

Abstract
Adenosine-to-inosine RNA editing by ADARs affects thousands of adenosines in an organism's transcriptome. However, adenosines are not edited at equal levels nor do these editing levels correlate well with ADAR expression levels. Therefore, additional mechanisms are utilized by the cell to dictate the editing efficiency at a given adenosine. To examine cis- and trans-acting factors that regulate A-to-I editing levels specifically in neural cells, we utilized the model organism Caenorhabditis elegans. We demonstrate that a double-stranded RNA (dsRNA) binding protein, ADR-1, inhibits editing in neurons, which is largely masked when examining editing levels from whole animals. Furthermore, expression of ADR-1 and mRNA expression of the editing target can act synergistically to regulate editing efficiency. In addition, we identify a dsRNA region within the Y75B8A.8 3′ UTR that acts as a cis-regulatory element by enhancing ADR-2 editing efficiency. Together, this work identifies mechanisms that regulate editing efficiency of noncoding A-to-I editing sites, which comprise the largest class of ADAR targets.
Keywords: ADAR, RNA editing, double-stranded RNA, neurons, regulation of RNA editing
INTRODUCTION
Post-transcriptional modification of RNA bases is one mechanism that organisms utilize to diversify the transcripts produced within different cell types from a static genomic sequence (Li and Mason 2014). With over 100 million predicted occurrences in the human transcriptome, one of the most prevalent RNA base modifications is the deamination of adenosine (Bazak et al. 2014). This modification, commonly referred to as RNA editing, is catalyzed by a family of proteins called adenosine deaminases that act on RNA or ADARs (Savva et al. 2012). This type of RNA editing is limited to structured RNA as ADARs only target regions of dsRNA for deamination (Nishikura 2010).
ADARs convert adenosine to inosine, which is a biological mimic of guanosine (Pullirsch and Jantsch 2010). ADAR editing activity has the potential to alter mRNA coding potential, affect alternative splicing, and influence miRNA biogenesis and targeting (Hundley and Bass 2010; Rieder and Reenan 2012; Rosenthal and Seeburg 2012; Wulff and Nishikura 2012). Moreover, the editing levels of individual sites can vary considerably during development and rarely reach 100% editing efficiency demonstrating the dynamic effect of ADARs on the transcriptome (Wahlstedt et al. 2009).
While much recent work has been dedicated to identifying new editing sites, understanding the factors that influence how efficiently an adenosine is deaminated stands out as an area of the RNA editing field with many unanswered questions. ADARs prefer an adenosine, uridine or cytidine 5′ of the target adenosine while guanosine is favored 3′ to the adenosine (Lehmann and Bass 2000; Eggington et al. 2011; Bahn et al. 2012; Bazak et al. 2014). However, editing sites with the same surrounding nucleotide context can exhibit drastically different editing levels, suggesting this is not the only determinant of editing efficiency (Kawahara et al. 2008; Tian et al. 2011; Wheeler et al. 2015). Alternative splicing of ADAR transcripts (Lai et al. 1997; Rueter et al. 1999) and post-transcriptional modification (Desterro et al. 2005) have both been observed to cause decreased ADAR activity. In addition, RNA-binding proteins can both promote and repress editing of specific adenosines (Bhogal et al. 2011; Tariq et al. 2013; Washburn et al. 2014). However, how much these mechanisms interact to determine overall editing efficiency and whether these mechanisms are active in all cell types is poorly understood. Furthermore, it has been suggested that levels of A-to-I editing do not always directly correlate with ADAR expression levels (Wahlstedt et al. 2009). For example, the editing efficiency of the Filamin A mRNA in mice can be drastically different in various tissues even though these tissues express the same level of ADAR and Filamin A mRNA (Stulić and Jantsch 2013). However, this does not address if altering the expression levels within the same tissue will have an influence on editing efficiency. Increasing the expression of ADAR1 in cell culture has been observed to increase the editing efficiency of hNeil1 (Daniel et al. 2014). This suggests that altering the ratio of ADARs and the editing target within a singular tissue context can influence editing efficiency; however, this has not yet been examined in vivo. Therefore, understanding the cellular mechanisms that dictate editing efficiency and how these mechanisms are regulated is paramount to understanding ADAR editing activity.
Recently, local dsRNA structures in close proximity, but not encompassing the ADAR editing site, have been observed to influence site-specific editing of coding sequences. In the Drosophila editing target para, dsRNA elements, including a predicted pseudoknot, created by the intron downstream from exonic editing sites, influence editing efficiency even though these regions lack editing sites (Rieder et al. 2013). Similarly, downstream dsRNA hairpins within the introns of the human Gabra-3 and Neil1 transcripts have also been shown to increase editing efficiency of adjacent editing sites in coding regions (Daniel et al. 2012, 2014). While all of these examples involved regulating editing efficiency in coding regions, it is unclear whether this mechanism also extends to editing events in noncoding regions, which harbor the vast majority of editing sites identified to date (Bazak et al. 2014; Washburn et al. 2014; Whipple et al. 2015).
To examine mechanisms that regulate noncoding editing efficiency in vivo, we utilized the model organism Caenorhabditis elegans. In C. elegans, two ADAR proteins, ADR-1 and ADR-2, are present in the genome (Tonkin et al. 2002). We previously demonstrated that ADR-1 does not edit RNA, but it can function to regulate the extent to which specific adenosines are edited by ADR-2 (Washburn et al. 2014). In addition, we identified a novel C. elegans editing target, the mRNA Y75B8A.8, and characterized the pattern of editing events within its 3′ UTR (Wheeler et al. 2015). As this 3′ UTR contains two highly edited sites, we utilized this gene to generate a reporter system to examine cis- and trans-acting factors that affect editing efficiency in vivo. In our prior approaches, we utilized RNA collected from whole animals, which could make examination of editing regulation difficult if distinct regulatory mechanisms are present in different tissues.
Using our reporter system, we demonstrate that the extent of editing of the Y75B8A.8 3′ UTR is significantly different in neurons compared to editing levels of Y75B8A.8 observed from whole animal RNA. We demonstrate that the major determinant in this difference is the presence of ADR-1. In addition, the mRNA expression level of the editing target can act synergistically with ADR-1 expression to alter editing efficiency. Last, removing an adjacent unedited region of dsRNA significantly decreases ADR-2 editing efficiency, suggesting that this region acts to recruit ADR-2 to the downstream editing sites.
RESULTS AND DISCUSSION
Tissue-specific reporter reveals distinct editing efficiency in neurons
To determine cis- and trans-acting factors that affect editing efficiency and exclude differences in editing levels due to distinct modes of regulation across tissues, a reporter system to examine editing of the Y75B8A.8 3′ UTR exclusively in neurons was developed. Briefly, the dsRNA region of the Y75B8A.8 3′ UTR was cloned into a GFP vector under the control of the pan-neuronal rab3 promoter. The reporter also includes the 3′ UTR of unc-54, which is not edited, but promotes transcript stability (Fig. 1A). This neuronal reporter construct together with a control construct expressing RFP, but lacking dsRNA, was introduced into wild-type worms via microinjection. To avoid cross-contamination in analyzing expression and editing of the endogenous and reporter transcripts, specific primers were designed to amplify only the endogenous Y75B8A.8 3′ UTR or the neuronal reporter. Compared to the endogenous Y75B8A.8 3′ UTR collected from total worm RNA, editing in the neuronal reporter was significantly lower with a 28% reduction in editing at site 227 and a 40% reduction at site 228 (Fig. 1B).
FIGURE 1.
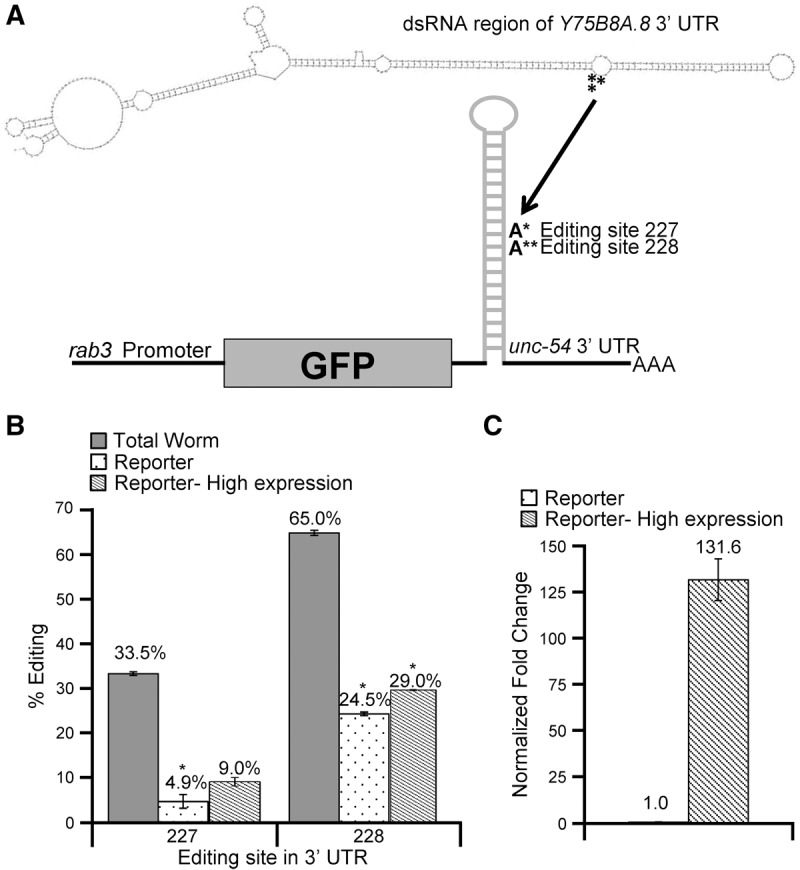
Y75B8A.8 3′ UTR neuronal editing reporter. (A) Schematic of the Y75B8A.8 3′ UTR neuronal reporter. The dsRNA region of the Y75B8A.8 3′ UTR (322 nt) was cloned into a vector downstream from the GFP coding region and upstream of the unc-54 3′ UTR. The pan-neuronal rab3 promoter drove reporter expression. Secondary structure of Y75B8A.8 dsRNA region was generated with mfold (Zuker 2003). Asterisks denote editing sites 227 (*) and 228 (**) in the mfold structure. (B, C) Error bars represent standard error of the mean (SEM). Significant changes (P ≤ 0.0005) by Student's t-test are marked with an asterisk. (B) Editing levels for both the endogenous Y75B8A.8 3′ UTR (gray), the Y75B8A.8 neuronal reporter (black dots), and the highly expressed Y75B8A.8 3′ UTR neuronal reporter (hatched) from three biological replicates of staged young-adult wild-type worms. (C) Relative expression of the Y75B8A.8 3′ UTR neuronal reporter (black dots) or the highly expressed Y75B8A.8 3′ UTR neuronal reporter (hatched). Fold change was determined by normalizing to the relative GFP expression of the Y75B8A.8 3′ UTR neuronal reporter (black dots).
One possibility for the reduced editing of the Y75B8A.8 3′ UTR reporter compared to endogenous Y75B8A.8 mRNA could be that the reporter mRNA expression is significantly lower than the endogenous mRNA expression, thus preventing significant amounts of ADR-2 from binding to the reporter mRNA. Another possibility for the reduced editing of the reporter might be an inability of ADR-2 to compete with neural RNA-binding factors for binding to the reporter mRNA. To examine these possibilities, we sought to increase the mRNA available to ADR-2 by creating additional worm lines that exhibited higher neural mRNA expression. This was accomplished by varying the concentration of the Y75B8A.8 neuronal reporter DNA in the microinjection mix. For each strain, mRNA expression level of the neuronal reporter and a rab3::RFP control, which was maintained at a constant concentration in all of the injection mixes, was determined by qRT-PCR. By comparing the expression of GFP, which is harbored by the neuronal reporter, to the control RFP, the relative mRNA expression of the neuronal reporter constructs was calculated. Using this method, we were able to increase expression of the neuronal reporter by more than 100-fold (Fig. 1C). However, when comparing editing levels at sites 227 and 228 of this highly expressed neuronal reporter to the neuronal reporter that was expressed 100-fold lower, editing levels only differed at site 228 by 5.5% (Fig. 1B). This indicates that differences in mRNA expression between the endogenous Y75B8A.8 mRNA and the neuronal reporter are not the sole determinant of the reduced editing levels observed in neurons.
ADR-1 represses editing efficiency of the Y75B8A.8 3′ UTR in neurons
To gain insight into the difference in editing levels of Y75B8A.8 in neurons compared to the whole animal, we next focused on ADR-1. We previously identified ADR-1 as a major regulator of editing in C. elegans (Washburn et al. 2014), and ADR-1 is known to be highly expressed in C. elegans neurons (Tonkin et al. 2002). To assess if ADR-1 regulates editing levels of the endogenous Y75B8A.8 3′ UTR, we examined editing efficiency in adr-1(-) null worms. When compared to wild-type, editing levels of the endogenous 3′ UTR are repressed by ADR-1 as editing efficiency increases nearly 6% at site 227 and 14% at site 228 in the absence of ADR-1 expression (Fig. 2). This increased editing level is due to loss of adr-1 and not other differences in the strain backgrounds, as reintroduction of an ADR-1 transgene restores editing levels of the endogenous Y75B8A.8 mRNA to wild-type levels (Supplemental Fig. 1). To examine if ADR-1 expression also affects editing efficiency of the Y75B8A.8 3′ UTR in neurons, the transgenic animals harboring the Y75B8A.8 3′ UTR neuronal reporters were crossed to worms lacking ADR-1 expression to generate animals expressing the Y75B8A.8 3′ UTR neuronal reporter in the adr-1(-) background. Interestingly, adr-1(-) males were unable to successfully mate (data not shown); therefore, adr-1(-);adr-2(-) males were utilized for the cross. Consistent with the effects of adr-1 on endogenous Y75B8A.8, editing at both sites 227 and 228 in the neuronal reporter is significantly higher in animals lacking adr-1 (Fig. 2). However, in neurons, ADR-1 expression has a much greater influence with a 20% increase in editing at site 227 and a nearly 33% increase at site 228. This demonstrates that in the tissue-specific context of neurons, ADR-1 repression of editing efficiency at these sites has a more pronounced effect. This suggests that a portion of endogenous Y75B8A.8 transcripts is located in cells lacking ADR-1 or with lower expression of ADR-1.
FIGURE 2.
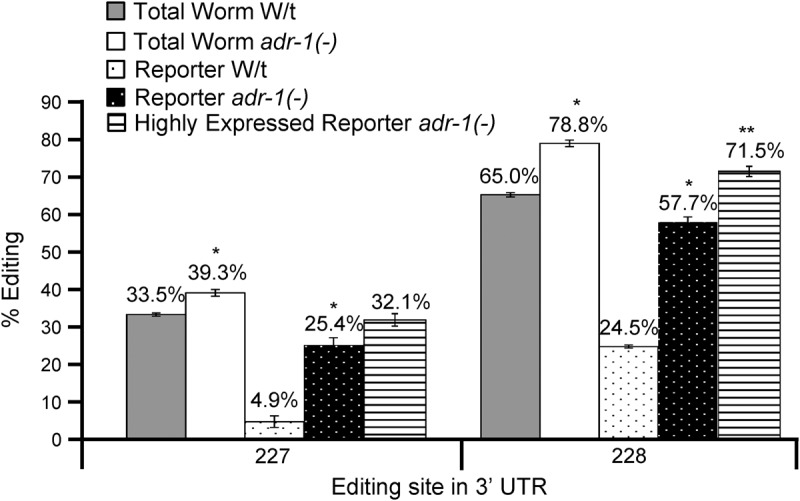
ADR-1 has a greater effect on repressing editing efficiency in neurons. (A) Editing levels for sites 227 and 228 in the endogenous Y75B8A.8 3′ UTR from wild-type worms (gray), endogenous Y75B8A.8 3′ UTR from adr-1(-) worms (white), the Y75B8A.8 3′ UTR neuronal reporter in wild-type worms (black dots), the Y75B8A.8 3′ UTR neuronal reporter in adr-1(-) worms (white dots), and the highly expressed Y75B8A.8 3′ UTR neuronal reporter in adr-1(-) worms (horizontal lines) were determined from three biological replicates of staged young-adult worms. Error bars represent SEM. Significant changes (P ≤ 0.005) between the respective wild-type and adr-1 editing levels are marked with an asterisk, and significant changes (P ≤ 0.005) between the highly expressed reporter and the reporter in adr-1 are marked by two asterisks.
Because ADR-1 represses editing in the Y75B8A.8 3′ UTR and ADR-1 is a known dsRNA binding protein, it is possible that ADR-1 binds to the Y75B8A.8 3′ UTR in neurons and blocks ADR-2 access to the target adenosines resulting in decreased editing. Additionally, as ADR-1 is known to be expressed approximately 10-fold higher than ADR-2 in the worm (Hundley et al. 2008), this ADR-1 repression of the editing model is consistent with the fact that increasing mRNA expression had a limited influence on editing efficiency in wild-type worms. To examine this, editing levels of the highly expressed neuronal reporters in the adr-1(-) background were determined (Fig. 2). In agreement with wild-type worms, mRNA expression does not significantly affect editing at site 227 in the absence of adr-1. However, at site 228, editing efficiency increases nearly 14% with increased mRNA expression in the absence of adr-1, which is greater than the increase observed in wild-type worms. This suggests that in the absence of ADR-1, mRNA expression has a greater impact on influencing editing levels at site 228. Furthermore, this demonstrates that while altering mRNA expression alone may have only a slight impact on editing levels, combining high mRNA expression with a loss of ADR-1 expression has an additive effect on increasing editing efficiency.
A dsRNA region within the Y75B8A.8 3′ UTR enhances editing efficiency
After determining that both mRNA and ADR-1 expression influence editing of the Y75B8A.8 3′ UTR neuronal reporter, we wanted to determine the minimal region of the Y75B8A.8 3′ UTR required for ADR-1 editing repression. Within the Y75B8A.8 3′ UTR, a dsRNA region adjacent to editing sites 227 and 228 contains no detectable editing sites (Wheeler et al. 2015). As ADR-1 likely competes with ADR-2 for access to the dsRNA surrounding the editing sites to repress editing, we speculated this region was not required ADR-1 editing repression. To assess this possibility, we generated a truncated version of the Y75B8A.8 neuronal reporter (Fig. 3A). Interestingly, the truncated reporter exhibited surprisingly lower editing levels at both editing sites 227 and 228 (Fig. 3B). In wild-type worms, editing at site 227 is undetectable in the truncated reporter and reduced by almost 20% at site 228. Furthermore, greater truncations of the Y75B8A.8 3′ UTR did not result in further decreases in editing efficiency (data not shown). This suggests that we serendipitously identified a region of the Y75B8A.8 that enhances editing efficiency. As the region absent in the truncated reporter does not contain any editing sites, this reduction in editing efficiency also cannot be attributed to loss of downstream editing sites.
FIGURE 3.

A dsRNA region within the Y75B8A.8 3′ UTR enhances editing efficiency. (A) Schematic showing the folded secondary structure of the full-length Y75B8A.8 neuronal reporter and the secondary structure of the truncated Y75B8A.8 neuronal reporter as predicted by mfold (Zuker 2003). Arrows indicate the location of editing sites 227 and 228 in each construct. The unedited region removed in the truncated reporter is indicated on the full-length reporter. (B) Editing levels for sites 227 and 228 in the full-length Y75B8A.8 reporter from wild-type worms (light gray), the truncated Y75B8A.8 reporter in wild-type worms (black), the full-length reporter in adr-1(-) worms (dark gray), and the truncated Y75B8A.8 reporter in adr-1(-) worms (white) were determined from three biological replicates of staged young-adult worms. Error bars represent SEM. Significant changes between the full-length and truncated reporter in each respective genetic background (P ≤ 0.01) by Student's t-test are marked with an asterisk. (C) Relative expression of the full-length Y75B8A.8 3′ UTR neuronal reporter in wild-type worms (light gray), the full-length Y75B8A.8 3′ UTR neuronal reporter in adr-1(-) (dark gray), the truncated Y75B8A.8 3′ UTR neuronal reporter in wild-type worms (black), and the truncated Y75B8A.8 3′ UTR neuronal reporter in adr-1(-) worms (white). Fold change was determined by normalizing to the relative GFP expression of the full-length Y75B8A.8 3′ UTR neuronal reporter in wild-type worms (gray). Error bars represent standard error of the mean (SEM). (D, left panel) Worm lysates of the indicated strains expressing the truncated Y75B8A.8 reporter were subjected to immunoblotting for the Flag epitope and actin as a loading control. (Right panel) Total RNA from the two biological replicates of the indicated strains expressing the truncated Y75B8A.8 reporter was reverse transcribed, PCR amplified with reporter specific primers and Sanger sequenced. Sequence chromatograms of the PCR products from one biological replicate are shown. The bold A indicates editing sites 227 and 228. (E) Proposed model of how the unedited region promotes editing efficiency of ADR-2 and how ADR-1 represses editing in the Y75B8A.8 3′ UTR. The unedited region acts to recruit ADR-2 to the Y75B8A.8 3′ UTR thereby increasing editing efficiency. If ADR-1 is expressed, it competes with ADR-2 for access to editing sites 227 and 228 resulting in repressing of editing efficiency.
One possibility for the reduction of editing observed in the truncated neuronal reporter could be reduced mRNA expression. However, in comparison to the full-length Y75B8A.8 neuronal reporter, the truncated reporter has a similar level of expression (Fig. 3C). Another possibility is that the truncation could disrupt the structure of the dsRNA encompassing the editing sites. In this regard, we did not observe any alterations in the predicted mfold structure surrounding the editing sites for the truncated reporter (Fig. 3A; Zuker 2003). If removing this region simply causes enhanced ADR-1 repression of editing, then editing levels of the truncated reporter should return to full-length levels in the absence of ADR-1. To assess this possibility, we examined editing efficiency of the truncated reporter in the absence of ADR-1 (Fig. 3B). Similar to the full-length reporter, editing efficiency of the truncated reporter increases at both sites 227 (over 20% increase) and 228 (over 40% increase) in the absence of ADR-1. This effect on editing can be directly attributed to loss of adr-1, as reintroduction of an adr-1 transgene expressing Flag-tagged ADR-1 (Fig. 3D, left panels) restores editing of the truncated Y75B8A.8 reporter to wild-type levels (Fig. 3D, right panels). However, despite the increased editing of the truncated reporter in the absence of adr-1(-), editing levels do not return to those observed for the full-length reporter. This suggests that this unedited region acts to increase ADR-2 editing efficiency at sites 227 and 228.
Based on these observations, we propose a model where the unedited dsRNA region serves as a cis-acting element to recruit ADR-2 to the Y75B8A.8 3′ UTR, thus increasing editing of sites 227 and 228 (Fig. 3E). It is also possible that truncating the Y75B8A.8 3′ UTR results in less stable base-pairing of the remaining stem or increases the reporter mRNA export from the nucleus, thus indirectly reducing editing levels. However, as editing of the truncated reporter in the absence of adr-1 exceeds the editing levels of the full-length reporter in wild-type cells by over 20%, the truncation does not appear to significantly disrupt the dsRNA structure around the editing site. Future experiments to address this model should include tests of whether transferring this sequence to other 3′ UTRs can function to enhance editing and whether this sequence increases editing by ADR-2 in vitro. Furthermore, as fusing the 3′ UTR of Y75B8A.8 to the GFP reporter has lower editing levels than endogenous Y75B8A.8, it is interesting to speculate whether other upstream cis-acting elements could also enhance editing of the 3′ UTR. While there are no other predicted double-stranded regions or editing sites present in the endogenous 3′ UTR and lacking in our reporter, editing sites in the first intron of endogenous Y75B8A.8 mRNA, located 2000 nucleotides upstream of the 3′ UTR, have been previously identified (Zhao et al. 2014). It would be interesting to determine whether this intronic region could influence editing of the downstream 3′ UTR region in endogenous Y75B8A.8 mRNA. Perhaps by analyzing worm strains that lack factors important for splicing or mutants with reduced speed of splicing, effects of the intron on 3′ UTR editing could be observed.
Conclusions
Using a reporter system to examine factors that affect RNA editing levels in a singular tissue, we have demonstrated three major determinants of editing efficiency for the Y75B8A.8 3′ UTR. Foremost, we observed that the dsRBP ADR-1 is the major factor influencing editing efficiency in neurons, which is largely absent when examining RNA from total worms. In neurons, we propose that ADR-1 binds to the 3′ UTR of the Y75B8A.8 mRNA, which blocks access of ADR-2 to the target adenosine and reduces editing (Fig. 3E). In the future, it will be critical to determine whether this function of ADR-1 is specific to neurons and if it is extends to other transcripts, as ADR-1 is known to be a major regulator of editing in 3′ UTRs. In addition, we observe that altering mRNA expression of the neuronal reporter can slightly influence editing efficiency. However, this influence is increased in the absence of ADR-1 suggesting these two factors have a combinatorial effect on regulating editing efficiency. Finally, we identify a region of the Y75B8A.8 3′ UTR that acts to increase ADR-2 editing efficiency. This demonstrates that unedited dsRNA regions of an editing substrate play important roles in regulating editing efficiency of noncoding regions of mRNA.
MATERIALS AND METHODS
Maintenance of worm strains and transgenics
Worm strains were maintained by growth on NGM plates seeded with Escherichia coli OP50. Transgenic worm lines were generated by microinjection into the gonads of young-adult worms of the appropriate genetic background. The injection mix used for generating transgenics contained the following: 1, 10, 20, or 40 ng/μL of the transgene of interest, 20 ng/μL of the dominant marker, and 1 kb DNA ladder (NEB) to raise the final concentration of the injection mix to 100 ng/μL. Transgenic strains were maintained by passaging only worms with the dominant marker. The dominant marker used in this study was rab3::rfp::unc-54 (HH plasmid #22). The transgenes expressing the full-length Y75B8A.8 3′ UTR (HH #340) or the truncated version of the Y75B8A.8 3′ UTR (HH#344) were injected in a modified version of the rab3::rfp::unc-54 (#HH21) plasmid, previously published (Hundley et al. 2008). Briefly, the full-length Y75B8A.8 3′ UTR and the truncated version were amplified by PCR from N2 genomic DNA with primers designed to add on Sal I and Pst I restriction sites that were introduced into the vector between the stop codon of GFP and the unc-54 3′ UTR.
To obtain young adults used in experiments, embryos were isolated from gravid worms grown on 6 cm plates using standard hypochlorite treatment. Young adults were obtained by hatching eggs overnight in M9 buffer, transferring synchronized L1 worms to NGM plates seeded with Escherichia coli OP50, and incubating at 20°C for 48–54 h.
The following strains were utilized in this study: Bristol strain N2 (HH#1), BB19 adr-1(tm668), BB21 adr-1(tm668);adr-2(ok735)(HH#7), BB21 adr-1(tm668) + blmEx1 [3XFLAG-adr-1 genomic, rab3::gfp::unc-54] (HH#76), N2 + blmEx5 [1 ng/μL full-length Y75B8A.8 3′ UTR transgene, 20 ng/μL rab3::rfp::unc-54] (HH# 179), N2 + blmEx5 (HH #180), BB19 adr-1(tm668) + blmEx5 (HH#181), N2 + blmEx6 [10 ng/μL full-length Y75B8A.8 3′ UTR transgene, 20 ng/μL rab3::rfp::unc-54] (HH#178), N2 + blmEx6 (HH#182), BB19 adr-1(tm668) + blmEx6 (HH#183), N2 + blmEx7 [40 ng/μL full-length Y75B8A.8 3′ UTR transgene, 20 ng/μL rab3::rfp::unc-54] (HH#177), N2 + blmEx7 (HH#184), BB19 adr-1(tm668) + blmEx7 (HH#184), N2 + blmEx8 [20 ng/μL truncated Y75B8A.8 3′ UTR transgene, 20 ng/μL rab3::rfp::unc-54] (HH#168), N2 + blmEx8 (HH#186), BB19 adr-1(tm668) + blmEx8 (HH#187).
RNA isolation and editing assays
Total RNA was isolated from young-adult worms using TRIzol (Invitrogen). RNA was further treated with Turbo DNase (Ambion) and then isolated using the RNA Easy Extraction kit (Qiagen). Editing assays were performed using 1 μg of DNase treated RNA, Thermoscript RT (Invitrogen) for reverse transcription and PFX Platinum DNA Polymerase (Invitrogen) for PCR amplification with either endogenous Y75B8A.8 or transgene-specific primers. PCR products were gel purified and 5 ng of purified DNA was subjected to Sanger sequencing. Editing was quantified by measuring the adenosine and guanosine peak heights in Photoshop. For all editing assays, negative controls were conducted without Thermoscript RT to ensure that all DNA subjected to Sanger sequencing resulted from cDNA amplification.
Transgene quantification
Utilizing DNase treated RNA obtained from young-adult worms, the transgene expression was determined by quantitate real-time PCR. For reverse transcription, 1 μg of total RNA was reverse transcribed with Superscript III (Invitrogen) with random hexamers. Following cDNA synthesis, two microliters of cDNA were analyzed per qRT-PCR in a Mastercycler Realplex2 instrument (Eppendorf) using the 2X KAPA SYBR FAST qPCR Master Mix (Kapa Biosystems). GFP and RFP were quantified using gene-specific primers. Expression of the neuronal reporter was determined by RT-qPCR for GFP present in the Y75B8A.8 neuronal reporters. Relative GFP expression levels were determined by correcting with the control rab3:RFP.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Indiana University School of Medicine (start-up funds to H.A.H.), a Research Scholar Grant (15-051-RMC) to H.A.H. from the American Cancer Society, and a National Institutes of Health predoctoral training grant to M.C.W. (T32 GM007757). We thank Sarah Deffit for assistance in editing the manuscript.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.055079.115.
REFERENCES
- Bahn JH, Lee JH, Li G, Greer C, Peng G, Xiao X. 2012. Accurate identification of A-to-I RNA editing in human by transcriptome sequencing. Genome Res 22: 142–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazak L, Haviv A, Barak M, Jacob-Hirsch J, Deng P, Zhang R, Isaacs FJ, Rechavi G, Li JB, Eisenberg E, et al. 2014. A-to-I RNA editing occurs at over a hundred million genomic sites, located in a majority of human genes. Genome Res 24: 365–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhogal B, Jepson JE, Savva YA, Pepper AS, Reenan RA, Jongens TA. 2011. Modulation of dADAR-dependent RNA editing by the Drosophila fragile X mental retardation protein. Nat Neurosci 14: 1517–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel C, Veno MT, Ekdahl Y, Kjems J, Ohman M. 2012. A distant cis acting intronic element induces site-selective RNA editing. Nucleic Acids Res 40: 9876–9886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel C, Silberberg G, Behm M, Ohman M. 2014. Alu elements shape the primate transcriptome by cis-regulation of RNA editing. Genome Biol 15: R28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desterro JM, Keegan LP, Jaffray E, Hay RT, O'Connell MA, Carmo-Fonseca M. 2005. SUMO-1 modification alters ADAR1 editing activity. Mol Biol Cell 16: 5115–5126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggington JM, Greene T, Bass BL. 2011. Predicting sites of ADAR editing in double-stranded RNA. Nat Commun 2: 319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hundley HA, Bass BL. 2010. ADAR editing in double-stranded UTRs and other noncoding RNA sequences. Trends Biochem Sci 35: 377–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hundley HA, Krauchuk AA, Bass BL. 2008. C. elegans and H. sapiens mRNAs with edited 3′ UTRs are present on polysomes. RNA 14: 2050–2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara Y, Megraw M, Kreider E, Iizasa H, Valente L, Hatzigeorgiou AG, Nishikura K. 2008. Frequency and fate of microRNA editing in human brain. Nucleic Acids Res 36: 5270–5280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai F, Chen CX, Carter KC, Nishikura K. 1997. Editing of glutamate receptor B subunit ion channel RNAs by four alternatively spliced DRADA2 double-stranded RNA adenosine deaminases. Mol Cell Biol 17: 2413–2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann KA, Bass BL. 2000. Double-stranded RNA adenosine deaminases ADAR1 and ADAR2 have overlapping specificities. Biochemistry 39: 12875–12884. [DOI] [PubMed] [Google Scholar]
- Li S, Mason CE. 2014. The pivotal regulatory landscape of RNA modifications. Annu Rev Genomics Hum Genet 15: 127–150. [DOI] [PubMed] [Google Scholar]
- Nishikura K. 2010. Functions and regulation of RNA editing by ADAR deaminases. Annu Rev Biochem 79: 321–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pullirsch D, Jantsch MF. 2010. Proteome diversification by adenosine to inosine RNA editing. RNA Biol 7: 205–212. [DOI] [PubMed] [Google Scholar]
- Rieder LE, Reenan RA. 2012. The intricate relationship between RNA structure, editing, and splicing. Semin Cell Dev Biol 23: 281–288. [DOI] [PubMed] [Google Scholar]
- Rieder LE, Staber CJ, Hoopengardner B, Reenan RA. 2013. Tertiary structural elements determine the extent and specificity of messenger RNA editing. Nat Commun 4: 2232. [DOI] [PubMed] [Google Scholar]
- Rosenthal JJ, Seeburg PH. 2012. A-to-I RNA editing: effects on proteins key to neural excitability. Neuron 74: 432–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueter SM, Dawson TR, Emeson RB. 1999. Regulation of alternative splicing by RNA editing. Nature 399: 75–80. [DOI] [PubMed] [Google Scholar]
- Savva YA, Rieder LE, Reenan RA. 2012. The ADAR protein family. Genome Biol 13: 252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stulić M, Jantsch MF. 2013. Spatio-temporal profiling of Filamin A RNA-editing reveals ADAR preferences and high editing levels outside neuronal tissues. RNA Biol 10: 1611–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tariq A, Garncarz W, Handl C, Balik A, Pusch O, Jantsch MF. 2013. RNA-interacting proteins act as site-specific repressors of ADAR2-mediated RNA editing and fluctuate upon neuronal stimulation. Nucleic Acids Res 41: 2581–2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian N, Yang Y, Sachsenmaier N, Muggenhumer D, Bi J, Waldsich C, Jantsch MF, Jin Y. 2011. A structural determinant required for RNA editing. Nucleic Acids Res 39: 5669–5681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonkin LA, Saccomanno L, Morse DP, Brodigan T, Krause M, Bass BL. 2002. RNA editing by ADARs is important for normal behavior in Caenorhabditis elegans. EMBO J 21: 6025–6035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlstedt H, Daniel C, Enstero M, Ohman M. 2009. Large-scale mRNA sequencing determines global regulation of RNA editing during brain development. Genome Res 19: 978–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washburn MC, Kakaradov B, Sundararaman B, Wheeler E, Hoon S, Yeo GW, Hundley HA. 2014. The dsRBP and inactive editor ADR-1 utilizes dsRNA binding to regulate A-to-I RNA editing across the C. elegans transcriptome. Cell Rep 6: 599–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler EC, Washburn MC, Major F, Rusch DB, Hundley HA. 2015. Noncoding regions of C. elegans mRNA undergo selective adenosine to inosine deamination and contain a small number of editing sites per transcript. RNA Biol 12: 162–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whipple JM, Youssef OA, Aruscavage PJ, Nix DA, Hong C, Johnson WE, Bass BL. 2015. Genome-wide profiling of the C. elegans dsRNAome. RNA 21: 786–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wulff BE, Nishikura K. 2012. Modulation of microRNA expression and function by ADARs. Curr Top Microbiol Immunol 353: 91–109. [DOI] [PubMed] [Google Scholar]
- Zhao HQ, Zhang P, Gao H, He X, Dou Y, Huang AY, Liu X, Ye AY, Dong MQ, Wei L. 2014. Profiling the RNA editomes of wild-type C. elegans and ADAR mutants. Genome Res 25: 66–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M. 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31: 3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.