

Abstract
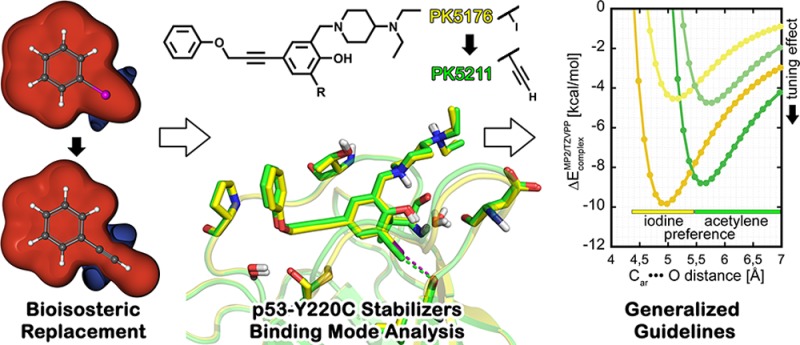
Bioisosteric replacements are widely used in medicinal chemistry to improve physicochemical and ADME properties of molecules while retaining or improving affinity. Here, using the p53 cancer mutant Y220C as a test case, we investigate both computationally and experimentally whether an ethynyl moiety is a suitable bioisostere to replace iodine in ligands that form halogen bonds with the protein backbone. This bioisosteric transformation is synthetically feasible via Sonogashira cross-coupling. In our test case of a particularly strong halogen bond, replacement of the iodine with an ethynyl group resulted in a 13-fold affinity loss. High-resolution crystal structures of the two analogues in complex with the p53-Y220C mutant enabled us to correlate the different affinities with particular features of the binding site and subtle changes in ligand binding mode. In addition, using QM calculations and analyzing the PDB, we provide general guidelines for identifying cases where such a transformation is likely to improve ligand recognition.
A frequent task faced by medicinal chemists is to replace chemical moieties with known liabilities in a lead molecule. Such bioisosteric replacements are often used to modulate physicochemical properties of lead compounds, mitigate compound metabolism issues, or tune activities.1 In addition to the more straightforward “classical” bioisosteric replacements, such as hydrogen to fluorine for increased metabolic stability, “nonclassical” bioisosteric replacements involve group exchanges that exceed simple one-atom substitutions while retaining biological activities (mostly by emulating spatial and electronic properties of the original group).1,2
The ethynyl (or “acetylene CH”) group is an interesting example of a nonclassical bioisostere because of its versatility: its π cloud is useful for mimicking aromatic systems,3,4 and its polarized −CH moiety is a weak hydrogen bond donor and is a replacement for an iodine atom in a study on p53-HDM2 inhibitors.5,6 The molecular electrostatic potentials for halobenzenes (Cl, Br, I) and phenylacetylene are remarkably similar, both bearing an area of positive charge at the tip of the C-X/H bond (where X = Cl, Br, I, or ethynyl) as well as an area of negative charge perpendicular to the C-X/H bond (Figure 1a). The paradigm of halogen bonding, which is based on this anisotropic electron density distribution (the σ-hole7,8), has attracted attention in life sciences9−11 and drug discovery.12−16
Figure 1.

Potential for bioisosterism between ethynyl and halogen substituents. (a) Electrostatic potentials plotted onto the isodensity surfaces at 0.003 au for chlorobenzene, bromobenzene, iodobenzene, and phenylacetylene. Color ranges of energies in atomic units are also shown. Calculations were done at the MP2/TZVPP level of theory. (b) Structural formulas for gefitinib (left) and erlotinib (right) with the chlorine-to-ethynyl substitution highlighted. (c) Cocrystal structure of gefitinib bound to EGFR (PDB: 2ITY) in an overlay with the binding mode of erlotinib from PDB 4HJO. The geometry of the Cl···O halogen bond is highlighted in yellow.
Intriguingly, there is bioisosteric replacement of a chlorine moiety with an ethynyl group for two marketed drugs sharing the same scaffold, the EGFR inhibitors gefitinib (iressa) and erlotinib (tarceva).17 The chloro moiety in gefitinib is involved in a weak halogen bond12 with the backbone carbonyl oxygen of Leu788 in the back pocket of the ATP-binding site of the kinase,18 and this contact is mimicked by the ethynyl group in the cocrystal structure of erlotinib19 (Figure 1c).
Inactivation of the tumor suppressor protein p53 by destabilizing mutations is frequently a crucial step in oncogenesis, and p53 is mutated in approximately half of human cancers.20 However, reversing the effects of such mutations is difficult, and p53 itself is often considered undruggable, although its interactions with its negative regulators MDM2 and MDM4 can be targeted.21 We have previously designed a novel class of biologically active small-molecule stabilizers that bind to a mutation-induced surface crevice in the DNA-binding domain of the p53 cancer mutant Y220C14 and inhibit mutant aggregation.22 A key feature of this class is a central iodophenol moiety that forms a halogen bond with the carbonyl oxygen of Leu145 at the bottom of the binding pocket. Systematic analysis of the PDB and evaluation of all attractive iodine···carbonyl contacts by our QM-based scoring function XBScore(23) revealed that the representatives of this series of p53 stabilizers consistently show one of the most favorable interaction geometries reported to date. Here, we used this ligand class to theoretically and experimentally study the effect of iodine-ethynyl substitution and its implications for the bioisosterism of both groups.
Results
Biophysical Studies
To assess the suitability of ethynyl substituents as bioisosteres of aromatic iodo moieties, we compared the binding affinities of the previously published p53-Y220C stabilizers PK5174 (tert-butyl N-[3-(3-{[4-(diethylamino)piperidin-1-yl]methyl}-4-hydroxy-5-iodophenyl)prop-2-yn-1-yl]carbamate; 1) and PK5176 (2-((4-(diethylamino)piperidin-1-yl)methyl)-6-iodo-4-(3-phenoxyprop-1-ynyl)phenol; 2)14 with the ethynyl analogue of 2, PK5211 (2-((4-(diethylamino)piperidin-1-yl)methyl)-6-ethynyl-4-(3-phenoxyprop-1-ynyl)phenol; 3) (structures in Scheme 1). 3 was synthesized from compound 2 in a Sonogashira cross-coupling reaction with trimethylsilylacetylene (Pd(PPh3)4, CuI, Et3N, THF, rt) followed by removal of the trimethylsilyl group with TBAF and LC/MS purification.
Scheme 1. Structural Formulas of the Three Compounds Used in This Work.

Affinities for p53-Y220C core domain as measured by (direct) ITC are KD (PK5174) = 15.5 μM and KD (PK5176) = 20.6 μM.14 The affinity of PK5211 (3) was determined indirectly as KD (PK5211) = 271 μM by competition ITC with PK5174 (1); see also Figure 2.
We assessed the binding of 3 to the stabilized DNA-binding domain of the p53 mutant Y220C (residues 94–312) using an array of biophysical techniques. In DSF experiments (e.g., Figure 2a), 3 behaved as a mutant p53 stabilizer, inducing a concentration-dependent increase of the protein melting point (Tm). Protein-observed 2D NMR (HSQC) experiments showed binding of 3 to the protein with chemical shift perturbation (CSP) and quenching (line broadening) of particular signals from within or close to the Y220C-induced surface crevice. The iodinated analogue 2 binds to the Y220C mutant in an intermediate exchange regime on the NMR time scale with quenching of residue signals consistent with its ∼20 μM KD. 3 showed CSP for only some signals while quenching others, as it did not bind as tightly as 2 (Figure 2b). We failed to obtain suitable profiles for analysis from direct ITC titration experiments for 3 and therefore determined its KD indirectly using competition ITC titrations with a suitable ligand (1) that has a large enthalpy of binding. In the presence of 500 μM of compound 3, the ITC binding affinity of 1 was shifted from 15.5 to 44.2 μM (Figure 2c, d). Using the competition binding equation of Zhang and Zhang,24 we obtained an estimated binding affinity of ∼270 μM for 3. Because the competition titration could not be performed at a c-value of 2 or higher, it was not possible to reliably decompose ΔG into ΔH and ΔS.25
Figure 2.

Biophysical characterization of PK5211 (3) binding to p53-Y220C DNA-binding domain. (a) Differential scanning fluorimetry (DSF) shows concentration-dependent thermal stabilization of the mutant protein. (b) 1H/15N-HSQC NMR shows that 3 perturbs and quenches specific residue signals upon binding, consistent with its binding to the mutation-induced surface crevice. (c, d) Competition ITC experiments: PK5174 (1) binds to p53-Y220C with KD = 15.5 μM; its binding affinity is shifted to KD = 44.2 μM upon addition of 500 μM of compound 3. The resulting affinity of 3 for p53-Y220C was then calculated as KD = 271 μM.24
Crystal Structures of Ligand Complexes
We determined the crystal structure of the p53-Y220C-3 complex at a resolution of 1.4 Å. The two acetylenic groups of the ligand fulfill two different roles: (i) the central ethynyl moiety provides a linear rigid linker and (ii) the terminal ethynyl substituent acts as a hydrogen bond donor and iodo bioisostere. Overall, the binding mode is essentially the same as for the parent compound (2, Figure 3). The phenol moiety binds to the center of the mutation-induced cavity, sandwiched between several prolines and a valine of loops S3/S4 and S7/S8. The phenol hydroxyl forms a hydrogen bond with a structural water molecule and an intramolecular hydrogen bond with the piperidine amino group. Via its acetylene linker, the ligand reaches into a different subsite of the cavity where the benzamine moiety formed a CH···π interaction with Pro153. As anticipated, the terminal acetylene group forms a CH···O hydrogen bond with the main chain oxygen of Leu145 at the bottom of the largely hydrophobic central cavity, thus mimicking the halogen bond in 2. The distance between the terminal carbon and the oxygen is 3.0 and 2.9 Å in chains A and B, respectively, corresponding to a hydrogen bond of moderate strength. As a result of the different distance between the donor and the closest carbon in the aromatic ring of the ligand (Csp2-Csp-Csp = 2.6 Å versus Csp2-I = 2.1 Å), 3 is slightly shifted as a rigid body by ∼0.5 Å along the longitudinal axis of the cavity compared to 2, accompanied by similar small shifts of Pro153 and Pro222 in the flanking loops to accommodate the ligand. The Csp-H···O angles in chains A and B are 164.0° and 170.4°, respectively, a near-linear arrangement in accordance with that previously observed for the halogen bond formed by 2.
Figure 3.
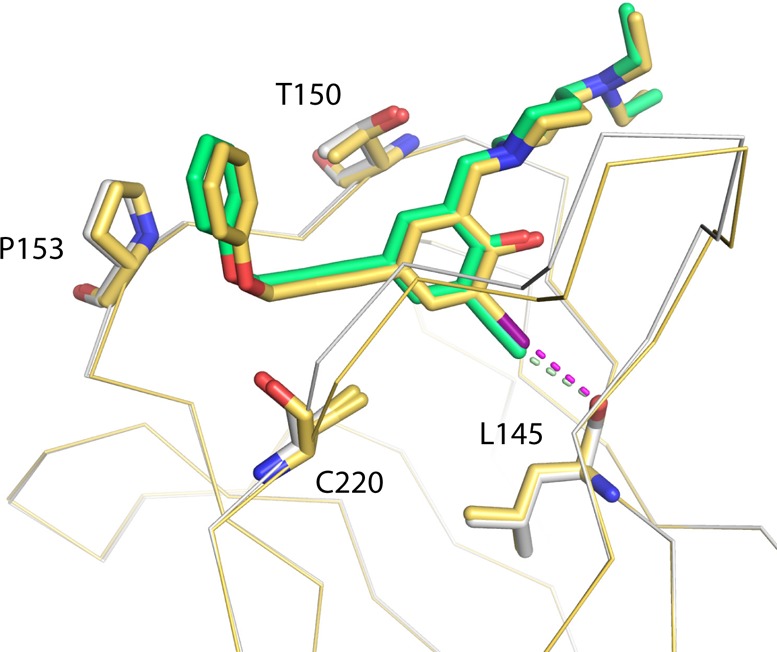
Binding modes of PK5176 (2, yellow carbons) and PK5211 (3, green carbons) in complex with the p53-Y220C DNA-binding domain by X-ray crystallography. Both compounds share a nearly identical binding mode, with 3 shifted rigidly upward away from Leu145 because of the increased length of its terminal ethynyl group. The I···O halogen bond between 2 and the protein is shown as a purple broken line, and the CH···O hydrogen bond between 3 and the protein is shown as a light green broken line.
In summary, exchanging the iodo moiety in 2 by ethynyl in 3 led to an approximately 13-fold loss in affinity for the p53-Y220C mutant. Given the similarity in the electrostatic potentials of iodine and ethynyl groups, and data from previous studies,5 this drastic drop in affinity was somewhat surprising. X-ray crystallography revealed a very similar binding mode for 2 and 3, the only difference being a small rigid-body shift of the entire compound 3, owing to the slightly larger size of the ethynyl moiety. To understand better the inherent reasons for the weaker potency of compound 3, we conducted QM calculations on model systems as a next step.
Quantum Chemical Calculations
Constrained Binding Site Optimization
For feasibility purposes, the binding sites of 2 and 3 in both chain A and B of the asymmetric unit were reduced to residues within 5 Å of the respective ligands, including two structural water molecules (Figure 4). The C- and N-termini of the selected amino acids were saturated with a methyl group. For residues with alternative conformations (Pro150, Glu221, and Thr230), the conformation with the highest occupancy in the crystal structure was used. The protein was prepared at standard settings with MOE2012.10.26 After adding hydrogen atoms, the binding sites comprised 343 atoms for both chains of the p53-Y220C-2 structure (PDB ID: 4AGP) and 345 atoms for both chains of p53-Y220C-3 (PDB ID: 5A7B), spanning residues 144–156, 219–224, and 228–231. Keeping the coordinates of all heavy atoms frozen, the positions of the hydrogen atoms were optimized at the TPSS-D3/def2-SV(P) level. Subsequently, the ligands’ complex formation energies were calculated. Using the optimized binding site of chain A of 4AGP as a reference (set to ΔΔE = 0.0 kcal/mol), the complex formation energy of chain B only differed by ΔΔE = +0.1 kcal/mol. In contrast, the complex formation energy of 3 is considerably disfavored with ΔΔE = +5.0 kcal/mol (chain A of 5A7B) and +5.3 kcal/mol (chain B of 5A7B), which is in agreement with the biophysical studies shown to favor 2 over 3. TPSS-D3/def2-SV(P) was chosen for this initial energy assessment because of previous favorable experience with its performance combined with Grimme’s dispersion correction (-D) in a systematic study on halogen bonds in small model systems.27,28 We additionally decided to forego the use of the computationally more demanding triple-ζ basis sets in favor of modeling the full binding site enclosing the ligand to capture all key protein–ligand interactions.
Figure 4.

Optimized binding sites of PK5176 (2) and PK5211 (3). (a) Binding site of 2 with two structural waters comprising 343 atoms. (b) Binding site of 3 with two structural waters comprising 345 atoms.
We performed further ab initio calculations on a MP2/def2-TZVPP level of theory using smaller model systems to investigate in more detail whether the ethynyl···carbonyl contact is disfavored in general or whether the particular binding geometry plays a role.
Distance Scans
To understand better the differences in interaction strength, we focused on the key interaction between the ligand and the carbonyl oxygen of Leu145 (halogen bond vs CH···O hydrogen bond). In our distance scans, Leu145 was represented as N-methylacetamide. For the ligands, we chose two model systems of different sizes and properties, representing substructures of the original ligands in their physiological protonation states: (i) 2-iodo-4,6-dimethylphenol or 2-ethynyl-4,6-dimethylphenol as “capped methyl”, the smallest substituted and uncharged fragments, and (ii) 1-(5-ethynyl-2-hydroxy-3-iodophenyl)-N,N-dimethylmethanaminium or 1-(3,5-diethynyl-2-hydroxyphenyl)-N,N-dimethylmethanaminium as “capped amine”, the more realistic, positively charged model systems including the protonated amine side chain (Figure 5a). Comparison of (i) and (ii) provides information on the tuning effects of the protonated amine on the interaction with the carbonyl oxygen. We have used this approach and similar model systems to systematically characterize halogen bonds with various interaction partners in recent years.27,29,30 All interaction geometries were extracted from chains A and B of the respective crystal structures of the complexes formed by 2 (4AGP) and 3 (5A7B). Hydrogen atoms were added to the model systems and optimized using the MP2/def2-TZVPP level of theory. Coordinates of heavy atoms were kept frozen. Starting from the optimized geometries, the distance between ligand and carbonyl oxygen was changed in steps of 0.1 Å, and interaction energies were calculated. Figure 5b shows interaction energies as a function of the distance between the iodo- or ethynyl-substituted aromatic carbon atom of the scaffold and the carbonyl oxygen.
Figure 5.

Distance scans of ligands derived from PK5176 (2) and PK5211 (3) with N-methylacetamide. (a) Ligand and carbonyl oxygen model systems used for the distance scans. (b) Distance scan plots for the four ligand models with N-methylacetamide. Labels next to the plotted curves denote whether the starting geometries were derived from chain A or B of the respective crystal structures. Black dots indicate the bond distance observed in the crystal structure.
For the smaller model systems, “capped methyl” 2 and 3, we found minimum/optimal geometries with very similar complex formation energies (ΔE = −4.5 kcal/mol (chain A) and −4.8 kcal/mol (chain B) for 2, ΔE = −4.7 kcal/mol for 3, Table 1), albeit at different equilibrium distances. Comparison of the computed equilibrium distances for both compounds with the actual distances found in the crystal structures (annotated by black dots in Figure 5b) revealed that the ethynyl···carbonyl contact of 3 is disfavored because the ligand is forced into a slightly too close C–H···O contact due to the spatial constraints of the binding site. For the larger model systems, “capped amine” 2 and 3, we observed several important differences. The positive charge located at the protonated amine withdraws electron density from the halogen and ethynyl group and significantly increases the overall interaction energy for both systems. This effect is further amplified by a reinforced intramolecular hydrogen bond between the protonated amine and the phenol oxygen, which in turn donates a weak hydrogen bond onto the negative electrostatic belt of either the iodine or the ethynyl π electron density (see also Figures 1a and 4). Interestingly, this tuning effect is much stronger for the I···O halogen bond; here, the interaction energy ΔE at equilibrium distance is approximately −10 kcal/mol, whereas the C–H···O contact is “only” tuned to approximately −8.8 kcal/mol. In line with the increase in interaction energy, the equilibrium distances are reduced by approximately 0.1–0.2 Å for both systems and in both chains. In conclusion, our QM studies indicate that the I···O halogen bond in the complex of 2 is favored compared with the “nonclassical hydrogen bond” in the complex of 3, most likely through a combination of its superior polarizability (tuning effect), its more suitable interaction geometry, and the better overall accommodation of the ligand within the binding pocket.
Table 1. Complex Formation Energies and Distances for Ligand Models Interacting with N-Methylacetamide (“Leu145”).
| structure (chain) | ΔE (kcal/mol) of crystal structure geometry | distance Caro···O [Å] in crystal structure | best overall ΔE (kcal/mol) | ideal distance Caro···O [Å] |
|---|---|---|---|---|
| 5176 capped methyl (A) | –4.5 | 5.18 | –4.5 | 5.08 |
| 5176 capped methyl (B) | –4.8 | 5.10 | –4.8 | 5.10 |
| 5211 capped methyl (A) | –4.5 | 5.54 | –4.7 | 5.77 |
| 5211 capped methyl (B) | –3.4 | 5.45 | –4.7 | 5.75 |
| 5176 capped amine (A) | –9.2 | 5.18 | –9.8 | 4.98 |
| 5176 capped amine (B) | –9.9 | 5.10 | –10.3 | 4.90 |
| 5211 capped amine (A) | –8.8 | 5.54 | –8.8 | 5.57 |
| 5211 capped amine (B) | –8.1 | 5.45 | –8.7 | 5.65 |
Conclusions and Discussion
We have used 2, a stabilizing small molecule that binds to the p53 cancer mutant Y220C via a halogen bond,14 to evaluate whether ethynyl is a suitable halogen bioisostere in cases where the halogen forms a halogen bond with a backbone oxygen. Similar halogen-to-hydrogen bond substitutions replacing bromine/iodine with an amine group have very recently been reported for inhibitors of human aldose reductase.31 Although iodine-to-ethynyl transformation appeared to maintain the affinity reasonably well in a series of 1,4-benzodiazepine-2,5-dione inhibitors of the HDM2-p53 interaction,5 we have found that in our test case, the transformation led to an approximately 13-fold loss in affinity. Our QM calculations suggest that the most probable reason for this affinity loss is the larger extent of the ethynyl moiety compared to iodine, which cannot be properly accommodated in the p53-Y220C binding site without causing a significant shift in the overall placement of the ligand scaffold. In addition, the larger polarizability of iodine allows its halogen bond to be tuned more strongly by the proximal protonated amine moiety. It should be noted that our QM calculations only represent adduct formation energies (ΔE) and do not take into account solvation/desolvation, relative conformational energies, or entropy.
QM distance scans indicated that comparable interaction energies can be achieved for the halogen bond and the nonclassical hydrogen bond if the interaction distances approach their optimal value. The ideal distance between the substituted aromatic carbon atom of the scaffold and the carbonyl oxygen is ∼0.7 Å longer for the ethynyl system than for the iodinated system (Table 1). Accordingly, an ethynyl moiety can be equally good or even superior when replacing iodine (or chlorine or bromine) bound in a suboptimal geometry with a too long halogen bond distance. It is also worthwhile to note that from a certain Caro···O distance (of ∼5.5 Å) onward, the ethynyl moiety is always energetically favored (see intersection points in Figure 5). In addition, the ethynyl moiety bears a much stronger electronegative belt than the halogens (Cl/Br/I), which can be targeted by orthogonal “side-on” interactions from adjacent residues in the binding site. Another possible advantage of an ethynyl moiety is the reduced directionality compared with a halogen bond. The polarized hydrogen is less shielded than the σ-hole by a proximal negative electrostatic belt. Deviations from the optimal linear hydrogen bond angle αC–H···O are therefore better tolerated than deviations from the optimal σ-hole angle αC-X···O (see Figure S1).
In terms of synthetic accessibility, an ethynyl moiety can be easily introduced starting from an iodinated or brominated precursor in a Sonogashira cross-coupling with trimethylsilylacetylene followed by deprotection, which makes comparisons of Br and I with ethynyl a straightforward task to tune affinities and physicochemical properties of tool compounds. Although there may still be some concerns regarding the incorporation of ethynyl groups (and also iodines) in druglike molecules, the erlotinib example shows that replacing a halogen with an ethynyl moiety can be used successfully for lead optimization.17−19 Halogen to acetylene bioisosteric transformation should therefore be explored, especially in cases where weak halogen bonds (at suboptimal distances and σ-hole angles) could be improved by the acetylene moiety without perturbing or disrupting other key interactions caused by a shift of the ligand scaffold.
Experimental Section
Molecular Modeling
The model systems for the binding site and the distance scans were extracted from crystal structures 4AGL and 5A7B. Hydrogen atoms were then added to the molecules, and their orientations were optimized using the Protonate3D feature within Molecular Operating Environment (MOE) 2012.1026 using default parameters. All quantum mechanical calculations were conducted using the TURBOMOLE 6.4 suite of programs.32,33 Relativistic effects for iodine were considered by an effective core potential (ECP) in the respective basis set. The calculations were done in combination with the resolution of identity (RI) technique.34 For MP2, the frozen core approximation was used. The frozen core orbitals were attributed by the default setting in TURBOMOLE, by which all orbitals possessing energies below 3.0 au are considered core orbitals. The SCF convergence criterion was increased to 10–8 Hartree for all calculations.
Constrained Binding Site Optimization
Optimization was performed using TPSS-D3/def2-SV(P),35−38 and the coordinates of the heavy atoms were kept frozen. Complex formation energies were calculated as ΔE = E(complex) – E(binding site) – E(ligand).
Distance Scans
Geometries for the distance scans were optimized using the MP2/def2-TZVPP38,39 level of theory. During the optimization, heavy atoms were kept frozen. Starting from these optimized geometries, the distance of the ligand (neighboring aromatic carbon atom of the halogen bond donating iodine) to the oxygen of N-methylacetamide was altered in steps of 0.1 Å. For each step, single point calculations were performed using MP2/def2-TZVPP. Interaction energies were calculated as ΔE = E(complex) – E(N-methylacetamide) – E(ligand).
Protein Expression and Purification
The stabilized DNA-binding domain of the p53 mutant Y220C, T-p53-Y220C, was expressed and purified as described.40 For the expression of 15N-labeled protein for NMR experiments, M9 minimal medium with 15NH4Cl (1 g/L) as the sole nitrogen source was used.
Chemical Compounds
Compounds PK5174 (1), PK5176 (2), and PK5211 (3) were synthesized by Roowin (Romainville, France). For all compounds, compound identity and >95% purity by LC–MS and NMR were guaranteed by the supplier.
Differential Scanning Fluorimetry (DSF)
The effect of compounds on the melting temperature of T-p53C-Y220C were monitored using SYPRO Orange (Invitrogen, USA) as a fluorescent probe, which quantitatively binds to hydrophobic protein patches exposed upon thermal denaturation. Real-time melt analysis was performed using a Corbett Rotor-Gene 6000 real-time qPCR thermocycler. Excitation and emission filters were set to 460 and 510 nm, respectively. Heating from 28 to 60 °C, a constant heating rate of 270 K/h was applied. The protein (final concentration of 10 μM) was briefly mixed with SYPRO orange (10x) in buffer (25 mM KPi pH 7.2, 150 mM NaCl, 1 mM TCEP), and compound (5 mM) dissolved in DMSO was added to give a final compound concentration of 250 μM in 5% (v/v) DMSO. The melting temperature (Tm) of the protein (10 μM) in the presence of compounds was determined from the inflection point of the melting curve. Melting temperatures were compared with control samples without compound (yielding ΔTm DSF). All samples were measured in triplicate.
NMR Spectroscopy
1H/15N-HSQC spectra of uniformly 15N-labeled T-p53-Y220C (75 μM) with and without compounds were acquired at 20 °C on a Bruker Avance-800 spectrometer using a 5 mm inverse cryogenic probe. Samples were prepared by adding dilutions of compound from stock solutions in DMSO-d6 to a final concentration of 5% (v/v) DMSO-d6 in buffer. All HSQC spectra were acquired with 8 transients per t1 data point, 1024 data points in t2, and 64 complex data points in t1, with spectral widths of 11.0 kHz for 1H and 2.7 kHz for 15N, and a recycle delay of 800 ms. After zero filling, forward complex linear prediction in f1 and Fourier transformation, the digital resolution was 0.01 ppm/point for 1H and 0.13 ppm/point for 15N. Chemical shifts were considered significant if the average weighted 1H/15N chemical shift difference
was greater than 0.04 ppm.
Isothermal Titration Calorimetry (ITC)
All ITC experiments were conducted using a Microcal iTC200 calorimeter as previously described.14 Briefly, protein and compounds were dissolved in 25 mM KPi, pH 7.2, 150 mM NaCl, 1 mM TCEP in 5% (v/v) DMSO. The cell unit contained 80 μM protein, and 2 mM compound was used for the syringe. The ITC competition experiment was conducted using 500 μM of compound 3 and 50 μM protein for the cell sample and 2 mM of compound 1 in the syringe. The actual and apparent binding constants for 1 were calculated with MicroCal Origin software using a standard one-site model. The binding constant of 3 was then calculated using an equation for ITC displacement titrations described by Zhang and Zhang24
X-Ray Crystallography
Crystals of T-p53C-Y220C were grown using the sitting-drop vapor diffusion technique as described previously.41 They were soaked for 3 h in a solution of 30 mM ligand in cryo buffer (19% polyethylene glycol 4000, 20% glycerol, 10 mM sodium phosphate, pH 7.2, 100 mM Hepes, pH 7.2, 150 mM KCl) and flash frozen in liquid nitrogen. An X-ray data set was collected at 100 K at beamline I03 of the Diamond Light Source, Oxford, and processed with XDS42 and SCALA.43 The structure of 3 bound to p53-Y220C was solved with difference Fourier techniques using the structure of the ligand-free mutant (PDB ID: 2J1X) as the starting model. After an initial round of refinement with PHENIX,44 water molecules were added to the structures using the same program. At this stage, the compound was built into the structure using Coot,45 and the resulting models were further refined with PHENIX and Coot. Data collection and refinement statistics are summarized in Table 2. The coordinates and structure factors are deposited in the Protein Data Bank (PDB ID: 5A7B).
Table 2. X-Ray Data Collection and Refinement Statistics of p53-Y220C-3 Complex.
| Data Collection | |
| space group | P212121 |
| a, b, c (Å) | 65.12, 71.11, 105.11 |
| molecules (AU) | 2 |
| resolution (Å)a | 29.6–1.40 (1.48–1.40) |
| unique reflections | 94,496 |
| completeness (%)a | 99.9 (99.9) |
| multiplicitya | 5.3 (5.2) |
| Rmerge (%)a,b | 6.4 (50.7) |
| <I/σI>a | 13.0 (3.2) |
| Wilson B value (Å2) | 14.8 |
| Refinement | |
| no. of protein atomsc | 3162 |
| no. of water atoms | 615 |
| no. of zinc atoms | 2 |
| no. of ligand atoms | 62 |
| overall B value (Å2) | 19.5 |
| Rcryst (%)d | 17.6 |
| Rfree (%)d | 19.2 |
| RMSD bonds (Å) | 0.006 |
| RMSD angles (deg) | 1.1 |
| PDB ID | 5A7B |
Values in parentheses are for the highest-resolution shell.
Rmerge = ∑(Ih,i – <Ih>)/∑Ih,i
Number includes alternative conformations.
Rcryst and Rfree = ∑∥Fobs| – |Fcalc∥/∑|Fobs|, where Rfree was calculated over 5% of the amplitudes chosen at random and not used in the refinement.
Acknowledgments
We thank the staff at Beamline I03 of Diamond Light Source for technical assistance during data collection. High performance computing resources of the bwGRID were provided by the federal state of Baden-Wuerttemberg. This work was funded by the ERC Advanced Grant “Tumour suppressor p53: structure, stability and novel anti-cancer drug development” and the federal state of Baden-Wuerttemberg, Germany.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschembio.5b00515.
σ-hole scans of iodobenzene and phenylacetylene with N-methylacetamide (PDF)
Author Contributions
⊥ These authors contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Meanwell N. A. (2011) Synopsis of Some Recent Tactical Application of Bioisosteres in Drug Design. J. Med. Chem. 54, 2529–2591. 10.1021/jm1013693. [DOI] [PubMed] [Google Scholar]
- Patani G. A.; LaVoie E. J. (1996) Bioisosterism: A Rational Approach in Drug Design. Chem. Rev. 96, 3147–3176. 10.1021/cr950066q. [DOI] [PubMed] [Google Scholar]
- Lenz C.; Boeckler F.; Hübner H.; Gmeiner P. (2005) Fancy bioisosteres: Synthesis, SAR, and pharmacological investigations of novel nonaromatic dopamine D3 receptor ligands. Bioorg. Med. Chem. 13, 4434–4442. 10.1016/j.bmc.2005.04.047. [DOI] [PubMed] [Google Scholar]
- Lenz C.; Haubmann C.; Hübner H.; Boeckler F.; Gmeiner P. (2005) Fancy bioisosteres: synthesis and dopaminergic properties of the endiyne FAUC 88 as a novel non-aromatic D3 agonist. Bioorg. Med. Chem. 13, 185–191. 10.1016/j.bmc.2004.09.044. [DOI] [PubMed] [Google Scholar]
- Parks D. J.; LaFrance L. V.; Calvo R. R.; Milkiewicz K. L.; Gupta V.; Lattanze J.; Ramachandren K.; Carver T. E.; Petrella E. C.; Cummings M. D.; Maguire D.; Grasberger B. L.; Lu T. (2005) 1,4-Benzodiazepine-2,5-diones as small molecule antagonists of the HDM2–p53 interaction: discovery and SAR. Bioorg. Med. Chem. Lett. 15, 765–770. 10.1016/j.bmcl.2004.11.009. [DOI] [PubMed] [Google Scholar]
- Bissantz C.; Kuhn B.; Stahl M. (2010) A medicinal chemist’s guide to molecular interactions. J. Med. Chem. 53, 5061–5084. 10.1021/jm100112j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark T.; Hennemann M.; Murray J. S.; Politzer P. (2007) Halogen bonding: the sigma-hole. J. Mol. Model. 13, 291–296. 10.1007/s00894-006-0130-2. [DOI] [PubMed] [Google Scholar]
- Politzer P.; Murray J. S.; Clark T. (2013) Halogen bonding and other sigma-hole interactions: a perspective. Phys. Chem. Chem. Phys. 15, 11178–11189. 10.1039/c3cp00054k. [DOI] [PubMed] [Google Scholar]
- Auffinger P.; Hays F. A.; Westhof E.; Ho P. S. (2004) Halogen bonds in biological molecules. Proc. Natl. Acad. Sci. U. S. A. 101, 16789–16794. 10.1073/pnas.0407607101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voth A. R.; Khuu P.; Oishi K.; Ho P. S. (2009) Halogen bonds as orthogonal molecular interactions to hydrogen bonds. Nat. Chem. 1, 74–79. 10.1038/nchem.112. [DOI] [PubMed] [Google Scholar]
- Parisini E.; Metrangolo P.; Pilati T.; Resnati G.; Terraneo G. (2011) Halogen bonding in halocarbon-protein complexes: a structural survey. Chem. Soc. Rev. 40, 2267–2278. 10.1039/c0cs00177e. [DOI] [PubMed] [Google Scholar]
- Wilcken R.; Zimmermann M. O.; Lange A.; Joerger A. C.; Boeckler F. M. (2013) Principles and Applications of Halogen Bonding in Medicinal Chemistry and Chemical Biology. J. Med. Chem. 56, 1363–1388. 10.1021/jm3012068. [DOI] [PubMed] [Google Scholar]
- Zimmermann M. O.; Lange A.; Wilcken R.; Cieslik M. B.; Exner T. E.; Joerger A. C.; Koch P.; Boeckler F. M. (2014) Halogen-enriched fragment libraries as chemical probes for harnessing halogen bonding in fragment-based lead discovery. Future Med. Chem. 6, 617–639. 10.4155/fmc.14.20. [DOI] [PubMed] [Google Scholar]
- Wilcken R.; Liu X.; Zimmermann M. O.; Rutherford T. J.; Fersht A. R.; Joerger A. C.; Boeckler F. M. (2012) Halogen-enriched fragment libraries as leads for drug rescue of mutant p53. J. Am. Chem. Soc. 134, 6810–6818. 10.1021/ja301056a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanfrlik J.; Kolar M.; Kamlar M.; Hurny D.; Ruiz F. X.; Cousido-Siah A.; Mitschler A.; Rezac J.; Munusamy E.; Lepsik M.; Matejicek P.; Vesely J.; Podjarny A.; Hobza P. (2013) Modulation of Aldose Reductase Inhibition by Halogen Bond Tuning. ACS Chem. Biol. 8, 2484–2492. 10.1021/cb400526n. [DOI] [PubMed] [Google Scholar]
- Hardegger L. A.; Kuhn B.; Spinnler B.; Anselm L.; Ecabert R.; Stihle M.; Gsell B.; Thoma R.; Diez J.; Benz J.; Plancher J.-M.; Hartmann G.; Banner D. W.; Haap W.; Diederich F. (2011) Systematic Investigation of Halogen Bonding in Protein–Ligand Interactions. Angew. Chem., Int. Ed. 50, 314–318. 10.1002/anie.201006781. [DOI] [PubMed] [Google Scholar]
- Ciardiello F.; Tortora G. (2001) A Novel Approach in the Treatment of Cancer: Targeting the Epidermal Growth Factor Receptor. Clin. Cancer Res. 7, 2958–2970. [PubMed] [Google Scholar]
- Yun C.-H.; Boggon T. J.; Li Y.; Woo M. S.; Greulich H.; Meyerson M.; Eck M. J. (2007) Structures of Lung Cancer-Derived EGFR Mutants and Inhibitor Complexes: Mechanism of Activation and Insights into Differential Inhibitor Sensitivity. Cancer Cell 11, 217–227. 10.1016/j.ccr.2006.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J. H.; Liu Y.; Lemmon; Mark A.; Radhakrishnan R. (2012) Erlotinib binds both inactive and active conformations of the EGFR tyrosine kinase domain. Biochem. J. 448, 417–423. 10.1042/BJ20121513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joerger A. C.; Fersht A. R. (2010) The Tumor Suppressor p53: From Structures to Drug Discovery. Cold Spring Harbor Perspect. Biol. 2, a000919. 10.1101/cshperspect.a000919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown C. J.; Lain S.; Verma C. S.; Fersht A. R.; Lane D. P. (2009) Awakening guardian angels: drugging the p53 pathway. Nat. Rev. Cancer 9, 862–873. 10.1038/nrc2763. [DOI] [PubMed] [Google Scholar]
- Wilcken R.; Wang G.; Boeckler F. M.; Fersht A. R. (2012) Kinetic mechanism of p53 oncogenic mutant aggregation and its inhibition. Proc. Natl. Acad. Sci. U. S. A. 109, 13584–13589. 10.1073/pnas.1211550109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M. O.; Lange A.; Boeckler F. M. (2015) Evaluating the Potential of Halogen Bonding in Molecular Design: Automated Scaffold Decoration Using the New Scoring Function XBScore. J. Chem. Inf. Model. 55, 687–699. 10.1021/ci5007118. [DOI] [PubMed] [Google Scholar]
- Zhang Y.-L.; Zhang Z.-Y. (1998) Low-Affinity Binding Determined by Titration Calorimetry Using a High-Affinity Coupling Ligand: A Thermodynamic Study of Ligand Binding to Protein Tyrosine Phosphatase 1B. Anal. Biochem. 261, 139–148. 10.1006/abio.1998.2738. [DOI] [PubMed] [Google Scholar]
- Wätzig H.; Oltmann-Norden I.; Steinicke F.; Alhazmi H.; Nachbar M.; El-Hady D.; Albishri H.; Baumann K.; Exner T.; Böckler F.; El Deeb S. (2015) Data quality in drug discovery: the role of analytical performance in ligand binding assays. J. Comput.-Aided Mol. Des. 1–19. 10.1007/s10822-015-9851-6. [DOI] [PubMed] [Google Scholar]
- Molecular Operating Environment (MOE), 2012.10; Chemical Computing Group Inc.: 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2009. [Google Scholar]
- Wilcken R.; Zimmermann M. O.; Lange A.; Zahn S.; Boeckler F. M. (2012) Using halogen bonds to address the protein backbone: a systematic evaluation. J. Comput.-Aided Mol. Des. 26, 935–945. 10.1007/s10822-012-9592-8. [DOI] [PubMed] [Google Scholar]
- Wilcken R. (2011) Nonclassical interactions in molecular recognition and their implications for targeted mutant rescue of the tumor suppressor protein p53. Ph.D. Thesis, Eberhard Karls Universitaet Tuebingen, Germany. [Google Scholar]
- Wilcken R.; Zimmermann M. O.; Lange A.; Zahn S.; Kirchner B.; Boeckler F. M. (2011) Addressing Methionine in Molecular Design through Directed Sulfur–Halogen Bonds. J. Chem. Theory Comput. 7, 2307–2315. 10.1021/ct200245e. [DOI] [PubMed] [Google Scholar]
- Lange A.; Zimmermann M. O.; Wilcken R.; Zahn S.; Boeckler F. M. (2013) Targeting Histidine Side Chains in Molecular Design through Nitrogen–Halogen Bonds. J. Chem. Inf. Model. 53, 3178–3189. 10.1021/ci4004305. [DOI] [PubMed] [Google Scholar]
- Fanfrlik J.; Ruiz F. X.; Kadlcikova A.; Rezac J.; Cousido-Siah A.; Mitschler A.; Haldar S.; Lepsik M.; Kolar M. H.; Majer P.; Podjarny A. D.; Hobza P. (2015) The Effect of Halogen-to-Hydrogen Bond Substitution on Human Aldose Reductase Inhibition. ACS Chem. Biol. 10, 1637. 10.1021/acschembio.5b00151. [DOI] [PubMed] [Google Scholar]
- Ahlrichs R.; Bar M.; Haser M.; Horn H.; Kolmel C. (1989) Electronic-Structure Calculations on Workstation Computers - the Program System Turbomole. Chem. Phys. Lett. 162, 165–169. 10.1016/0009-2614(89)85118-8. [DOI] [Google Scholar]
- Jones G.; Willett P.; Glen R. C. (1995) Molecular recognition of receptor sites using a genetic algorithm with a description of desolvation. J. Mol. Biol. 245, 43–53. 10.1016/S0022-2836(95)80037-9. [DOI] [PubMed] [Google Scholar]
- Weigend F.; Häser M. (1997) RI-MP2: first derivatives and global consistency. Theor. Chem. Acc. 97, 331–340. 10.1007/s002140050269. [DOI] [Google Scholar]
- Tao J.; Perdew J. P.; Staroverov V. N.; Scuseria G. E. (2003) Climbing the Density Functional Ladder: Nonempirical Meta\char21{}Generalized Gradient Approximation Designed for Molecules and Solids. Phys. Rev. Lett. 91, 146401. 10.1103/PhysRevLett.91.146401. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Antony J.; Ehrlich S.; Krieg H. (2010) A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104. 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- Schäfer A.; Horn H.; Ahlrichs R. (1992) Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J. Chem. Phys. 97, 2571–2577. 10.1063/1.463096. [DOI] [Google Scholar]
- Weigend F.; Ahlrichs R. (2005) Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 7, 3297–3305. 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
- Weigend F.; Haser M.; Patzelt H.; Ahlrichs R. (1998) RI-MP2: optimized auxiliary basis sets and demonstration of efficiency. Chem. Phys. Lett. 294, 143–152. 10.1016/S0009-2614(98)00862-8. [DOI] [Google Scholar]
- Boeckler F. M.; Joerger A. C.; Jaggi G.; Rutherford T. J.; Veprintsev D. B.; Fersht A. R. (2008) Targeted rescue of a destabilized mutant of p53 by an in silico screened drug. Proc. Natl. Acad. Sci. U. S. A. 105, 10360–10365. 10.1073/pnas.0805326105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joerger A. C.; Ang H. C.; Fersht A. R. (2006) Structural basis for understanding oncogenic p53 mutations and designing rescue drugs. Proc. Natl. Acad. Sci. U. S. A. 103, 15056–15061. 10.1073/pnas.0607286103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. (2010) XDS. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66, 125–132. 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr., Sect. D: Biol. Crystallogr. 62, 72–82. 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- Adams P. D.; Grosse-Kunstleve R. W.; Hung L.-W.; Ioerger T. R.; McCoy A. J.; Moriarty N. W.; Read R. J.; Sacchettini J. C.; Sauter N. K.; Terwilliger T. C. (2002) PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr., Sect. D: Biol. Crystallogr. 58, 1948–1954. 10.1107/S0907444902016657. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. (2010) Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66, 486–501. 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.