

Abstract
RNA modifications confer complexity to the 4-nucleotide polymer; nevertheless, their exact function is mostly unknown. rRNA 2′-O-ribose methylation concentrates to ribosome functional sites and is important for ribosome biogenesis. The methyl group is transferred to rRNA by the box C/D RNPs: The rRNA sequence to be methylated is recognized by a complementary sequence on the guide RNA, which is part of the enzyme. In contrast to their eukaryotic homologs, archaeal box C/D enzymes can be assembled in vitro and are used to study the mechanism of 2′-O-ribose methylation. In Archaea, each guide RNA directs methylation to two distinct rRNA sequences, posing the question whether this dual architecture of the enzyme has a regulatory role. Here we use methylation assays and low-resolution structural analysis with small-angle X-ray scattering to study the methylation reaction guided by the sR26 guide RNA from Pyrococcus furiosus. We find that the methylation efficacy at sites D and D′ differ substantially, with substrate D′ turning over more efficiently than substrate D. This observation correlates well with structural data: The scattering profile of the box C/D RNP half-loaded with substrate D′ is similar to that of the holo complex, which has the highest activity. Unexpectedly, the guide RNA secondary structure is not responsible for the functional difference at the D and D′ sites. Instead, this difference is recapitulated by the nature of the first base pair of the guide-substrate duplex. We suggest that substrate turnover may occur through a zip mechanism that initiates at the 5′-end of the product.
Keywords: RNA modification, 2′-O methylation, box C/D RNP, regulatory mechanism
INTRODUCTION
Eukaryotic rRNA is highly modified to provide additional complexity. Modifications include 2′-O-ribose methylation, the isomerization of uracil to pseudo-uracil and a number of modifications of particular bases at distinct positions. The role of this chemical complexity is largely unknown and may range from the stabilization of RNA structures and assistance in the folding process (Liu et al. 2008) to eliciting selective interactions with protein readers (Fu et al. 2014). 2′-O-ribose methylation is one of the most abundant modifications, totaling 54 rRNA sites in Saccharomyces cerevisiae and approximately twice as many in humans (Piekna-Przybylska et al. 2008; Machnicka et al. 2013). In rRNA, 2′-O-ribose methylations cluster in functionally conserved regions such as intersubunit interfaces, decoding and peptidyltransferase centers (Decatur and Fournier 2002; Polikanov et al. 2015). This suggests a critical role of 2′-O-ribose methylation in the assembly of functional ribosomes, which remains to be discovered.
In eukaryotes and Archaea, 2′-O-ribose methylation is carried out by the box C/D small nucleolar RNA–protein complex [s(no)RNP]. The guide s(no)RNAs target the methylation machinery to the sites of modification, due to their complementarity to the RNA sequences to be modified. In addition to the guide sites, which base pair with the substrates, s(no)RNAs have particular structural motifs: box C/D and box C′/D′. These elements build assembly sites for four proteins in yeast (Snu13, Nop56, Nop58, and Nop1), while in Archaea the complexity is reduced to three proteins (L7Ae, Nop5, and Fibrillarin).
To interrogate the cells on the role of methylation at specific rRNA sites, it is key to understand the catalytic mechanism of the box C/D methylation machinery. Studying the function of eukaryotic snoRNP complexes remains challenging: The enzymes cannot be reconstituted in vitro in functional form, and their biogenesis is a complex multistep process requiring several cis and trans factors in vivo. Fortunately, their archaeal counterparts, the box C/D sRNPs, are readily assembled in vitro into functional enzymes and provide a tool for mechanistic studies.
In Archaea, the assembly of the box C/D sRNP complex is initiated by binding of the L7Ae protein to the K-turn and K-loop elements (Fig. 1A; Kuhn et al. 2002). The L7Ae-sRNA complex recruits Nop5, a scaffold protein consisting of three domains (Omer et al. 2002; Aittaleb et al. 2003). The C-terminal domain (CTD) of Nop5 binds to the L7Ae–RNA complex, the coiled-coil (CC) domain is responsible for self-dimerization, and the N-terminal domain (NTD) interacts with the third protein, Fibrillarin (Aittaleb et al. 2003). Fibrillarin is the methylation enzyme in the complex and uses the cofactor S-adenosyl-L-methionine (SAM) as methyl group donor (Wang et al. 2000). The guide sRNA pairs with two different substrate RNAs (10–21 base pairs [bp] long) and selects the methylation site, which is the fifth nucleotide upstream of box D (or D′) (Reichow et al. 2007).
FIGURE 1.

The structure of the box C/D RNP and the sequential methylation mechanism. (A) The components of the box C/D sRNP enzyme. L7Ae recognizes the conserved K-loop/K-turn elements of the sRNA. L7Ae binds the C-terminal domain of Nop5, which recruits the methyltransferase Fib. Fib methylates the substrate pre-rRNAs at the fifth base upstream of box D/D′, marked with a star. (B) The apo and holo (substrate-bound) states of the box C/D complex. In the apo state the Nop5 scaffold of the complex (gray) adopts a flat, square-like conformation in which Fib (blue) is positioned away from the sRNAs by the Nop5-NTD (purple). L7Ae (green) is bound to the sRNA (orange). In the holo sate, the complex adopts an extended conformation, which allows Fib to be positioned onto the two substrate D′ (or D) methylation sites. Two copies of Fib remain in the off position, thereby allowing only one substrate type to be methylated at any given time. (C) The mono- and di-RNP models for the box C/D enzyme. (D) NMR-detected methylation assay. Substrate RNAs are added to a 3× excess to the box C/D complex in the presence of 6× excess of 13C-labeled SAM; the release of methylated product is monitored. The upper panels show that the addition of substrate D′ stimulates the release of methylated substrate D. The lower panels show that substrate D′ is methylated and released efficiently in isolation. Nevertheless, addition of substrate D enhances the activity of the complex and results in the release of both products. ([Upper panels] 12.5 μM box C/D enzyme, 75 μM substrate RNAs, 150 μM 13C-SAM; [lower panels] 14.5 μM box C/D enzyme, 87 μM substrate RNAs, 174 μM 13C-SAM).
Interestingly, all guide sRNAs display two distinct guide sequences and direct methylation to two correspondingly distinct target sites, despite the fact that the proteins assembled around the box C/D and box C′/D′ elements are identical. This architecture suggests that the methylation of the two sites addressed by the same guide sRNA might be functionally coupled.
Structural information on the box C/D methylation enzyme has been obtained exclusively from reconstituted archaeal sRNP complexes from Methanocaldococcus jannashii (Mj), Sulfolobus solfataricus (Ss), and Pyrococcus furiosus (Pf). In 2013, we solved the structure of the box C/D sRNP complex from P. furiosus in solution with a combination of solution-state NMR and small-angle X-ray and neutron scattering (SAXS and SANS) (Fig. 1B; Lapinaite et al. 2013). Both the SAXS profile of the particle and size exclusion chromatography confirmed that the complex has a molecular weight of ∼400 kDa, namely consists of two copies of guide sRNA, two copies of each substrate D and D′, and four copies of each protein (di-RNP). The structures of the apo- and holo-enzymes (in the absence and in the presence of substrate RNAs), both of which differ from the ones reported previously by X-ray crystallography or EM (Bleichert et al. 2009; Lin et al. 2011), allowed us to reconstitute the mechanistic pathway of rRNA methylation: In the absence of substrate RNA, the four Fibrillarin molecules are in the “off” position; binding of substrate RNA triggers a substantial conformational change, leading to complete reshaping of the Nop5 components and to binding of only two Fibrillarin molecules to two of the four substrate RNAs (Fig. 1B). Methylation of the other two substrate RNAs then requires an additional conformational rearrangement. These results finally linked the asymmetry of all naturally occurring box C/D sRNAs with the enzyme structure, showing that substrate D and substrate D′ occupy two functionally and structurally different environments in the di-RNP.
In addition, using an NMR-based functional assay with 13C-labeled cofactor, we could show that the unequal environment of the four Fibrillarin copies leads to functional differences at substrate D and D′: In the sR26 RNA of Pf, substrate D can be methylated and released only in the presence of substrate D′, while for substrate D′ turnover occurs also in the absence of substrate D (Fig. 1D). Two different scenarios can explain this observation: (i) Substrate D is not bound by the enzyme in the absence of substrate D′; (ii) substrate D is bound and methylated but cannot be released from the enzyme in the absence of substrate D′. This unexpected regulation mechanism suggests a sequentially ordered methylation process at the two sites targeted by the same guide sRNA; the functional significance of this regulation is still unclear.
Here, we use in vitro functional methylation assays and low-resolution structural analysis by SAXS to explore the difference in turnover efficiency at the substrate D and D′ sites. We find that the nature of the first base pair at the 5′-end of the substrate has a large influence on turnover efficiency. On the basis of this, we propose that the product dissociates from the enzyme via an unzipping mechanism that initiates at the 5′-end of the substrate and propagates to the 3′-end. In addition, we validate our structural model of the catalytically active di-RNP by testing the activity of enzymes reconstituted with mutant Nop5 and Fibrillarin proteins. The reduced activity of the mutants is compatible with the di-RNP model, while it is not recapitulated by the mono-RNP structure of Lin et al. (2011) (Fig. 1C).
RESULTS
Methylation assays: impact of salt, magnesium, and RNA secondary structure
In all in vitro methylation assays we utilize two guide RNA (gRNA) constructs: the wild-type sR26 sequence from Pf and the engineered st-sR26 (stabilized sR26) gRNA, where the loop between box D′ and C′ has been substituted by a short helix capped with a GAAA tetraloop (Fig. 2). A similar construct to st-sR26 has been used for structural studies (Lapinaite et al. 2013), because of the superior stability and homogeneity of the complex assembled with it. In addition, the st-sR26 allows us to test the influence of the structure of the box C′/D′ motif on methylation efficiency (K-loop in sR26 versus K-turn in st-sR26).
FIGURE 2.

In vitro methylation assays on the box C/D RNP complex. (A) Addition of 0.5 μM of individual substrates to 0.15 μM box C/D complex in the presence of 1 μM [3H]-SAM shows a disparity in methylation activity of substrate D and substrate D′. Furthermore, substrate D′ is turned over during the course of the assay, while substrate D is not. (B) Assay performed as in A until 110 min, at which point substrate D is added to the substrate D′ reaction (and vice versa). This leads to an increase in methylation of the original substrate. Diamonds and circles indicate the same substrate, before (diamond) and after (circle) addition of the other substrate. (C–E) Activity assays on box C/D complexes reconstituted using sR26, st-sR26, and inv-sR26, respectively. Assays were performed in the presence (square) and absence (diamond) of 1 mM MgCl2. Substrate D′ remains preferentially methylated in all conditions.
As described in the Materials and Methods section, our in vitro assay detects the total amount of methylated product, independent of whether it has been released or it is still in complex with the enzyme. In addition, we design the assay in such a way that one of the two products can be selectively monitored in the presence of the other. To this end, we use a combination of biotinylated/nonbiotinylated substrate D/D′ or D′/D; after the reaction with tritiated SAM, we isolate the biotinylated product on NeutrAvidin beads for the radioactivity count.
The highest activity of thermophile box C/D enzymes has been observed at temperatures > 70°C (Lin et al. 2011). Here, we run the methylation reaction at 50°C, to ensure compatibility with both previous structural data and the SAXS analysis, which cannot be performed at higher temperature. All methylation reactions are performed at two salt concentrations: 500 (as in our previous structural analysis) and 150 mM NaCl.
Throughout our methylation assays, we find that the final concentration of products in the reaction strongly depends on an intact SAM cofactor. We observe differences as high as 0.5–1 orders of magnitude, depending on the SAM batch and age. The degraded cofactor competes with SAM for Fibrillarin binding, thereby inhibiting the reaction. Thus, the final concentration of the product is not comparable between assays that use different SAM batches.
First, we perform the in vitro methylation assays described in the Materials and Methods section in the same buffer as the NMR-detected assay of Figure 1D (500 mM NaCl, 0 mM MgCl2, 50°C) and in conditions of multiple turnover (substrate concentration, 0.5 μM; enzyme concentration, 0.15 μM) (Fig. 2A).
Separate addition of substrate D or D′ to the box C/D enzyme assembled around sR26 shows that the enzyme methylates substrate D also in the absence of substrate D′ (red diamonds); however, in the conditions of multiple turnover under which we run the assay, the methylation of substrate D′ is much more efficient (blue diamonds). This result confirms the observation of the NMR-detected assay; both assays taken together attribute the modest methylation of substrate D in the absence of D′ to inefficient release of product D. Addition of the second substrate stimulates turnover (Figs. 1D, 2B), without altering the ratio of methylation efficiency at the two sites.
In the presence of magnesium (Fig. 2C, square symbols) the initial velocity of the reaction and the final quantity of the products increases at both sites; however, the difference in turnover efficiency at the D and D′ sites persists.
To verify whether the higher efficiency of methylation at substrate D′ depends on the presence of the K-loop element, we construct the artificial st-sR26 (stabilized sR26) gRNA. In st-sR26, the loop between box D′ and C′ is substituted by a short helix capped with a GAAA tetraloop, effectively turning the K-loop element into a K-turn. For the enzyme reconstituted with st-sR26, we observe the same difference in the methylation efficiency of substrate D and D′ as for the enzyme reconstituted with sR26. This result excludes that a putative interaction of the 5′-end overhang of substrate D′ with the flexible K-loop makes the D′ site more competent for methylation (Fig. 2D).
Next, we test whether the position of the two guide-substrate helices in the complex determines the efficiency in methylation. To this end, we invert the position of the two guide sequences, to generate the inv-sR26 gRNA (Fig. 2E). Also in this case the efficiency of methylation at substrate D′ remains superior to that at substrate D, with the difference between the methylation levels at the two sites increasing even further. Addition of magnesium does not change the picture.
All assays are repeated in a low salt buffer with similar results (Supplemental Fig. S1), confirming that the superior efficiency of methylation at the substrate D′ site is not an artifact of the high salt content of the buffer. A quantification of the apparent turnover rates, extracted from the initial slopes of the methylation time courses, is given in Supplemental Table S1.
The sequence of the substrate-guide helix determines the efficiency of methylation
After excluding the influence of the structural elements around the guide-substrate helices, we turn our attention to the substrate sequences. Both substrates bind to the two respective guide sequences forming 11 bp, with equal G,C content. In addition, both substrates are modified at a cytosine ribose. These facts suggest that neither the nature of the target nucleotide nor the composition of the substrate-guide helix recapitulate the different reaction efficiency at sites D and D′.
The most prominent difference between the two substrate-guide helices is observed at the 5′-end of the substrate, where the substrate-guide D′ and D helices are closed by a U–A and a G–C base pair, respectively. To test the relevance of the substrate RNA sequence at the 5′-end of the substrate-guide duplex, we invert both the fourth and the fifth nucleotides of substrate D′ and the corresponding nucleotides on the guide D′ sequence (zip-sR26), so as to generate a substrate-guide D′ sequence with a terminal C–G base pair (Fig. 3A). The enzyme assembled with the zip-sR26 gRNA shows a dramatic decrease of turnover efficiency at the D′ site and methylates substrate D′ even less efficiently than substrate D (Fig. 3B).
FIGURE 3.
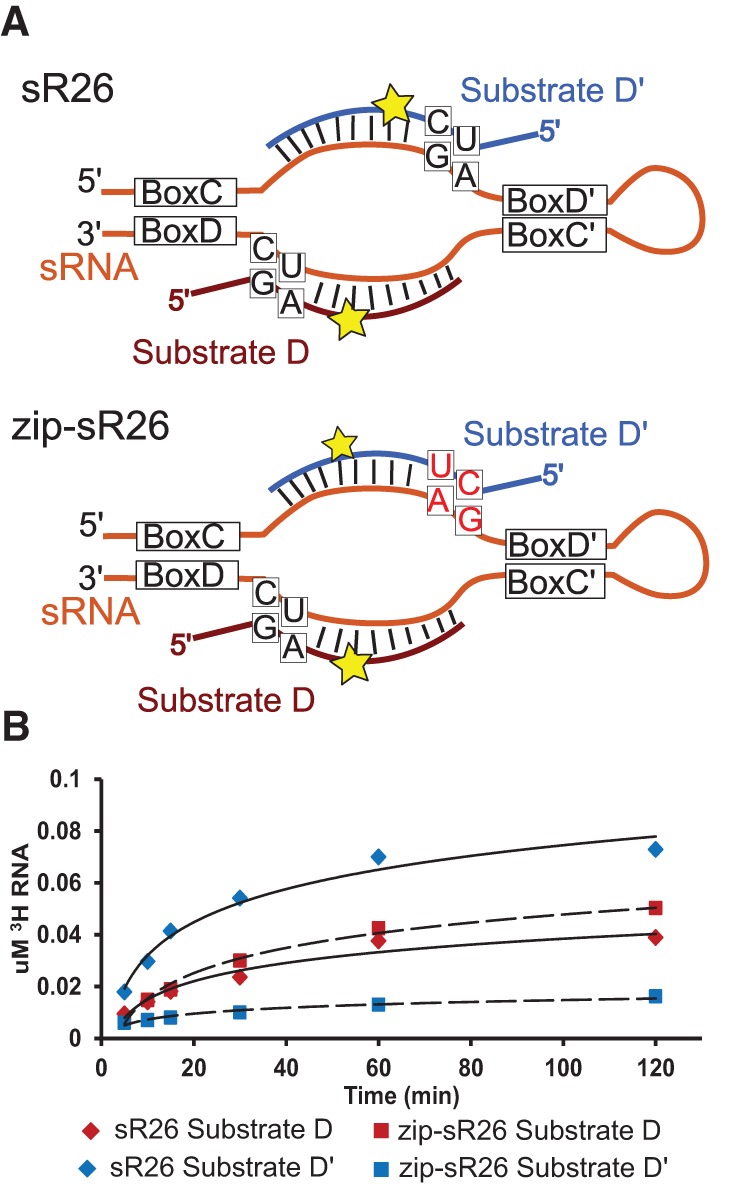
Product release is initiated at the 5′-end of the substrate RNAs. (A) In the zip-sR26 construct, the order of the base pairs at the 5′-end of the substrate D′-binding region is inverted in order to stabilize the end of the guide-substrate duplex. (B) Methylation assays of zip-sR26 and sR26 box C/D complexes show that this mutation greatly reduces the methylation efficiency of substrate D′.
SAXS analysis of half-loaded complexes
In order to verify whether the difference in the methylation activity at the D and D′ sites corresponds to differences in the structures of the box C/D enzyme loaded with either substrate D or D′, we compare small-angle X-ray scattering (SAXS) curves of the apo (without substrates), half-loaded (+ substrate D or D′) and fully loaded (+ both substrate D and D′) complexes. SAXS curves report on the shape of the complex at low resolution. Figure 4 shows that the shape of the complex half-loaded with substrate D′ is close to that of the fully loaded complex, where activity is optimal (Fig. 2A). In contrast, the complex half-loaded with substrate D displays parameters between those of the apo and holo complexes. The data indicate that the box C/D enzyme is very flexible and can adopt different conformations depending on the substrate RNA binding to it. Moreover, one conformation is optimal for efficient catalysis and it is adopted by the fully loaded complex, as well as by the complex half-loaded with substrate D′. In contrast, the complex half-loaded with substrate D assumes a less active conformation.
FIGURE 4.

SAXS profiles of box C/D enzyme bound to different substrate RNAs. Titration of substrate RNAs onto 5 mg/mL box C/D complex reconstituted using st-sR26 guide RNA. Curves are scaled to match I(0) for the purpose of comparison. (A) The profiles show that the complex adopts similar conformations when bound to substrate D′ and in the fully loaded holo state, while it adopts a third distinct conformation when bound to substrate D. The radius of gyration Rg is calculated using the Guinier approximation within the ATSAS package PRIMUS. (B) The pairwise-distance distribution function P(r) was extracted from the scattering curves using the program GNOM. The P(r) functions show a marked difference between the enzymes bound to substrate D or D′.
Effect of mutations in the Nop5 α3-α4 loop
In our structure of the fully loaded di-RNP (Lapinaite et al. 2013), the loop residues 118–127, connecting the α3 helix of the Nop5 NTD domain with the α4 helix of the Nop5-CC domain in Pf, assume an extended conformation to allow Fibrillarin to reach the methylation site (Fig. 5B). In the structure of the mono-RNP (Lin et al. 2011), instead, this nonconserved stretch of the Nop5 of Solpholobus solfataricus, forms the C-terminal part of the α3 helix and can tolerate shortening without impairing the recognition of the substrate by Fibrillarin (Fig. 5A). To prove the importance of the length of the α3–α4 loop of Nop5, we designed four deletion mutants (Δ119–123, Δ121–125, Δ121–122, Δ122) and measured their methylation activity (Fig. 5C). Interestingly, even the deletion of one amino acid substantially impairs activity (Δ122, Fig. 5C), supporting the structure of the substrate-loaded di-RNP (Lapinaite et al. 2013).
FIGURE 5.
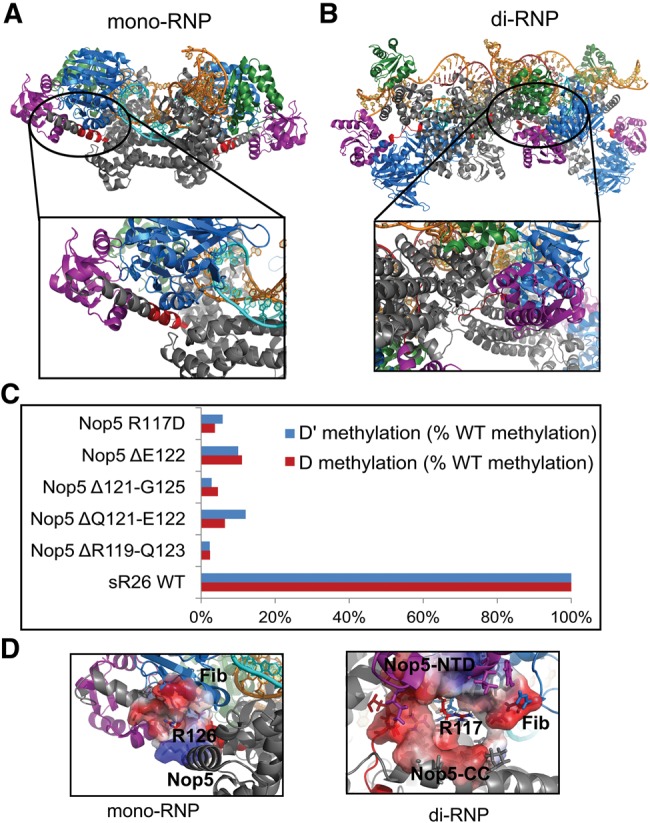
The role of the Nop5 α3–α4 loop. Color scheme as in Figure 1. (A) The Nop5 α3–α4 loop responsible for positioning the Nop5-NTD/Fib subcomplex adopts an α−helical conformation in the mono-RNP crystallographic structure from Ss. (B) In the Pf di-RNP structure, the α3–α4 loop is disordered and extended, which allows it to shuttle the Nop5-NTD/Fib to and from the guide-substrate duplex. (C) Mutational analysis of the α3–α4 loop. In each case, the mutant complex was reconstituted with sR26 RNA and measured together with the wild-type box C/D sR26 complex. The amount of methylated product after 120 min is shown relative to the wild-type box C/D enzyme. Mutations affecting the length and charge of this region severely impair the methylation capability of the box C/D RNP. (D) In the di-RNP structure, R117 in the α3–α4 loop is placed in a negatively charged environment, in agreement with the critical role of the charge of this residue highlighted by mutational analysis. Conversely, in the mono-RNP structure, the homologous residue R126 of Ss Nop5 does not make any critical interaction and points to the solvent.
Next, we tested the relevance of the neighboring R117 for activity. In agreement with the conservation of this residue throughout all species, the Nop5 R117D mutant is incapable of methylation (Fig. 5C). In the di-RNP structure, R117 interacts with a negatively charged surface of both Fib and the Nop5-CC (Fig. 5D). In contrast, the homologous R126 in the mono-RNP structure from Ss is exposed to the solvent and does not make any side-chain specific interaction.
DISCUSSION
All archaeal box C/D sRNAs target two different rRNA sites for methylation. The significance of combining methylation sites pairwise, by linking them to the same enzyme particle, is not understood. However, it is reasonable to hypothesize that this arrangement is functionally relevant, for example, by providing a means to regulate the methylation state of one site in dependence of the methylation state of the other site.
Intriguingly, the crystal structure of the Ss box C/D sRNP, which was reconstituted using an artificial two-piece RNA and consisted of one copy of guide sRNA and two copies of each protein (mono-RNP, Fig. 1C), displays a twofold symmetry (Lin et al. 2011). The structure places the different substrate D and D′ sequences in identical environments, thus suggesting that the two substrates can be methylated independently of each other.
Prior to the crystallographic structure, a low-resolution model of the apo state (without substrate RNAs) of a box C/D sRNP from Mj had been obtained by electron microscopy (Bleichert et al. 2009). This study proposed the concept of the box C/D di-RNP, that is of an enzymatic particle consisting of two copies of guide sRNA and four copies of each protein. The model was derived in the absence of the substrate RNAs and the guide sRNA was not visible in the microscopy images, thus preventing any conclusion on the location of the substrates D and D′. Later, the same group demonstrated that all box C/D sRNPs from Archaea are di-RNPs, while a mono-RNP is obtained only when assembling the complex around an artificial two-piece guide sRNA (Bower-Phipps et al. 2012).
Recently, we conducted a combined NMR and SANS study of the box C/D sRNP from Pf assembled around a one-piece, native-like guide sRNA and obtained the structure of the substrate-loaded (holo) sRNP in solution. Our complex structure confirmed the di-RNP model and showed that the complex undergoes a large conformational change upon substrate binding (Fig. 1B). In the substrate-bound conformation the environment of substrates D and D′ are profoundly different: The two copies of one substrate type (either D or D′) are in contact with Fibrillarin and can be readily methylated; the two copies of the other substrate are far away from Fibrillarin, in an environment that is incompetent for methylation. The structure suggested that the two substrates D and D′ can be methylated only one after the other and that methylation of the other substrate type requires a large conformational conversion that exchanges the position of the substrates D and D′ and of the related proteins. This conformational change is likely to require dissociation of the products and transition through a more apo-like, flexible conformation.
In our previous work, we designed an NMR-detected activity assay, to verify whether methylation at the D and D′ sites proceeded with equal efficiency. The assay recorded the methylated and released product D or D′ under conditions of multiple turnover (excess of substrate). The different chemical shifts of the methyl groups of product D and D′, obtained using 13C-labeled SAM as cofactor, allowed us to distinguish between methylation reactions at the two sites. Interestingly, we found that efficient turnover occurs at the D′ site, independently of the presence of substrate D, while turnover at site D requires the D′ site to be occupied (Fig. 1D). This result confirmed the existence of a regulatory interaction between the methylation at the two sites.
In the present work, we address the differences in the methylation efficiency at the D and D′ sites systematically, using in vitro methylation assays. We confirm that in sR26 the D′ site is methylated more efficiently than the D site in a wide range of salt concentrations, both in the presence and in the absence of magnesium. The D site binds the substrate and is capable of methylation, suggesting that its low efficiency is due to slower release of the product.
Interestingly, addition of the other substrate simulates turnover at both sites D and D′ (Fig. 2B). Knowing that each guide-substrate pairing results in an A-form helix (Appel and Maxwell 2007), we propose that this effect is due to the strain imposed on the guide-substrate duplexes by the loading of all four substrates. Upon binding of the substrates the structure of the sRNP transitions from a loose, square-shaped complex to a tightly packed elongated complex (Fig. 1B). The holo structure can be relaxed by disruption of the duplex helices, after Fibrillarin is released from the methylated double-stranded RNA. In our structure, the unwinding of the guide-product helix can be aided by Nop5 helix α9A, situated in the C-terminal domain of the protein. This helix has been previously implicated in positioning the guide-substrate duplex with respect to the NC stem of the K-turn element (Xue et al. 2010). In our structure, stretches 289–292 and 296–298 of α9A contact the major groove of the guide-substrate duplex (Supplemental Fig. S2). Bulky hydrophobic side-chains of leucine and aromatic as well as arginine residues penetrate the RNA helix, potentially perturbing its local geometry and favoring its unwinding.
If the elongated holo structure promotes turnover, we expect the conformation of the D′-loaded enzyme, which displays efficient methylation, to be similar to that of the fully loaded holo enzyme. Nicely, the SAXS curves confirm that the overall shape of the sRNP half-loaded with substrate D′ is very similar to that of the holo enzyme, while the conformation of the sRNP half-loaded with D, which does not perform efficient methylation, is different (Fig. 4). Thus, the SAXS data support that efficient turnover is promoted by the elongated, strained conformation of the holo sRNP.
The difference between D and D′ sites observed throughout our work is consistent with biochemical data available for the Mj box C/D complex, where deoxynucleotide substitutions within the D or D′ substrate sequences yielded different results (Appel and Maxwell 2007). Specifically, a larger number of ribonucleotides (i.e., a longer A-helix region) is required for methylation of substrate D when compared to substrate D′, supporting a difference both in the structures of the substrate D-loaded and substrate D′-loaded complexes, and in the environments around the substrate RNAs, which is not consistent with the mono-RNP model.
Unexpectedly, the position of the guide D and D′ sequences does not influence turnover efficiency, indicating that the different architectures of the K-turn (box C/D) and K-loop (box C′/D′) elements are not the determinants of function at the corresponding substrates (Fig. 2). We find that the higher turnover at the D′ site is caused by the nature of the base pair at the 5′-end of the substrate. The less stable A:U base pair at the beginning of the D′ guide-substrate duplex recapitulates the higher methylation efficiency at the D′ site when compared to the D site, which displays a C:G base pair at the same position. This observation suggests that product release is initiated at the 5′-end of the product, possibly through a zip-like mechanism. Likely, the interactions with Nop5 helix α9A help stabilize a partially unwound conformation, which then leads to complete unwinding of the guide-product helix at the high temperature of the methylation reaction. This process cannot occur as long as Fibrillarin binds the guide-substrate duplex. However, our previous NMR analysis demonstrated that the 2′-O-methylation mark considerably reduces the affinity of Fibrillarin for the RNA duplex, thus allowing for product release. This hypothesis is also in agreement with previous biochemical results, indicating that distortion or melting of the A-form guide-substrate duplex is tolerated upstream of the methylated nucleotide, i.e., toward the 5′-end of the substrate (Appel and Maxwell 2007).
In conclusion, we show that the box C/D enzyme in Archaea methylates substrates D and D′ with different efficiency, while binding of substrate RNA to the opposite site stimulates turnover. The fully loaded enzyme displays the highest levels of methylation, followed by the enzyme half-loaded with substrate D′. In contrast, the enzyme half-loaded with substrate D is the least efficient. This scale correlates with the global form of the complex: The elongated, compact shape of both the fully loaded and D′ half-loaded enzymes methylates substrate RNAs more efficiently than the more apo-like shape of the enzyme half-loaded with substrate D. Our data suggest that the discrepancy in methylation levels at the D and D′ sites originates from differences in turnover rate. Product release seems to initiate at the 5′-end: A weaker base pair at this position could favor the unwinding of the duplex and results in higher turnover rates.
In the context of the rRNA, the differential efficiency of product release at site D and D′ of the box C/D enzymes provides a mechanism of communication between the two sites. In the specific case of sR26, the rRNA sequence complementary to guide D can be methylated and released only after binding and methylation of the rRNA sequence complementary to guide D′. On the other hand, the sequence of the rRNA complementary to guide D′ can be methylated and released also before recognition of the sequence complementary to guide D. The consequence of this regulation process is a ribosomal rRNA where either only site D′ or both sites D′ and D are methylated; site D, however, would never be methylated alone. The significance of such a regulation mechanism for ribosome folding, stability and function is still unknown.
Our model of sequential, conditional methylation would predict that the methylation levels at site D and D′ are different, with site D′ being methylated more often than site D for the sR26 gRNA. Interestingly, quantitation of 2′-O-methylation levels in the eukaryotic organism S. cerevisiae showed that rRNA sites targeted by box C/D snoRNAs are not 100% modified; nucleoside A100 of the 18S rRNA, for example, carries a 2′-O-methylation marker only in 65% of the population (Buchhaupt et al. 2014). A100 ribose methylation is catalyzed by snR51, which has a second methylation site within the 25S rRNA at position U2729. The question arises whether the two sites addressed by snR51 are methylated at different levels and whether this can be attributed to the mechanism of controlled methylation found for the archaeal box C/D complex. To date, eukaryotic snoRNP complexes have not been demonstrated to assemble in di-RNP; furthermore, the presence of two different Nop5 orthologs, Nop56, and Nop58, associated with the box C′/D′ and box C/D elements, respectively, provides a means to structurally distinguish the D and D′ sites also in the context of a mono-RNP. Structural information on the eukaryotic complex has to be awaited to confirm or disprove similarity between the regulation of rRNA ribose methylation in eukaryotes and Archaea.
MATERIALS AND METHODS
Protein expression and purification
The DNA sequences for L7Ae (UniProtKb accession code Q8U160) and Fibrillarin (Fib, Q8U4M2) were amplified from Pyrococcus furiosus genomic DNA and cloned into the pETM-11 expression vector (EMBL collection) using BamHI and NcoI restriction sites. Both proteins were expressed with an N-terminal His6 fusion tag carrying a TEV (tobacco etch virus) protease site in Rosetta 2 (Novagen) and BL21 DE3 (EMBL collection) Escherichia coli strains, respectively. We expressed Nop5 (Q8U4M1) from a commercial synthetic gene (GeneArt) designed with optimized codon usage for E. coli expression. The construct was also cloned into pETM-11 and two mutations (L148K and V223E) were introduced in order to avoid aggregation. All three proteins were expressed in LB media and induced at OD600∼0.7 with 1 mM IPTG. After induction, cells were incubated for 18–22 h at 22°C.
Cells expressing all three proteins were resuspended in lysis buffer (50 mM Tris, 10 mM imidazole, 1 M NaCl, 5 mM β-mercaptoethanol, pH 7.5). EDTA-free protease inhibitors (Roche), DNase I and 0.25 mg/mL lysozyme were added to the lysate, which was incubated for 30 min at room temperature. The cells were also lysed by sonication. The lysate was then spun down.
Nop5 and Fib lysates were then incubated at 80°C for 15 min to remove endogenous proteins and spun down. The soluble fractions were purified by loading onto a HisTrap FF column (GE Healthcare), eluting with a gradient of elution buffer (50 mM Tris, 300 mM imidazole, 1 M NaCl, 2 M LiCl, 5 mM β-mercaptoethanol, pH 7.5). The elute was then exchanged back into lysis buffer using a HiPrep 26/10 desalting column (GE Healthcare) and the presence of RNases was checked using the RNase Alert Kit (Ambion). If no RNases were detected, the His-Tag was cleaved by addition of 1 mg TEV protease/50 mg protein and incubation overnight at room temperature. The TEV and the His-Tag were finally removed by reverse purification on a HisTrap FF column.
For L7Ae, the soluble fraction was directly bound to a HisTrap FF column equilibrated in lysis buffer. The bound lysate was washed several (4–7) times with gradients of LiCl buffer (50 mM Tris, 10 mM imidazole, 1 M NaCl, 2 M LiCl, 5 mM β-mercaptoethanol, pH 7.5) to remove RNA. After equilibration back into lysis buffer, the sample was eluted with elution buffer. The eluate was exchanged into a low salt buffer (25 mM Tris, 150 mM NaCl, 2 mM DTT, pH 7.5) using a HiPrep 26/10 desalting column. The protein was then loaded onto a HiTrap Q FF column (GE Healthcare) and the residual bound RNA was eluted with a salt gradient. The flow-through was tested for RNases and purification proceeded as for Nop5 and Fib.
All protein mutations were introduced with the QuikchangeXL protocol (Agilent Technologies). Mutated constructs were confirmed by sequencing, expressed and purified as above.
Reconstitution of the full box C/D RNP complex
First, the guide RNA:L7Ae subcomplex was prepared by mixing the guide RNA with L7Ae (in low salt buffer) in a molar ratio of 2.2:1 L7Ae:RNA. Separately, the Fib:Nop5 subcomplex was prepared by mixing the Fib:Nop5 proteins with a ratio of 1.1:1. In both cases, the subcomplexes were incubated for 15 min at 80°C and allowed to gently cool down to room temperature. Each subcomplex was then purified by size exclusion chromatography using a Superdex S200 10/300 GL column (GE Healthcare) previously equilibrated in sample buffer (50 mM NaPi, 500 mM NaCl, pH 6.6).
The full box C/D complex was reconstituted by mixing the two previously assembled subcomplexes at a L7Ae/RNA:Nop5/Fib ratio of 1:2.2. After incubation for 15 min at 80°C, the sample was allowed to cool down and purified as described above. For complexes in 20 mM HEPES, 150 mM NaCl, pH 7.5 the gel filtration peak was repurified by a further gel filtration column in the buffer of choice.
RNA synthesis
The following RNAs were prepared by in vitro transcription using T7-polymerase produced in-house, unlabeled rNTPs (Sigma-Aldrich), and double-stranded plasmid DNA template:
Wild-type guide RNA (sR26 gRNA): 5′-GCGAGCAAUGAUGAGUGAUGGGCGAACUGAAAUAGUGAUGACGGAGGUGAUCUCUGAGCUCGC-3′
Inverted wild-type guide RNA (inv-sR26 gRNA): 5′-GCGAGCAAUGAUGACGGAGGUGAUCACUGAAAUAGUGAUGAGUGAUGGGCGAUCUGAGCUCGC-3′
Stabilized guide RNA (st-sR26 gRNA): 5′-GCGAGCAAUGAUGAGUGAUGGGCGAACUGAGCUCGAAAGAGCAAUGAUGACGGAGGUGAUCACUGAGCUCGC-3′
Inverted stabilized guide RNA (inv-st-sR26 gRNA): 5′-GCGAGCAAUGAUGACGGAGGUGAUCACUGAGCUCGAAAGAGCAAUGAUGAGUGAUGGGCGAACUGAGCUCGC-3′
Target RNAs were produced in the same way, with synthetic oligonucleotides used as templates:
Substrate D′: 5′-GCUUCGCCCAUCAC-3′
Substrate D: 5′-GUAGAUCACCUCCG-3′
The RNAs were purified by denaturing 12% or 20% polyacrylamide gel electrophoresis as run-off constructs.
In vitro activity assays
For activity measurements, reconstituted box C/D complexes (either in 50 mM NaPi buffer, 500 mM NaCl, pH 6.6, or in 20 mM HEPES, 150 mM NaCl, pH 7.5) were mixed with 5′-end biotinylated substrate D′ or substrate D (or both substrates, of which only one contained the biotin residue; Microsynth) in ∼3:1 molar ratio guide:substrate RNA (typically 0.50:0.15 μM). Reactions were initiated by addition of the methyl-3H-SAM (sixfold molar excess over guide RNA, 15 Ci/mmol; Hartmann Analytic) and samples were transferred to a 50°C heat block. Aliquots (10 µL) were removed at specified time points and mixed with 15 µL of stop buffer (0.1 mg/mL proteinase K [Thermo Scientific], 20 mM Tris pH 7.8, 20 mM NaCl, 0.1% SDS, 20 mM EDTA). Samples were then incubated at 50°C for one additional hour. Next, 5 µL aliquots were removed and added to the NeutrAvidin Agarose beads (20 µL bed volume; Thermo Scientific) previously equilibrated in low salt buffer (20 mM HEPES, 150 mM NaCl, pH 7.5). Samples were incubated at 25°C for 1 h. Next, beads were washed 3–4 times with low salt buffer and transferred to the scintillation vials. 3H-SAM incorporation into substrate RNA was determined by scintillation counting (Tri-Carb Liquid Scintillation Counter, PerkinElmer). Two independent aliquots were taken at a given time point and experiments were carried out in duplicates.
Small-angle X-ray scattering (SAXS) data collection and processing
Solution scattering data was acquired at the beamline BM29 of ESRF, Grenoble. Box C/D complexes were reconstituted with st-sR26 RNA to a final concentration of 5.0 mg/mL in sample buffer. Substrate RNAs were added to a guide:substrate molar ratio of 1:1.25. 2 mM dithiothreitol (DTT) was added to the sample to reduce radiation damage. The measurements were carried out at 40°C. The samples were exposed for 10 frames of 1 s each. The curves were compared, merged, and buffer subtracted by the beamline software BsxCube. For the apo complex, only seven frames were merged, because of radiation damage. Scattering curves were displayed and analyzed using the ATSAS package (Petoukhov et al. 2012). The final figure was generated using the R package ggplot2.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by DFG grant CA294/3-2, by EU FP7 ITN project RNPnet (contract number 289007), and by the EMBL. For the SAXS experiments we thank the ESRF, BAG MX1695.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.054320.115.
REFERENCES
- Aittaleb M, Rashid R, Chen Q, Palmer JR, Daniels CJ, Li H. 2003. Structure and function of archaeal box C/D sRNP core proteins. Nat Struct Biol 10: 256–263. [DOI] [PubMed] [Google Scholar]
- Appel CD, Maxwell ES. 2007. Structural features of the guide:target RNA duplex required for archaeal box C/D sRNA-guided nucleotide 2′-O-methylation. RNA 13: 899–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleichert F, Gagnon KT, Brown BA, Maxwell ES, Leschziner AE, Unger VM, Baserga SJ. 2009. A dimeric structure for archaeal box C/D small ribonucleoproteins. Science 325: 1384–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bower-Phipps KR, Taylor DW, Wang HW, Baserga SJ. 2012. The box C/D sRNP dimeric architecture is conserved across domain Archaea. RNA 18: 1527–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchhaupt M, Sharma S, Kellner S, Oswald S, Paetzold M, Peifer C, Watzinger P, Schrader J, Helm M, Entian KD. 2014. Partial methylation at Am100 in 18S rRNA of baker's yeast reveals ribosome heterogeneity on the level of eukaryotic rRNA modification. PLoS One 9: e89640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decatur WA, Fournier MJ. 2002. rRNA modifications and ribosome function. Trends Biochem Sci 27: 344–351. [DOI] [PubMed] [Google Scholar]
- Fu Y, Dominissini D, Rechavi G, He C. 2014. Gene expression regulation mediated through reversible m6A RNA methylation. Nat Rev Genet 15: 293–306. [DOI] [PubMed] [Google Scholar]
- Kuhn JF, Tran EJ, Maxwell ES. 2002. Archaeal ribosomal protein L7 is a functional homolog of the eukaryotic 15.5kD/Snu13p snoRNP core protein. Nucleic Acids Res 30: 931–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapinaite A, Simon B, Skjaerven L, Rakwalska-Bange M, Gabel F, Carlomagno T. 2013. The structure of the box C/D enzyme reveals regulation of RNA methylation. Nature 502: 519–523. [DOI] [PubMed] [Google Scholar]
- Lin JZ, Lai SM, Jia R, Xu AB, Zhang LM, Lu J, Ye KQ. 2011. Structural basis for site-specific ribose methylation by box C/D RNA protein complexes. Nature 469: 559–563. [DOI] [PubMed] [Google Scholar]
- Liu B, Liang XH, Piekna-Przybylska D, Liu Q, Fournier MJ. 2008. Mis-targeted methylation in rRNA can severely impair ribosome synthesis and activity. RNA Biol 5: 249–254. [DOI] [PubMed] [Google Scholar]
- Machnicka MA, Milanowska K, Oglou OO, Purta E, Kurkowska M, Olchowik A, Januszewski W, Kalinowski S, Dunin-Horkawicz S, Rother KM, et al. 2013. MODOMICS: a database of RNA modification pathways—2013 update. Nucleic Acids Res 41: D262–D267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omer AD, Ziesche S, Ebhardt H, Dennis PP. 2002. In vitro reconstitution and activity of a C/D box methylation guide ribonucleoprotein complex. Proc Natl Acad Sci 99: 5289–5294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petoukhov MV, Franke D, Shkumatov AV, Tria G, Kikhney AG, Gajda M, Gorba C, Mertens HDT, Konarev PV, Svergun DI. 2012. New developments in the ATSAS program package for small-angle scattering data analysis. J Appl Cryst 45: 342–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piekna-Przybylska D, Decatur WA, Fournier MJ. 2008. The 3D rRNA modification maps database: with interactive tools for ribosome analysis. Nucleic Acids Res 36: D178–D183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polikanov YS, Melnikov SV, Söll D, Steitz TA. 2015. Structural insights into the role of rRNA modifications in protein synthesis and ribosome assembly. Nat Struct Mol Biol 22: 342–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichow SL, Hamma T, Ferré-D'Amaré AR, Varani G. 2007. The structure and function of small nucleolar ribonucleoproteins. Nucleic Acids Res 35: 1452–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Boisvert D, Kim KK, Kim R, Kim SH. 2000. Crystal structure of a fibrillarin homologue from Methanococcus jannaschii, a hyperthermophile, at 1.6 A resolution. EMBO J 19: 317–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue S, Wang R, Yang F, Terns RM, Terns MP, Zhang X, Maxwell ES, Li H. 2010. Structural basis for substrate placement by an archaeal box C/D ribonucleoprotein particle. Mol Cell 39: 939–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.