1 h MAPK activation requires both ongoing protein synthesis (Fig. 2A) and TGFβ signaling (Fig. 3A), we next asked whether the protein synthesis requirement for persistent MAPK activation was upstream of TGFβ signaling (e.g., involving synthesis and/or activation of TGFβ ligand), or downstream from TGFβ (e.g., involving protein synthesis-dependent actions downstream from TGFβ receptor signaling). We explored this question by treating isolated ganglia with recombinant TGFβ ligand, as above, but in the presence of emetine. Although we previously found that persistent MAPK activation by TNSs is significantly disrupted by emetine treatment (Fig. 2A), persistent MAPK activation induced by recombinant TGFβ was not disrupted by emetine (Fig. 3C; [TGFβ + ASW]: n = 7, 131 ± 12% ASW only control; [TGFβ + emetine]: n = 13, 138 ± 11% emetine only control; TGFβ + ASW versus TGFβ + emetine, P = 0.73). These findings suggest that the protein synthesis requirement for TGFβ-dependent 1 h MAPK activation may lie upstream of the recruitment of TGFβ signaling (possibly in the synthesis of TGFβ ligand itself) and does not require de novo gene expression or protein synthesis downstream from TGFβ receptor signaling. However, these data do not exclude the possibility that there are additional protein synthesis-dependent mechanisms upstream of MAPK activation that are independent of TGFβ signaling.
LTM requires TGFβ signaling in the consolidation window
In a final series of experiments, we explored the requirement of TGFβ signaling during the consolidation window for LTM. Using the semi-intact preparation (Fig. 1C), animals were trained (4 TSs, ITI = 10 min) and then the CNS was exposed for 3 h to the TGFβsR-Fc chimera to disrupt extracellular TGFβ signaling specifically during the post-training consolidation window. A third group of animals was exposed to the TGFβsR-Fc chimera for the equivalent treatment, but in the absence of training, to control for any nonspecific effects on baseline responding. Our results showed that TGFβ signaling during consolidation is critical for LTM formation. Specifically, a one-way ANOVA revealed a significant difference between groups (H = 6.598, P = 0.0369, Kruskal–Wallis test), with only those animals trained in the presence of vehicle showing significant LTM (Fig. 3D; [VEH, 4xTSs]: n = 12, median ± IQR, 139 ± 58%, P < 0.05; [TGFβsR-Fc, 4xTSs]: n = 8, 109 ± 28%, P = 0.33, NS). There was no effect of TGFβsR-Fc treatment on baseline responding at the 20-h test in untrained animals ([TGFβsR-Fc, untrained]: n = 6, 97 ± 33%, NS). Thus, consistent with its recruitment of persistent MAPK activation, these data reveal an important role for TGFβ signaling specifically within the consolidation window for LTM.
Discussion
The results of the present study support four principle conclusions (illustrated in Fig. 4): (i) The training requirements for the recruitment of a persistent phase of MAPK activation parallel those necessary for the induction of LTM, (ii) LTM consolidation is associated with at least two mechanistically distinct phases of post-training MAPK activation, (iii) the translation-dependent phase of MAPK activation is required for LTM consolidation, and (iv) TGFβ signaling during the consolidation window maintains MAPK activation to support LTM formation.
Figure 4.
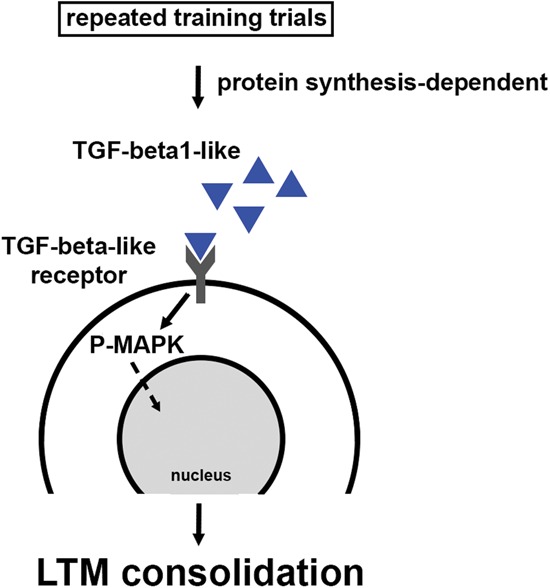
A model for post-training signaling during the consolidation of LTM. Repeated training trials recruit persistent MAPK activation beyond the training window to support LTM consolidation, and this requires protein synthesis, but not de novo gene expression. We now report that a critical effector of this protein synthesis is the up-regulation of extracellular TGFβ signaling, which may be supported through the production of TGFβ ligand, or its translation-dependent activation within the LTM consolidation window. Solid arrows indicate confirmed data and dashed arrows identify elements to be confirmed.
The training requirements for persistent MAPK activation share features in common with the requirements for LTM induction
Previously, we showed that the training pattern for two-trial LTM induction (two trials spaced by 45 min) shared the induction requirements for persistent MAPK activation (Philips et al. 2007; Kopec et al. 2015). We now demonstrate that the repeated-trial training pattern for LTM (which uses short 10–15-min ITIs), also mirrors the induction requirements for persistent MAPK activation in tail SNs. Specifically, four spaced TSs (ITI of 10 to 15 min) induce robust LTM and MAPK activation, whereas three (or less) TSs fail to induce both LTM and sustained MAPK activation. Thus we hypothesize that the fourth shock generates signaling events necessary to exceed a mechanistic threshold either by (i) activation of permissive factors (i.e., TGFβ), (ii) a relief of repressive factors (i.e., calcineurin [see Sharma et al. 2003a], or (iii) a combination of these two possibilities.
Intriguingly, in contrast to our behavioral observations of a sharp training threshold for the activation of sustained MAPK and LTM induction, previous findings show that there is a graded translocation of MAPK to the nucleus following one, three or five pulses of 5HT (Martin et al. 1997). Thus the behavioral threshold for LTM induction may be the result of different levels of mechanistic processing. For example, although each training trial (i.e., tail shock) may translocate increasing amounts of MAPK to the nucleus, the fourth stimulus may trigger the critical event (e.g., sustained nuclear PKA activity) to engage CREB/C/EBP-mediated gene expression necessary for LTM.
There are two mechanistically distinct phases of MAPK activation during LTM consolidation
Although post-training activation of MAPK has been observed in Aplysia and in other model systems (Swank and Sweatt 2001; Sharma et al. 2003b; Trifilieff et al. 2006), our identification of two mechanistically distinct phases of activation was unexpected. Specifically, we identified a protein synthesis- and TGFβ-independent, immediate phase of MAPK activity, which is followed at 1 h by a second phase of MAPK activity that is highly dependent upon both ongoing protein synthesis and on TGFβ signaling. These data are consistent with the findings of Lyons and colleagues (Michel et al. 2011), who also observed two mechanistically distinct phases of MAPK activation following behavioral training of Aplysia on a task that involves animals learning that food is inedible (LFI). They found that immediate and 1 h phases of MAPK activation were recruited in the buccal ganglion following LFI training, and that these two temporal phases were differentially sensitive to an inhibitor of the cGMP-dependent protein kinase (PKG): immediate MAPK was PKG-independent and 1 h MAPK activation was partially dependent on PKG. Collectively, these findings suggest that the recruitment of mechanistically distinct phases of sustained signaling for LTM consolidation is a shared theme in Aplysia, and would not be unexpected in other model systems as well.
TGFβ maintains persistent MAPK activity to support LTM consolidation
As discussed above, our results suggest that sensitization training recruits two temporally and mechanistically distinct phases of MAPK activity. Restricting the application of U0126 to a period after training (which spares the immediate phase of MAPK activation) significantly impaired LTM for sensitization, highlighting the importance of the late phase of MAPK activity to LTM consolidation. Moreover, pharmacological blockade of TGFβ signaling similarly spared immediate MAPK activation, but significantly disrupted both 1 h MAPK as well as consolidation of LTM. These findings strongly support the view that a TGFβ-dependent sustained MAPK activity is critical for the consolidation of LTM. Intriguingly, our data also suggest that the protein synthesis requirement for sustained MAPK activation is upstream of extracellular TGFβ signaling, since recombinant TGFβ ligand was able to activate MAPK in the presence of an inhibitor of protein synthesis. Collectively, these data suggest a model in which there is a requirement for the continued synthesis or protein synthesis-dependent activation (Liu et al. 1997) of an endogenous TGFβ ligand that promotes persistent MAPK activity and LTM consolidation (Fig. 4).
The importance of post-training growth factor signaling in LTM consolidation and maintenance has also been established for brain derived neurotrophic factor (BDNF) (Bekinschtein et al. 2007; Katche et al. 2013; Bambah-Mukku et al. 2014). In rodents, BDNF is important for LTM consolidation at multiple time points following training and this is regulated through a BDNF autoregulatory feedback loop (Bambah-Mukku et al. 2014). In Aplysia, a well-established downstream target of growth factor signaling is CREB-dependent gene expression, which supports the consolidation of LTF at sensorimotor synapses through a positive CREB autoregulatory feedback loop lasting for several hours following training (Mohamed et al. 2005; Liu et al. 2008). Since MAPK can phosphorylate the CREB inhibitor, CREB2, in vitro (Michael et al. 1998), our results are consistent with an intriguing model in which sustained TGFβ-dependent MAPK activity is required to help promote and sustain the positive CREB feedback loop that is important for the LTM consolidation window through the persistent phosphorylation and inactivation of CREB2. Importantly, as the molecular mechanisms underlying LTM consolidation are identified, it will be important to understand how long these persistent signaling activities are required after training. In the present study, LTM consolidation was disrupted by (i) inhibitors of protein synthesis, (ii) MAPK activation, and (iii) extracellular TGFβ signaling introduced immediately following training. However, the duration of the critical temporal window (or windows) following training remains to be established. An advanced understanding of the temporal and mechanistic features of the LTM consolidation may provide novel therapeutic targets for the strengthening or weakening of memories.
In conclusion, the results of the present study are consistent with data from many model systems, and provide strong support for a model wherein consolidation occurs through a highly conserved mechanism that involves the sustained presence of extracellular growth factors that drive persistent kinase activity and extend the activity of effector proteins (e.g., transcription factors) to consolidate synaptic strengthening and memory.
Materials and Methods
Behavioral procedures
Reduced behavioral preparations of Aplysia californica were used in all behavioral experiments (described in Sutton et al. 2001). Briefly, wild-caught Aplysia (250–400 g; supplied by Marinus, Long Beach, CA) were anesthetized by injection of isotonic MgCl2 (∼100 mL/100 g body weight). The tail and mantle were surgically removed along with the ring ganglia, abdominal ganglion, and their peripheral connections. The siphon artery was cannulated with silastic tubing (ID = 0.25 in) and perfused at ∼5 mL/min with cooled seawater (Instant Ocean, 15°C), to keep the siphon inflated throughout the experiment. The tail was bisected and manually perfused (∼15 mL) via a 22-guage needle inserted into the internal cavity. The tail and mantle were pinned to the Sylgard-coated floor of a chamber containing circulating tank seawater (15°C), while the ring ganglia (comprised of the cerebral and two paired pleural-pedal ganglia) were pinned in a separate Sylgard-coated subchamber that was independently perfused with RT (20°C–22°C) artificial seawater (ASW) containing in mM: 460 NaCl, 55 MgCl2, 11 CaCl2 10 KCl, 10 Tris pH 7.6 (Fig. 1C). The P9 nerves and abdominal pleural-abdominal nerves exited the subchamber through small slits that were sealed with Vaseline. Preparations were allowed at least 60 min to recover prior to pretest measurements.
Prior to training, 3–4 pretests (intertest interval [ITI] = 15 min) were conducted to establish a baseline of tail-elicited siphon withdrawal (T-SW) reflex duration. The right and left half of the tail (∼1 cm anterior from its tip, and midway between the lateral and medial margins) was stimulated with a mild shock (12 mA - 0.5 sec duration) from a hand-held bipolar electrode (alternating sides). The tail hemi-segment that produced a stable baseline T-SW was identified and trained, which consisted of four strong tail shocks (120 mA and 1.5-sec duration) delivered to the training site (see Fig. 1C) at 10 min intervals. Importantly, our prior work has shown that behavioral sensitization of the T-SW circuit, as well as serotonin release, is restricted within the CNS to the side ipsilateral to training shocks (Bristol et al. 2004; Sutton et al. 2004; Marinesco et al. 2006). Three post-tests of the T-SW (elicited in the same fashion as the pretests described above; ITI = 15 min) were performed the next day ∼18–20 h after the final pretest.
All drug treatments were restricted to the ring ganglia subchamber. Following pretests, drugs were applied for 3 h (static bath), which represents the time period during which MAPK remains persistently active following TS. After incubation, the drugs were perfused out of the subchamber and preparations remained overnight with circulating seawater (15°C). Control and experimental groups were always interleaved. All drugs were obtained from Calbiochem. Recombinant human TGFβ1 and TGFβsR-Fc were obtained from R&D Systems.
Western blot analysis
For experiments examining the activation of MAPK in the intact animal, 2–4 shocks (AC, 1.5-sec duration) were delivered with an ITI of 10 min to one side of the tail through a hand-held bipolar electrode (shock area = 9 mm). Animals were anesthetized by injection of isotonic MgCl2, ganglia were removed and SN clusters were excised from the pleural ganglia at indicated time points following the last shock.
To assess the activation of MAPK in the presence of pharmacological agents (the tail-nerve shock [TNS] experiments), the ring ganglia (containing the SNs) with the tail nerves were removed (following anesthetization as described) and placed into a small dish suitable for experimental manipulation. TNS, like TS, activates a small fraction of cells within the ipsilateral SN cluster (∼10% of the Vc cluster) and releases serotonin throughout the ipsilateral CNS (Marinesco and Carew 2002; Marinesco et al. 2006). After desheathing the pleural and pedal ganglia (to expose SN and MN cells) in 50:50 MgCl2:ASW, the ganglia were perfused with 30 min of 100% ASW at 5 mL/min through a 4-mL perfusion dish. Training consisted of four spaced TNSs (40 Hz, 50 V, 1.5 sec; ITI = 10 min). Drugs were added into a static bath either 1 h before training (throughout), or immediately after training (after). We did two types of experiments with exogenous TGF ligand. For the experiment reported in Figure 3B, 100 ng/mL TGFβ1 was exogenously applied for 1.5 h to either the right or left pleural-pedal pair (randomized) and we used the other side as the vehicle treated control (0.1% BSA in ASW). In Figure 3C, we repeated this experiment in the presence of either emetine or its vehicle (ASW) applied to both ganglia pairs. Emetine or vehicle was applied beginning 30 min prior to TGFβ/vehicle exposure and remained until collection of sensory clusters at 1.5 h into TGFβ/vehicle treatment.
All SN clusters were harvested in the MgCl2 solution and lysed immediately in 20 µL of lysis buffer [50 mM Tris–Cl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 50 mM sodium fluoride, 1 mM sodium orthovanadate, 10 mM sodium β-glycerophosphate, 4 mM para-nitrophenylphosphate, 2% sodium dodecyl sulfate, and a protease inhibitor cocktail (one tablet of Roche Diagnostics GmbH protease inhibitor cocktail/25 mL)] by mechanical agitation. Samples were then stored at −80°C until use. Samples were resolved using SDS-PAGE (4%–12% Bis–Tris Gel in MES buffer) and transferred to nitrocellulose using standard procedures. Blots were initially probed with rabbit anti-phospho-p44/p42 MAPK primary antibodies (Cell Signaling, 1:2000), then stripped with buffer (62.5 mM Tris–Cl, pH 6.8, 100 mM β-mercaptoethanol, 2% SDS) at 65°C for 45 min (confirmed by probing the blots without the primary antibody), and probed with rabbit anti-total MAPK antibody (Cell Signaling, 1:1000) (Sharma et al. 2003b). After each primary antibody incubation, blots were incubated with anti-rabbit HRP-conjugated secondary antibodies and exposed to film in the presence of enhanced chemiluminescent substrate (ECL; Thermo Fisher Scientific). Exposure of the film was kept in the linear range and band intensity was quantified using NIH Image software (NIH). Approximately equal amounts of tissue were taken for the control and experimental samples. To account for small differences in the protein amount, the phospho-MAPK signal was normalized to the total MAPK signal in each lane (Patterson et al. 2001).
Data analysis and statistics
Behavioral
An observer, blind to both the training and drug treatment, measured the duration of T-SW, the elapsed time from stimulus onset to the initial relaxation of the siphon from the contracted position (Sutton et al. 2001). The average of three pretests was used to determine the baseline. We performed nonparametric statistical analyses, due to a non-normal distribution of the data. Median LTM scores (next day) were normalized to the averaged baseline measurement (pretests) and plotted as median ± interquartile range (IQR). Mann–Whitney U-tests were used for between group comparisons and Wilcoxon Signed Rank tests were used for within-group comparisons.
Molecular
Each animal contains a pair of sensory clusters (left and right). The experimental side was shocked (either TS or TNS) or received exogenous TGFβ ligand and the other always served as a naïve within animal control (untrained or vehicle treated). For data presentation, the phospho-MAPK/total-MAPK ratio from the experimental sensory cluster (right or left) was normalized to the control side for that animal to generate a normalized MAPK activation ratio. Group data are presented in the figures as mean ± SEM. For statistical analyses, planned comparisons were then made across treatment groups using an unpaired t-test on the normalized ratios (obtained using above calculation) between treatment groups. The TGFβ experiment in Figure 3B was designed after Chin et al. (2002) and used a within animal design, therefore the planned comparison reported here was a paired t-test. We always ran controls groups (vehicle alone) and experimental groups in parallel.
Acknowledgements
This work was supported by National Institutes of Health grants MH094792 and MH041083 to T.J.C. We thank Carew Laboratory members for comments on an earlier draft of this manuscript, and we thank Heather Boyle and Vickie Hsuan for their excellent technical assistance.
Footnotes
Article is online at http://www.learnmem.org/cgi/doi/10.1101/lm.040915.115.
References
- Abel T, Martin KC, Bartsch D, Kandel ER. 1998. Memory suppressor genes: inhibitory constraints on the storage of long-term memory. Science 279: 338–341. [DOI] [PubMed] [Google Scholar]
- Alberini CM, Kandel ER. 2015. The regulation of transcription in memory consolidation. Cold Spring Harb Perspect Biol 7: a021741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bambah-Mukku D, Travaglia A, Chen DY, Pollonini G, Alberini CM. 2014. A positive autoregulatory BDNF feedback loop via C/EBPβ mediates hippocampal memory consolidation. J Neurosci 34: 12547–12559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartsch D, Ghirardi M, Skehel PA, Karl KA, Herder SP, Chen M, Bailey CH, Kandel ER. 1995. Aplysia CREB2 represses long-term facilitation: relief of repression converts transient facilitation into long-term functional and structural change. Cell 83: 979–992. [DOI] [PubMed] [Google Scholar]
- Bekinschtein P, Cammarota M, Igaz LM, Bevilaqua LR, Izquierdo I, Medina JH. 2007. Persistence of long-term memory storage requires a late protein synthesis- and BDNF- dependent phase in the hippocampus. Neuron 53: 261–277. [DOI] [PubMed] [Google Scholar]
- Bristol AS, Sutton MA, Carew TJ. 2004. Neural circuit of tail-elicited siphon withdrawal in Aplysia. I. Differential lateralization of sensitization and dishabituation. J Neurophysiol 91: 666–677. [DOI] [PubMed] [Google Scholar]
- Chain DG, Casadio A, Schacher S, Hegde AN, Valbrun M, Yamamoto N, Goldberg AL, Bartsch D, Kandel ER, Schwartz JH. 1999. Mechanisms for generating the autonomous cAMP-dependent protein kinase required for long-term facilitation in Aplysia. Neuron 22: 147–156. [DOI] [PubMed] [Google Scholar]
- Chin J, Angers A, Cleary LJ, Eskin A, Byrne JH. 1999. TGF-β1 in Aplysia: role in long-term changes in the excitability of sensory neurons and distribution of TβR-II-like immunoreactivity. Learn Mem 6: 317–330. [PMC free article] [PubMed] [Google Scholar]
- Chin J, Angers A, Cleary LJ, Eskin A, Byrne JH. 2002. Transforming growth factor β1 alters synapsin distribution and modulates synaptic depression in Aplysia. J Neurosci 22: RC220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin J, Liu RY, Cleary LJ, Eskin A, Byrne JH. 2006. TGF-β1-induced long-term changes in neuronal excitability in aplysia sensory neurons depend on MAPK. J Neurophysiol 95: 3286–3290. [DOI] [PubMed] [Google Scholar]
- Costa-Mattioli M, Sossin WS, Klann E, Sonenberg N. 2009. Translational control of long-lasting synaptic plasticity and memory. Neuron 61: 10–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farah CA, Weatherill D, Dunn TW, Sossin WS. 2009. PKC differentially translocates during spaced and massed training in Aplysia. J Neurosci 29: 10281–10286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katche C, Cammarota M, Medina JH. 2013. Molecular signatures and mechanisms of long-lasting memory consolidation and storage. Neurobiol Learn Mem 106: 40–47. [DOI] [PubMed] [Google Scholar]
- Kopec AM, Philips GT, Carew TJ. 2015. Distinct growth factor families are recruited in unique spatiotemporal domains during long-term memory formation in Aplysia californica. Neuron 86: 1228–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu QR, Hattar S, Endo S, MacPhee K, Zhang H, Cleary LJ, Byrne JH, Eskin A. 1997. A developmental gene (Tolloid/BMP-1) is regulated in Aplysia neurons by treatments that induce long-term sensitization. J Neurosci 17: 755–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu RY, Fioravante D, Shah S, Byrne JH. 2008. cAMP response element-binding protein 1 feedback loop is necessary for consolidation of long-term synaptic facilitation in Aplysia. J Neurosci 28: 1970–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinesco S, Carew TJ. 2002. Serotonin release evoked by tail nerve stimulation in the CNS of aplysia: characterization and relationship to heterosynaptic plasticity. J Neurosci 22: 2299–2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinesco S, Wickremasinghe N, Carew TJ. 2006. Regulation of behavioral and synaptic plasticity by serotonin release within local modulatory fields in the CNS of Aplysia. J Neurosci 26: 12682–12693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin KC, Michael D, Rose JC, Barad M, Casadio A, Zhu H, Kandel ER. 1997. MAP kinase translocates into the nucleus of the presynaptic cell and is required for long-term facilitation in Aplysia. Neuron 18: 899–912. [DOI] [PubMed] [Google Scholar]
- McGaugh JL. 2000. Memory–a century of consolidation. Science 287: 248–251. [DOI] [PubMed] [Google Scholar]
- Menges SA, Riepe JR, Philips GT. 2015. Latent memory facilitates relearning through molecular signaling mechanisms that are distinct from original learning. Neurobiol Learn Mem 123: 35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael D, Martin KC, Seger R, Ning MM, Baston R, Kandel ER. 1998. Repeated pulses of serotonin required for long-term facilitation activate mitogen-activated protein kinase in sensory neurons of Aplysia. Proc Natl Acad Sci 95: 1864–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel M, Green CL, Eskin A, Lyons LC. 2011. PKG-mediated MAPK signaling is necessary for long-term operant memory in Aplysia. Learn Mem 18: 108–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohamed HA, Yao W, Fioravante D, Smolen PD, Byrne JH. 2005. cAMP-response elements in Aplysia creb1, creb2, and Ap-uch promoters: implications for feedback loops modulating long term memory. J Biol Chem 280: 27035–27043. [DOI] [PubMed] [Google Scholar]
- Muller U, Carew TJ. 1998. Serotonin induces temporally and mechanistically distinct phases of persistent PKA activity in Aplysia sensory neurons. Neuron 21: 1423–1434. [DOI] [PubMed] [Google Scholar]
- Patterson SL, Pittenger C, Morozov A, Martin KC, Scanlin H, Drake C, Kandel ER. Some forms of cAMP-mediated long-lasting potentiation are associated with release of BDNF and nuclear translocation of phospho-MAP kinase. Neuron 32: 123–140. [DOI] [PubMed] [Google Scholar]
- Philips GT, Tzvetkova EI, Carew TJ. 2007. Transient mitogen-activated protein kinase activation is confined to a narrow temporal window required for the induction of two-trial long-term memory in Aplysia. J Neurosci 27: 13701–13705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philips GT, Ye X, Kopec AM, Carew TJ. 2013. MAPK establishes a molecular context that defines effective training patterns for long-term memory formation. J Neurosci 33: 7565–7573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santini E, Huynh TN, Klann E. 2014. Mechanisms of translation control underlying long-lasting synaptic plasticity and the consolidation of long-term memory. Prog Mol Biol Transl Sci 122: 131–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma SK, Bagnall MW, Sutton MA, Carew TJ. 2003a. Inhibition of calcineurin facilitates the induction of memory for sensitization in Aplysia: requirement of mitogen-activated protein kinase. Proc Natl Acad Sci 100: 4861–4866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma SK, Sherff CM, Shobe J, Bagnall MW, Sutton MA, Carew TJ. 2003b. Differential role of mitogen-activated protein kinase in three distinct phases of memory for sensitization in Aplysia. J Neurosci 23: 3899–3907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton MA, Masters SE, Bagnall MW, Carew TJ. 2001. Molecular mechanisms underlying a unique intermediate phase of memory in aplysia. Neuron 31: 143–154. [DOI] [PubMed] [Google Scholar]
- Sutton MA, Ide J, Masters SE, Carew TJ. 2002. Interaction between amount and pattern of training in the induction of intermediate- and long-term memory for sensitization in Aplysia. Learn Mem 9: 29–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton MA, Bagnall MW, Sharma SK, Shobe J, Carew TJ. 2004. Intermediate-term memory for site-specific sensitization in Aplysia is maintained by persistent activation of protein kinase C. J Neurosci 24: 3600–3609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swank MW, Sweatt JD. 2001. Increased histone acetyltransferase and lysine acetyltransferase activity and biphasic activation of the ERK/RSK cascade in insular cortex during novel taste learning. J Neurosci 21: 3383–3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trifilieff P, Herry C, Vanhoutte P, Caboche J, Desmedt A, Riedel G, Mons N, Micheau J. 2006. Foreground contextual fear memory consolidation requires two independent phases of hippocampal ERK/CREB activation. Learn Mem 13: 349–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wainwright ML, Zhang H, Byrne JH, Cleary LJ. 2002. Localized neuronal outgrowth induced by long-term sensitization training in aplysia. J Neurosci 22: 4132–4141. [DOI] [PMC free article] [PubMed] [Google Scholar]