

Abstract
Decay of mRNA is essential for the efficient regulation of gene expression. A major pathway of mRNA degradation is initiated by the shortening of the poly(A) tail via the CCR4/NOT deadenylase complex. Deadenylation is followed by removal of the 5′ cap (i.e., decapping) and then 5′ to 3′ exonucleolytic decay of the message body. The highly conserved CCR4/NOT deadenylase complex consists of the exonucleases CCR4 and POP2/CAF1, as well as a group of four or five (depending on organism) accessory factors of unknown function, i.e., the NOT proteins. In this study, we find that Saccharomyces cerevisiae Not2p, Not3p, and Not5p (close paralogs of each other) are involved in promoting mRNA decapping. Furthermore, we find that Not3p and Not5p bind to the decapping activator protein Pat1p. Together, these data implicate the deadenylase complex in coordinating the downstream decapping reaction via Not2p, Not3p, and Not5p. This suggests that the coupling of deadenylation with decapping is, in part, a direct consequence of coordinated assembly of decay factors.
Keywords: deadenylation, decapping, mRNA decay, mRNA stability
INTRODUCTION
The process of mRNA degradation is vital for controlling the overall level of gene expression within the cell. In yeast, mRNA degradation is initiated by the shortening of the 3′ polyadenosine [poly(A)] tail. For many transcripts, poly(A) tail shortening allows for the removal of the 5′ cap structure by the Dcp2p/Dcp1p holoenzyme. This ultimately results in degradation of the transcript body by the 5′ to 3′ exonuclease activity, Xrn1p (Coller and Parker 2004; Ghosh and Jacobson 2010).
In Saccharomyces cerevisiae, there are two distinct deadenylase complexes, the Pan2p/Pan3p complex and the Ccr4p/Not1p–5p complex. The Pan2p/Pan3p complex is hypothesized to be involved in the initial trimming of the poly(A) tail, while the Ccr4p/Not1p–5p complex is the major deadenylase activity degrading the bulk of the poly(A) tail (Tucker et al. 2001, 2002; Wolf and Passmore 2014). The Ccr4p/Not1p-5p deadenylase complex is composed of two enzymatic activities, Ccr4p and Caf1p (Pop2p). Both of these proteins have been documented to have deadenylase activity (Daugeron et al. 2001; Tucker et al. 2002). Ccr4p and Pop2p form a complex with each other and numerous other factors, most notably the NOT proteins. There are five NOT proteins (Not1p–5p), and their function in controlling mRNA degradation is somewhat enigmatic. Not1p is a large protein (240 kDa) that coordinates the binding of Ccr4p/Pop2p to other factors, including Not2p–5p (Basquin et al. 2012). The roles of Not2p–5p in mRNA metabolism are largely unknown but they have been implicated in mRNA deadenylation (Tucker et al. 2002). Not4p is a putative ubiquitin-ligase of unknown function and is species-specific. Not2p, Not3p, and Not5p are close homologs of each other and share similarity through a common motif referred to as the NOT box (Bhaskar et al. 2013). Not2p is relatively small and is comprised of mainly the NOT box region, while Not3p and Not5p have N-terminal extensions of significant similarity. Not3p and Not5p are most likely a manifestation of fungal genomic duplication, as only one copy of NOT3/5 is found in metazoans. In metazoans, NOT3/5 is largely referred to as NOT3 although it bears greater homology with the yeast Not5p protein.
In metazoans, NOT3/5 has been shown to be essential for embryonic development, heart function, and metabolism (Neely et al. 2010; Morita et al. 2011). Previous studies of NOT2, NOT3, and NOT5 have shown that these proteins have closely related activities (Bai et al. 1999; Bawankar et al. 2013). Moreover, Not2p may be a core member of the Ccr4p/Not1p–5p deadenylase complex that recruits Not3p and/or Not5p into the complex (Bhaskar et al. 2013).
In this study, we demonstrate that Saccharomyces cerevisiae Not2p, Not3p, and Not5p, stable components of the Ccr4p/Not1p-5p deadenylase complex, are involved in promoting mRNA decapping. First, we show that loss of Not2p or Not5p has a general influence on reporter and endogenous mRNA decapping. Second, tethering experiments demonstrate that Not3p also promotes mRNA decapping. Furthermore, we demonstrate that the decapping function of Not3p and Not5p maps to a highly conserved region in their C-terminal domain. Using yeast two-hybrid analysis, we demonstrate that the C-terminal domain of Not3p and Not5p binds to the decapping activator protein Pat1p. Together, our data suggest that the coupling of deadenylation and decapping occurs in part through specific recruitment of decapping activators by the deadenylase complex itself.
RESULTS
Not2p and Not5p promote mRNA decapping
Not2p and Not5p have been documented to stimulate mRNA deadenylation. Specifically, deletion of NOT2 or NOT5 resulted in significantly slower removal of the MFA2 mRNA poly(A) tail (Tucker et al. 2002). Importantly, deletion of NOT3 only marginally affected the stability of MFA2 mRNA. We confirmed these previous analyses using a transcriptional shut-off approach. Specifically, we used a construct expressing the MFA2 gene under the control of the inducible GAL1 promoter. This construct also contains a polyguanosine [poly(G)] stretch of 18 residues in the reporter gene's 3′ untranslated region (UTR) which allows for the detection of decay intermediates resulting from deadenylation-dependent decapping and exonucleolytic decay. Cells were grown in the presence of galactose to induce expression of the reporter mRNA. At mid-log phase, transcription of the MFA2 reporter was inhibited by shifting the cells from galactose media to glucose media. Following the inhibition of transcription, time points were taken and RNA was extracted and analyzed by PAGE followed by Northern blot visualization. The reporter was detected using a probe specific for the poly(G) tract. We observed that in wild-type cells, the MFA2 reporter mRNA is quickly degraded upon transcriptional shut-off with an observed half-life of 4 min. Importantly, when NOT2 or NOT5 are deleted, the MFA2 mRNA reporter is stabilized threefold (Fig. 1A). Since these mRNAs were resolved on polyacrylamide gels, we were able to monitor deadenylation following inhibition of transcription. For both not2Δ and not5Δ cells, there is a mild defect in deadenylation consistent with previous observations. Importantly, however, we also observed that compared to wild-type cells, the MFA2 mRNA in these deletion strains remains stable even after deadenylation—a phenotype hallmarked by other gene mutants known to be required for efficient mRNA decapping. Deletion of the NOT3 gene mildly affects the half-life and deadenylation of the MFA2 reporter, but no gross defect in decapping is observed (Fig. 1A).
FIGURE 1.

Deletion of NOT2 or NOT5 results in an mRNA decapping defect. (A) Transcriptional shut-off analysis of the indicated strains expressing an MFA2 reporter containing a poly(G) tract in its 3′ UTR. Time points above each lane represent time (in minutes) after transcriptional repression. Half-life was determined after quantifying MFA2 mRNA levels relative to an SCR1 RNA loading control (Supplemental Fig. S1). A80 and A0 refer to the position of MFA2 mRNA that is fully adenylated or fully deadenylated, respectively. (B) Steady-state mRNA isolated from the indicated strains was analyzed by Northern blot to detect EDC1 mRNA and CYH2 mature and pre-mRNA. The 18S rRNA ethidium stain is shown as a loading control. EDC1 mRNA levels normalized to SCR1 RNA are graphically represented for each strain relative to the wild-type strain, which is set to 100.
To confirm a role for Not2p and Not5p in mRNA decapping, we analyzed deletion mutants for the expression of endogenous EDC1 mRNA. The EDC1 mRNA is exquisitely sensitive to loss of decapping complex function because of sequence elements embedded within its 3′ UTR that render its decay independent of prior deadenylation (Muhlrad and Parker 2005). Thus, EDC1 mRNA levels are a strong indicator of genes required for normal mRNA decapping. Consistent with this, we observe a 6.8-fold increase in EDC1 mRNA levels in cells deleted for the major decapping enzyme gene, DCP2. Deletion of either NOT2 or NOT5 also results in a 4.7 to 6.8-fold increase in EDC1 mRNA levels, demonstrating that these two genes are required for mRNA decapping (Fig. 1B). This effect on EDC1 levels is consistent with previous observations (Muhlrad and Parker 2005). Loss of the NOT3 gene does not impact EDC1 mRNA decapping. However, we demonstrate that it can impact decapping when tethered to a reporter mRNA (see below). We do not observe an additive effect on mRNA decapping when NOT2, NOT3, and NOT5 are deleted, suggesting that they work through a similar pathway (Fig. 1A,B).
In yeast, the process of nonsense-mediated mRNA decay (NMD) occurs by deadenylation-independent mRNA decapping (Smith and Baker 2015). The CYH2 pre-mRNA is a natural substrate for NMD in yeast. Factors involved in the catalytic activity of the decapping holoenzyme are predicted to affect both normal mRNA decay and NMD in yeast. Importantly, we observe that loss of Not2p, Not3p, or Not5p has no effect on CYH2 pre-mRNA levels, while loss of Upf1p (a critical effector of NMD) significantly enhances its steady-state levels (Fig. 1B). These data suggest that the role of Not2p, Not3p, and Not5p in decapping does not involve the catalytic activity of Dcp2p and that they are regulators of decapping activity similar to Pat1p, Lsm1-7p, and Dhh1p (Coller and Parker 2004).
Tethered Not3p dramatically destabilizes an mRNA reporter
Given the similarity between Not3p and Not5p, and since deletion of NOT2 and NOT5 has a dramatic effect on mRNA decapping, we hypothesized that Not3p might also be involved in promoting mRNA decapping but perhaps its effects are restricted to specific mRNAs. Indeed, deletion of NOT3 only mildly affects the decay of MFA2 mRNA and has no effect on EDC1 mRNA steady-state levels (Fig. 1A,B). Without knowledge of the mRNA target(s) of Not3p, we elected to study its effects on message stability using a tethered function assay (Coller and Wickens 2007). A tethered approach allows for the assay of an RNA binding protein's function without knowledge of its real targets or the constraint of its RNA binding activity. We tested the function of Not3p on mRNA decay by fusing the full-length protein (amino acids 1–836) to the MS2 bacteriophage coat protein. The effect of the MS2-Not3p (1–836) chimera was then monitored using a PGK1 reporter harboring RNA recognition elements in its 3′ UTR that are bound by the MS2 coat protein. The insertion of tandem MS2 coat protein RNA binding sites does not alter the stability of this reporter (data not shown). Expression of the MS2 coat protein alone also results in a half-life that is similar to that of the endogenous transcript (25 min; Muhlrad et al. 1995). Dramatically, expression of MS2-Not3p (1–836) in wild-type cells results in a twofold destabilization of the PGK1 reporter mRNA (Fig. 2A). These data demonstrate that Not3p is a potent regulator of mRNA decay when tethered.
FIGURE 2.
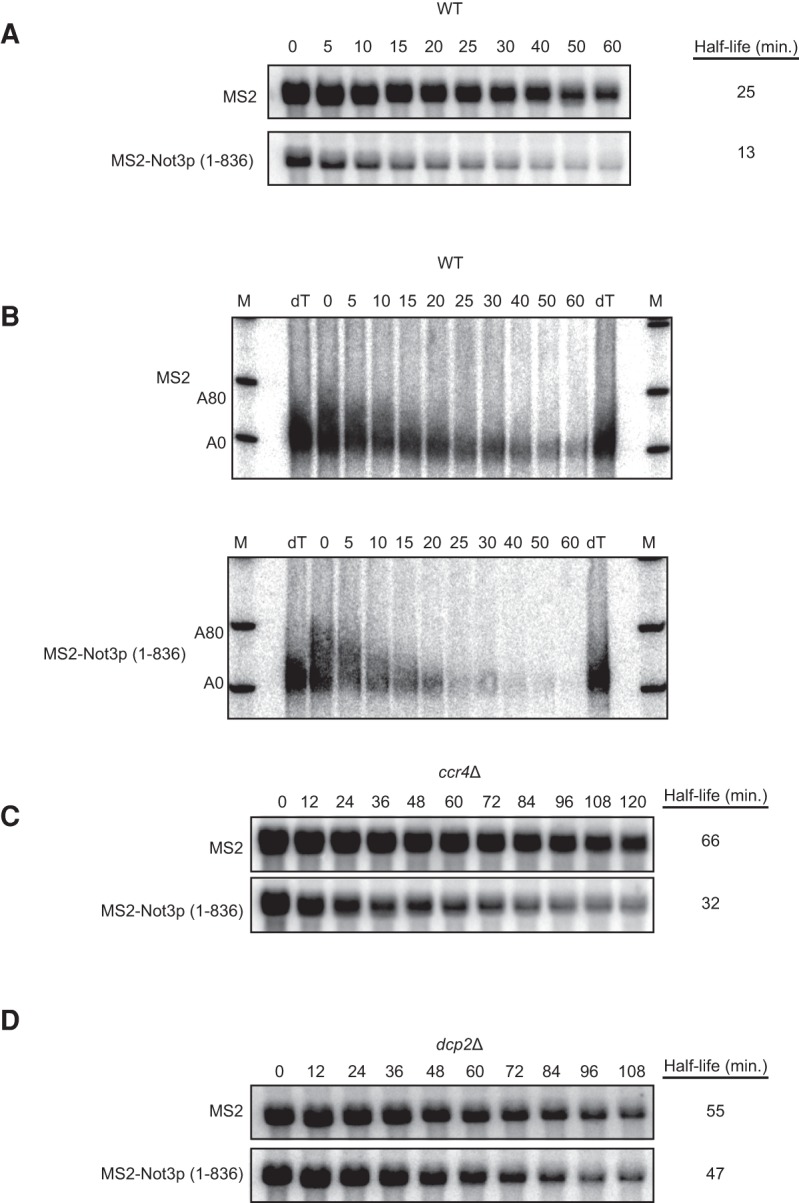
Not3p stimulates decapping of a PGK1 reporter when tethered. (A) Transcriptional shut-off followed by half-life analysis was performed using a tethering approach in which a PGK1 reporter containing MS2 binding sites in its 3′ UTR was coexpressed with either MS2 alone or an MS2-Not3p (1–836) fusion protein in wild-type cells. Time points above each lane represent time (in minutes) after transcriptional repression. Half-life was determined after quantifying PGK1 mRNA levels relative to an SCR1 RNA loading control (Supplemental Fig. S2A). (B) Analysis of A by high-resolution polyacrylamide Northern. dT lanes represent RNA cleaved with RNase H/oligo(dT) to indicate deadenylated PGK1 mRNA. A80 and A0 refer to the position of MFA2 mRNA that is fully adenylated or fully deadenylated, respectively. The SCR1 RNA loading control is shown in Supplemental Figure S2B. (C,D) Transcriptional shut-off followed by half-life analysis was performed as in A using ccr4Δ (C) and dcp2Δ (D) cells expressing the PGK1 reporter containing MS2 binding sites in its 3′ UTR, as well as either MS2 alone or MS2-Not3p (1–836). The SCR1 RNA loading controls for Figure 2C and D are shown in Supplemental Figure S2C and D, respectively.
Tethered Not3p stimulates mRNA decapping
We tested the effect of tethered Not3p (1–836) on deadenylation and/or decapping by repeating the transcriptional shut-off experiment and monitoring deadenylation by analysis of the 3′ end of the PGK1 reporter using high-resolution PAGE. To allow for sufficient resolution, the reporter was reduced in size by in vitro cleavage of the 3′ UTR by hybridizing a DNA oligo directed against a 23-nucleotide (nt) region near the termination codon in the presence of RNase H. The 3′ UTR fragment was then resolved on a 6% denaturing polyacrylamide gel and Northern analysis was conducted using a probe specific to the MS2 RNA recognition elements. As seen before, tethering of Not3p (1–836) to the PGK1 reporter in wild-type cells results in significant destabilization of the reporter mRNA (Fig. 2B). The PGK1 reporter, which has a maximum poly(A) tail length of 80 nt, reaches an oligoadenylated length by 15 min following transcriptional shut-off in both the MS2 alone and MS2-Not3p (1–836) expressing cells. Importantly, the PGK1 mRNA has a slow rate of decapping which manifests as a persistent, largely deadenylated species from 20 to 60 min (Muhlrad et al. 1995). Tethering of Not3p (1–836) mildly affects deadenylation but strongly results in the precocious destabilization of the capped, deadenylated species (Fig. 2B). These data suggest that tethered Not3p predominantly alters the decapping rate of mRNA.
Our data suggest that Not3p affects mRNA decapping rates. To verify this hypothesis, we repeated our transcriptional shut-off analysis of tethered Not3p (1–836) in strains mutant for the major deadenylase enzyme (i.e., ccr4Δ) or mutant for the major decapping enzyme (i.e., dcp2Δ). In the absence of the deadenylase Ccr4p, the reporter mRNA is stabilized as compared to a wild-type strain (i.e., a half-life of 66 min versus 25 min; Fig. 2A,C). This is to be expected since the reporter is metabolized like a normal mRNA and therefore requires Ccr4p for its decay. Importantly, expression of MS2-Not3p (1–836) still results in a significant decrease in PGK1 mRNA half-life even in the absence of the deadenylase (Fig. 2C). In contrast, tethered Not3p (1–836) has little appreciable effect on half-life in strains lacking the decapping enzyme (Fig. 2D). Together, these data demonstrate that the primary role of tethered Not3p is to stimulate mRNA decapping but not deadenylation since it requires Dcp2p but not Ccr4p for its activity.
Identification of a conserved amino acid motif required to elicit Not3p-dependent decapping
We have demonstrated that tethered Not3p stimulates the decapping of a reporter mRNA. Not3p is 836 amino acids long and has regions of evolutionary conservation both at the N-terminal and C-terminal ends. To determine the region of Not3p required for decapping activation, we performed structure function analysis of truncated polypeptides. Specifically, steady-state mRNA analysis was performed to determine what effect tethering Not3p truncations has on PGK1 mRNA levels (Fig. 3A,B). We find that the N-terminal end of Not3p is dispensable since deletion of the first 234 amino acids [MS2-Not3p (235–836)] still results in a fivefold decrease in PGK1 mRNA levels, similar to full-length Not3p [MS2-Not3p (1–836)]. In contrast, deletion of the C-terminal 144 amino acids of Not3p [MS2-Not3p (1–692)] significantly diminishes the ability of truncated Not3p to reduce PGK1 mRNA levels (70% mRNA remaining compared to 20% with full-length Not3p; Fig. 3A,B). To more precisely define the region of Not3p involved in mRNA destabilization, we made two smaller C-terminal truncations, deleting the last 96 [MS2-Not3p (1–740)] or 48 [MS2-Not3p (1–788)] amino acids of Not3p. We observed that whereas tethering Not3p (1–788) and full-length Not3p are indistinguishable in their ability to reduce PGK1 mRNA levels, tethered Not3p (1–740) is impaired (Fig. 3A,B). The above results suggest that the region between amino acids 741 and 788 in Not3p is critical for its role in mRNA destabilization.
FIGURE 3.

Conserved amino acid residues in the C terminus of Not3p are required for stimulation of mRNA decay. (A) Schematic representation of full-length Not3p [Not3p (1–836)], an N-terminal Not3p truncation [Not3p (235–836)] and three C-terminal truncations [Not3p (1–692), Not3p (1–740), and Not3p (1–788)], that were fused to the bacteriophage MS2 coat protein. (B) Steady-state Northern analysis of RNA isolated from wild-type cells coexpressing a PGK1 reporter containing MS2 binding sites in its 3′ UTR and either MS2 alone, MS2 fused to full-length Not3p, or MS2 fused to the Not3p truncations depicted in A. PGK1 mRNA levels were normalized to SCR1 RNA levels and are represented graphically relative to MS2 alone. (C) Bioinformatic analysis in various organisms of the region between amino acids 741 and 788 in the yeast Not3p protein reveals several conserved amino acid residues. The conserved amino acids at positions 742, 743, 745, 757, and 764 (highlighted in orange) in yeast Not3p were mutated to alanine residues. (D) Transcriptional shut-off followed by half-life analysis was performed using wild-type cells coexpressing the PGK1 reporter with MS2 recognition elements in its 3′ UTR and either MS2 alone, MS2 fused to wild-type Not3p, or MS2 fused to Not3p (W742A, F743A, R745A, E757A, F764A). Time after transcriptional shut-off is indicated in minutes above each lane. Half-life was determined after normalizing PGK1 mRNA levels to an SCR1 RNA loading control.
NOT3 is conserved from yeast to humans. Bioinformatic comparison of several NOT3 homologs reveals evolutionary conservation of 10 amino acid residues within the portion of the NOT box identified above to be important for destabilization of the PGK1 mRNA reporter by tethered Not3p (between amino acids 741 and 788 in the yeast protein; Fig. 3C). Considering this conservation, we reasoned that these residues may be important for the ability of tethered Not3p to drive mRNA decapping. Amino acids W742, F743, R745, E757, and F764 were mutated to alanine and expression of this mutant form of Not3p was confirmed by Western blot analysis (Supplemental Fig. S3). We repeated the transcriptional shut-off experiment and found that tethering Not3p (W742A, F743A, R745A, E757A, and F764A) results in a PGK1 mRNA half-life that is essentially identical to the half-life that results when MS2 alone is tethered (Fig. 3D). We conclude, therefore, that a conserved five amino acid patch is required for Not3p-mediated stimulation of decapping.
Not3p and Not5p promote mRNA decapping through a conserved motif
We have shown that five amino acids in the NOT box of Not3p are required for its ability to promote mRNA decapping. Interestingly, both Not2p and Not5p bear identical or similar amino acids in these positions (Fig. 4A). We tested whether this motif was also required for Not2p and Not5p to promote mRNA decapping. We mutated these residues to alanine in both Not2p and Not5p and tested the effect of these mutations on decay of the MFA2 mRNA. Importantly, alteration of this conserved domain has no effect on the ability of Not2p to promote mRNA decay, but strongly impedes the ability of Not5p to promote decay (Fig. 4B). This suggests that Not3p and Not5p promote mRNA decapping through a common mechanism (see below).
FIGURE 4.

Amino acids required for Not3p-mediated stimulation of decapping are conserved in Not2p and Not5p and are required for Not5p-mediated decapping stimulation. (A) Schematic representation of Not2p, Not3p, and Not5p, with colors representing regions of similarity between the NOT proteins. The purple region is indicative of the NOT box. Sequence alignment of the region surrounding the five amino acid patch in Not3p found to be required for promoting mRNA decapping and the equivalent region in Not2p and Not5p is shown. These five amino acids in Not3p, as well as the amino acids in the equivalent positions in Not2p and Not5p, are in red. (B) Transcriptional shut-off followed by half-life analysis of an MFA2 reporter containing a poly(G) tract in its 3′ UTR harbored in not2Δ cells expressing either wild-type Not2p or Not2p (W146A, L147A, K149A, E164A, F171A) (top two panels), or harbored in not5Δ cells expressing either wild-type Not5p or Not5p (W512A, Y513A, K515A, E528A, F535A) (bottom two panels). Time after transcriptional shut-off is indicated in minutes above each lane. Half-life was determined after normalizing the MFA2 mRNA levels to an SCR1 loading control (Supplemental Fig. S4). A80 and A0 refer to the position of MFA2 mRNA that is fully adenylated or fully deadenylated, respectively.
Not3p and Not5p bind to the decapping activator protein Pat1p
We have demonstrated that Not3p stimulates decapping and that a conserved patch of amino acids in the C terminus is required for this function. Importantly, however, Not3p is not rigorously documented to interact with the decapping complex. We sought, therefore, to identify a factor(s) required for Not3p decapping function. More precisely, we sought to identify factors that interact with full-length wild-type Not3p but fail to interact when mutations are made in the conserved domain we found to be required for the stimulation of decay. Not3p protein partners were identified using a traditional yeast two-hybrid approach. Specifically, a vector expressing full-length wild-type Not3p fused to the DNA binding domain of Gal4p [Gal4p-BD-Not3p (1–836)] was transformed into Saccharomyces cerevisiae strain yJC518 along with a full-length ORF library fused to the Gal4p activation domain. Protein–protein interactions were assayed through transcriptional activation of a HIS3 reporter resulting in an ability to grow on media lacking histidine and supplemented with 3-amino-1,2,4-triazole (3-AT). We screened over 500,000 colonies (thereby covering the complete ORF library 80 times). From this initial pool, we identified 50 colonies that grew on –His media (supplemented with 3-AT) within the first six days following transformation. Of these 50, only 19 isolates were found to robustly activate a second reporter, an ADE2 gene under the control of the GAL2 upstream activating sequence.
We next sought to identify binding partners required for the decapping function of Not3p. We isolated plasmid DNA from the 19 positive isolates and tested for interaction between these 19 isolates and a Gal4p DNA binding domain-Not3p fusion lacking the C terminus [i.e., Gal4p-BD-Not3p (1–692)] which fails to stimulate decapping when tethered. Of the original 19 isolates, 13 isolates were identified that interact with full-length Not3p but fail to interact with the C-terminal truncation Not3p (1–692). Sequencing identified that two of these plasmids contain the PAT1 gene, which encodes a decapping activator (Hatfield et al. 1996). The remaining 11 plasmids were found to contain ATG17, a gene involved in autophagy. We investigated the role of Atg17p in mRNA decay and found no discernible phenotype. Since Pat1p has well-documented links to mRNA decay and decapping, we continued our yeast two-hybrid analysis using this protein. We tested for interaction between the Gal4p activation domain-Pat1p fusion and the Gal4p DNA binding domain-Not3p fusion harboring alanine mutations in the five conserved amino acids required for decapping stimulation and observed no detectable interaction (Fig. 5A). Western blot analysis confirmed that both Gal4p-BD-Not3p (1–692) and Gal4p-BD-Not3p (W742A, F743A, R745A, E757A, and F764A) are expressed (Supplemental Fig. S5). Taken together, our yeast two-hybrid analysis shows that Pat1p interacts with full-length wild-type Not3p, and this interaction is lost with either of the decapping-deficient NOT3 alleles.
FIGURE 5.

Not3p and Not5p interact with the decapping activator protein Pat1p through a patch of five conserved amino acid residues. (A) Yeast two-hybrid analysis of yJC518 cells expressing the indicated proteins. Growth of cells on media lacking histidine and supplemented with 3-AT is indicative of an interaction between proteins. (B) Results from a yeast two-hybrid assay performed as in A using yJC518 cells coexpressing either Pat1p or Dhh1p fused to the Gal4p activation domain and the indicated NOT proteins fused to the Gal4p DNA binding domain. An interaction between two proteins is indicated by a (+) sign. No interaction is indicated by a (−) sign. ND, not determined.
Having documented that Not3p binds the decapping activator protein Pat1p and that this interaction is mediated via the conserved amino acids required for Not3p to elicit mRNA decay, we directly tested whether Gal4p-AD-Pat1p could interact with Gal4p-BD-Not2p and Gal4p-BD-Not5p. Not2p failed to interact with Pat1p while Not5p interacted strongly. Moreover, we find that mutating the amino acids in Not5p that correspond to the amino acids required for Not3p to interact with Pat1p results in a loss of interaction (Fig. 5B). Expression of Gal4p-BD-Not2p, as well as wild-type and mutant Gal4p-BD-Not5p, was confirmed by Western blot analysis (Supplemental Fig. S5). As a control, we tested whether Gal4p-BD-Not2p, Gal4p-BD-Not3p, and Gal4p-BD-Not5p interact with another decapping activator protein, Dhh1p (Gal4p-AD-Dhh1p), which has been shown to interact with Pat1p (Coller et al. 2001). We did not detect an interaction between Dhh1p and Not2p, Not3p, or Not5p (Fig. 5B). Together, our yeast two-hybrid analysis demonstrates that Not3p and Not5p specifically bind to the decapping activator protein Pat1p. Moreover, these data demonstrate that the conserved domain required for Not3p and Not5p to stimulate decapping is also required for Pat1p interaction.
DISCUSSION
Deadenylation represents a critical first step in the decay of most eukaryotic mRNAs (Chen and Shyu 2011; Norbury 2013). Loss of the poly(A) tail allows for the rapid removal of the 5′ cap structure. Coupling between deadenylation and decapping is thought to occur, in part, through the poly(A) binding protein (Pab1p) (Caponigro and Parker 1995). Pab1p ubiquitously binds mRNA poly(A) tails and physically interacts with the cap through the translational initiation factor eIF-4Gp. The Pab1p/eIF-4Gp interaction creates a “closed-loop” that is thought to explain why the poly(A) tail both stimulates mRNA translation and protects the mRNA from decapping (Amrani et al. 2008). Deadenylation, therefore, serves to break the closed-loop through loss of Pab1p binding and thereby exposes the 5′ cap to the decapping enzyme complex Dcp2p/Dcp1p. Consistent with this, genetic disruption of Pab1p allows mRNA decapping to occur, at some level, independent of prior deadenylation (Caponigro and Parker 1995). Moreover, tethering of Pab1p stabilizes mRNAs by blocking mRNA decapping (Coller et al. 1998). Despite these data, however, depletion of eIF-4Gp does not uncouple deadenylation-dependent mRNA decapping (Schwartz and Parker 1999). Moreover, nontranslating mRNA reporters are still degraded by deadenylation-dependent mRNA decapping (Beelman and Parker 1994; Muhlrad et al. 1995). Collectively, these data argue that other features of the decay machinery are involved in promoting the coupling of deadenylation with decapping independent of the known functions of Pab1p.
The coordination of mRNA decapping with prior deadenylation is the hallmark of eukaryotic mRNA decay (Decker and Parker 1993). Here we show that some of this coordination may be through direct recruitment of decapping factors by the deadenylase. First, we see that Not2p and Not5p, components of the Ccr4p/Not1p-5p deadenylase complex, inhibit mRNA decapping when mutated. Second, tethering of Not3p stimulates the robust decapping of a reporter substrate. Third, Not3p and Not5p specifically interact with the decapping regulator Pat1p. Finally, the interaction of Not3p and Not5p with Pat1p occurs through a conserved amino acid motif. Together, these data suggest a model where components of the deadenylase may recruit and aid in the assembly of the decapping complex. Thus, at some level, the coupling of deadenylation and decapping might be a simple result of a linear order of assembly of processing factors on the mRNA.
MATERIALS AND METHODS
Yeast strains
The genotypes of the yeast strains used in this study are listed in Table 1.
TABLE 1.
Yeast strains

Plasmids
The plasmids and oligonucleotides used in this study are listed in Tables 2 and 3, respectively.
TABLE 2.
Plasmids

TABLE 3.
Oligonucleotides

Tethered function constructs
Full-length NOT3 and NOT3 (codons 235–836) were amplified using oJC1148/oJC1149 and oJC1052/oJC1252, respectively, for cloning downstream from MS2 in pJC398 using NheI and XhoI sites to create pJC435 [MS2-NOT3 (1–836)] and pJC482 [MS2-NOT3 (235–836)]. oJC1253/oJC1254, oJC1326/oJC1327, and oJC1328/oJC1329 were used to introduce a stop codon followed by an XhoI site after codons 692, 740, and 788, respectively, in NOT3 within pJC435. These plasmids were then digested with XhoI to release the C-terminal NOT3 fragments and self-ligated to create pJC483 [MS2-NOT3 (1–692)], pJC490 [MS2-NOT3 (1–740)], and pJC491 [MS2-NOT3 (1–788)]. Point mutations W742A, F743A, R745A, E757A, F764A were introduced in NOT3 within pJC435 using oJC1384/oJC1385 and oJC1386/oJC1387 to create pJC506.
NOT2 and NOT5 expression constructs
Full-length NOT2 in addition to 500 bp of 5′ UTR sequence and 460 bp of 3′ UTR sequence was amplified using oJC2067/oJC2068 (BamHI and SphI sites) and oJC2067/oJC2098 (BamHI and SpeI sites) for cloning into pJC70 and pJC387, respectively, to create pJC613 and pJC617, respectively. oJC2078/oJC2079 and oJC2080/oJC2081 were used to introduce W146A, L147A, K149A, E164A, and F171A point mutations in NOT2 within pJC613 to create pJC622. pJC622 was then used as a template for amplification of NOT2 (W146A, L147A, K149A, E164A, F171A) in addition to 500 bp of 5′ UTR sequence and 460 bp of 3′ UTR sequence using oJC2067/oJC2098 for cloning into pJC387 using BamHI and SpeI sites to create pJC627.
Full-length NOT5 in addition to 500 bp of 5′ UTR sequence and 460 bp of 3′ UTR sequence was amplified using oJC2069/oJC2070 (BamHI and SphI sites) and oJC2069/oJC2099 (BamHI and SpeI sites) for cloning into pJC70 and pJC387, respectively, to create pJC616 and pJC618, respectively. oJC2082/oJC2083 and oJC2084/oJC2085 were used to introduce W512A, Y513A, K515A, E528A, and F535A point mutations in NOT5 within pJC616 to create pJC626. NOT5 (W512A, Y513A, K515A, E528A, F535A) in addition to 500 bp of 5′ UTR sequence and 460 bp of 3′ UTR sequence was amplified from pJC626 using oJC2069/oJC2099 for cloning into pJC387 using BamHI and SpeI sites to create pJC628.
Yeast two-hybrid constructs
oJC1465/oJC1466 were used to replace a portion of the EcoRI and SmaI/XmaI sites within the multiple cloning site of pJC351 (pGBKT7) with an SpeI site to create pJC513. Full-length NOT3 and NOT3 (codons 1–692) were amplified using oJC1467/oJC1468 and oJC1467/oJC1469, respectively, for cloning into pJC513 using NcoI and SpeI sites to create pJC514 [GAL4-BD-NOT3 (1–836)] and pJC515 [GAL4-BD-NOT3 (1–692)]. NOT3 (W742A, F743A, R745A, E757A, F764A) was amplified from pJC506 using oJC1467/oJC1468 for cloning into pJC513 using NcoI and SpeI sites to create pJC516.
Full-length NOT2 was amplified using oJC2011/oJC2012 (SpeI and BamHI sites) and cloned into pJC513. oJC2977/oJC2978 were subsequently used to mutate the SpeI site upstream of NOT2 from ACTAGT to ACAAGT to create pJC818 (GAL4-BD-NOT2). Full-length NOT5 was amplified using oJC2015/oJC2016 (NcoI and SpeI sites) and cloned into pJC513 to create pJC600 (GAL4-BD-NOT5). oJC2015/oJC2016 were used to amplify NOT5 (W512A, Y513A, K515A, E528A, F535A) from pJC628 for cloning into pJC513 using NcoI and SpeI sites to create pJC644. pJC361 (GAL4-AD-PAT1) and pJC362 (GAL4-AD-DHH1) were created by cloning full-length PAT1 (amplified with oJC759/oJC760) and full-length DHH1 (amplified with oJC720/oJC721) into pJC352 using BamHI and SacI sites.
Transcriptional shut-off and steady-state RNA analysis
Cells expressing the MFA2 reporter harbored in pJC312 and containing a poly(G) tract in its 3′ UTR or the PGK1 reporter harbored in pJC441 and containing MS2 binding sites in its 3′ UTR, both of which are under the control of the inducible GAL1 promoter, were grown at 24°C in synthetic media (pH 6.5) containing 2% galactose, 1% sucrose, and lacking the appropriate amino acids. Once the cells reached mid-log phase, transcription was repressed by transferring the cells into media containing 4% glucose. Aliquots were harvested and immediately frozen on dry ice at the time points indicated in each figure. Total RNA was isolated as previously described (Geisler et al. 2012). Up to 40 µg of RNA from each time point was run on either 1.4% agarose formaldehyde gels or 6% denaturing polyacrylamide gels and the RNA was transferred to nylon membranes. Northern analysis was performed using radiolabeled oligonucleotide probes that are complementary to either the poly(G) tract in the MFA2 reporter (oJC168), the MS2 binding sites in the PGK1 reporter (oJC1006), or SCR1 RNA as a loading control (oJC306).
For the RNase H cleavage experiment shown in Figure 2B, a transcriptional shut-off experiment was performed as above using wild-type cells coexpressing the PGK1 reporter (pJC441) and either MS2 or MS2-Not3p (1–836). Of note, 35 µg of the isolated total RNA was hybridized to oJC591, which binds just upstream of the PGK1 stop codon, and the DNA–RNA hybrid was digested with RNase H. In addition, RNase H cleavages of oJC591- and oligo(dT)-treated samples were prepared to indicate the migration of a completely deadenylated PGK1 mRNA. The cleavage products were subsequently resolved on 6% denaturing polyacrylamide gels and the 3′ end of the PGK1 reporter was detected by Northern analysis using radiolabeled oJC1006.
For the steady-state RNA analyses in Figures 1B and 3B, cells were grown to mid-log phase at 24°C in synthetic media (pH 6.5) containing 2% glucose (Fig. 1B) or 2% galactose and 1% sucrose (Fig. 3B) and lacking the appropriate amino acids. Of note, 35 μg of isolated total RNA was run on 1.4% agarose formaldehyde gels, transferred to nylon membranes, and probed with radiolabeled oligonucleotide probes complementary to EDC1 (oJC221) and CYH2 mature and pre-mRNA (oKB147) for Figure 1B, and complementary to the MS2 binding sites in the 3′ UTR of the PGK1 reporter (oJC1006) for Figure 3B. Radiolabeled oligonucleotide oJC306 was used to detect SCR1 RNA as a loading control.
Yeast two-hybrid assay
yJC518 cells expressing full-length wild-type Not3p fused to the Gal4p DNA binding domain were grown to mid-log phase before transformation with a Saccharomyces cerevisiae ORF library fused to the Gal4p activation domain as described in the Matchmaker GAL4 Two-Hybrid System 3 & Libraries User Manual (Clontech). At least 500,000 colonies were screened and transformants were plated onto synthetic media lacking tryptophan, leucine, and histidine, and supplemented with 5 mM 3-amino-1,2,4-triazole (3-AT), to assay for protein–protein interactions that result in transcriptional activation of a HIS3 reporter. These and all subsequent yeast two-hybrid assay plates were incubated at 30°C. Fifty colonies were observed during the first six days following transformation. These 50 colonies were next streaked onto synthetic media lacking tryptophan, leucine, and adenine, to assay for activation of ADE2, a second reporter in this system. Robust growth of cream-colored colonies was observed for 19 out of the 50 isolates screened, indicating strong activation of the ADE2 gene. Plasmid DNA was isolated from these 19 isolates and transformed into yJC518 cells expressing a C-terminally truncated version of Not3p [Not3p (1–692)] fused to the Gal4p DNA binding domain to determine which plasmids express proteins that require the C-terminal end of Not3p for interaction. Of the 19 isolates expressing proteins that interacted strongly with full-length Not3p, 13 isolates were nonviable on synthetic media plates lacking tryptophan, leucine, and histidine, and containing 5 mM 3-AT when Not3p was C-terminally truncated. Following sequencing of these 13 plasmids, it was determined that two of the plasmids contain the PAT1 gene. One of these Gal4p activation domain-Pat1p fusion constructs (pJC526) was next transformed into yJC518 cells expressing Not3p (W742A, F743A, R745A, E757A, F764A) fused to the Gal4p DNA binding domain and transformants were again plated on synthetic media lacking tryptophan, leucine, and histidine, and supplemented with 5 mM 3-AT.
For the directed yeast two-hybrid plating assays in Figure 5, yJC518 cells expressing the indicated proteins were patched onto synthetic media plates that lack tryptophan and leucine, and either contain histidine or lack histidine and contain 5 mM 3-AT (Not2p, wild-type and mutant Not3p, and mutant Not5p assays) or 15 mM 3-AT (wild-type Not5p assay). Growth on the plates lacking histidine, which is indicative of protein–protein interactions that lead to activation of the HIS3 reporter, was analyzed three to five days after patching.
Western analysis
Cells were grown at 24°C (Supplemental Fig. S3) or 30°C (Supplemental Fig. S5) to mid-log phase in synthetic media (pH 6.5) containing 2% galactose and 1% sucrose (Supplemental Fig. S3) or 2% glucose (Supplemental Fig. S5). Proteins were isolated and Western blot analysis was performed as previously described (Geisler et al. 2012). MS2 fusion proteins were detected using Millipore anti-MS2, Gal4p DNA binding domain fusion proteins were detected using Roche anti-c-myc, and Pgk1p (used as a loading control) was detected using Thermo Fisher Scientific anti-Pgk1p.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank the members of the Coller and Baker laboratories for advice and input into this work. Furthermore, we thank Dr. Marv Wickens for the gift of the yeast ORF-AD library. This work was funded by National Institutes of Health grant GM080465 to J.C.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.054742.115.
REFERENCES
- Amrani N, Ghosh S, Mangus DA, Jacobson A. 2008. Translation factors promote the formation of two states of the closed-loop mRNP. Nature 453: 1276–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Y, Salvadore C, Chiang YC, Collart MA, Liu HY, Denis CL. 1999. The CCR4 and CAF1 proteins of the CCR4-NOT complex are physically and functionally separated from NOT2, NOT4, and NOT5. Mol Cell Biol 19: 6642–6651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basquin J, Roudko VV, Rode M, Basquin C, Séraphin B, Conti E. 2012. Architecture of the nuclease module of the yeast Ccr4-not complex: the Not1-Caf1-Ccr4 interaction. Mol Cell 48: 207–218. [DOI] [PubMed] [Google Scholar]
- Bawankar P, Loh B, Wohlbold L, Schmidt S, Izaurralde E. 2013. NOT10 and C2orf29/NOT11 form a conserved module of the CCR4-NOT complex that docks onto the NOT1 N-terminal domain. RNA Biol 10: 228–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beelman CA, Parker R. 1994. Differential effects of translational inhibition in cis and in trans on the decay of the unstable yeast MFA2 mRNA. J Biol Chem 269: 9687–9692. [PubMed] [Google Scholar]
- Bhaskar V, Roudko V, Basquin J, Sharma K, Urlaub H, Séraphin B, Conti E. 2013. Structure and RNA-binding properties of the Not1-Not2-Not5 module of the yeast Ccr4-Not complex. Nat Struct Mol Biol 20: 1281–1288. [DOI] [PubMed] [Google Scholar]
- Brachmann CB, Davies A, Cost GJ, Caputo E, Li J, Hieter P, Boeke JD. 1998. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 14: 115–132. [DOI] [PubMed] [Google Scholar]
- Caponigro G, Parker R. 1995. Multiple functions for the poly(A)-binding protein in mRNA decapping and deadenylation in yeast. Genes Dev 9: 2421–2432. [DOI] [PubMed] [Google Scholar]
- Chen CY, Shyu AB. 2011. Mechanisms of deadenylation-dependent decay. Wiley Interdiscip Rev RNA 2: 167–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coller J, Parker R. 2004. Eukaryotic mRNA decapping. Annu Rev Biochem 73: 861–890. [DOI] [PubMed] [Google Scholar]
- Coller J, Wickens M. 2007. Tethered function assays: an adaptable approach to study RNA regulatory proteins. Methods Enzymol 429: 299–321. [DOI] [PubMed] [Google Scholar]
- Coller JM, Gray NK, Wickens MP. 1998. mRNA stabilization by poly(A) binding protein is independent of poly(A) and requires translation. Genes Dev 12: 3226–3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coller JM, Tucker M, Sheth U, Valencia-Sanchez MA, Parker R. 2001. The DEAD box helicase, Dhh1p, functions in mRNA decapping and interacts with both the decapping and deadenylase complexes. RNA 7: 1717–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daugeron MC, Mauxion F, Seraphin B. 2001. The yeast POP2 gene encodes a nuclease involved in mRNA deadenylation. Nucleic Acids Res 29: 2448–2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker CJ, Parker R. 1993. A turnover pathway for both stable and unstable mRNAs in yeast: evidence for a requirement for deadenylation. Genes Dev 7: 1632–1643. [DOI] [PubMed] [Google Scholar]
- Geisler S, Lojek L, Khalil AM, Baker KE, Coller J. 2012. Decapping of long noncoding RNAs regulates inducible genes. Mol Cell 45: 279–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Jacobson A. 2010. RNA decay modulates gene expression and controls its fidelity. Wiley Interdiscip Rev RNA 1: 351–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz RD, Sugino A. 1988. New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene 74: 527–534. [DOI] [PubMed] [Google Scholar]
- Hatfield L, Beelman CA, Stevens A, Parker R. 1996. Mutations in trans-acting factors affecting mRNA decapping in Saccharomyces cerevisiae. Mol Cell Biol 16: 5830–5838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W, Sweet TJ, Chamnongpol S, Baker KE, Coller J. 2009. Co-translational mRNA decay in Saccharomyces cerevisiae. Nature 461: 225–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita M, Oike Y, Nagashima T, Kadomatsu T, Tabata M, Suzuki T, Nakamura T, Yoshida N, Okada M, Yamamoto T. 2011. Obesity resistance and increased hepatic expression of catabolism-related mRNAs in Cnot3+/− mice. EMBO J 30: 4678–4691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhlrad D, Parker R. 2005. The yeast EDC1 mRNA undergoes deadenylation-independent decapping stimulated by Not2p, Not4p, and Not5p. EMBO J 24: 1033–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhlrad D, Decker CJ, Parker R. 1995. Turnover mechanisms of the stable yeast PGK1 mRNA. Mol Cell Biol 15: 2145–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neely GG, Kuba K, Cammarato A, Isobe K, Amann S, Zhang L, Murata M, Elmén L, Gupta V, Arora S, et al. 2010. A global in vivo Drosophila RNAi screen identifies NOT3 as a conserved regulator of heart function. Cell 141: 142–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norbury CJ. 2013. Cytoplasmic RNA: a case of the tail wagging the dog. Nat Rev Mol Cell Biol 14: 643–653. [DOI] [PubMed] [Google Scholar]
- Schwartz DC, Parker R. 1999. Mutations in translation initiation factors lead to increased rates of deadenylation and decapping of mRNAs in Saccharomyces cerevisiae. Mol Cell Biol 19: 5247–5256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JE, Baker KE. 2015. Nonsense-mediated RNA decay—a switch and dial for regulating gene expression. Bioessays 37: 612–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweet T, Kovalak C, Coller J. 2012. The DEAD-box protein Dhh1 promotes decapping by slowing ribosome movement. PLoS Biol 10: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker M, Valencia-Sanchez MA, Staples RR, Chen J, Denis CL, Parker R. 2001. The transcription factor associated Ccr4 and Caf1 proteins are components of the major cytoplasmic mRNA deadenylase in Saccharomyces cerevisiae. Cell 104: 377–386. [DOI] [PubMed] [Google Scholar]
- Tucker M, Staples RR, Valencia-Sanchez MA, Muhlrad D, Parker R. 2002. Ccr4p is the catalytic subunit of a Ccr4p/Pop2p/Notp mRNA deadenylase complex in Saccharomyces cerevisiae. EMBO J 21: 1427–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf J, Passmore LA. 2014. mRNA deadenylation by Pan2-Pan3. Biochem Soc Trans 42: 184–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.