

Abstract
Pre-mRNA splicing must occur with high fidelity and efficiency for proper gene expression. The spliceosome uses DExD/H box helicases to promote on-pathway interactions while simultaneously minimizing errors. Prp8 and Snu114, an EF2-like GTPase, regulate the activity of the Brr2 helicase, promoting RNA unwinding by Brr2 at appropriate points in the splicing cycle and repressing it at others. Mutations linked to retinitis pigmentosa (RP), a disease that causes blindness in humans, map to the Brr2 regulatory region of Prp8. Previous in vitro studies of homologous mutations in Saccharomyces cerevisiae show that Prp8-RP mutants cause defects in spliceosome activation. Here we show that a subset of RP mutations in Prp8 also causes defects in the transition between the first and second catalytic steps of splicing. Though Prp8-RP mutants do not cause defects in splicing fidelity, they result in an overall decrease in splicing efficiency. Furthermore, genetic analyses link Snu114 GTP/GDP occupancy to Prp8-dependent regulation of Brr2. Our results implicate the transition between the first and second catalytic steps as a critical place in the splicing cycle where Prp8-RP mutants influence splicing efficiency. The location of the Prp8-RP mutants, at the “hinge” that links the Prp8 Jab1–MPN regulatory “tail” to the globular portion of the domain, suggests that these Prp8-RP mutants inhibit regulated movement of the Prp8 Jab1/MPN domain into the Brr2 RNA binding channel to transiently inhibit Brr2. Therefore, in Prp8-linked RP, disease likely results not only from defects in spliceosome assembly and activation, but also because of defects in splicing catalysis.
Keywords: retinitis pigmentosa, splicing fidelity, splicing mechanism
INTRODUCTION
Most eukaryotic genes contain introns, which must be efficiently and precisely removed in order to ensure fidelity in gene expression (Lander et al. 2001; Wahl et al. 2009). Splicing occurs via two sequential transesterification reactions, first between the branch site (BS) adenosine and 5′ splice site (SS), and then between the 5′SS and 3′SS. These reactions are catalyzed by the spliceosome. The spliceosome is an RNA–protein machine composed of approximately 80 conserved proteins and five small nuclear RNAs (snRNAs): U1, U2, U4, U5, and U6 (Wahl et al. 2009). The spliceosome must assemble anew on each pre-mRNA substrate. Assembly and catalysis occur via an intricate pathway that involves striking RNA and protein rearrangements, many of which require ATP hydrolysis by DExD/H-box ATPases to proceed (Staley and Guthrie 1998; Wahl et al. 2009). These DExD/H box ATPases have also been shown to promote the fidelity of splicing through kinetic proofreading mechanisms (Semlow and Staley 2012).
Three U5 snRNP proteins, Ski2-family ATPase Brr2, GTPase Snu114, and Prp8, play key roles both in spliceosome assembly and in formation of the catalytic core central to splicing catalysis (Wahl et al. 2009). Brr2 was initially shown to unwind the U4/U6 snRNA duplex (Laggerbauer et al. 1998; Raghunathan and Guthrie 1998), required for formation of the spliceosome catalytic core (Madhani and Guthrie 1994a). Brr2 has also been implicated genetically in the second step of splicing (Hahn et al. 2012), and also in spliceosome disassembly and recycling (Small et al. 2006). Snu114 regulates Brr2 activity in a nucleotide-dependent manner, repressing Brr2 activity in its GDP-bound form and activating in its GTP-bound form (Small et al. 2006; Bellare et al. 2008). Prp8, which accommodates the catalytic snRNAs and pre-mRNA substrate in a central cavity (Newman et al. 1995; Teigelkamp et al. 1995; Galej et al. 2013; Yan et al. 2015), has been shown to regulate Brr2 activity through its RNaseH (Mozaffari-Jovin et al. 2012) and Jab1/MPN domains (Maeder et al. 2009; Mozaffari-Jovin et al. 2013). These proteins show multiple strong genetic (Kuhn et al. 2002; Bottner et al. 2005; Grainger and Beggs 2005; Liu et al. 2006), yeast-two-hybrid (van Nues and Beggs 2001), and physical interactions (Achsel et al. 1998), indicating a high degree of coordination between them.
Initial genetic evidence indicated that the Prp8 C-terminal Jab1/MPN domain negatively regulated Brr2 unwinding during splicing (Kuhn and Brow 2000; Kuhn et al. 2002). Later biochemical studies revealed that the Prp8 Jab1/MPN domain also stimulates Brr2 helicase activity while suppressing Brr2 ATPase activity (Pena et al. 2007; Maeder et al. 2009; Mozaffari-Jovin et al. 2014). Further studies have revealed additional complexity. A long “tail” extending from the Jab1/MPN domain has been shown to directly inhibit RNA binding by Brr2 (Mozaffari-Jovin et al. 2013). A series of mutations associated with retinitis pigmentosa (RP) have been mapped to the Prp8 Jab1/MPN domain and its C-terminal extension (Daiger et al. 2013). These mutations target highly conserved amino acids in Prp8, and Saccharomyces cerevisiae (yeast) carrying homologous mutations exhibit growth defects, especially at elevated temperatures (Boon et al. 2007; Pena et al. 2007; Maeder et al. 2009; Mozaffari-Jovin et al. 2013). These Prp8-RP mutants directly affect U5 snRNP and tri-snRNP maturation (Boon et al. 2007), Brr2 helicase, and ATPase activities (Pena et al. 2007; Maeder et al. 2009; Mozaffari-Jovin et al. 2013), and yeast-two-hybrid interactions between Brr2, Prp8, and Snu114 (Table 1; Pena et al. 2007). Thus Prp8-RP mutations provide a potent tool to understand how deregulation of Brr2 by Prp8 impacts splicing.
TABLE 1.
Summary of previously published yeast Prp8-RP data

Here we present evidence that Prp8-RP mutants deregulate Brr2 activity both in vitro and in vivo. Splicing reporters revealed that splicing fidelity is unaffected in Prp8-RP mutant yeast. Rather, microarray and RT-qPCR analysis show that Prp8-RP mutants cause a genome-wide decrease in splicing efficiency. In vitro splicing assays show that the decrease in splicing efficiency impacts both catalytic steps of splicing, not only spliceosome activation as had previously been suggested (Boon et al. 2007; Pena et al. 2007; Maeder et al. 2009; Mozaffari-Jovin et al. 2013). The second step of splicing is particularly affected, as shown by negative genetic interactions between Prp8-RP mutants and multiple second step splicing proteins. Finally, we link Snu114 GTPase activity in the second step of splicing. Together these data implicate proper regulation of Brr2 activity by the Prp8 Jab1/MPN domain as essential for the transition between the first and second steps of splicing, and implicate aberrant Brr2 activity late in the splicing cycle as a significant contributing factor to retinitis pigmentosa.
RESULTS
Selection of Prp8-RP mutants to use in this study
Prp8 is a highly conserved (Hodges et al. 1995), multidomain protein (Galej et al. 2013) considered to be the master regulator of splicing (Grainger and Beggs 2005). All retinitis pigmentosa (RP) mutants found in Prp8 are highly conserved and localize to its C-terminal Jab1–MPN domain (Daiger et al. 2014). In this study, we have chosen four RP-associated point mutations: Prp8-S2197F (S. cerevisiae numbering is used throughout this paper), Prp8-H2387P, Prp8-R2388G, and Prp8-R2388K (Fig. 1A). Homologous yeast mutations have previously been studied by multiple groups (Boon et al. 2007; Pena et al. 2007; Maeder et al. 2009; Mozaffari-Jovin et al. 2013), revealing that different Prp8-RP mutants have distinct phenotypes (Table 1). For example, both in vitro expression level and solubility are known to be decreased for the Prp8-H2387P mutant, but not its neighboring mutant Prp8-R2388G/K (Maeder et al. 2009; Mozaffari-Jovin et al. 2013). However, both Prp8-H2387P and Prp8-R2388G/K exhibit reduced tri-snRNP stability (Mozaffari-Jovin et al. 2013). The mutants also differ in their abilities to stimulate Brr2 helicase activity (Maeder et al. 2009), and interact with GTPase Snu114 (Table 1; Pena et al. 2007). These previous data have been taken in support of the model that Prp8-RP mutants cause problems with spliceosome assembly and activation, largely by disruption of the tri-snRNP (Boon et al. 2007; Pena et al. 2007; Maeder et al. 2009; Mozaffari-Jovin et al. 2013). However, because Brr2 and Prp8 are present throughout the splicing cycle and not just during assembly and activation, we reasoned that Prp8-RP mutants might also impact the catalytic steps of splicing. To test this hypothesis we chose four Prp8-RP mutants for further study: Prp8-S2197F, Prp8-H2387P, Prp8-R2388G, and Prp8-R2388K. We chose these particular RP mutants of the many available for multiple reasons (Table 1). (i) All four Prp8-RP mutants retain the ability to interact with Brr2 in vitro (Boon et al. 2007; Pena et al. 2007; Maeder et al. 2009). (ii) We wanted to test Prp8-RP mutants that affected Brr2 activity in distinct ways. For example, Prp8-R2388G affects both Brr2 helicase and ATPase activities, while Prp8-H2387P only impacts Brr2 ATPase activity (Maeder et al. 2009). (iii) We wanted Prp8-RP mutants that showed different interaction patterns with other spliceosome components. For instance, Prp8-H2387P retains the ability to interact with Snu114, while Prp8-R2388G does not (Pena et al. 2007). Our characterization of these four Prp8-RP mutants during the transition between the first and second step of splicing provides an overall picture of how Prp8-RP mutants affect splicing, and informs on the molecular pathology of retinitis pigmentosa.
FIGURE 1.

Splicing-specific microarrays reveal a broad splicing defect in Prp8-RP mutants. (A) Representative image from the human Prp8 Jab1/MPN domain Brr2 co-crystal (PDB:4KIT) (Mozaffari-Jovin et al. 2013). Prp8 Jab1/MPN is shown in tan, with the Prp8-RP mutants highlighted in red. The Brr2 NTD, which contains Brr2 helicase and ATPase activities, is shown in cyan, while the inactive CTD, thought to be a protein–protein interaction domain, is colored gray. (B) Microarray. Total transcript levels (T) were assessed by a probe in the 3′ exon. Pre-mRNA levels (P) were monitored by a probe complementary to the intron, and mature mRNA levels (M) were monitored by a probe that spanned the exon–exon junction. Heat map showing relative levels of total, pre- and mature mRNA for all intron-containing genes in yeast in the Prp8-S2197F mutant relative to a wild-type Prp8 strain, both grown at 37°C. Yellow indicates a relative accumulation while blue represents a relative depletion in the Prp8-S2197F mutant relative to wild type of the target of a particular probe. Values represented are log2 ratios plotted on a −2.0 to 2.0 scale. Averaged values from three distinct biological replicates are presented. All microarray data have been uploaded to the GEO database (GSE75081). The inset shows the heat map for the ACT1 gene, which is expressed at high abundance in log phase yeast and was used to confirm the array. (C) RT-qPCR validation of the array. Values represent averages and error bars are standard error of the mean for three independent biological replicates. In addition to Prp8-S2197F, relative pre-mRNA accumulation is depicted for the other Prp8-RP mutants studied. See Table 2 for more RT-qPCR data.
Prp8 retinitis pigmentosa mutants have a broad splicing defect
We initially wanted to determine how Prp8-RP mutants affected the splicing of all intron-containing genes in S. cerevisiae. Toward this goal, we performed splicing-specific microarrays (Fig. 1; Pleiss et al. 2007). These splicing-specific microarrays allow simultaneous monitoring of the cellular abundances of total mRNA (T in Fig. 1B), pre-mRNA (P in Fig. 1B) and spliced, mature mRNA (M in Fig. 1B) for all known intron-containing genes. We examined total RNA prepared from actively growing yeast cultures shifted to 37°C for 3–4 doublings, as Prp8-RP yeast often show a temperature-sensitive phenotype (Table 1). Experiments were performed as a competitive hybridization wherein results are reported as the log2 fold change in mutant RNA relative to wild type (Pleiss et al. 2007). Yellow coloring in the heat map (Fig. 1B) indicates that the cellular levels of a particular feature are increased in the Prp8-RP mutant relative to wild type, while blue represents a decrease. Prp8-S2197F shows a broad splicing defect, as indicated by the relative abundance of pre-mRNA (yellow, Fig. 1B) and lower overall level of mature mRNA (blue, Fig. 1B) in Prp8-S2197F relative to wild type. We performed RT-qPCR on ACT1 to confirm the splicing defect seen on our microarrays. Primers within the 3′ exon were designed to determine total transcript levels, while a primer set that spanned the intron–exon border assessed levels of unspliced pre-mRNA. Validating our array (inset, Fig. 1B), Prp8-S2197F shows a 2.5-fold increase in intron retention relative to wild type by RT-qPCR (Fig. 1C). We also noted that a subset of Prp8-S2197F mRNA transcripts are more abundant. We analyzed these further, looking for potential structural or functional links between these overabundant transcripts, but were unable to decipher any. All microarray data have been uploaded to the GEO database (GSE75081).
We also performed microarray analyses on three other Prp8-RP mutants, Prp8-H2387P, Prp8-R2388G, and Prp8-R2388K. Unlike Prp8-S2197F these mutants did not show a clear splicing defect on microarrays. As Prp8-S2197F shows one of the strongest temperature-sensitive growth defects among Prp8-RP mutants (Maeder et al. 2009), we hypothesized that our inability to detect a defect with the other Prp8-RP mutants was because the splicing defect in these mutants is too slight to detect on a microarray. To address this, we performed RT-qPCR on a large number of yeast transcripts present in total RNA samples isolated from Prp8-RP mutant yeast strains. RT-qPCR is a highly sensitive technique for quantifying relative transcript abundances (Wong and Medrano 2005). Consistent with our prediction that a slight splicing defect is present, Prp8-H2387P, Prp8-R2388G, and Prp8-R2388K all showed a modest (approximately 1.6-fold) splicing defect on the ACT1 transcript (Fig. 1C).
We expanded our RT-qPCR analyses to additional gene products to establish the generality of the splicing defect in Prp8-H2387P, Prp8-R2388G, and Prp8-R2388K. Results are shown in Table 2. In addition to ACT1, we tested NSP1 and BET1, all genes with canonical 5′ splice site (5′SS), branch site (BS), and 3′ splice-site (3′SS) sequences. All Prp8-RP mutants showed a splicing defect. As in the case of ACT1, the defect was more pronounced for the Prp8-S2197F mutant relative to the other Prp8-RP mutants. We further examined transcripts made from genes with noncanonical 5′SS (HOP2), BS (ERV1 and CIN2), and 3′SS (SEC17 and UBC5), as well as transcripts from a gene with a short BS–3′SS distance (HMRA1) and a gene with a very large intron (DBP2). All followed a similar pattern: Prp8-S2197F had the strongest defect, with the other Prp8-RP mutants showing a less pronounced defect. Taken together, our microarray and RT-qPCR analyses show that Prp8-RP mutants cause a significant splicing defect in yeast on a wide variety of pre-mRNA transcripts in vivo.
TABLE 2.
Summary of splicing efficiency of multiple endogenous transcripts as assessed by RT-qPCR

ACT1-CUP1 splicing reporter analyses indicate that Prp8-RP mutants perform both catalytic steps of splicing inefficiently
We wished to examine the Prp8-RP splicing defect further by determining if Prp8-RP mutants alter splicing accuracy. We used the well-established ACT1-CUP1 splicing reporter assay (Lesser and Guthrie 1993a; Stutz and Rosbash 1994; Libri et al. 1995; Query and Konarska 2004) to assess splicing fidelity and efficiency. The Cup1 protein, which under normal growth conditions is not essential, chelates copper in a concentration-dependent manner, allowing yeast to survive on otherwise lethal copper-containing media (Hamer et al. 1985). Relative yeast growth in copper-containing media therefore directly reflects the level of Cup1 protein present in the cell (Fogel et al. 1983; Hamer et al. 1985). In the ACT1-CUP1 reporter system, the well-characterized ACT1 intron is fused upstream of the coding sequence of CUP1 (Fig. 2A) such that the production of Cup1, and thus the ability of yeast to survive on growth media containing increasing concentrations of copper, directly reflects the efficiency of splicing (Lesser and Guthrie 1993a). Fidelity can also be assessed by measuring relative growth on copper in the presence of an ACT1-CUP1 reporter containing a mutation to the canonical 5′SS, BS, or 3′SS sequences (Fig. 2A). Yeast strains that show decreased fidelity display increased growth relative to wild-type yeast on copper-containing media, while hyperaccurate or inefficient splicing is revealed as poorer growth relative to wild type (Lesser and Guthrie 1993a).
FIGURE 2.

ACT1-CUP1 reporter assay shows decreased splicing efficiency in Prp8-RP mutants, especially at elevated temperatures. (A) Schematic of the ACT1-CUP1 reporter, with mutations made to the canonical splice sites of the ACT1 intron shown in red. (B) Representative copper growth of a subset of Prp8-RP mutants. Growth on copper was assessed by serial dilution onto minimal media containing 0–1.5 mM CuSO4 at 17°C, 25°C, 30°C, and 37°C. (C) Quantitation of ACT1-CUP1 reporter data. The maximum concentration of CuSO4 that supported growth was determined for wild-type and all Prp8-RP mutants at 17°C, 25°C, 30°C, and 37°C. Values shown were calculated by log2 ([Cumax Prp8-RP]/[Cumax Prp8-WT]), and colored such that worse growth than WT is shown by blue intensity. (D) Primer extension analysis of Prp8-RP mutants carrying WT, BSC, or U301G ACT1-CUP1 reporters. Total RNA was prepared from log phase cultures growing at 37°C. Primer is complementary to the 3′ exon, and U1 was used as a loading control. (E) Quantification of primer extensions. Values represent the average and standard error of the mean for three distinct biological replicates.
We incorporated the Prp8-RP mutants into the ACT1-CUP1 reporter strain background and assayed the fidelity and efficiency of splicing of the wild-type reporter, three 5′SS mutants (G1A, U2A, G5A), three BS mutants (C256A, BSC, BSG), and three 3′SS mutants (U301G, A302G, G303/4C) at 17°C, 25°C, 30°C, and 37°C on media containing 0–1.5 mM copper sulfate (Fig. 2). We then determined the maximum concentration of copper that supported growth for each Prp8-RP mutant in the presence of each ACT1-CUP1 reporter at 17°C, 25°C, 30°C, and 37°C. No Prp8-RP mutant showed decreased splicing fidelity, instead all showed hyperaccuracy/decreased efficiency especially at 37°C (Fig. 2C). The only mutant to show a significant growth defect on WT template at 37°C was Prp8-H2387P, which is likely due to the fact that this strain has previously been shown to be temperature sensitive when grown in minimal media at 37°C (Maeder et al. 2009). The ability of Prp8-RP mutants to splice ACT1-CUP1 reporters harboring mutated 5′SS, BS, or 3′SS sequences are discussed below.
The 5′SS first base pairs to the 5′ end of U1 snRNA (Zhuang and Weiner 1986; Séraphin et al. 1988; Siliciano and Guthrie 1988), which is then contacted by both the U6 (Kandels-Lewis and Séraphin 1993; Lesser and Guthrie 1993b; Sontheimer and Steitz 1993; Kim and Abelson 1996; Ryan et al. 2004) and U5 (Newman and Norman 1992; Newman et al. 1995; O'Keefe et al. 1996) snRNAs. These interactions are important for spliceosome assembly as well as splicing catalysis (for review, see Wahl et al. 2009). We tested three different 5′SS reporters, G1A, U2A, and G5A. These 5′SS mutations have previously been shown to affect the second step of splicing (exon ligation), causing an accumulation of lariat-3′exon intermediate (Fouser and Friesen 1986; Vijayraghavan et al. 1989; Lesser and Guthrie 1993a). Prp8-H2387P exhibited decreased survival on copper when compared to wild-type Prp8 when challenged with a G1A or U2A at 30°C and 37°C. In contrast, Prp8-S2197F, Prp8-R2388G, and Prp8-R2388K all showed decreased survival on copper at all temperatures when challenged with a G5A 5′SS reporter (Fig. 2B,C). Because the block to the second step of splicing is less severe with a G5A mutant when compared to G1A or U2A mutations (Lesser and Guthrie 1993a), the difference in reporter sensitivity between different Prp8-RP point mutants likely reflects a specific RP point mutation effect, rather than simply the relative strength of the block to exon ligation caused by the 5′SS reporter mutation, and is consistent with Prp8-RP mutants causing a decrease in exon ligation, the second catalytic step of splicing.
We next examined ACT1-CUP1 reporters containing mutations to the pre-mRNA BS. The pre-mRNA branch site is inspected at multiple points during the splicing cycle, both by the Msl5-Mud2 heterodimer (Berglund et al. 1997; Wang et al. 2008) during assembly and by the U2 snRNA (Black et al. 1985; Parker et al. 1987; Wu and Manley 1989) during catalysis. Branch site mutation C256A affects spliceosome assembly and the first step of splicing causing the accumulation of pre-mRNA (Fouser and Friesen 1986; Vijayraghavan et al. 1989; Burgess and Guthrie 1993), while a BSG mutation affects exon ligation, resulting in accumulation of lariat-3′exon intermediate (Bellare et al. 2008). A BSC mutation affects both catalytic steps (Fouser and Friesen 1986; Vijayraghavan et al. 1989; Burgess et al. 1990; Query and Konarska 2004). As with the 5′SS reporter mutants, different Prp8-RP point mutations showed differential copper growth effects with individual BS reporters (Fig. 2C). The BSG reporter had no effect upon the ability of Prp8-RP mutants to grow on copper (Fig. 2C). In contrast, all Prp8-RP mutants showed reduced growth on copper when coupled with a BS C256A mutant (Fig. 2B,C), especially at higher temperatures. The effect was most severe for Prp8-H2387P (Fig. 2C). Prp8-RP mutants interacted differently with the BSC reporter. Prp8-S2197F showed no reduction in growth on copper (Fig. 2B,C), while Prp8-H2387P, Prp8-R2388G, and Prp8-R2388K all exhibited reduced growth on copper at multiple temperatures (Fig. 2B,C). We confirmed this result by primer extension analysis of the BSC mutant pre-mRNA template (Fig. 2D). Prp8-H2387P, Prp8-R2388G, and Prp8-R2388K all showed increased pre-mRNA abundance relative to wild-type yeast (Fig. 2E). Together, these BS reporters indicate that, in addition to the previously documented defect in spliceosome assembly (Pena et al. 2007; Mozaffari Jovin 2013), a splicing defect at the first and second catalytic steps.
3′SS mutations U301G, A302G, and G303-4C cause a block to exon ligation, resulting in the accumulation of lariat-3′exon intermediate (Fouser and Friesen 1987; Vijayraghavan et al. 1989). As for 5′SS and BS ACT1-CUP1 reporter mutations, the Prp8-RP point mutants showed differential copper growth effects in the presence of different 3′SS mutations. Only at 37°C, Prp8-S2197F, Prp8-R2388G, and Prp8-R2388K exhibited reduced growth on copper in the presence of an A302G reporter (Fig. 2C). In contrast, Prp8-H2387P, Prp8-R2388G, and Prp8-R2388K grew less well on copper in the presence of a U301G reporter at 17°C, 25°C, 30°C, and 37°C (Fig. 2B,C), though higher temperatures exacerbated the growth defect (Fig. 2C). These results were confirmed by primer extension of a U301G pre-mRNA template (Fig. 2D), which showed a decrease in mature mRNA formation coupled with an increase in lariat-3′exon intermediate for Prp8-RP mutants (Fig. 2E). Together, results obtained from analysis of 3′SS reporter templates are consistent with the hypothesis that the Prp8-RP mutants show a decrease in efficiency at the second step of splicing.
Together, our ACT1-CUP1 reporter data reveal a complex interplay between the identity of a pre-mRNA reporter and individual Prp8-RP point mutations. Consistent with a tendency toward temperature sensitivity (Boon et al. 2007; Pena et al. 2007; Maeder et al. 2009), Prp8-RP mutations show exacerbated growth defects on copper-containing media at higher temperatures (Fig. 2C). Furthermore, Prp8-RP point mutants exhibited poor growth on copper in the presence of reporters with mutations that affect both steps of splicing, indicating that the Prp8-RP-mediated decrease in splicing efficiency revealed in our RT-qPCR (Table 2) and microarray (Fig. 1) analyses is not simply due to a defect in tri-snRNP formation or stability, or in spliceosome assembly as previously hypothesized (Boon et al. 2007; Pena et al. 2007; Maeder et al. 2009; Ferrari et al. 2011; Mozaffari Jovin 2013).
In vitro determination of first and second step catalysis rates in RP mutants
We explored the Prp8-RP splicing efficiency defects revealed by the ACT1-CUP1 reporter assay in more detail by examining the kinetics of the first and second steps of splicing using in vitro splicing assays (Lin et al. 1987) with splicing extracts (Dunn and Rader 2014) made from Prp8-RP mutant yeast. To facilitate characterization of the first step of splicing, we performed an established bimolecular splicing assay (Anderson and Moore 1997; Schellenberg et al. 2013). Instead of using a full-length actin pre-mRNA, this assay uses an actin pre-mRNA transcript truncated just downstream from the BS, which causes the splicing reaction to stall after formation of lariat intermediate (Anderson and Moore 1997). Gel electrophoresis of samples isolated from multiple time points during the splicing reaction allows for quantification of the fraction of first step lariat product (Fig. 3A). All Prp8-RP mutants showed decreased efficiency (less lariat first step product) in performing the first step of splicing (Fig. 3A,B). This decrease in first step splicing efficiency could result, in part, from a defect in tri-snRNP formation or stability, as has been reported for Prp8-RP mutants previously (Pena et al. 2007; Mozaffari-Jovin et al. 2013, 2014). Overall these data are consistent with our BSC ACT1-CUP1 reporter data (Fig. 2), as the BSC reporter affects both catalytic steps of splicing (Fouser and Friesen 1986; Vijayraghavan et al. 1989; Burgess et al. 1990; Query and Konarska 2004).
FIGURE 3.

In vitro splicing assays reveal Prp8-RP mutants show decreased first and second step splicing efficiencies. (A) Representative gel showing a splicing time course using a splicing template containing the 5′ exon and intron through the branch point in extracts made from log phase Prp8 WT and Prp8-R2388G grown at 30°C. Unlabeled lanes show control reactions with a full-length actin pre-mRNA template. Templates are body-labeled with Cy5. (B) Quantitation of the level of first step product (lariat) at 30 min. (C) splicing time course using a full-length actin pre-mRNA template and extracts from A. (D) Quantitation of second step products [(flariat + fmRNA)/(flariat-int + flariat + fmRNA)]. Values in B and D represent the average and standard error of the mean for three distinct biological replicates. The gel in Figure 5B shows that the Prp8-S2197F extract does not complete the second step of splicing.
We initially attempted to use the bimolecular splicing assay to monitor the second catalytic step of splicing as well, adding in a 3′ exon in trans to monitor formation of pre-mRNA product (Anderson and Moore 1997). However, this reaction is very low efficiency (Anderson and Moore 1997, 2000; Schellenberg et al. 2013), and coupled with the decrease in splicing efficiency caused by the Prp8-RP mutation, we were unable to obtain reliable data with this method. Instead, we chose to use a full-length actin pre-mRNA template, monitoring the second step by the appearance of both free lariat and mature mRNA (Fig. 3). These splicing time courses performed in wild-type extracts show an almost immediate appearance of second step products (mRNA and free lariat) upon the appearance of first step product (lariat-intermediate), whereas the appearance of second step products is delayed in the Prp8-RP mutant (compare 5 min time points in Fig. 3C). We observed a significant decrease in second step efficiency for all Prp8-RP mutants (Fig. 3C,D), consistent with our ACT1-CUP1 reporter data (Fig. 2). Prp8-S2197F was unable to perform the second step at all. Together these data show that the Prp8-RP decrease in splicing efficiency revealed through microarrays, RT-qPCR, and in the ACT1-CUP1 reporters is due not only to problems with tri-snRNP stability and spliceosome activation as previously thought (Boon et al. 2007; Maeder et al. 2009; Tanackovic et al. 2011; Mozaffari Jovin 2013), but also due to decreased efficiency during splicing catalysis, particularly at the second catalytic step.
Prp8-RP mutants are synthetic sick with Prp16 ATPase mutants prp16-1 and prp16-302
We found the decreased second step catalytic efficiency revealed by our ACT1-CUP1 and in vitro bimolecular splicing assays to be of particular interest because it indicates that proper control of Brr2 by Prp8 is disrupted by the Prp8-RP mutants at this later step, not simply during splicing assembly as has been previously demonstrated (Boon et al. 2007; Maeder et al. 2009; Tanackovic et al. 2011; Mozaffari Jovin 2013). Prp8, which directly regulates Brr2 activity (Maeder et al. 2009; Nielsen and Staley 2012; Mozaffari-Jovin et al. 2013; Nguyen et al. 2013), has also been implicated in 3′SS selection (Umen and Guthrie 1995a,b, 1996; Siatecka et al. 1999) as well as the transition between the first and second catalytic steps (Frank et al. 1992; Query and Konarska 2004; Liu et al. 2007). Brr2 activity has been shown to be required for unwinding the U4/U6 snRNA duplex during spliceosome activation (Raghunathan and Guthrie 1998; Zhang et al. 2015) and for spliceosome disassembly (Small et al. 2006). In addition, a Brr2 mutant has been isolated that has second step defects and shows negative genetic interactions with multiple second step factors (Hahn et al. 2012). Brr2 has also been shown to interact physically with second step factor Prp16 (van Nues and Beggs 2001; Cordin et al. 2014). Together these data led us to investigate whether the Prp8-RP mutants interacted genetically with DEAH box helicase Prp16.
We examined three different well-characterized Prp16 point mutations, prp16-1 (Burgess et al. 1990), prp16-101 (Burgess and Guthrie 1993), and prp16-302 (Madhani and Guthrie 1994b; Noble and Guthrie 1996). All three of these mutations localize to canonical DExD/H motifs in Prp16 and decrease splicing fidelity, allowing splicing of pre-mRNA branch-site adenosine to cytosine mutations (Burgess et al. 1990; Burgess and Guthrie 1993; Madhani and Guthrie 1994b). In addition, prp16-1 and prp16-302 are cold sensitive (Burgess et al. 1990; Burgess and Guthrie 1993; Noble and Guthrie 1996) and exhibit reduced RNA helicase and ATPase activity, stall before the second step of splicing at low temperatures in vitro, and are improperly retained on the spliceosome after the completion of the first step of splicing (Schwer and Guthrie 1992b; Burgess and Guthrie 1993; Wang et al. 1998; Villa and Guthrie 2005). We crossed strains containing these Prp16 mutants with our Prp8-RP mutants and examined the resultant double mutants for growth defects at 17°C, 25°C, 30°C, and 37°C. Strikingly, all Prp8-RP mutants showed synthetic sickness at 37°C when combined with either the prp16-1 or prp16-302 mutant background strain (Fig. 4). The strength of this effect is Prp8-RP mutant specific, with Prp8-R2388G > Prp8-H2387P > Prp8-R2388K > Prp8-S2197F. None of the Prp8-RP mutants showed genetic interactions with prp16-101 (Fig. 4), the only Prp16 branch-site suppressor mutant that is not cold sensitive and has been hypothesized to affect splicing fidelity more than it affects the rearrangements to the spliceosome promoted by Prp16 (Umen and Guthrie 1995b). This split between prp16-101 and both prp16-1 and prp16-302 has been noted previously with prp8-101, a Prp8 mutant that causes defects at the second step of splicing (Umen and Guthrie 1995b). These data are consistent with a model wherein deregulation of Brr2 activity by Prp8-RP mutants compounds the splicing defect that results from the defects in RNA rearrangements caused by ATPase-deficient Prp16 mutants during the transition between the first and second catalytic steps of splicing.
FIGURE 4.

Prp16 ATPase mutants show temperature-sensitive synthetic sick phenotypes when combined with Prp8-RP mutants. Growth of double mutants carrying a Prp8-RP mutant and a Prp16 mutant were assessed by serial dilution at 17°C, 25°C, 30°C, and 37°C.
Prp8-RP mutants are synthetic sick in combination with slu7-1 or prp18Δ mutants
Prp18 and Slu7 act in concert to restructure the spliceosome between the first and second steps of splicing (Vijayraghavan and Abelson 1990; Frank et al. 1992; Zhang and Schwer 1997; Chua and Reed 1999a; James et al. 2002). Slu7 interacts directly with the 3′SS after Prp16 hydrolyzes ATP (Umen and Guthrie 1995b) and mediates 3′SS selection (Frank and Guthrie 1992; Chua and Reed 1999b), especially in introns with long BS to 3′SS distances (Brys and Schwer 1996; Clark et al. 2002). Nonessential Prp18 (Horowitz and Abelson 1993) stabilizes the association of pre-mRNA exons with the U5 loop 1 (Bacíková and Horowitz 2005; Aronova et al. 2007; Crotti et al. 2007) and further stabilizes Slu7 association with the spliceosome (Zhang and Schwer 1997; Bacíková and Horowitz 2002; James et al. 2002). Slu7, Prp18, and Prp16 interact genetically with a subset of Prp8 alleles as well as with each other (Frank et al. 1992; Umen and Guthrie 1995b; Zhang and Schwer 1997; Aronova et al. 2007). We hypothesized that if Prp8-RP mutants caused defects in positioning of the exons for exon ligation, they would show synthetic sick interactions when combined with Slu7 and/or Prp18 mutants.
We created double-mutant prp18Δ (Vijayraghavan et al. 1989) Prp8-RP and slu7-1 (Frank et al. 1992) Prp8-RP strains and assayed them for growth at 17°C, 25°C, 30°C, and 37°C. slu7-1 by itself is temperature sensitive (Frank et al. 1992), and prp18Δ is both cold and temperature sensitive (Bacíková and Horowitz 2005). As seen with Prp16 ATPase mutants, different Prp8-RP mutants showed different patterns of genetic interactions with slu7-1 and prp18Δ (Fig. 5A). Prp8-H2387P slu7-1 double mutants exhibited slow growth at all temperatures tested, whereas slu7-1 Prp8-R2388K double mutants were cold sensitive (Fig. 5A, top row). Neither Prp8-S2197F nor Prp8-R2388G interacted genetically with slu7-1. Prp8-H2387P prp18Δ double mutants exhibited slow growth at all temperatures tested, similar to Prp8-H2387P slu7-1 double mutants. Prp8-S2197F prp18Δ double mutants were extremely temperature sensitive, and Prp8-R2388G and Prp8-R2388K prp18Δ double mutants exhibited a slight but repeatable temperature-sensitive phenotype (Fig. 5A, bottom row). These data are consistent with the hypothesis that Prp8-RP mutants inhibit conformational rearrangements required for exon ligation during the second catalytic step of splicing.
FIGURE 5.

Prp8-RP mutants show synthetic sickness when combined with mutations in Slu7 or Prp18. (A) Growth of double mutants carrying a Prp8-RP mutant in a slu7-1 (top) or prp18Δ background were assessed by serial dilution at 17°C, 25°C, 30°C, and 37°C. (B) In vitro splicing assays showing splicing of actin pre-mRNA templates with different BS to 3′SS distances, 38 nt, 7 nt, or 9 nt. The band representing the lariat is difficult to resolve for the templates with 7 or 9 nt between the BS and 3′SS. (C) Quantification of the fraction of mRNA (relative to total counts in each lane) for each Prp8-RP mutant splicing each actin pre-mRNA template. BS to 3-SS distance has no effect on splicing efficiency in these strains. Values represent the average and standard error of the mean for three distinct biological replicates.
As Slu7 and Prp18 are less stringently required for the splicing of templates with short BS to 3′SS distances (Brys and Schwer 1996; Clark et al. 2002), presumably because the shorter distance decreases the search area for the 3′SS making the activities of Slu7 and Prp18 less critical (Brys and Schwer 1996), we assessed if our Prp8-RP mutants also showed a negative relationship between BS to 3′SS distance and splicing efficiency. To this end, we performed in vitro splicing assays using extracts made from our Prp8-RP mutants on labeled actin pre-mRNA templates wherein the distance between the BS and 3′SS was shortened from the wild-type distance of 38 nucleotides to seven nucleotides (Brys and Schwer 1996), and compared the splicing efficiency between these different pre-mRNA templates. We observed no difference in the efficiency of splicing when the distance between the BS and 3′SS was altered (Fig. 5B,C), consistent with our microarray and RT-qPCR data, which did not show any pattern of decreased efficiency in splicing templates with long BS to 3′SS distances (Table 2; Fig. 1). Together, these genetic and biochemical data indicate that the defect caused by Prp8-RP mutants is not alleviated by a short pre-mRNA BS to 3′SS distance, and is likely distinct from that caused by slu7-1 and prp18Δ.
Prp8-RP mutants interact genetically with Snu114 GTPase mutants
Snu114 is an essential U5 component (Fabrizio et al. 1997) that interacts physically (Achsel et al. 1998; van Nues and Beggs 2001) and genetically (Brenner and Guthrie 2005; Costanzo et al. 2010) with Prp8 and Brr2. Like the Prp8 Jab1/MPN domain (Maeder et al. 2009; Mozaffari-Jovin et al. 2013), Snu114 has also been shown to regulate Brr2 activity (Small et al. 2006) and has been implicated in the second catalytic step of splicing (Liu et al. 1997) in addition to its well-characterized role in spliceosome activation (Bartels et al. 2002, 2003; Brenner and Guthrie 2005). Snu114 is a GTPase with strong homology with the ribosomal translocase EF-2/EF-G (Fabrizio et al. 1997), and its GTPase activity correlates with several important steps in spliceosome assembly (Bartels et al. 2002; Brenner and Guthrie 2005, 2006), catalysis (Bartels et al. 2003; Bellare et al. 2008), and disassembly (Small et al. 2006). Multiple mutations designed to affect Snu114's ability to bind GTP, hydrolyze GTP, and act as a guanine exchange factor (GEF) have been developed (Bartels et al. 2003; Brenner and Guthrie 2005), based on Snu114 homology with EF-2/EF-G. The snu114-12 mutation is presumed to decrease the rate of GTP hydrolysis by Snu114 and is temperature sensitive. The snu114-14 mutation is presumed to cause decreased GEF activity and is both cold and temperature sensitive, and the snu114-15 mutation is presumed to decrease nucleotide binding and is temperature sensitive (Brenner and Guthrie 2005, 2006).
Because of its genetic, physical, and regulatory interactions with both Brr2 and Prp8 (Table 1), we hypothesized that Snu114 mutants with compromised GTPase activity might exacerbate the defects caused by deregulation of Brr2 by Prp8-RP mutants. To this goal, we created a series of double mutants wherein we combined a Snu114 mutant with a Prp8-RP mutant and assessed double mutant growth at multiple temperatures. As in all of our previous double mutant genetics, the strength of the effect varied by RP mutant. H2387P showed the strongest temperature-sensitive growth defects when combined with snu114-12, snu114-14, or snu114-15 at 37°C (Fig. 6). Prp8-S2197F showed a temperature-sensitive growth defect when combined with Snu114 mutants that affect GTP binding or hydrolysis (snu114-12 or snu114-15). Double mutants snu114-15 Prp8-R2388G and snu114-15 Prp8-R2388K exhibited temperature-sensitive growth at 37°C (Fig. 6). Together these data show that impaired Snu114 GTPase activity exacerbates the Prp8-RP phenotype and is consistent with regulation of Brr2 by both Prp8 and Snu114.
FIGURE 6.

Prp8-RP mutants show synthetic sickness when combined with Snu114 GTPase mutants. Growth of double mutants carrying a Prp8-RP mutant and a Snu114 mutant were assessed by serial dilution at 17°C, 25°C, 30°C, and 37°C.
DISCUSSION
Using microarrays and RT-qPCR, we have shown that Prp8-RP mutants cause an overall decrease in splicing efficiency, irrespective of pre-mRNA template consensus site strength. We have further shown, using the ACT1-CUP1 reporter system, that Prp8-RP mutants do not cause a defect in splicing fidelity. Instead, through the strong negative growth effects Prp8-RP mutants exhibited in the presence of the U301G ACT1-CUP1 reporter, this assay hinted that Prp8-RP mutants caused a splicing defect at the second catalytic step, a result we clarified using in vitro splicing assays made from Prp8-RP mutants. Consistent with this, we show synthetic sickness/lethality between double mutants of Prp8-RP mutants and mutants of multiple second step splicing factors, including Prp16, Slu7, and Prp18. Finally, we show synthetic sickness between double mutants of Snu114 GTPase mutants and Prp8-RP mutants, suggesting a potential means for regulation. Together, our results indicate that Prp8-RP mutants cause deregulation of Brr2 at multiple points in the splicing cycle to cause a decrease in overall splicing efficiency.
Characterization of a Prp8-RP splicing defect
Previously, Tanackovic et al. (2011) demonstrated a decrease in splicing efficiency of over 50 housekeeping genes in lymphocytes isolated from humans carrying Prp8-RP mutants. This is consistent with previous data showing that distinct Prp8 mutants affect different pools of pre-mRNA transcripts in both yeast (Pleiss et al. 2007) and humans (Ivings et al. 2008). Here we present microarray data (Fig. 1) that show Prp8-S2197F causes a broad splicing defect in yeast. In addition, we show by RT-qPCR (Table 2) that Prp8-RP mutants H2387P, R2388G, and R2388K also have a splicing defect on a wide variety of endogenous pre-mRNA templates. The degree of splicing defect varies both by Prp8-RP mutant as well as by pre-mRNA template, consistent with previous observations for other Prp8 mutants (Pleiss et al. 2007; Ivings et al. 2008). We examined transcripts with both consensus and nonconsensus 5′SS, BS, and 3′SS and found no correlation between how strongly the splice-sites matched consensus and the relative defect in splicing seen in Prp8-RP mutants for a given transcript.
Prp8 Jab1/MPN domain regulation of Brr2 activity at multiple points in the splicing cycle: movement of the Jab1/MPN tail
Regulation of Brr2 by the Jab1/MPN domain is complex. Mutational data initially indicated that the interaction of Prp8 Jab1/MPN with Brr2 was repressive to Brr2 activity (Kuhn et al. 2002), though later biochemical evidence showed that the Jab1/MPN domain in fact activates Brr2 helicase activity (Pena et al. 2007; Maeder et al. 2009) while repressing futile ATPase cycling (Maeder et al. 2009) to make Brr2 a more efficient enzyme. Later structural and biochemical data complicated things further. A human Prp8 Jab1/MPN domain Brr2 co-crystal revealed a previously unresolved conserved “tail” extending from the globular portion of the Prp8 Jab1/MPN domain into the RNA binding pocket of Brr2 (Fig. 1A; Mozaffari-Jovin et al. 2013). Biochemical analyses confirmed that the Prp8 Jab1/MPN domain tail inhibited Brr2 binding to RNA, and removal of the tail restored binding (Mozaffari-Jovin et al. 2013). Consistent with this, Brr2 is bound to the U4 snRNA in the yeast tri-snRNP structure (Nguyen et al. 2015), and in this structure, the Prp8 Jab1/MPN tail is not present, as the presence of RNA precludes the occupation of the Brr2 RNA binding pocket by the Prp8 Jab1/MPN tail. However, interpretation of Brr2 regulation based on the tri-snRNP crystal structure is complicated by the fact that though Brr2 is loaded onto U4 and in an active conformation (i.e., the Prp8-Jab1/MPN tail is not bound), addition of ATP causes the tri-snRNP to dissociate to U4/U6 di-snRNP and free U5. This is an interesting result, as Brr2 is expected to unwind U4/U6 in the context of the tri-snRNP (Laggerbauer et al. 1998; Raghunathan and Guthrie 1998), dissociating it into three individual snRNPs (free U4, U5, and U6), and suggests that more is required to activate Brr2 to unwind U4/U6. Thus, it is likely that, though the binding affinity between Brr2 and the Prp8 Jab1/MPN domain is high (Mozaffari-Jovin et al. 2014), the Prp8 Jab1/MPN tail can dissociate from the Brr2 RNA binding pocket upon minute conformational changes. This is consistent with a previous Brr2 Prp8 Jab1/MPN domain co-crystal (Nguyen et al. 2013), where the density corresponding to the Prp8 Jab1/MPN tail is absent.
A Brr2 Prp8-Jab1/MPN co-crystal (Mozaffari-Jovin et al. 2013) shows that histidine 2387 of Prp8 is located precisely at the hinge connecting the Jab1/MPN tail to the globular portion of the domain (Fig. 1A). Conversion of histidine to proline in the Prp8-H2387P mutation both removes the potential for stabilizing hydrogen bonds to form and, perhaps more importantly, the proline would restrict hinge flexibility (Hovmöller et al. 2002), potentially making it much more difficult for the Prp8 Jab1/MPN tail to insert into the Brr2 RNA binding pocket to inhibit RNA binding by Brr2, resulting in an increased likelihood of aberrant unwinding. Increasing temperature would exacerbate this effect. Prp8-S2197 caps an α-helix in the globular portion of the Prp8 Jab1/MPN domain (Fig. 1) and is likely important to the fold stability of this portion of the protein (Mozaffari-Jovin et al. 2013). Prp8-R2388G and Prp8-R2388K showed differential effects in many of our genetic assays. In the Brr2 Prp8 Jab1/MPN domain co-crystal, one of the Prp8-R2388 side-chain amino groups is within three angstroms of the backbone of a conserved stretch of prolines in the Brr2 ratchet domain (Mozaffari-Jovin et al. 2013). R2388 is not in a normal extended conformation, rather its side chain is bent toward Brr2 (Fig. 1; Mozaffari-Jovin et al. 2013). Mutation of arginine to glycine would remove this contact due to shortening the side chain as well as removing the positive charge, while mutation of arginine to lysine preserves overall side chain length and charge, but removes an amino group. Phenotypes of the Prp8-R2388K and Prp8-R2388G mutations can potentially be attributed to loss of this or other electrostatic interactions in the spliceosome.
In addition to the Prp8 Jab1/MPN domain, Brr2 activity is also auto-regulated by an extension from the N-terminal domain of Brr2 (Absmeier et al. 2015; Zhang et al. 2015). Deletion of the N-terminal extension of Brr2 is lethal in yeast (Zhang et al. 2015), while more modest truncations cause defects in splicing with the extent of intron retention occurring proportionally to the extent of the truncation (Absmeier et al. 2015). Furthermore, the Brr2 N-terminal extension affects Brr2 interaction with RNA, where the truncations actually increase the affinity of Brr2 for RNA, and stimulates Brr2 helicase and ATPase activities above those of the WT protein (Absmeier et al. 2015). Importantly, the Brr2 N-terminal extension impacts Brr2 function in the tri-snRNP, as rather than unwinding the U4/U6 snRNAs in the tri-snRNP, truncated Brr2 instead stimulates release of free U5 and U4/U6 di-snRNP (Absmeier et al. 2015). These data suggest Brr2 also contains the ability to regulate splicing via promotion of an off-pathway discard of U5.
Combining these data, a model (Fig. 7) emerges. We speculate that after association of the U5 snRNP to the U4/U6 di-snRNP to form the tri-snRNP, Brr2 binds to the U4 snRNA (Hahn et al. 2012; Nguyen et al. 2015) and its N-terminal extension rearranges to promote Brr2 activity (Absmeier et al. 2015). Then, after unwinding the U4/U6 snRNA duplex during spliceosome activation, the Prp8 Jab1/MPN domain tail inserts into the Brr2 RNA binding channel, and the Brr2 N-terminal extension rearranges, inhibiting Brr2 from aberrantly unwinding RNA prior to the first step of splicing. As genetic data indicate that Brr2 is important for the second step of splicing (Hahn et al. 2012), after the first transesterification reaction the Prp8 Jab1/MPN domain tail must then be removed from the Brr2 RNA binding pocket so it can unwind RNA duplexes to rearrange the spliceosome for the second catalytic step. The Prp8 Jab1/MPN tail would then reinsert into the Brr2 RNA binding pocket to inhibit Brr2 from inappropriately unwinding RNA until after completion of the second step, where Brr2 is again required to disassemble the spliceosome (Small et al. 2006). It is important to note that Brr2 has been shown to function in numerous critical protein–protein interactions throughout the splicing cycle (van Nues and Beggs 2001; Cordin et al. 2014), and therefore the Prp8-Jab1/MPN tail may not be regulating Brr2 helicase activity per se as we hypothesize above, rather it might be affecting Brr2 conformation and thus its ability to serve as a binding platform for other spliceosomal proteins.
FIGURE 7.
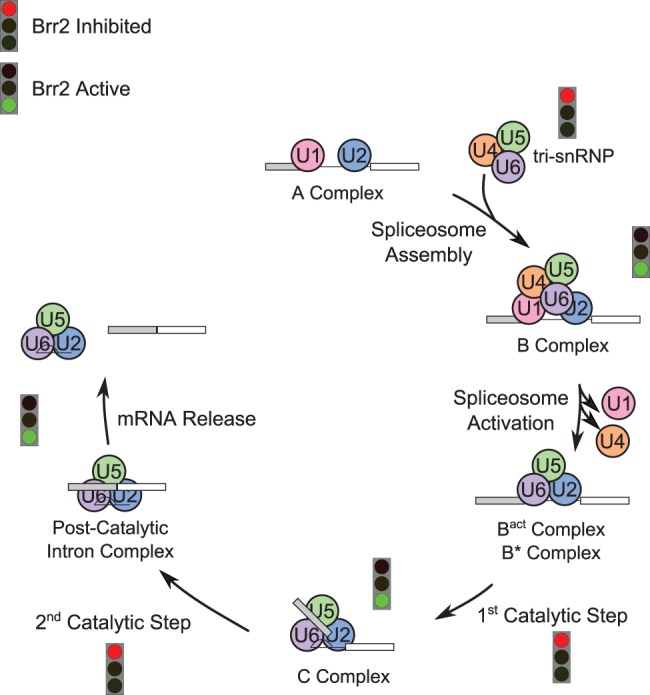
Model of Brr2 activity at different points in the splicing cycle. Pathways for assembly and catalysis of the spliceosome (Wahl et al. 2009) are shown. Points when Brr2 is expected to be active and unwinding RNA are indicated by a green light, while points where Brr2 will be repressed by the Prp8 Jab1/MPN domain are indicated by a red light.
Regulation via the GTP/GDP binding state of Snu114 and/or regulation via ubiquitin binding of the Prp8 Jab1/MPN domain (Bellare et al. 2006, 2008) are appealing options for what controls the movement of the Prp8 Jab1/MPN domain tail into the Brr2 RNA binding pocket. It has been previously shown that GDP is repressive to spliceosome disassembly, and that GTP alleviates GDP-dependent repression (Small et al. 2006). In our model above, the GTP-bound form of Snu114 would be required for Brr2 activity, while the GDP-bound form of Snu114 would be repressive. Consistent with this idea, in the recent structure of a post-catalytic spliceosome, wherein Brr2 activity is presumed to be repressed, Snu114 is found in a GDP-bound form (Yan et al. 2015). If Brr2 activity was required for the second step, Snu114 would thus have to rebind GTP. The Prp8 Jab1/MPN domain has been shown to have ubiquitin binding activity in vitro (Bellare et al. 2006), and has been shown to regulate U4/U6 snRNA unwinding by Brr2 (Bellare et al. 2008). It is possible that the presence or absence of ubiquitin on the Jab1/MPN domain regulates the Prp8 C-terminal tail, thus regulating Brr2 activity, however no biochemical or crystallographic evidence currently exists to support this idea.
This model (Fig. 7) nicely reconciles our data and previous data in the literature about how Prp8-RP mutants affect the ability of the Jab1/MPN domain to regulate Brr2 activity (Table 1). The defect in tri-snRNP assembly and stability results from decreased Prp8 protein stability caused by the Prp8-RP mutants (Pena et al. 2007; Maeder et al. 2009; Mozaffari-Jovin et al. 2013) as well as U5 maturation defects (Boon et al. 2007) that make it less likely that intact tri-snRNPs will form. Cells with Prp8-RP mutations also show decreased levels of other tri-snRNP components, including Snu114 and Brr2 (Tanackovic et al. 2011), further decreasing tri-snRNP levels. The well-documented spliceosome activation defect (Boon et al. 2007; Tanackovic et al. 2011; Mozaffari-Jovin et al. 2013) seen in the presence of Prp8-RP mutants, commonly attributed to the negative effects on Brr2 helicase and ATPase coupling activity caused by Prp8-RP mutants (Pena et al. 2007; Maeder et al. 2009) and reflected in the decreased efficiency of both catalytic steps in our in vitro splicing assays (Fig. 3A), would then further impact splicing efficiency in RP.
Prp8-RP mutants show negative genetic interactions with second step splicing factors
Prp16 has been shown to interact directly with the C-terminal helicase cassette of Brr2 (Cordin et al. 2014), potentially linking its activity to Brr2 activity. We tested this idea genetically using Prp16 mutants (Fig. 4). Cold-sensitive Prp16 mutants, prp16-1 and prp16-302, affect the fidelity (Burgess et al. 1990; Burgess and Guthrie 1993), ATPase (Schwer and Guthrie 1991, 1992a,b) activity, and dwell time on the spliceosome (Schwer and Guthrie 1991, 1992a,b; Villa and Guthrie 2005; Tseng et al. 2011) of Prp16, whereas prp16-101 only affects Prp16 fidelity (Burgess et al. 1990). When combined with either prp16-1 or prp16-302, Prp8-H2387P showed the strongest temperature sensitivity, followed by Prp8-R2388G, Prp8-R2388K, and finally Prp8-S2197F. However, in combination with wild-type Prp16, Prp8-H2387P has the strongest temperature-sensitive phenotype in this background, followed by Prp8-S2197F. These two Prp8-RP mutants show the strongest (Prp8-H2387P) and weakest (Prp8-S2197F) phenotypes, respectively, when combined with Prp16 ATPase mutants, indicating that the degree of temperature sensitivity of the Prp8-RP mutant is not the primary determinant. Prp8-H2387P and Prp8-R2388G/K both localize to the hinge region of the Prp8 Jab1/MPN tail, supporting disruption of tail movement as a potential explanation for the relative strength differences. No phenotype was observed when Prp8-RP mutants were combined with prp16-101, leading us to hypothesize that it is the disruption of the RNA rearrangements catalyzed by Prp16 ATPase activity and/or Prp16 retention on the spliceosome that causes the synthetic phenotype with Prp8-RP mutants, not the relaxed fidelity these mutants show.
Slu7 and Prp18 function as a heterodimer (Zhang and Schwer 1997) and are important for positioning the 3′ exon for exon ligation (James et al. 2002; Aronova et al. 2007; Crotti et al. 2007), especially for pre-mRNA transcripts that have long BS to 3′SS distances (Brys and Schwer 1996). Prp8-H2387P showed the strongest overall temperature-sensitive growth defect when combined with either the slu7-1 point mutation or deletion of Prp18 (Fig. 5A). This could be because Prp8-H2387P exhibits the strongest growth defect at elevated temperatures, which likely reflects the difficulty of controlling Brr2 activity in this mutant. Coupling increased the likelihood of Brr2 unwinding RNA structures inappropriately with difficulties in positioning the 5′ and 3′ exons for the second step caused by deletion of Prp18 or the slu7-1 point mutation likely makes it harder for the spliceosome to form a correct second step conformation.
The slu7-1 phenotype itself is strong enough that no growth for either wild-type Prp8 or the Prp8-RP mutants could be observed at 37°C (Fig. 5A). Though the prp18Δ mutation causes slow growth at 37°C, the combination of prp18Δ with Prp8-S2197F showed an even more severe temperature-sensitive phenotype, while Prp8-R2388G/K showed similar levels of growth in combination with prp18Δ as wild-type Prp8 (Fig. 5A). Because the S2197F mutation is thought to destabilize the fold of the Prp8 Jab1/MPN domain (Mozaffari-Jovin et al. 2013), the severity of the temperature sensitivity in this double mutant might reflect either direct or indirect weakening of the interaction of Slu7 with the spliceosome as a compounding consequence of Prp8 Jab1/MPN destabilization and Prp18 deletion.
Oddly, the combination of temperature-sensitive mutants Prp8-H2387P and Prp8-R2388K with temperature-sensitive slu7-1 resulted in a cold-sensitive phenotype at 17°C (Fig. 5A). Cold exacerbates the effects of stalls in a biochemical pathway, so one potential explanation for this is that the combination of these individual point mutations affects a particular conformational rearrangement that requires both Slu7 and Brr2 activity for forward progression through the splicing cycle. Because we saw no difference in the efficiency of splicing of pre-mRNA templates with different BS-3′SS distances, we assume that Prp8-RP mutants are not directly involved in the same step of 3′ exon positioning as slu7-1 and prp18Δ (Fig. 5B,C), rather that these Prp8-RP mutants affect a different aspect of exon ligation.
Inefficient splicing: consequences for retinitis pigmentosa
The human retina is a highly metabolically active tissue and consequently must correctly splice a large volume of pre-mRNAs in order to meet demand (Dezso et al. 2008; Tanackovic et al. 2011). Spliceosome snRNAs, and presumably other spliceosome components, have been shown to be highly expressed in retinal cells (Tanackovic et al. 2011). Given the demand for splicing in the retina, it is not surprising that defects in pre-mRNA splicing factors can lead to diseases like RP. In addition to Prp8, RP mutations have been mapped to spliceosome proteins Brr2, Prp16, Prp31, Prp3, and Prp6 (Ajmal et al. 2014; Daiger et al. 2014). With the exception of Prp16, all of the other spliceosome targets of RP are tri-snRNP components (Wahl et al. 2009). Previous studies of RP homologs in yeast have demonstrated defects in U5 biogenesis (Boon et al. 2007) and tri-snRNP assembly and/or stability (Pena et al. 2007; Mozaffari-Jovin et al. 2013). Similarly, studies of cells from RP patients as well as mouse models of RP have pointed to defects in tri-snRNP joining and spliceosome activation (Graziotto et al. 2011; Tanackovic et al. 2011; Farkas et al. 2014). These data have led to a molecular model for RP pathogenesis in which inefficient tri-snRNP joining at the step of spliceosome activation leads to an overall decrease in splicing efficiency such that the spliceosome cannot meet demand in the retina, causing RP.
Our data expand on and complement this model. We show that, in yeast, Prp8-RP mutants show defects at both catalytic steps of splicing, not simply at the step of tri-snRNP joining. This implies that the overall RP-related splicing defect seen both in yeast (this work) and humans (Tanackovic et al. 2011) reflects not only problems with initial assembly and activation of the spliceosome, but throughout the catalytic steps of the splicing cycle as well. The recent discovery of a family with Prp16 related RP (Ajmal et al. 2014) supports this model, as Prp16 is only a second step factor and is not present during spliceosome assembly and activation.
Conclusions and future directions
Together, our data have important implications for both the regulation and mechanism of the catalytic steps of splicing as well as the molecular pathology of Prp8-linked retinitis pigmentosa. We have shown that Prp8-RP mutants cause deregulation of Brr2 throughout the splicing cycle, not simply at the early steps of splicing assembly and activation as previously proposed (Boon et al. 2007; Pena et al. 2007; Maeder et al. 2009; Mozaffari-Jovin et al. 2013). Based on the fact that Prp8-RP mutants show synthetic sick/lethal interactions with multiple step two factors, this deregulation likely manifests as inappropriate unwinding of RNA helices between the first and second catalytic steps of splicing. Future studies should focus on precisely defining the nature of the spliceosome rearrangements undergone between the catalytic steps of splicing.
MATERIALS AND METHODS
Plasmids
Plasmids used in this study are indicated in Table 3. ACT-CUP reporter plasmids have been described previously (Burgess and Guthrie 1993; Lesser and Guthrie 1993a; Query and Konarska 2004; Liu et al. 2007). Plasmid pPrp8WT is the PRP8 gene in a pRS313 plasmid (Kuhn et al. 2002). Mutations S2197F, H2387P, R2388G, and R2388K were made in pRS313-PRP8 using Quikchange Mutagenesis (Agilent Technologies). Mutants prp16-1, prp16-101, and prp16-302 were made by mutagenesis of plasmid pRS314-PRP16. Snu114 plasmids have been described previously (Brenner and Guthrie 2005, 2006).
TABLE 3.
Plasmids used in this study

Strains
Strains were constructed using standard genetic methods (Guthrie and Fink 2004). A list of strains used in this study is available in Table 4. Strain yMeg035 is identical to strain yTB72, a gift from Tamara Brenner. Strain yMeg030 was isolated from a cross between yMeg035 and yTV311 (Villa and Guthrie 2005). Strain yMeg365 is identical to strain yMeg030, except DBR1 was deleted and replaced by NatNT2 (Janke et al. 2004) in the genome. Strain yTT120 was a gift from Argenta Price. Strain yMeg251 was isolated from a cross of A7C8Aa (Frank and Guthrie 1992) with yMeg030. Strain yMeg212 was a generous gift of Haralambos Hadjivassiliou.
TABLE 4.
Strains included in this work

Double mutants were made using 5FOA shuffling (Guthrie and Fink 2004). Strains used for Figures 1 and 2 were created by shuffling in plasmids from Table 3 column Prp8-RP into strain yMeg035. Strains used in Figure 2 were created by shuffling in plasmids from Table 3 column Prp8-RP into strain yMeg030 (growth assays) or yMeg365 (primer extensions). Strains used in Figure 4 were a matrix made by shuffling plasmids from columns Prp8 and Prp16 into strain yTT120. Strains used for Figure 4 were created by shuffling in plasmids from Table 3 column Prp8-RP into strain yMeg410. Strains used for Figure 5 were created by shuffling in plasmids from Table 3 column Prp8-RP into strain yMeg251 or yMeg212. Strains used in Figure 6 were a matrix made by shuffling plasmids from columns Prp8 and Snu114 into strain yTB108.
Microarrays
Microarrays were designed by Jeffrey Pleiss and have been described previously (Pleiss et al. 2007). Total RNA was made using a previously described hot acid phenol extraction protocol (DeRisi et al. 1997) from strains grown overnight to saturation in YEPD media at 30°C and then diluted to OD600 ≈ 0.1 and allowed to grow to an OD600 ≈ 0.6 at 37°C. cDNA synthesis was performed as described previously (Pleiss et al. 2007). Briefly, 50 μg of total RNA was primed with 5 mg/mL random dN9 primers and reverse transcribed in the presence of amino-allyl UTP to enable later dye labeling. Reactions were left for at least 2 h at 42°C. RNA was subsequently removed by treatment with NaOH, and cDNA was split into two samples, one labeled with Cy3 and the other with Cy5 to enable comparison of dye-flipped samples. Labeled cDNAs were hybridized to the arrays at 65°C for 16 h and washed as previously described (Pleiss et al. 2007).
Microarray fluorescence was measured at 532 and 635 nm with an Axon Instruments GenePix 4000B, and resultant images processed with GenePixPro v. 5.1. Six technical replicates were combined and normalized as log2 ratios that were averaged and subjected to centroid-linkage hierarchical means clustering using Cluster 3.0 (de Hoon et al. 2004). Results are presented as a heat map generated using Java Treeview (Saldanha 2004) showing the averaged values of three independent biological replicates.
ACT-CUP assay
ACT-CUP reporter plasmids (Burgess and Guthrie 1993; Lesser and Guthrie 1993a; Query and Konarska 2004; Liu et al. 2007) were transformed into strains yMeg031, yMeg032, yMeg132, yMeg033, and yMeg034 using standard methods (Guthrie and Fink 2004). Saturated overnight cultures grown in –Leu media at 30°C were diluted to OD600 ≈ 0.1 and allowed to grow to an OD600 ≈ 0.6. Cultures were then diluted back to an OD600 = 0.1 and spotted onto –Leu plates supplemented with 0–1.5 mM CuSO4. Plates were placed at 30°C, 37°C, 25°C, and 17°C and were grown for 2 (30°C), 3 (25°C and 37°C), or 7 (17°C) days and photographed (Lesser and Guthrie 1993a). The maximum concentration a specific strain could grow at for each temperature and ACT1-CUP1 reporter was assessed visually and a score was assessed using the formula log2 ([Cumax Prp8-RP]/[Cumax Prp8-WT]) for comparisons between strains.
Primer extensions
Cultures were grown overnight to saturation in –Leu media at 30°C and then diluted to OD600 ≈ 0.1 and allowed to grow to an OD600 ≈ 0.6 at 37°C. Cells were pelleted, washed with water, and frozen at −80°C until isolation. Total cellular RNA was isolated using hot acid phenol followed by isopropanol precipitation as described previously (DeRisi et al. 1997). Primer extensions were performed as previously described (Frank and Guthrie 1992), except the U1 (Lesser and Guthrie 1993a) and YAC6 (Query and Konarska 2004) primers were end-labeled with Cy5 to facilitate detection using a Typhoon Phosphorimager (GE Healthcare).
To quantify the primer extension gels, we first calculated the intensity of the bands corresponding to the pre-mRNA (fpre), lariat intermediate (flariat-int), and mature mRNA (fmRNA) individually, and summed them to determine ftotal = fpre + flariat-int + fmRNA. We then calculated relative level of the pre-mRNA, lariat intermediate, and mature mRNA as Relpre = fpre/ftotal, Rellariat-int = flariat-int/ftotal, and RelmRNA = fmRNA/ftotal. The U1 band was used only to aid in identification of the individual bands on the gel.
RNA isolation and RT-qPCR
Cultures were grown overnight to saturation in YEPD media at 30°C and then diluted to OD600 ≈ 0.1 and allowed to grow to an OD600 ≈ 0.6 at 37°C. Cells were pelleted, washed briefly with water, and then snap frozen at −80°C until need for isolation. Total cellular RNA was isolated using hot acid phenol followed by isopropanol precipitation as described previously (DeRisi et al. 1997). 20 µg total RNA was combined with 15 µL RNase Free DNase I (NEB) in a total volume of 250 µL for 1 h at 37°C, and then phenol–chloroform extracted again.
RT-qPCR was performed as described previously (Pleiss et al. 2007). Briefly, for reverse transcription 2 µg of DNase-treated total RNA was incubated with 0.5 µL of 1 mg/mL dN9 primers (Life Technologies) in a total volume of 10 µL at 65°C for 5 min and then chilled on ice for 5 min. Four microliters of 5× First Strand Synthesis Buffer (Invitrogen), 1 µL 10 mM dNTPs (Fermentas), 1 µL 0.1 M DTT (Invitrogen), 1 µL Superscript III Reverse Transcriptase (Invitrogen), and 3 µL of water were added to bring the total reaction volume to 20 µL. Reverse transcription was allowed to proceed for 30 min at 50°C. Reactions were then diluted between 1:75 and 1:250 with water prior to analysis by qPCR. For qPCR, 10 µL of diluted cDNA from reverse transcription was combined with 0.25 mM each gene-specific primer, 1× SYBR green (Roche), 0.625 µL dimethyl sulfoxide (Invitrogen), 0.4 mM dTNPs (Fermentas), 1× Standard Taq Buffer (NEB) and 0.25 µL Standard Taq (NEB) and water to a total volume of 25 µL. qPCR cycle conditions were 95°C 3 min, 39 cycles of 95°C 15 sec, 55°C 30 sec, 72°C 15 sec, followed by one cycle at 72°C for 3 min. Exon primers were designed to be within the 3′ exon of all genes tested and were used to measure total RNA levels. pre-mRNA levels were assessed using intron primers. The product of these primes spanned an intron–exon junction to ensure that only pre-mRNA was measured. Splicing efficiency was calculated as the level of pre-mRNA divided by the total RNA for each gene. Fold change relative to wild type was determined by dividing the splicing efficiency of a given gene in a Prp8-RP mutant strain by its splicing efficiency in the isogenic Prp8-WT strain. Three biological replicates using total RNA isolated from cultures grown on different days were each assayed in technical triplicate (Table 5).
TABLE 5.
qPCR primers used

Preparation of splicing extracts
Cultures to be used in the making of splicing extracts were grown in rich media at 30°C until reaching an OD600 of approximately 1.0. Under these conditions, all RP mutants showed identical doubling times to WT yeast. Splicing extracts were prepared as described previously (Umen and Guthrie 1995a) except that frozen cell pellets were ground with a ball mill (Retch MM 301 mixer-mill; 3×3 min at 11 Hz and 2×3 min at 12 Hz). Prepared extracts were made into aliquots and stored at −80°C until used.
In vitro splicing assays and determination of rates of first and second catalytic steps of splicing
Pre-mRNA templates were transcribed with Cy5-UTP (5% of total UTP). In vitro splicing reactions consisted of 40% vol/vol splicing extract combined with a pre-mRNA template and 3% PEG 8000, 60 mM potassium phosphate pH 7.0, 2.5 mM MgCl2, 2 mM ATP, and 10 units of RNasin. Reactions were performed for indicated lengths and temperatures (usually 30 min at 30°C). Reactions were stopped by the addition of SD to 2.5% and 1 mM EDTA. Ten units of proteinase K (Invitrogen) were added, and reactions were digested at 60°C for 10 min and then loaded onto a 6% polyacrylamide, 6 M urea, TBE gel (Hadjivassiliou et al. 2014).
Quantitation of the level of first step product [(flariat-int)/total)] and second step product [(flariat + fmRNA)/(flariat-int + flariat + fmRNA)] was performed at the 30 and 10 min time points, respectively. Values reported represent the average and standard error of the mean for three distinct biological replicates using three distinct preparations of splicing extract.
DATA DEPOSITION
All data sets have been submitted to the GEO database (GSE75081) and are available for download.
ACKNOWLEDGMENTS
We thank Argenta Price, Sarah Ledoux, Haralambos Hadjivassiliou, Beate Schwer, Raymond O'Keefe, Martin Kupiec, Jonathan Staley, Tamara Brenner, and Corina Maeder for strains and/or plasmids. We thank Erica Moehle, Jaclyn Greimann, and Argenta Price for critical assistance with microarray printing and analysis. We also thank Anne de Bruyn Kops, John Abelson, and the rest of the Guthrie laboratory for helpful discussions and comments on this manuscript. This work was supported by grants RO1GM21119-42 (C.G.) and F32GM109512 (M.M.) from the National Institutes of Health National Institute of General Medical Sciences. C.G. is an American Cancer Society Research Professor of Molecular Genetics.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.055459.115.
REFERENCES
- Absmeier E, Wollenhaupt J, Mozaffari-Jovin S, Becke C, Lee C-T, Preussner M, Heyd F, Urlaub H, Lührmann R, Santos KF, et al. 2015. The large N-terminal region of the Brr2 RNA helicase guides productive spliceosome activation. Genes Dev 29: 2576–2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Achsel T, Ahrens K, Brahms H, Teigelkamp S, Lührmann R. 1998. The human U5-220kD protein (hPrp8) forms a stable RNA-free complex with several U5-specific proteins, including an RNA unwindase, a homologue of ribosomal elongation factor EF-2, and a novel WD-40 protein. Mol Cell Biol 18: 6756–6766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ajmal M, Khan MI, Neveling K, Khan YM, Azam M, Waheed NK, Hamel CP, Ben-Yosef T, Baere ED, Koenekoop RK, et al. 2014. A missense mutation in the splicing factor gene DHX38 is associated with early-onset retinitis pigmentosa with macular coloboma. J Med Genet 51: 444–448. [DOI] [PubMed] [Google Scholar]
- Anderson K, Moore MJ. 1997. Bimolecular exon ligation by the human spliceosome. Science 276: 1712–1716. [DOI] [PubMed] [Google Scholar]
- Anderson K, Moore MJ. 2000. Bimolecular exon ligation by the human spliceosome bypasses early 3′ splice site AG recognition and requires NTP hydrolysis. RNA 6: 16–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronova A, Bacikova D, Crotti LB, Horowitz DS, Schwer B. 2007. Functional interactions between Prp8, Prp18, Slu7, and U5 snRNA during the second step of pre-mRNA splicing. RNA 13: 1437–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacíková D, Horowitz DS. 2002. Mutational analysis identifies two separable roles of the Saccharomyces cerevisiae splicing factor Prp18. RNA 8: 1280–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacíková D, Horowitz DS. 2005. Genetic and functional interaction of evolutionarily conserved regions of the Prp18 protein and the U5 snRNA. Mol Cell Biol 25: 2107–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartels C, Klatt C, Lührmann R, Fabrizio P. 2002. The ribosomal translocase homologue Snu114p is involved in unwinding U4/U6 RNA during activation of the spliceosome. EMBO Rep 3: 875–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartels C, Urlaub H, Luhrmann R, Fabrizio P. 2003. Mutagenesis suggests several roles of Snu114p in pre-mRNA splicing. J Biol Chem 278: 28324–28334. [DOI] [PubMed] [Google Scholar]
- Bellare P, Kutach AK, Rines AK, Guthrie C, Sontheimer EJ. 2006. Ubiquitin binding by a variant Jab1/MPN domain in the essential pre-mRNA splicing factor Prp8p. RNA 12: 292–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellare P, Small EC, Huang X, Wohlschlegel JA, Staley JP, Sontheimer EJ. 2008. A role for ubiquitin in the spliceosome assembly pathway. Nat Struct Mol Biol 15: 444–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berglund JA, Chua K, Abovich N, Reed R, Rosbash M. 1997. The splicing factor BBP interacts specifically with the pre-mRNA branchpoint sequence UACUAAC. Cell 89: 781–787. [DOI] [PubMed] [Google Scholar]
- Black DL, Chabot B, Steitz JA. 1985. U2 as well as U1 small nuclear ribonucleoproteins are involved in premessenger RNA splicing. Cell 42: 737–750. [DOI] [PubMed] [Google Scholar]
- Boon K-L, Grainger RJ, Ehsani P, Barrass JD, Auchynnikava T, Inglehearn CF, Beggs JD. 2007. prp8 mutations that cause human retinitis pigmentosa lead to a U5 snRNP maturation defect in yeast. Nat Struct Mol Biol 14: 1077–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottner CA, Schmidt H, Vogel S, Michele M, Käufer NF. 2005. Multiple genetic and biochemical interactions of Brr2, Prp8, Prp31, Prp1 and Prp4 kinase suggest a function in the control of the activation of spliceosomes in Schizosaccharomyces pombe. Curr Genet 48: 151–161. [DOI] [PubMed] [Google Scholar]
- Brenner TJ, Guthrie C. 2005. Genetic analysis reveals a role for the C terminus of the Saccharomyces cerevisiae GTPase Snu114 during spliceosome activation. Genetics 170: 1063–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner TJ, Guthrie C. 2006. Assembly of Snu114 into U5 snRNP requires Prp8 and a functional GTPase domain. RNA 12: 862–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brys A, Schwer B. 1996. Requirement for SLU7 in yeast pre-mRNA splicing is dictated by the distance between the branchpoint and the 3′ splice site. RNA 2: 707–717. [PMC free article] [PubMed] [Google Scholar]
- Burgess SM, Guthrie C. 1993. A mechanism to enhance mRNA splicing fidelity: the RNA-dependent ATPase Prp16 governs usage of a discard pathway for aberrant lariat intermediates. Cell 73: 1377–1391. [DOI] [PubMed] [Google Scholar]
- Burgess S, Couto JR, Guthrie C. 1990. A putative ATP binding protein influences the fidelity of branchpoint recognition in yeast splicing. Cell 60: 705–717. [DOI] [PubMed] [Google Scholar]
- Chua K, Reed R. 1999a. Human step II splicing factor hSlu7 functions in restructuring the spliceosome between the catalytic steps of splicing. Genes Dev 13: 841–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua K, Reed R. 1999b. The RNA splicing factor hSlu7 is required for correct 3′ splice-site choice. Nature 402: 207–210. [DOI] [PubMed] [Google Scholar]
- Clark TA, Sugnet CW, Ares M. 2002. Genomewide analysis of mRNA processing in yeast using splicing-specific microarrays. Science 296: 907–910. [DOI] [PubMed] [Google Scholar]
- Cordin O, Hahn D, Alexander R, Gautam A, Saveanu C, Barrass JD, Beggs JD. 2014. Brr2p carboxy-terminal Sec63 domain modulates Prp16 splicing RNA helicase. Nucleic Acids Res 42: 13897–13910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo M, Baryshnikova A, Bellay J, Kim Y, Spear ED, Sevier CS, Ding H, Koh JLY, Toufighi K, Mostafavi S, et al. 2010. The genetic landscape of a cell. Science 327: 425–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crotti LB, Bacíková D, Horowitz DS. 2007. The Prp18 protein stabilizes the interaction of both exons with the U5 snRNA during the second step of pre-mRNA splicing. Genes Dev 21: 1204–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daiger SP, Sullivan LS, Bowne SJ. 2013. Genes and mutations causing retinitis pigmentosa. Clin Genet 84: 132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daiger SP, Bowne SJ, Sullivan LS. 2014. Genes and mutations causing autosomal dominant retinitis pigmentosa. Cold Spring Harb Perspect Med 5: a017129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Hoon MJL, Imoto S, Nolan J, Miyano S. 2004. Open source clustering software. Bioinforma Oxf Engl 20: 1453–1454. [DOI] [PubMed] [Google Scholar]
- DeRisi JL, Iyer VR, Brown PO. 1997. Exploring the metabolic and genetic control of gene expression on a genomic scale. Science 278: 680–686. [DOI] [PubMed] [Google Scholar]
- Dezso Z, Nikolsky Y, Sviridov E, Shi W, Serebriyskaya T, Dosymbekov D, Bugrim A, Rakhmatulin E, Brennan RJ, Guryanov A, et al. 2008. A comprehensive functional analysis of tissue specificity of human gene expression. BMC Biol 6: 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn EA, Rader SD. 2014. Preparation of yeast whole cell splicing extract. Methods Mol Biol 1126: 123–135. [DOI] [PubMed] [Google Scholar]
- Fabrizio P, Laggerbauer B, Lauber J, Lane WS, Lührmann R. 1997. An evolutionarily conserved U5 snRNP-specific protein is a GTP-binding factor closely related to the ribosomal translocase EF-2. EMBO J 16: 4092–4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farkas MH, Lew DS, Sousa ME, Bujakowska K, Chatagnon J, Bhattacharya SS, Pierce EA, Nandrot EF. 2014. Mutations in pre-mRNA processing factors 3, 8, and 31 cause dysfunction of the retinal pigment epithelium. Am J Pathol 184: 2641–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari S, Di Iorio E, Barbaro V, Ponzin D, Sorrentino FS, Parmeggiani F. 2011. Retinitis pigmentosa: genes and disease mechanisms. Curr Genomics 12: 238–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogel S, Welch JW, Cathala G, Karin M. 1983. Gene amplification in yeast: CUP1 copy number regulates copper resistance. Curr Genet 7: 347–355. [DOI] [PubMed] [Google Scholar]
- Fouser LA, Friesen JD. 1986. Mutations in a yeast intron demonstrate the importance of specific conserved nucleotides for the two stages of nuclear mRNA splicing. Cell 45: 81–93. [DOI] [PubMed] [Google Scholar]
- Fouser LA, Friesen JD. 1987. Effects on mRNA splicing of mutations in the 3′ region of the Saccharomyces cerevisiae actin intron. Mol Cell Biol 7: 225–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank D, Guthrie C. 1992. An essential splicing factor, SLU7, mediates 3′ splice site choice in yeast. Genes Dev 6: 2112–2124. [DOI] [PubMed] [Google Scholar]
- Frank D, Patterson B, Guthrie C. 1992. Synthetic lethal mutations suggest interactions between U5 small nuclear RNA and four proteins required for the second step of splicing. Mol Cell Biol 12: 5197–5205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galej WP, Oubridge C, Newman AJ, Nagai K. 2013. Crystal structure of Prp8 reveals active site cavity of the spliceosome. Nature 493: 638–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grainger RJ, Beggs JD. 2005. Prp8 protein: at the heart of the spliceosome. RNA 11: 533–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graziotto JJ, Farkas MH, Bujakowska K, Deramaudt BM, Zhang Q, Nandrot EF, Inglehearn CF, Bhattacharya SS, Pierce EA. 2011. Three gene-targeted mouse models of RNA splicing factor RP show late-onset RPE and retinal degeneration. Invest Ophthalmol Vis Sci 52: 190–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guthrie C, Fink GR. 2004. Guide to yeast genetics and molecular and cell biology. Gulf Professional Publishing, Oxford, UK. [Google Scholar]
- Hadjivassiliou H, Rosenberg OS, Guthrie C. 2014. The crystal structure of S. cerevisiae Sad1, a catalytically inactive deubiquitinase that is broadly required for pre-mRNA splicing. RNA 20: 656–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn D, Kudla G, Tollervey D, Beggs JD. 2012. Brr2p-mediated conformational rearrangements in the spliceosome during activation and substrate repositioning. Genes Dev 26: 2408–2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamer DH, Thiele DJ, Lemontt JE. 1985. Function and autoregulation of yeast copperthionein. Science 228: 685–690. [DOI] [PubMed] [Google Scholar]
- Hodges PE, Jackson SP, Brown JD, Beggs JD. 1995. Extraordinary sequence conservation of the PRP8 splicing factor. Yeast Chichester Engl 11: 337–342. [DOI] [PubMed] [Google Scholar]
- Horowitz DS, Abelson J. 1993. A U5 small nuclear ribonucleoprotein particle protein involved only in the second step of pre-mRNA splicing in Saccharomyces cerevisiae. Mol Cell Biol 13: 2959–2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovmöller S, Zhou T, Ohlson T. 2002. Conformations of amino acids in proteins. Acta Crystallogr D Biol Crystallogr 58: 768–776. [DOI] [PubMed] [Google Scholar]
- Ivings L, Towns KV, Matin MA, Taylor C, Ponchel F, Grainger RJ, Ramesar RS, Mackey DA, Inglehearn CF. 2008. Evaluation of splicing efficiency in lymphoblastoid cell lines from patients with splicing-factor retinitis pigmentosa. Mol Vis 14: 2357–2366. [PMC free article] [PubMed] [Google Scholar]
- James S-A, Turner W, Schwer B. 2002. How Slu7 and Prp18 cooperate in the second step of yeast pre-mRNA splicing. RNA 8: 1068–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janke C, Magiera MM, Rathfelder N, Taxis C, Reber S, Maekawa H, Moreno-Borchart A, Doenges G, Schwob E, Schiebel E, et al. 2004. A versatile toolbox for PCR-based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes. Yeast Chichester Engl 21: 947–962. [DOI] [PubMed] [Google Scholar]
- Kandels-Lewis S, Séraphin B. 1993. Involvement of U6 snRNA in 5′ splice site selection. Science 262: 2035–2039. [DOI] [PubMed] [Google Scholar]
- Kim CH, Abelson J. 1996. Site-specific crosslinks of yeast U6 snRNA to the pre-mRNA near the 5′ splice site. RNA 2: 995–1010. [PMC free article] [PubMed] [Google Scholar]
- Kuhn AN, Brow DA. 2000. Suppressors of a cold-sensitive mutation in yeast U4 RNA define five domains in the splicing factor Prp8 that influence spliceosome activation. Genetics 155: 1667–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn AN, Reichl EM, Brow DA. 2002. Distinct domains of splicing factor Prp8 mediate different aspects of spliceosome activation. Proc Natl Acad Sci 99: 9145–9149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laggerbauer B, Achsel T, Lührmann R. 1998. The human U5-200kD DEXH-box protein unwinds U4/U6 RNA duplices in vitro. Proc Natl Acad Sci 95: 4188–4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. 2001. Initial sequencing and analysis of the human genome. Nature 409: 860–921. [DOI] [PubMed] [Google Scholar]
- Lesser CF, Guthrie C. 1993a. Mutational analysis of pre-mRNA splicing in Saccharomyces cerevisiae using a sensitive new reporter gene, CUP1. Genetics 133: 851–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesser CF, Guthrie C. 1993b. Mutations in U6 snRNA that alter splice site specificity: implications for the active site. Science 262: 1982–1988. [DOI] [PubMed] [Google Scholar]
- Libri D, Stutz F, McCarthy T, Rosbash M. 1995. RNA structural patterns and splicing: molecular basis for an RNA-based enhancer. RNA 1: 425–436. [PMC free article] [PubMed] [Google Scholar]
- Lin RJ, Lustig AJ, Abelson J. 1987. Splicing of yeast nuclear pre-mRNA in vitro requires a functional 40S spliceosome and several extrinsic factors. Genes Dev 1: 7–18. [DOI] [PubMed] [Google Scholar]
- Liu ZR, Laggerbauer B, Lührmann R, Smith CW. 1997. Crosslinking of the U5 snRNP-specific 116-kDa protein to RNA hairpins that block step 2 of splicing. RNA 3: 1207–1219. [PMC free article] [PubMed] [Google Scholar]
- Liu S, Rauhut R, Vornlocher H-P, Lührmann R. 2006. The network of protein-protein interactions within the human U4/U6.U5 tri-snRNP. RNA 12: 1418–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Query CC, Konarska MM. 2007. Opposing classes of prp8 alleles modulate the transition between the catalytic steps of pre-mRNA splicing. Nat Struct Mol Biol 14: 519–526. [DOI] [PubMed] [Google Scholar]
- Madhani HD, Guthrie C. 1994a. Dynamic RNA-RNA interactions in the spliceosome. Annu Rev Genet 28: 1–26. [DOI] [PubMed] [Google Scholar]
- Madhani HD, Guthrie C. 1994b. Genetic interactions between the yeast RNA helicase homolog Prp16 and spliceosomal snRNAs identify candidate ligands for the Prp16 RNA-dependent ATPase. Genetics 137: 677–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeder C, Kutach AK, Guthrie C. 2009. ATP-dependent unwinding of U4/U6 snRNAs by the Brr2 helicase requires the C-terminus of Prp8. Nat Struct Mol Biol 16: 42–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozaffari Jovin S. 2013. Mechanism of regulation of spliceosome activation by Brr2 and Prp8 and links to retinal disease. https://ediss.uni-goettingen.de/handle/11858/00-1735-0000-0001-BC3E-4 (accessed November 22, 2013).
- Mozaffari-Jovin S, Santos KF, Hsiao H-H, Will CL, Urlaub H, Wahl MC, Lührmann R. 2012. The Prp8 RNase H-like domain inhibits Brr2-mediated U4/U6 snRNA unwinding by blocking Brr2 loading onto the U4 snRNA. Genes Dev 26: 2422–2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozaffari-Jovin S, Wandersleben T, Santos KF, Will CL, Lührmann R, Wahl MC. 2013. Inhibition of RNA helicase Brr2 by the C-terminal tail of the spliceosomal protein Prp8. Science 341: 80–84. [DOI] [PubMed] [Google Scholar]
- Mozaffari-Jovin S, Wandersleben T, Santos KF, Will CL, Lührmann R, Wahl MC. 2014. Novel regulatory principles of the spliceosomal Brr2 RNA helicase and links to retinal disease in humans. RNA Biol 11: 298–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman AJ, Norman C. 1992. U5 snRNA interacts with exon sequences at 5′ and 3′ splice sites. Cell 68: 743–754. [DOI] [PubMed] [Google Scholar]
- Newman AJ, Teigelkamp S, Beggs JD. 1995. snRNA interactions at 5′ and 3′ splice sites monitored by photoactivated crosslinking in yeast spliceosomes. RNA 1: 968–980. [PMC free article] [PubMed] [Google Scholar]
- Nguyen THD, Li J, Galej WP, Oshikane H, Newman AJ, Nagai K. 2013. Structural basis of Brr2-Prp8 interactions and implications for U5 snRNP biogenesis and the spliceosome active site. Structure 21: 910–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen THD, Galej WP, Bai X-C, Savva CG, Newman AJ, Scheres SHW, Nagai K. 2015. The architecture of the spliceosomal U4/U6.U5 tri-snRNP. Nature 523: 47–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen KH, Staley JP. 2012. Spliceosome activation: U4 is the path, stem I is the goal, and Prp8 is the keeper. Let's cheer for the ATPase Brr2! Genes Dev 26: 2461–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble SM, Guthrie C. 1996. Identification of novel genes required for yeast pre-mRNA splicing by means of cold-sensitive mutations. Genetics 143: 67–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Keefe RT, Norman C, Newman AJ. 1996. The invariant U5 snRNA loop 1 sequence is dispensable for the first catalytic step of pre-mRNA splicing in yeast. Cell 86: 679–689. [DOI] [PubMed] [Google Scholar]
- Parker R, Siliciano PG, Guthrie C. 1987. Recognition of the TACTAAC box during mRNA splicing in yeast involves base pairing to the U2-like snRNA. Cell 49: 229–239. [DOI] [PubMed] [Google Scholar]
- Pena V, Liu S, Bujnicki JM, Lührmann R, Wahl MC. 2007. Structure of a multipartite protein-protein interaction domain in splicing factor prp8 and its link to retinitis pigmentosa. Mol Cell 25: 615–624. [DOI] [PubMed] [Google Scholar]
- Pleiss JA, Whitworth GB, Bergkessel M, Guthrie C. 2007. Transcript specificity in yeast pre-mRNA splicing revealed by mutations in core spliceosomal components. PLoS Biol 5: e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Query CC, Konarska MM. 2004. Suppression of multiple substrate mutations by spliceosomal prp8 alleles suggests functional correlations with ribosomal ambiguity mutants. Mol Cell 14: 343–354. [DOI] [PubMed] [Google Scholar]
- Raghunathan PL, Guthrie C. 1998. RNA unwinding in U4/U6 snRNPs requires ATP hydrolysis and the DEIH-box splicing factor Brr2. Curr Biol 8: 847–855. [DOI] [PubMed] [Google Scholar]
- Ryan DE, Kim CH, Murray JB, Adams CJ, Stockley PG, Abelson J. 2004. New tertiary constraints between the RNA components of active yeast spliceosomes: a photo-crosslinking study. RNA 10: 1251–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldanha AJ. 2004. Java Treeview—extensible visualization of microarray data. Bioinformatics 20: 3246–3248. [DOI] [PubMed] [Google Scholar]
- Schellenberg MJ, Wu T, Ritchie DB, Fica S, Staley JP, Atta KA, LaPointe P, MacMillan AM. 2013. A conformational switch in PRP8 mediates metal ion coordination that promotes pre-mRNA exon ligation. Nat Struct Mol Biol 20: 728–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwer B, Guthrie C. 1991. PRP16 is an RNA-dependent ATPase that interacts transiently with the spliceosome. Nature 349: 494–499. [DOI] [PubMed] [Google Scholar]
- Schwer B, Guthrie C. 1992a. A conformational rearrangement in the spliceosome is dependent on PRP16 and ATP hydrolysis. EMBO J 11: 5033–5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwer B, Guthrie C. 1992b. A dominant negative mutation in a spliceosomal ATPase affects ATP hydrolysis but not binding to the spliceosome. Mol Cell Biol 12: 3540–3547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semlow DR, Staley JP. 2012. Staying on message: ensuring fidelity in pre-mRNA splicing. Trends Biochem Sci 37: 263–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Séraphin B, Kretzner L, Rosbash M. 1988. A U1 snRNA:pre-mRNA base pairing interaction is required early in yeast spliceosome assembly but does not uniquely define the 5′ cleavage site. EMBO J 7: 2533–2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siatecka M, Reyes JL, Konarska MM. 1999. Functional interactions of Prp8 with both splice sites at the spliceosomal catalytic center. Genes Dev 13: 1983–1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siliciano PG, Guthrie C. 1988. 5′ splice site selection in yeast: genetic alterations in base-pairing with U1 reveal additional requirements. Genes Dev 2: 1258–1267. [DOI] [PubMed] [Google Scholar]
- Small EC, Leggett SR, Winans AA, Staley JP. 2006. The EF-G-like GTPase Snu114p regulates spliceosome dynamics mediated by Brr2p, a DExD/H box ATPase. Mol Cell 23: 389–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sontheimer EJ, Steitz JA. 1993. The U5 and U6 small nuclear RNAs as active site components of the spliceosome. Science 262: 1989–1996. [DOI] [PubMed] [Google Scholar]
- Staley JP, Guthrie C. 1998. Mechanical devices of the spliceosome: motors, clocks, springs, and things. Cell 92: 315–326. [DOI] [PubMed] [Google Scholar]
- Stutz F, Rosbash M. 1994. A functional interaction between Rev and yeast pre-mRNA is related to splicing complex formation. EMBO J 13: 4096–4104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanackovic G, Ransijn A, Thibault P, Abou Elela S, Klinck R, Berson EL, Chabot B, Rivolta C. 2011. PRPF mutations are associated with generalized defects in spliceosome formation and pre-mRNA splicing in patients with retinitis pigmentosa. Hum Mol Genet 20: 2116–2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teigelkamp S, Whittaker E, Beggs JD. 1995. Interaction of the yeast splicing factor PRP8 with substrate RNA during both steps of splicing. Nucleic Acids Res 23: 320–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng C-K, Liu H-L, Cheng S-C. 2011. DEAH-box ATPase Prp16 has dual roles in remodeling of the spliceosome in catalytic steps. RNA 17: 145–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umen JG, Guthrie C. 1995a. A novel role for a U5 snRNP protein in 3′ splice site selection. Genes Dev 9: 855–868. [DOI] [PubMed] [Google Scholar]
- Umen JG, Guthrie C. 1995b. Prp16p, Slu7p, and Prp8p interact with the 3′ splice site in two distinct stages during the second catalytic step of pre-mRNA splicing. RNA 1: 584–597. [PMC free article] [PubMed] [Google Scholar]
- Umen JG, Guthrie C. 1996. Mutagenesis of the yeast gene PRP8 reveals domains governing the specificity and fidelity of 3′ splice site selection. Genetics 143: 723–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Nues RW, Beggs JD. 2001. Functional contacts with a range of splicing proteins suggest a central role for Brr2p in the dynamic control of the order of events in spliceosomes of Saccharomyces cerevisiae. Genetics 157: 1451–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayraghavan U, Abelson J. 1990. PRP18, a protein required for the second reaction in pre-mRNA splicing. Mol Cell Biol 10: 324–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayraghavan U, Company M, Abelson J. 1989. Isolation and characterization of pre-mRNA splicing mutants of Saccharomyces cerevisiae. Genes Dev 3: 1206–1216. [DOI] [PubMed] [Google Scholar]
- Villa T, Guthrie C. 2005. The Isy1p component of the NineTeen complex interacts with the ATPase Prp16p to regulate the fidelity of pre-mRNA splicing. Genes Dev 19: 1894–1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl MC, Will CL, Lührmann R. 2009. The spliceosome: design principles of a dynamic RNP machine. Cell 136: 701–718. [DOI] [PubMed] [Google Scholar]
- Wang Y, Wagner JD, Guthrie C. 1998. The DEAH-box splicing factor Prp16 unwinds RNA duplexes in vitro. Curr Biol CB 8: 441–451. [DOI] [PubMed] [Google Scholar]
- Wang Q, Zhang L, Lynn B, Rymond BC. 2008. A BBP-Mud2p heterodimer mediates branchpoint recognition and influences splicing substrate abundance in budding yeast. Nucleic Acids Res 36: 2787–2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong ML, Medrano JF. 2005. Real-time PCR for mRNA quantitation. Biotechniques 39: 75–85. [DOI] [PubMed] [Google Scholar]
- Wu J, Manley JL. 1989. Mammalian pre-mRNA branch site selection by U2 snRNP involves base pairing. Genes Dev 3: 1553–1561. [DOI] [PubMed] [Google Scholar]
- Yan C, Hang J, Wan R, Huang M, Wong CCL, Shi Y. 2015. Structure of a yeast spliceosome at 3.6-angstrom resolution. Science 349: 1182–1191. [DOI] [PubMed] [Google Scholar]
- Zhang X, Schwer B. 1997. Functional and physical interaction between the yeast splicing factors Slu7 and Prp18. Nucleic Acids Res 25: 2146–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Li X, Hill RC, Qiu Y, Zhang W, Hansen KC, Zhao R. 2015. Brr2 plays a role in spliceosomal activation in addition to U4/U6 unwinding. Nucleic Acids Res 43: 3286–3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang Y, Weiner AM. 1986. A compensatory base change in U1 snRNA suppresses a 5′ splice site mutation. Cell 46: 827–835. [DOI] [PubMed] [Google Scholar]