

Abstract
Platelet G-protein–coupled receptors influence platelet function by mediating the response to various agonists, including ADP, thromboxane A2, and thrombin. Blockade of the ADP receptor, P2Y12, in combination with cyclooxygenase-1 inhibition by aspirin has been among the most widely used pharmacological strategies to reduce cardiovascular event occurrence in high-risk patients. The latter dual pathway blockade strategy is one of the greatest advances in the field of cardiovascular medicine. In addition to P2Y12, the platelet thrombin receptor, protease activated receptor-1, has also been recently targeted for inhibition. Blockade of protease activated receptor-1 has been associated with reduced thrombotic event occurrence when added to a strategy using P2Y12 and cyclooxygenase-1 inhibition. At this time, the relative contributions of these G-protein–coupled receptor signaling pathways to in vivo thrombosis remain incompletely defined. The observation of treatment failure in ≈10% of high-risk patients treated with aspirin and potent P2Y12 inhibitors provides the rationale for targeting novel pathways mediating platelet function. Targeting intracellular signaling downstream from G-protein–coupled receptor receptors with phosphotidylionisitol 3-kinase and Gq inhibitors are among the novel strategies under investigation to prevent arterial ischemic event occurrence. Greater understanding of the mechanisms of G-protein–coupled receptor–mediated signaling may allow the tailoring of antiplatelet therapy.
Keywords: blood platelet, coronary disease, GTP-binding proteins, purinerginc 2Y12 receptor agoists, receptors, thrombin
Rapid platelet activation and aggregation are crucial for the development of arterial thrombotic events. Platelets adhere to the injured vessel wall site after spontaneous plaque rupture during acute coronary syndrome (ACS) and during percutaneous coronary intervention (PCI). Adhered platelets undergo shape change, cytosolic Ca++ mobilization, and activation. Platelet activation leads to release of secondary agonists, thromboxane A2 and adenosine diphosphate (ADP). These agonists amplify the response to injury and produce sustained platelet aggregation in the presence of high arterial shear rates. Simultaneously, subpicomolar concentrations of thrombin are generated after exposure of blood to tissue factor–bearing cells in the subendothelial compartment and activate platelets by cleaving platelet protease activated receptors (PARs). Platelet activation, in turn, leads to the generation of larger amounts of thrombin on the procoagulant platelet surface and on released microparticles. Thrombin converts fibrinogen to fibrin to further stabilize the platelet–fibrin clot.1 A major area of controversy exists at this time about the relative contribution of each agonist-induced platelet activation pathway (ADP, thromboxane A2, and thrombin) to the genesis of an in vivo stable thrombus. The latter determination is critical in decision making for drug targeting.
Human genome analysis has demonstrated ≈1000 unique G-protein–coupled receptors (GPCRs) associated with a wide array of physiological functions.2 GPCRs regulate many of the cellular events in humans through signal transduction stimulated by various agonists. GPCRs are target of ≈30% to 50% of all commercially available drugs.3 Platelet function is influenced by soluble agonists that stimulate intracellular signaling through GPCRs; ADP through P2Y1 and P2Y12, thrombin through PAR-1 and PAR-4, thromboxane A2 through TP, epinephrine through the α-adrenergic receptor, and prostaglandin (PG)I2 through the IP.4,5 These signaling pathways are highly conserved as are regulatory mechanisms.
GPCRs consist of a single polypeptide chain with 7 transmembrane α-helices connected by three extracellular loops and 3 intracellular loops. The extracellular loop consists of an amino terminus and a ligand (agonist) binding site; the intracellular loop consists of a carboxyl-terminal domain associated with guanine nucleotide binding proteins (G proteins; Figure 1). A single GPCR can be associated with multiple functionally different G proteins that elicit specific intracellular responses to agonists. G proteins are heterotrimers with α, β, and γ subunits. Gα subunit in its inactivated state is bound to guanosine diphosphate (GDP) and tightly associated with βγ subunit. On activation by agonists, GDP is replaced by GTP, releasing α and βγ units for interactions with downstream effectors. Depending on the receptor type, the α subunit is associated with phospholipase C-β (PLC-β), Rho-GEF (guanine nucleotide exchange factor), or adenylyl cyclase activity, whereas the βγ subunit is associated with phosphotidylionisitol 3-kinase (PI3K) and PLC-β activity. Comparatively less is known about the function of the βγ subunit.4,5
Figure 1.
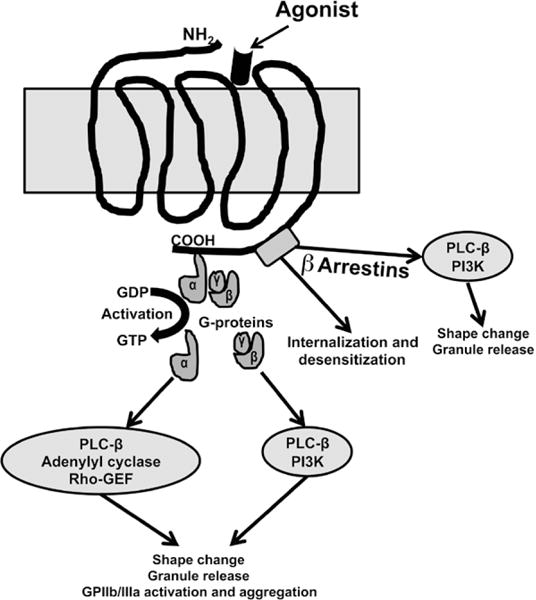
G-protein–coupled receptor (GPCR) signaling in platelets. Binding of an agonist on the extracellular loop of the GPCR is associated with the exchange of GTP for GDP on the α subunit resulting in the dissociation of the α subunit from βγ subunit. Depending on the receptor type, the α subunit activates phospholipase C-β (PLC-β), Rho-GEF (guanine nucleotide exchange factor), or adenylyl cyclase, whereas the βγ subunit activates phosphotidylionisitol 3-kinase (PI3K) and PLC-β. Protease activated receptor (PAR) is activated by thrombin by creating a tethered ligand or through a noncanonical mechanism where cleavage by a proteinase occurs at a site different from the canonical cleavage site. The tethered ligand can also stimulate signaling through a G-protein–independent pathway involving β-arrestin–mediated signaling scaffold. β-arrestin is also involved in the internalization and desensitization of the PAR receptors.
There are ≥10 forms of Gα in platelets that are members of the Giα, Gqα, G12/13α and Gsα families. G proteins are associated with redundancy in their responses (signaling pathways). Therefore, targeting >1 receptor is an attractive antiplatelet strategy. The direct inhibition of platelet-specific GPCRs (P2Y12 or PAR-1) and enzymes associated with platelet agonist release (COX-1) has been extensively exploited when compared with the direct inhibition of G proteins or the downstream signaling pathways.6 In contrast to the 2 state model of receptor activation (inactive versus single active conformation), it has been proposed that GPCRs exist in multiple ligand-specific active conformations that are associated with different downstream signaling events. The ability of an agonist to induce a response from a specific GPCR that varies across the signaling pathways is termed, biased agonism. Biased GPCR agonists have therapeutic potential by their ability to induce differential signaling associated with desirable effects.7–10
Short-term desensitization of GPCRs occurs through the phosphorylation by kinases that enhance the affinity for adapter proteins known as β-arrestins. Subsequently, uncoupling of the interaction between GPCR and G protein occurs leading to endocytosis via clathrin-coated pits. The GPCR–β-arrestin interaction, as a multifunctional scaffold and adapter, also activates signaling pathways. The role of arrestins in the regulation of platelet function has received increased attention (see below).8–10 Although a large body of information has been obtained in the past decade on GPCR-induced signaling and regulation, the function of specific GPCRs in atherothrombosis remains incompletely defined.
Purinergic Receptors
ADP is stored in platelet dense granules and is released on activation by agonists and high arterial shear. ADP acts on 2 GPCRs: P2Y1 and P2Y12. A third platelet purinergic receptor, P2X1, is a non-GPCR ligand-gated ion channel. Activation of P2X1 by ATP is associated with rapid influx of external Ca++ and platelet shape change.
P2Y1
P2Y1 is a 373 amino acid polypeptides coupled to the Gq protein. It is widely distributed in human tissues. Approximately 150 P2Y1 receptors are present per platelet. The α subunit of Gq is associated with PLC-β activity. Activation of PLC-β leads to phosphoinositide hydrolysis and cytosolic Ca++ mobilization through generated IP. The latter signaling event results in shape change, granule secretion, and PLA2 and integrin activation. Phospholipase A2 activation results in the production of arachidonic acid from membrane phospholipids, and the subsequent synthesis and release of thromboxane A2 by COX-1 and thromboxane synthase activity (Figure 2).11
Figure 2.
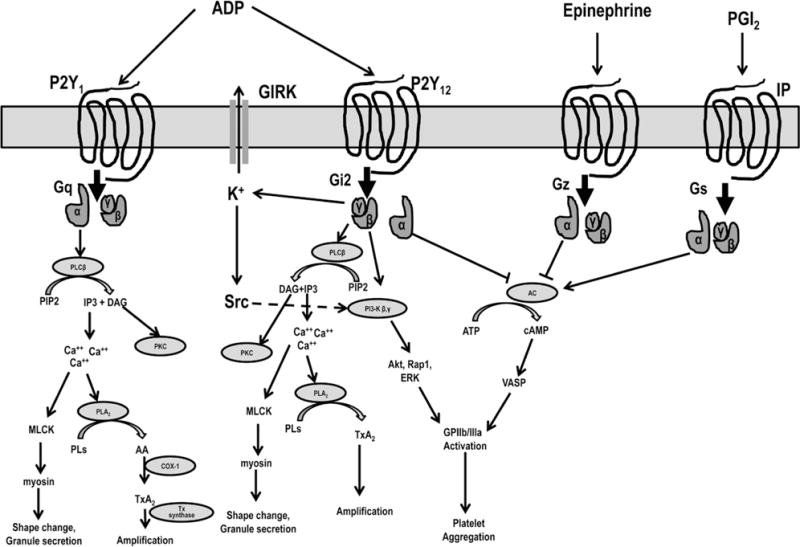
An overview of signaling pathways of major G-protein–coupled receptors. Adenosine diphosphate activates platelets through P2Y1 and P2Y12 receptors. P2Y1 is coupled to Gq, which activate phospholipase (PLC)-β through Gqα and its intracellular signaling leads to platelet shape change, granule secretion, and amplification of platelet activation through generation of thromboxane A2. P2Y12 is coupled to Gi. The α subunit of Gi is associated with adenylyl cyclase and vasodilator stimulated phosphoprotein phosphorylase activity. The βγ subunit is associated with PI3-kinase that activates Akt, Rap1, ERK, and Src family kinases and G-protein–gated inwardly rectifying potassium channels (GIRK) in addition to PLC-β activity as described above. Activation of P2Y12 leads to glycoprotein (GP) IIb/IIIa activation and stable platelet aggregation. Epinephrine is associated with Gz protein, which on activation inhibits adenylyl cyclase activity through α subunit. Prostaglandin I2 (PGI2) receptor is associated with Gs protein, which on activation stimulates adenylyl cyclase activity through α subunit. DAG indicates diacylglycerol; ERK, extracellular-signal-regulated kinase; IP3, inositol 1,4,5-triphosphate; MLCK, myosin light chain kinase; PLA2, phospholipase A2; Pls, phospholipids; Tx A2, thromboxane A2; and VASP, vasodilator-stimulated phosphoprotein.
Downstream signaling from both P2Y1 and P2Y12 is responsible for normal platelet aggregation. P2Y1 signaling is responsible for initial platelet activation, shape change, and transient platelet aggregation, whereas P2Y12 signaling more potently amplifies platelet aggregation induced by ADP and other agonists. Among the challenges for P2Y1 receptor inhibitors as antithrombotic agents is their widespread expression in human tissues. In an animal model using laser-induced injury, mesenteric arterial thrombosis was inhibited by a P2Y1 antagonist (MRS2500), and bleeding was moderately affected.12 At this time, P2Y1 antagonists, such as A3P5PS, MRS2500, and MRS2179, have not been studied in human trials.
P2Y12
The P2Y12 receptor contains 342 amino acids and is expressed mainly in platelets and to a lesser extent in the central nervous system.13 Approximately 600 P2Y12 receptors are present per platelet. When compared with P2Y1, the limited cell type expression of P2Y12 makes it an ideal antithrombotic target. Inhibition of platelet aggregation by P2Y12 blockers can be overcome by stimulating Gzα signaling with epinephrine.14 The P2Y12 receptor has ≈24% sequence homology with the PAR-1 receptor. Most recently, x-ray structural analysis of human P2Y12 reveals an unusual straight conformation of transmembrane helix V, differing from other class A GPCRs, with a long α-helical extension into the cytoplasm where it may form extensive interactions with G protein.15,16 Although speculative, this observation may explain why P2Y12 forms preferentially high-affinity interactions with Gαi at the expense of other Gαi couplers, such as PAR-1.17 On the basis of a thermostabilizing construct model, we reported that disulfide bonds bridging the amino terminus Cys17 with Cys270 of helix VII and the highly conserved disulfide bond bridging Cys97 of helix III and Cys175 of extracellular loop 2 were present. During agonist binding with 2MesADP, the main chain carbonyl of Cys97 forms a hydrogen bond with the 3′ hydroxyl group of ribose of 2MesADP. The main chain amino group of Cys175 interacts with the β-phosphate group. Binding with the antagonist, AZD1283, was associated with the disruption of the Cys97–Cys175 disulfide bond. However, the latter disulfide bond seems to be dynamic. The active thiolactone metabolites of thienopyridines are also predicted to destabilize interactions of the P2Y12 receptor with nucleotide agonists, such as ADP, by binding covalently to the free thiol of Cys97 and thereby irreversibly preventing ADP signaling. ATP derivatives, such as 2MesATP, ARC66096 (a noncleavable triphosphate mimetic), cangrelor (ARC69931MX), and ARC67085, also bind to the P2Y12 receptor in a manner similar to 2MesADP and prevent ADP binding. Because of a bulky N6 substituent, ticagrelor (AZD6140) cannot bind to the P2Y12 receptor in a conformation similar to 2MesADP. Moreover, non-nucleotide derivatives, such as AZD1283 and ticagrelor, behave as competitive antagonists. It was also reported that the R256 residue that potentially makes contact with 2MesADP is important for P2Y12 activation, whereas the R265 residue in extracellular loop 3 is also important for agonist activity but does not directly interact with 2MesADP.15,16
The Gi2α subunit coupled to P2Y12 is associated with suppression of adenylyl cyclase activity. Inhibition of cAMP-dependent protein kinase–mediated phosphorylation of vasodilator-stimulated phosphoprotein is associated with glycoprotein IIb/IIIa activation.14 As a marker of P2Y12 receptor signaling, vasodilator-stimulated phosphoprotein phosphorylation is used to monitor the antiplatelet response to P2Y12 receptor blockers.
Reducing cAMP by Giα-associated inhibition of adenylyl cyclase activity alone is not sufficient for normal aggregation; downstream signaling through the βγ subunit activity plays a major role. The βγ subunit is associated with PLC-β activation leading to Ca++ mobilization, shape change, and granule secretion through diacylglycerol and IP3 release from PIP2. βγ subunits are also associated with PI3K activity, which is linked to glycoprotein IIb/IIIa activation via catalytic modulation of Akt and Rap1b activities. It has been reported that Rap1b activation is important for the sustained activation of GPIIb/IIIa. PAR-1–induced Akt activation depends predominantly on Gi stimulation through secreted ADP. In addition, Gi signaling also leads to the activation of extracellular-signal-regulated kinase, myosin light chain kinase, Src family kinases, and G-protein–gated inwardly rectifying potassium channels14,18 (Figure 2).
P2Y12 activation is also associated with the phosphatidylserine exposure and procoagulant properties.19,20 Clopidogrel and the active metabolite of prasugrel have been shown to attenuate procoagulant activity.21,22 Moreover, P2Y12 receptor activation also influences the formation of collagen- and thrombin-activated platelets that express procoagulant factors, such as phosphatidylserine and activated factor V.23 In addition, P2Y12 activation is associated with P-selectin and CD-40 L expressions that influence platelet–leukocyte interactions and modulate inflammation and tissue factor exposure.1 At this time, P2Y12 intracellular signaling pathways remain incompletely defined.
P2Y12 Inhibition
A major objective of P2Y12 inhibitor therapy is the rapid establishment of high-level receptor blockade to prevent amplification of platelet activation induced by ADP and other agonists that degranulate the platelet. Reversibility of the P2Y12 inhibitor may confer benefit to reduce bleeding. P2Y12 inhibitors are the most widely prescribed antiplatelet agents in cardiovascular medicine after aspirin.
Thienopyridines
Ticlopidine, clopidogrel, and prasugrel are clinically available orally administered thienopyridine prodrugs that are metabolically activated by the cytochrome P450 pathway. Pharmacodynamic studies conclusively demonstrated wide response variability and nonresponsiveness to clopidogrel. The mechanisms of resistance to clopidogrel have been intensively investigated in the past 10 years. Suboptimal active metabolite generation and pharmacodynamic effects have been related to single nucleotide polymorphisms in genes encoding specific CYP P450 cytochromes associated with loss-of-function, drug–drug interactions between thienopyridines and other coadministered drugs that compete/inhibit specific CYPP450 cytochromes and demographic variables. Because P2Y12 inhibitors are coadministered with various pharmacological agents, an avoidance of pharmacological interactions is also important.24
The development of prasugrel was an advance in thienopyridine therapy. Prasugrel is more rapidly and efficiently metabolized than clopidogrel and is thus associated with a more rapid onset of action and greater inhibition of ADP-induced platelet aggregation resulting in less response variability and a lower prevalence of nonresponsiveness than clopidogrel.25 Prasugrel and clopidogrel active metabolites have similar in vitro P2Y12 binding affinities and resultant antiplatelet effects. Therefore, the enhanced antiplatelet effect of prasugrel is because of increased generation and exposure of prasugrel active metabolite compared with the clopidogrel.26
Ticagrelor
Ticagrelor, a recently approved cyclopentyltriazolopyrimidine, is a reversibly binding, selective, potent, direct acting, and orally administered P2Y12 receptor blocker.27 Ticagrelor does not prevent ADP binding to P2Y12, but instead reversibly inhibits the ADP-induced receptor conformational change and G-protein activation by binding to a site distinct from the ADP-binding site. These characteristics keep the receptor in an inactive state and after ticagrelor unbinding, the receptor can be reactivated by ADP. On the basis of modeling data, we suggested that because of its bulky 7-[2-(3,4-difluorophenyl)-cyclopropylamino] and 5-propylsulfanyl substituents, ticagrelor cannot be accommodated in the center of the transmembrane cage (in the same pocket), where ADP binds but rather binds to a second pocket consisting of the upper transmembrane domains (domains 1, 2, and 7), extracellular loop 2, and the N-terminal domain of P2Y12 (see above).28
Ticagrelor is metabolized rapidly by hepatic CYP3A4/5 to produce AR-C124910XX, the main metabolite of ticagrelor that is equipotent in inhibiting the P2Y12 receptor. Ticagrelor is associated with a rapid onset of action, a greater level of inhibition and a more rapid offset of pharmacodynamic action compared with clopidogrel.27
Ticagrelor and Inhibition of Adenosine Uptake Antiplatelet Effect
In addition to P2Y12 inhibition, ticagrelor inhibits adenosine reuptake in erythrocytes and other cells.29–32 The latter effect has been attributed to the inhibition of sodium-independent equilibrative nucleoside transporter. Unlike the strong adenosine reuptake inhibitor, dipyridamole, ticagrelor exhibited 16-fold lower affinity for equilibrative nucleoside transporter 1 (Ki=41 versus 2.6 nmol/L). However, other P2Y12 blockers such as the active metabolites of clopidogrel and prasugrel, cangrelor, and AR-C124910xx do not have significant influence on adenosine reuptake. Ticagrelor has ≈20-fold higher affinity for P2Y12 when compared with equilibrative nucleoside transporter 1 affinity. There was no direct influence of ticagrelor on adenosine receptors and ticagrelor is not metabolized to adenosine.29 In an in vitro study, in the presence of adenosine, the antiplatelet effect of ticagrelor was partially mediated by a drug-induced increase in extracellular adenosine levels and adenosine-mediated platelet inhibition via the A2A receptor.30 A significant higher plasma concentration of adenosine in blood collected from patients with ACS treated with ticagrelor than with clopidogrel has been reported. In addition, in vitro uptake of exogenous adenosine by erythrocytes was inhibited when incubated with serum from patients treated with ticagrelor but not with clopidogrel.31
Coronary Blood Flow and Infarct Size Effect
In an animal model, ticagrelor and dipyridamole dose dependently enhanced left anterior descending coronary artery blood flow after occlusion and after intracoronary adenosine administration.32 In a healthy volunteer study, ticagrelor increased coronary blood flow velocity that correlated with plasma ticagrelor levels. However, levels of AR-C124910xx did not correlate with enhanced blood flow indicating that adenosine-related blood flow effects were confined to ticagrelor.33 In a subsequent prospective crossover study of 56 patients with non–ST-segment–elevation ACS undergoing PCI, ticagrelor but not prasugrel was associated with greater coronary blood flow velocity when incremental doses of adenosine were coadministered.34 Ticagrelor, but not clopidogrel, has been shown to reduce the infarct size in a rat perfusion model that was completely reversed by adenosine receptor antagonist, suggesting that the latter effect is mediated by endogenous adenosine.35 Similarly, in a canine coronary thrombosis model, ticagrelor but not clopidogrel, significantly reduced infarct size and rapidly restored tissue perfusion when adjunctively added to tissue-type plasminogen activator.36 These off-target effects of ticagrelor may account for some of the clinical benefits associated with ticagrelor than with clopidogrel therapy observed in the Platelet Inhibition and Patient Outcomes (PLATO) trial.
Interaction Between Ticagrelor and Aspirin
In the PLATO trial, the absence of a beneficial effect of ticagrelor versus clopidogrel for treatment of ACS was observed in the North American subgroup when compared with rest of the world. The latter paradox has been attributed to a higher concomitant aspirin dose used in North America, but a play of chance may also explain the discordant outcomes.37 Of note, there has been no reported influence of aspirin dose on the clinical efficacy of either prasugrel or clopidogrel, P2Y12 inhibitors that produce less potent P2Y12 blockade than ticagrelor.38,39
It has been shown that the ADP-P2Y12 signaling pathway is important for the production of thromboxane A2 and for the irreversible aggregation associated with TP receptor activation.40,41 In the presence of a high concentration of ticagrelor in vitro, aspirin did not provide additional antiaggregatory effects in response to ADP, arachidonic acid, U46619 (TP receptor agonist), epinephrine, and thrombin receptor activator peptide.42 In an ex vivo study of the effect aspirin dose on platelet function, higher aspirin doses of 81 mg daily were associated with greater antiplatelet effects.43 It has been demonstrated that P2Y12 inhibitors reduce thromboxane A2–induced platelet aggregation and thromboxane A2 production and sensitize platelets to the antiaggregatory effects of PGI2 by blocking P2Y12 receptor-mediated inhibition of adenylyl cyclase. With this background, it was suggested that high-dose aspirin in the presence of potent P2Y12 blockade induced by ticagrelor may reduce the production of PGI2 and shift the influence of aspirin toward prothrombotic environment. Moreover, by inhibiting the production of gastroprotective prostanoids, the risk of gastrointestinal bleeding may also increase.44 It has also been reported that the high-dose but not the low-dose aspirin is associated with an impaired anticontractile effect of ticagrelor on ADP-induced vascular smooth muscle cell contraction in a rat model.45
Cangrelor
A limitation associated with all of the currently available oral P2Y12 inhibitors is the lack of an immediate and reversible pharmacodynamic effect. Although ticagrelor binds reversibly, its pharmacodynamic offset is slow.38 Because immediate P2Y12 inhibition is a major goal in the treatment of high-risk coronary artery disease, parenteral therapy is an attractive approach (Inhibition of PAR-1 and G Protein Interaction section of this article). Moreover, a rapid pharmacodynamic offset is also desirable in the setting of bleeding. Finally, in patients on chronic P2Y12 blockade who need surgery after coronary artery stenting, the administration of a bridging P2Y12 inhibitor with rapid offset effect may limit the vulnerability to thrombosis in the perioperative period.
Cangrelor (previously known as AR-C67085MX) is a direct-acting parenteral ATP analog; modification of the polyphosphate side chain of ATP prevents breakdown to ADP and substitution of the adenine moiety enhances P2Y12 receptor affinity and selectivity. ADP-induced platelet function is competitively inhibited by cangrelor, with a rapid onset and offset effect within minutes.46 In preclinical studies, thrombus formation was inhibited by cangrelor infusion.47 Cangrelor has been studied in the CHAMPION (Cangrelor versus Standard Therapy to Achieve Optimal Management of Platelet Inhibition) trials for acute therapy in patients undergoing PCI and also as a bridging strategy in patients previously treated with clopidogrel who require urgent surgery.48–50 Cangrelor is not yet clinically approved.
Pharmacodynamic Interactions Between P2Y12 Blockers
Expected levels of platelet inhibition induced by clopidogrel were not observed after simultaneous administration of cangrelor and clopidogrel indicating a drug–drug interaction. On the baiss of these observations, we suggested that cangrelor prevents thiol adduct formation by clopidogrel and prasugrel active metabolites in the extracellular region of P2Y12.51 It was further shown that preincubation of blood with cangrelor before addition of clopidogrel or prasugrel active metabolite reduced the ability of the metabolite to irreversibly antagonize P2Y12. However, irreversible inhibition was maintained when blood was preincubated with metabolites before the addition of cangrelor.52 In an ex vivo animal study and in a study of patients with stable coronary artery disease, a significant pharmacodynamic interaction was not demonstrated between ticagrelor and cangrelor.53,54 The interaction between cangrelor and thienopyridines may have important clinical implications when clopidogrel or prasugrel is loaded, and cangrelor is bound to P2Y12.
Switching from chronic clopidogrel therapy after an ACS event to either a prasugrel maintenance dose or loading dose was associated with a further reduction in platelet function.55 However, switching to prasugrel therapy 12 hours after the last dose of ticagrelor maintenance therapy was associated with an increase in platelet reactivity compared with continued ticagrelor therapy.39 Because thienopyridine active metabolites are only transiently exposed, pharmacodynamic interactions are more likely to occur after switching than for ticagrelor, a direct acting agent with a longer plasma half-life. The ticagrelor–prasugrel interaction was partially mitigated by the administration of a prasugrel loading dose. Waiting longer than 12 hours for more P2Y12 receptors to be unbound by ticagrelor before prasugrel loading or repetitive prasugrel loading may further mitigate the pharmacodynamic effect. The relative contribution of the ticagrelor active metabolite (AR-C124910XX) versus the parent agent to the interaction is unknown.39 Additional work is needed to define the binding sites of the different P2Y12 inhibitors and the dosing regimens that optimally attenuate pharmacodynamic interactions during switching.
Other Novel P2Y12 Receptor Antagonists
AZD1283
A novel series of ethyl 6-aminonicotinate acyl sulfonamides have been recently discovered as P2Y12 antagonists. Structure–activity relationship investigations demonstrated that replacement of the 5-chlorothienyl of ethyl 6-aminonicotinate acyl sulfonamide with a benzyl substituent is associated with an increased antiaggregatory effect, whereas introduction of substituents on the benzyl group led to higher microsomal clearance but no further increase in antiaggregatory potency. Among 6 compounds, 1 candidate compound, AZD1283, was associated with the strongest inhibition of ADP-induced platelet aggregation, increased blood flow, and a therapeutic index ≥10 in the separation of efficacy and bleeding time. AZD1283 has been selected for further clinical studies. Its binding properties with P2Y12 are discussed above.56
Diadenosine 5′,5″-P1,P4-Dithio-P2,P3-Chloromethylenetetraphosphate (Ap4A)
Diadenosine 5′,5″-P1,P4-dithio-P2,P3-chloromethylenetetraphosphate is the member of dinucleoside polyphosphates that are stored in platelet dense granules and released on activation along with ADP and ATP. It has been reported that Ap4A with the stereo configuration at P1 and P4 has strong plasma stability and an inhibitory effect on P2Y12 and a lesser effect on P2Y1. This compound remains under investigation.57
PARs
The potent P2Y12 receptor blockers, prasugrel and ticagrelor, are associated with lower ischemic event occurrence compared with clopidogrel in patients with ACS. However, a clinical unmet need still exists. The degree of adverse event reduction (≈20% relative and ≈2% absolute) compared with clopidogrel therapy is modest and treatment failure (≈10%) persists along with a significantly greater bleeding risk. The latter observations suggest that the strategy of COX-1 and potent P2Y12 inhibition is associated with ceiling of net clinical benefit. Inhibition of another GPCR-mediating platelet activation, namely PAR-1, has been explored as a novel strategy to reduce treatment failure associated with conventional dual antiplatelet therapy.58
PARs play important roles in normal and pathological states and have been targeted for therapy of a variety of diseases, including colitis, asthma, and tumor metastasis, in addition to cardiovascular disease. PAR-1, a major receptor for thrombin, contains 425 amino acids and is a high-affinity receptor with fast and transient downstream signaling. Approximately 1000 to 2000 PAR-1 receptors are present per platelet. In addition to platelets, the PAR-1 receptor is also expressed in other cells and plays a role in modulating inflammation and in the vascular remodeling processes in unstable atherosclerotic plaques and in restenosis.17,59 Therefore, PAR-1 inhibition may confer clinical benefits beyond platelet function modulation.
PARs are activated through a novel tethered ligand mechanism. The hirudin-like sequence of the N-terminal exodomain of the PAR-1 binds with high affinity to exosite-1 of thrombin (fibrinogen-binding site), allowing it to outcompete fibrinogen (present in large quantities in blood) for thrombin binding.58,60 Thrombin cleaves the bond between Arg 41 and Ser 42 of PAR-1 in the extracellular domain, thereby exposing a new N terminus with the initial sequence SFLLRN (serine–phenylalanine–leucine–leucine–arginine–asparagine). Once cleaved, this tethered ligand rapidly undergoes conformational change and binds to ligand-binding site-I located in the N-terminal exodomain. This leads to a conformational change in the transmembrane domains that is transmitted to intracellular domains and finally stimulates GDP–GTP exchange and activates G12/13, and Gq (Figure 3).
Figure 3.
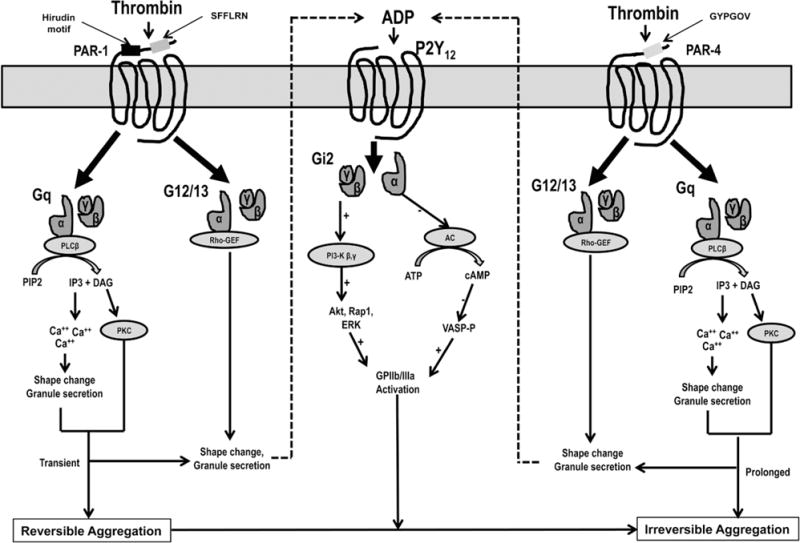
P2Y12, protease activated receptor (PAR)-1 and PAR-4 signaling pathways in platelets. Thrombin binds to the hirudin-like sequence of the N-terminal exodomain of the PAR-1 and cleaves extracellular domain, thereby exposing tethered ligand with SFLLRN sequence. The tethered ligand rapidly undergoes conformational change and binds to ligand-binding site I located in the N-terminal exodomain of the receptor. Subsequent conformational change in the PAR-1 receptor leads the activation of G12/13 and Gq that finally results in shape change, granule secretion and transient platelet aggregation through Rho-GEF (guanine nucleotide exchange factor) and phospholipase C-β (PLC-β) activities, respectively. Thrombin binds to the active site of the PAR-4 through a low-affinity, negatively charged cluster of amino acid residues and cleaves extracellular domain exposing tethered ligand with GYPGOV sequence. Similar to PAR-1, PAR-4 activation through tethered ligand leads to the activation of Rho-GEF and PLC-β that finally results in prolonged and irreversible platelet aggregation. PAR-1–mediated platelet aggregation may be transient and reversible unless strengthened by additional signaling from secreted ADP through P2Y12 receptor and from PAR-4 receptor. DAG indicates diacylglycerol; IP3, inositol 1,4,5-triphosphate; PI3K, phosphotidylionisitol 3-kinase; Tx A2, thromboxane A2; and VASP, vasodilator-stimulated phosphoprotein.
PAR-4, a lower affinity platelet thrombin receptor with a highly prolonged signal, contributes to irreversible platelet aggregation.17 PAR-4 is 385 amino acids in length and lacks a hirudin-like sequence and binds to the active site of thrombin through a negatively charged cluster of amino acid residues (anionic cluster) resulting in the prolongation of dissociation of the thrombin from the receptor. Despite lower affinity and slower intracellular signaling, PAR-4 activation leads to most of the calcium mobilization and sustained glycoprotein IIb/IIIa activation induced by thrombin. Unlike PAR-1, PAR-4 activation leads to irreversible platelet aggregation, even in the absence of the ADP-autocrine response.17 Using Western blotting and coimmunoprecipitation techniques, it has been shown that PAR-1 and PAR-4 present as a stable heterodimeric complex in human platelets and enable thrombin to act as a bivalent functional agonist and mediate intracellular signaling at both low and high concentrations. Available data suggest that after binding, thrombin cleaves PAR-1 and then cleaves PAR-4 while still bound to PAR-1.60 Activation of either PAR is sufficient to trigger platelet secretion and aggregation, whereas PAR-1 is likely to be the more physiologically relevant receptor for controlling hemostasis.
The mechanisms that regulate PAR activity are complex. PAR can be activated either by proteinases through the above tethered ligand mechanism or through a noncanonical mechanism where cleavage by a proteinase occurs at a site different from the canonical cleavage site. The proteases, activated protein C, plasmin, matrix metalloprotease-1 (MMP-1), MMP-13, and kallikrein cleave PAR-1 at noncanonical cleavage site(s) distinct from the Arg 41 target of thrombin, albeit at reduced efficiency when compared with thrombin.61 The tethered ligand can also stimulate signaling through a G-protein–independent pathway involving the formation of an internalized β arrestin–mediated signaling scaffold (see below).8,9 Cleavage of PAR to the C terminus of the classical tethered ligand sequence may permanently disarm the receptor or lead to alternative biased signaling. Thus, disarming of signaling induced by a specific protease may also be associated with activation via a MAP kinase pathway in the absence of Ca++ mobilization.8
In this regard, it has been demonstrated that PAR receptors have the ability to activate one signaling pathway in preference to another, a process known as functional selectivity or biased agonism. These biased receptor signaling pathways or functional selectivity associated with noncanonical cleavage of PAR-1 receptors are selective for intracellular signaling associated with either classic G-protein pathways or β-arrestins, which can activate mitogen-activated protein kinases or PI3Ks.8 Thus, in addition to internalization and desensitization of GPCRs, β-arrestins can initiate intracellular signaling events that are independent of G-protein coupling and activation.
The exposure of platelets to fibrillar collagen and subsequent conversion of platelet-bound pro–MMP-1 zymogen to active MMP-1 also promotes platelet aggregation through PAR-1 by exposure of a distinct ligand, PR-SFLLRN. The MMP1–PAR-1 interaction is a novel pathway of platelet activation that may play an important role during the early stages of platelet–vessel wall interactions. Analyses of G-protein signaling pathways suggest that MMP-1 and the PR-SFLLRN ligand are biased agonists that preferentially activate G12/13, and that thrombin preferentially activates Gq in human platelets.61 The activation of G12/G13 results in Rho-dependent platelet shape change and granule secretion. The intracellular signaling events after the activation Gq are described above. Unlike other GPCRs, which recycle after internalization, internalized PAR-1 receptor after single activation is targeted to lysozomes for degradation through β-arrestin.9
In several preclinical studies, PAR-1 antagonism did not prolong bleeding time or coagulation-related time (activated partial thromboplastin time, prothrombin time, or thrombin time), suggesting that PAR-1 inhibition does not seem to interfere with normal hemostasis.58 Despite coupling to similar G proteins, PAR-1 and PAR-4 play distinct roles in thrombosis and hemostasis. It has been shown that PAR-4–activating peptide-stimulated platelets release more microparticles and procoagulant factor V and are associated with a higher density of factor V on activated platelet surface when compared with PAR-1 stimulation.62 However, another study demonstrated that SFLLRN (PAR-1 agonist)–induced platelet activation is associated with more rapid stimulation of platelet procoagulant activity as measured by annexin V binding and clotting assays when compared with PAR-4 stimulation by GYPGQV.63 Therefore, at this time, it is still unclear which pathway, thrombin PAR-1/PAR-4, or ADP-P2Y12 pathway, predominates for the platelet procoagulant response and may vary between individuals and ethnicities/races.
Interaction Between PAR and P2Y12
Close synergistic interaction between PARs and P2Y12 in platelets may be critical for the development of stable thrombus generation and resultant adverse event occurrences. It has been shown that PAR-1–mediated platelet aggregation is transient unless strengthened by additional signaling input from P2Y12 or PAR-4.17 It is still not absolutely clear whether the cross talk of PAR-1 with Gi pathways is only through P2Y12 signaling.64 PAR-1 antagonism did not inhibit ADP-, U6441-, or collagen-induced platelet aggregation in animal studies, and there were no significant effects on thrombin receptor activator peptide–induced platelet aggregation after aspirin and clopidogrel loading dose administration in patients with coronary artery disease undergoing stenting.65,66
The mechanisms of Akt activation induced by thrombin receptors versus P2Y12 differ, but are synergistic and contribute to the stabilization of platelet-rich thrombi. In addition to heterodimeric interaction between PAR-1 and PAR-4 described earlier, it was recently demonstrated that P2Y12 signaling may be essential for PAR-4–mediated Akt activation and recruitment of arrestins to PAR-4/P2Y12 signaling complexes. Recruitment of arrestins to the latter complex leads to incorporation of Lyn, subsequent PI3K-dependent phosphorylation of Akt, glycoprotein IIb/IIIa activation, and stabilization of platelet thrombi.10 In the presence of potent P2Y12 blockade, such as induced by ticagrelor and prasugrel, and low thrombin concentrations, the above findings may have clinical relevance with respect to enhancement of antithrombotic efficacy by adding a PAR inhibitor.
PAR Inhibition
Inhibition of the interaction between thrombin and PAR-1 has been shown to attenuate ischemic events in selected patients treated with dual antiplatelet therapy. Several PAR-1 inhibitors have been developed including SCH 530348 (vorapaxar), E5555, FR-17113, F16618, F16357, PZ-128, RWJ-56110 and RWJ-58259, and BMS-200261.67 E5555 (atopaxar) development has been discontinued.
Tethered ligand antagonists of PAR-4, transcinnamoyl-Tyr-Pro-Gly-Lys-Phe-NH, and YD-3 (1-benzyl-3(ethoxycarbonylphenyl)-indazole) have been shown to inhibit GYPGKF (PAR-4 agonist)–induced aggregation of washed human platelets with no or little effect on platelet aggregation induced by thrombin, SFLLRN, collagen or U46619.68 RWJ-56110, a potent PAR-1 antagonist, inhibited low (0.05 U/mL) but not high concentration thrombin-induced platelet aggregation, whereas YD-3, alone, had little or no effect on thrombin-induced platelet aggregation, but significantly enhanced the antiaggregatory activity of a PAR-1 antagonist. These data indicate that both PAR-1 and PAR-4 inhibition are required for marked thrombin–platelet inhibition.69
A murine PAR knockout model may provide circumstantial evidence for the importance of PAR inhibition as an antithrombotic strategy. Murine platelets express PAR-3 and PAR-4 but not PAR-1. There is some evidence to suggest that PAR-3 knockout mice platelets may be analogous to PAR-1–inhibited human platelets. However, PAR-3–deficient mice are partially protected against thrombosis, whereas PAR-4–deficient mice are completely protected against thrombosis. These data suggest that incomplete blockade of thrombin-induced signaling may still provide an antithrombotic effect. In nonhuman primates that express PAR-1 and PAR-4 like humans, blockade of PAR-1 reduced carotid arterial thrombotic events.70 Taken together, these data may be relevant to the consideration of PAR-1 therapy alone as an antithrombotic strategy. The clinical use of dual inhibition of PAR-1 and PAR-4 compared with PAR-1 alone still remains unknown at this time.
Vorapaxar is a high-affinity PAR-1 antagonist that inhibits PAR-1 in a competitive and slowly reversible manner.58 On the basis of crystal structure analysis, we recently revealed that the vorapaxar binding site is located closely to the extracellular surface of the receptor unlike other ligands for GPCRs that penetrate more deeply into the transmembrane core of the receptor. It has been proposed that a unique interaction between vorapaxar and PAR-1 results in the closing of the binding pocket after vorapaxar binding; the latter may account for the low dissociation rate of vorapaxar and a delayed offset of pharamcodynamic properties.71 Vorapaxar prevents high-affinity binding of the tethered ligand to the ligand-binding site.
In phase I and II studies, vorapaxar specifically and effectively inhibited the platelet PAR-1 activity without affecting the response to other receptor agonists and had no influence on coagulation parameters. Vorapaxar significantly inhibited platelet function for ≤3 weeks (plasma half-life of 5–11 days) after a single loading dose.58 Recently, vorapaxar has been approved by the Food and Drug Admistration to reduce the risk of myocardial infarction, stroke, cardiovascular death, and revascularization in patients with a previous myocardial infarction or peripheral arterial disease.72 The thrombin–PAR-1 interaction may also be pivotal to maintain vascular integrity in the central nervous system, an organ rich in hemostasis-promoting tissue factor. Chronic inhibition of this innate defense mechanism in patients with previous central nervous system ischemic necrosis may have undesired consequences.
Inhibition of PAR-1 and G-Protein Interaction
Thrombin inhibitors such as bivalirudin effectively suppress PAR-1–dependent platelet activation; however, direct inhibition of thrombin may potentially increase bleeding in patients undergoing PCI because it also interferes with activation of the PAR-4 thrombin receptor and fibrinogen-dependent hemostasis.73,74 The ability to rapidly and reversibly inhibit PAR-1 signaling by a parenteral strategy seems ideal in the high-risk patient undergoing PCI and may be associated with less bleeding risk in the setting of unanticipated surgery.67 A fast-acting, short half-life pepducin, PZ-128, is currently being evaluated in a human phase I study (NCT01806077). Pepducins are lipidated peptides that target the cytoplasmic surface of their cognate receptor. PZ-128 consists of a 7 amino acid peptide derived from the third intracellular loop of PAR-1 and is conjugated to palmitate lipid.60,75,76 PZ-128 interrupts signaling to internally located G proteins. The structure of PZ-128 was found to mimic the off-state of the corresponding intracellular region of PAR-1 that is critical for coupling to G proteins. PZ-128 rapidly and reversibly inhibits platelet activation and arterial thrombosis in guinea pigs and primates without affecting bleeding or other coagulation parameters. Excellent dose- and concentration-dependent inhibition of PAR-1–induced platelet aggregation and arterial thrombosis was associated with pepducin administration in baboons.77 Inhibition of PAR-1 by PZ-128 was reversible, as evidenced by loss of inhibition with higher concentrations of SFLLRN agonist at both the 3 and 6 mg/kg doses. PZ-128 administration was not associated with significant inhibition of either ADP or PAR-4 platelet responses (0%–10%) at any time point including at 1, 2, or 24 hours.77 Dose-dependent protection against arterial thrombosis with an EC50 of 0.075 mg/kg PZ-128 was determined in guinea pig, and synergistic protective effects were observed with oral clopidogrel. The pharmacokinetic and anti-platelet pharmacodynamic properties of PZ-128 indicate that this lipopeptide reaches maximal activity during (<15 minutes) and immediately after intravenous infusion and is completely eliminated from plasma within 24 hours. PZ-128 had no effect on bleeding or coagulation parameters in primates or in blood from patients undergoing PCI.77
Other GPCR Receptors
The thromboxane A2 receptor (TP) is an another important platelet receptor associated with amplification of platelet activation and aggregation. TP consists of 342 amino acids and there are 1000 receptors per platelet. TP is coupled to Gq and G12 activation leading to intracellular Ca2+ mobilization, platelet shape change, and aggregation (see above). It has been reported that in the presence of potent P2Y12 inhibition, the added antiplatelet effect of aspirin is less pronounced (see above). In this context, there is current interest in deleting aspirin cotherapy to reduce bleeding potentiated by gastrointestinal COX-1 blockade and to reduce the potential prothrombotic effects of COX-1 blockade on PGI2 production when a potent P2Y12 inhibitor therapy is used. Blockade of thromboxane A2 synthesis and TP receptor are strategies that obviate the risks associated with COX-1 blockade.
Terutroban, a selective orally active TP antagonist, is in clinical development for use in secondary prevention of thrombotic events. Terutroban was associated with similar level of inhibition of arachidonic acid– and collagen-induced platelet aggregation as aspirin in patients with peripheral arterial disease and was superior to aspirin in inhibiting platelet aggregation and thrombus formation in an ex vivo model of thrombosis.78,79 However, the phase III clinical trial, PERFORM (Prevention of cerebrovascular and cardiovascular Events of ischemic origin with teRutroban in patients with a history oF ischemic strOke or tRansient ischeMic attack), failed to demonstrate superiority of terutroban than of aspirin in secondary prevention of cerebrovascular and cardiovascular events in ≈20 000 patients with stroke.80
Ridogrel, a combined thromboxane synthase and TP receptor antagonist, was associated with similar angiographic patency and the primary end point (TIMI [thrombolysis in myocardial infarction] flow grades 2 and 3) compared with aspirin in patients with myocardial infarction receiving streptokinase in the RAPT (Ridogrel Versus Aspirin Patency Trial) trial.81 Another new combined thromboxane A2 receptor and synthase inhibitor, terbogrel is being studied for long-term antithrombotic therapy.82
Downstream Intracellular Signaling Targets
Although intracellular signaling molecules may provide important targets for antithrombotic therapy, their wide spread presence in various cells and comparatively less available genetic knockout models has impeded research in this area versus the study of direct GPCR inhibitors. However, several factors are to be considered. In the case of PI3Kγ, expression is mainly on hematopoetic cells, and PI3Kγ may preferentially associate toward specific Gα and Gβγ subunits. Because platelet intracellular signaling has extensive redundancy, inhibition of PI3Kγ may confer a less aggressive antiplatelet approach than direct inhibition upstream at the GPCR itself.
PI3Kβ Inhibitors
PI3Kβ inhibitors have been shown to inhibit in vitro platelet aggregation and also in vivo thrombus generation.83,84 AZD6482, a PI3Kβ inhibitor was associated with a rapid onset of action and short half-life in inhibiting platelet aggregation in animal models and humans. In addition, a 3-hour parenteral infusion of AZD6482 in humans was well tolerated, and there was no change in bleeding time. However, the potential influence on insulin signaling is a major concern. It has been suggested that AZD6482 may be an effective reversible agent in the management of patients undergoing cardiopulmonary bypass surgery and stroke to inhibit platelet activation.85
Gq Inhibitors
Although, YM-254890, a novel inhibitor of Gq signaling, has been shown to inhibit ADP-induced platelet aggregation suggesting a potential clinical role for Gq signaling inhibitors, the latter approach may have adverse effects in multiple organs because of its widespread influence on many GPCR pathways.86
Conclusions
In the past 10 years, platelet receptor biology research has advanced significantly with greater understanding of signaling mechanisms induced by important platelet agonists. ADP-P2Y12, thromboxane A2-TP, and thrombin–PAR-1/-PAR-4 interactions are important GPCR-mediated pathways associated with platelet activation and aggregation. The relative contribution of these pathways to in vivo thrombosis remains an area of great interest and is directly relevant to the development of future antiplatelet strategies.
Simultaneous inhibition of the ADP-P2Y12 pathway with clopidogrel and the COX-1 pathway with aspirin has been a major strategy to treat patients with high-risk coronary artery disease. Accruing evidence has demonstrated that the platelet response to thromboxane A2 and thrombin is highly influenced by P2Y12 signaling. These data suggest a potential major role for potent P2Y12 inhibitor monotherapy in the treatment of high-risk cardiovascular disease.
A ceiling of net clinical benefit associated with aspirin and potent P2Y12 receptor blockers indicates a potential need for targeting alternative platelet activation pathways. One pathway that most recently has been targeted is the PAR-1 receptor by vorapaxar. Rapid inhibition of thrombin-induced platelet activation is likely important to attenuate platelet thrombus formation in high blood flow states present in the arterial bed. In vivo data from animal studies support an antithrombotic effect from PAR-1 inhibition alone and an enhancement to the antithrombotic effect of P2Y12 and COX-1 inhibition by the addition of PAR-1 blockade. PAR-1–mediated platelet aggregation is transient unless strengthened by additional signaling input from P2Y12 and PAR-4. These data suggest a potential future clinical role for combined PAR-1 and P2Y12 inhibition and combined PAR-1 and PAR-4 inhibition. Targeting thromboxane synthase or the TP receptor has not been convincingly more clinically effective than aspirin possibly because of beneficial non-COX-1–mediated effects of aspirin. Finally, experimental evidence supports a potential interaction between high-dose aspirin and the most recently approved potent P2Y12 inhibitor ticagrelor, an effect that may be explained by the actions of PGI2 and ticagrelor on their respective GPCRs. Targeting intracellular signaling downstream from GPCR receptors with PI3K and Gq inhibitors is among the novel strategies under investigation to prevent arterial ischemic event occurrence. Greater understanding of the mechanism of the GPCR signaling events in the individual patient may allow the tailoring of antiplatelet therapy.
Table.
Characteristics of the Important G-Protein–Coupled Receptor Blockers
| Clopidogrel | Prasugrel | Ticagrelor | Cangrelor | Vorapaxar | |
|---|---|---|---|---|---|
| Target | P2Y12 receptor | P2Y12 receptor | P2Y12 receptor | P2Y12 receptor | PAR-1 receptor |
| Structure | Thienopyridine | Thienopyridine | CPTP derivative | ATP analog | Tricyclic 3-phenylpyridine |
| Metabolism | Prodrug | Prodrug | Direct* | Direct | Direct |
| Administration | Oral, once daily | Oral, once daily | Oral, twice daily | Intravenous | Oral, once daily |
| Conversion to active metabolite, % | 15 | 85 | 90–100* | … | … |
| Type of action | Irreversible | Irreversible | Reversibly binding | Reversible | Competitive and slowly reversible |
| Elimination half-life | Active metabolite: after 75 mg: 0.3 h | Active metabolite: after 10 mg: 0.5 h; after 60 mg: 7.4 h | Parent compound: 7 h active metabolite: 9 h | 3–4 min | 8 d |
| Time to steady inhibition | 4–6 h/5 d | 1 h | 2 h | Immediate | 1 h |
| Offset | 5–7 d | ≤9 d | 5–7 d | Immediate | 2–3 wk |
| Level of inhibition at steady state | 40%–50% wide response variability | 65%–80% | 65%–80% | >90% | ≥80% |
| Elimination | Urine 40% Feces 60% | Urine 68% Feces 27% | Urine ≈30% Feces ≈60% | Urine | Feces |
CPTP indicates cyclopentyltriazolopyrimidine; and PAR-1, protease activated receptor-1.
Active metabolite is equally effective.
Significance.
Platelet G-protein–coupled receptors (GPCR) influence platelet function by mediating the response to various agonists including ADP, thromboxane A2, and thrombin. Dual antiplatelet therapy of blockade of the ADP receptor and cyclooxygenase-1 inhibition by aspirin is a well-established strategy to reduce arterial ischemic event occurrence in high-risk patients. However, persistence occurrence of recurrent ischemic events (≈10%) is a major concern. An inhibitor of protease activated receptor-1 (a thrombin receptor) has been recently approved to treat patients with cardiovascular disease in addition to dual antiplatelet therapy. Targeting biased GPCR agonists such as β-arrestins and intracellular signaling downstream from GPCR receptors with PI3K and Gq inhibitors are among the novel strategies under investigation to prevent arterial ischemic event occurrence. Because the relative contributions of these GPCR signaling pathways to in vivo thrombosis remain incompletely defined, greater understanding of the mechanism of the GPCR signaling events in the individual patient may allow the tailoring of antiplatelet therapy.
Acknowledgments
Sources of Funding
This study was supported by the grants from the Sinai Center for Thrombosis Research and from the National Institute of Health (NIH P50 HL110789).
Dr Gurbel reports serving as a consultant fees/receiving honoraria from Daiichi Sankyo, Lilly, Bayer, AstraZeneca, Merck, Boehringer, Janssen, and CSL; receiving grants from the National Institutes of Health, Daiichi Sankyo/Lilly, CSL, AstraZeneca, Harvard Clinical Research Institute, Bayer, Haemonetics, Duke Clinical Research Institute, Sinnowa, Coramed and Accumetrics. Dr Kuliopulos reports serving as a consultant for Eli Lilly.
Nonstandard Abbreviations and Acronyms
- ACS
acute coronary syndrome
- COX-1
cyclooxygenase-1
- GPCR
G-protein–coupled receptors
- MMP
matrix metalloprotease
- PARs
protease activated receptors
- PI3K
phosphotidylionisitol 3-kinase
- PLC-β
phospholipase C-β
Footnotes
Disclosures
Dr Tantry reports no conflicts.
References
- 1.Gurbel PA, Bliden KP, Hayes KM, Tantry U. Platelet activation in myocardial ischemic syndromes. Expert Rev Cardiovasc Ther. 2004;2:535–545. doi: 10.1586/14779072.2.4.535. [DOI] [PubMed] [Google Scholar]
- 2.Takeda S, Kadowaki S, Haga T, Takaesu H, Mitaku S. Identification of G protein-coupled receptor genes from the human genome sequence. FEBS Lett. 2002;520:97–101. doi: 10.1016/s0014-5793(02)02775-8. [DOI] [PubMed] [Google Scholar]
- 3.Salon JA, Lodowski DT, Palczewski K. The significance of G protein-coupled receptor crystallography for drug discovery. Pharmacol Rev. 2011;63:901–937. doi: 10.1124/pr.110.003350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Offermanns S. Activation of platelet function through G protein-coupled receptors. Circ Res. 2006;99:1293–1304. doi: 10.1161/01.RES.0000251742.71301.16. [DOI] [PubMed] [Google Scholar]
- 5.Smyth SS, Woulfe DS, Weitz JI, Gachet C, Conley PB, Goodman SG, Roe MT, Kuliopulos A, Moliterno DJ, French PA, Steinhubl SR, Becker RC. 2008 Platelet Colloquium Participants. G-protein-coupled receptors as signaling targets for antiplatelet therapy. Arterioscler Thromb Vasc Biol. 2009;29:449–457. doi: 10.1161/ATVBAHA.108.176388. [DOI] [PubMed] [Google Scholar]
- 6.Gurbel PA, Tantry US. Combination antithrombotic therapies. Circulation. 2010;121:569–583. doi: 10.1161/CIRCULATIONAHA.109.853085. [DOI] [PubMed] [Google Scholar]
- 7.Strachan RT, Sun JP, Rominger DH, Violin JD, Ahn S, Rojas Bie, Thomsen A, Zhu X, Kleist A, Costa T, Lefkowitz RJ. Divergent transducer-specific molecular efficacies generate biased agonism at a G protein-coupled receptor (GPCR) J Biol Chem. 2014;289:14211–14224. doi: 10.1074/jbc.M114.548131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gieseler F, Ungefroren H, Settmacher U, Hollenberg MD, Kaufmann R. Proteinase-activated receptors (PARs) - focus on receptor-receptor-interactions and their physiological and pathophysiological impact. Cell Commun Signal. 2013;11:86. doi: 10.1186/1478-811X-11-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ramachandran R, Noorbakhsh F, Defea K, Hollenberg MD. Targeting proteinase-activated receptors: therapeutic potential and challenges. Nat Rev Drug Discov. 2012;11:69–86. doi: 10.1038/nrd3615. [DOI] [PubMed] [Google Scholar]
- 10.Li D, D’Angelo L, Chavez M, Woulfe DS. Arrestin-2 differentially regulates PAR4 and ADP receptor signaling in platelets. J Biol Chem. 2011;286:3805–3814. doi: 10.1074/jbc.M110.118018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hechler B, Cattaneo M, Gachet C. The P2 receptors in platelet function. Semin Thromb Hemost. 2005;31:150–161. doi: 10.1055/s-2005-869520. [DOI] [PubMed] [Google Scholar]
- 12.Hechler B, Nonne C, Roh EJ, Cattaneo M, Cazenave JP, Lanza F, Jacobson KA, Gachet C. MRS2500 [2-iodo-N6-methyl-(N)-methanocarba-2′-deoxyadenosine-3′,5′-bisphosphate], a potent, selective, and stable antagonist of the platelet P2Y1 receptor with strong antithrombotic activity in mice. J Pharmacol Exp Ther. 2006;316:556–563. doi: 10.1124/jpet.105.094037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hollopeter G, Jantzen HM, Vincent D, Li G, England L, Ramakrishnan V, Yang RB, Nurden P, Nurden A, Julius D, Conley PB. Identification of the platelet ADP receptor targeted by antithrombotic drugs. Nature. 2001;409:202–207. doi: 10.1038/35051599. [DOI] [PubMed] [Google Scholar]
- 14.Kim S, Kunapuli SP. P2Y12 receptor in platelet activation. Platelets. 2011;22:56–60. doi: 10.3109/09537104.2010.497231. [DOI] [PubMed] [Google Scholar]
- 15.Zhang J, Zhang K, Gao ZG, et al. Agonist-bound structure of the human P2Y12 receptor. Nature. 2014;509:119–122. doi: 10.1038/nature13288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang K, Zhang J, Gao ZG, et al. Structure of the human P2Y12 receptor in complex with an antithrombotic drug. Nature. 2014;509:115–118. doi: 10.1038/nature13083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang P, Covic L, Kuliolulos A. Protease-activated receptors. In: Michelson AD, editor. Platelets. 3rd. San Diego, CA: Academic Press; 2013. pp. 249–259. [Google Scholar]
- 18.Brass LF, Newman DK, Wannemacher KM, Zhu L, Stalker TJ. Signal transduction during platelet plug formation. In: Michelson AD, editor. Platelets. San Diego, CA: Academic Press; 2013. pp. 367–398. [Google Scholar]
- 19.Gachet C. P2Y(12) receptors in platelets and other hematopoietic and non-hematopoietic cells. Purinergic Signal. 2012;8:609–619. doi: 10.1007/s11302-012-9303-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leon C, Ravanat C, Freund M, Cazenave JP, Gachet C. Differential involvement of the P2Y1 and P2Y12 receptors in platelet procoagulant activity. Arterioscler Thromb Vasc Biol. 2003;23:1941–1947. doi: 10.1161/01.ATV.0000092127.16125.E6. [DOI] [PubMed] [Google Scholar]
- 21.Gurbel PA, Bliden KP, Antonino MJ, Gesheff T, Cummings CC, Dubois BV, Herzog WR, Tantry US. Time dependence of clopidogrel loading effect: platelet activation versus platelet aggregation. Thromb Res. 2012;129:1–2. doi: 10.1016/j.thromres.2011.07.048. [DOI] [PubMed] [Google Scholar]
- 22.Judge HM, Buckland RJ, Sugidachi A, Jakubowski JA, Storey RF. The active metabolite of prasugrel effectively blocks the platelet P2Y12 receptor and inhibits procoagulant and pro-inflammatory platelet responses. Platelets. 2008;19:125–133. doi: 10.1080/09537100701694144. [DOI] [PubMed] [Google Scholar]
- 23.Kotova YN, Ataullakhanov FI, Panteleev MA. Formation of coated platelets is regulated by the dense granule secretion of adenosine 5′diphosphate acting via the P2Y12 receptor. J Thromb Haemost. 2008;6:1603–1605. doi: 10.1111/j.1538-7836.2008.03052.x. [DOI] [PubMed] [Google Scholar]
- 24.Gurbel PA, Tantry US. Do platelet function testing and genotyping improve outcome in patients treated with antithrombotic agents?: platelet function testing and genotyping improve outcome in patients treated with antithrombotic agents. Circulation. 2012;125:1276–87. doi: 10.1161/CIRCULATIONAHA.111.031195. discussion 1287. [DOI] [PubMed] [Google Scholar]
- 25.Jeong YH, Tantry US, Gurbel PA. Importance of potent P2Y(12) receptor blockade in acute myocardial infarction: focus on prasugrel. Expert Opin Pharmacother. 2012;13:1771–1796. doi: 10.1517/14656566.2012.704909. [DOI] [PubMed] [Google Scholar]
- 26.Payne CD, Li YG, Small DS, Ernest CS, II, Farid NA, Jakubowski JA, Brandt JT, Salazar DE, Winters KJ. Increased active metabolite formation explains the greater platelet inhibition with prasugrel compared to high-dose clopidogrel. J Cardiovasc Pharmacol. 2007;50:555–562. doi: 10.1097/FJC.0b013e3181492209. [DOI] [PubMed] [Google Scholar]
- 27.Gurbel PA, Kereiakes DJ, Tantry US. Ticagrelor for the treatment of arterial thrombosis. Expert Opin Pharmacother. 2010;11:2251–2259. doi: 10.1517/14656566.2010.511175. [DOI] [PubMed] [Google Scholar]
- 28.VAN Giezen JJ, Nilsson L, Berntsson P, Wissing BM, Giordanetto F, Tomlinson W, Greasley PJ. Ticagrelor binds to human P2Y(12) independently from ADP but antagonizes ADP-induced receptor signaling and platelet aggregation. J Thromb Haemost. 2009;7:1556–65. doi: 10.1111/j.1538-7836.2009.03527.x. [DOI] [PubMed] [Google Scholar]
- 29.Armstrong D, Summers C, Ewart L, Nylander S, Sidaway JE, van Giezen JJ. Characterization of the adenosine pharmacology of ticagrelor reveals therapeutically relevant inhibition of equilibrative nucleoside transporter 1. J Cardiovasc Pharmacol Ther. 2014;19:209–219. doi: 10.1177/1074248413511693. [DOI] [PubMed] [Google Scholar]
- 30.Nylander S, Femia EA, Scavone M, Berntsson P, Asztély AK, Nelander K, Löfgren L, Nilsson RG, Cattaneo M. Ticagrelor inhibits human platelet aggregation via adenosine in addition to P2Y12 antagonism. J Thromb Haemost. 2013;11:1867–1876. doi: 10.1111/jth.12360. [DOI] [PubMed] [Google Scholar]
- 31.Bonello L, Laine M, Kipson N, Mancini J, Helal O, Fromonot J, Gariboldi V, Condo J, Thuny F, Frere C, Camoin-Jau L, Paganelli F, Dignat-George F, Guieu R. Ticagrelor increases adenosine plasma concentration in patients with an acute coronary syndrome. J Am Coll Cardiol. 2014;63:872–877. doi: 10.1016/j.jacc.2013.09.067. [DOI] [PubMed] [Google Scholar]
- 32.van Giezen JJ, Sidaway J, Glaves P, Kirk I, Björkman JA. Ticagrelor inhibits adenosine uptake in vitro and enhances adenosine-mediated hyperemia responses in a canine model. J Cardiovasc Pharmacol Ther. 2012;17:164–172. doi: 10.1177/1074248411410883. [DOI] [PubMed] [Google Scholar]
- 33.Wittfeldt A, Emanuelsson H, Brandrup-Wognsen G, van Giezen JJ, Jonasson J, Nylander S, Gan LM. Ticagrelor enhances adenosine-induced coronary vasodilatory responses in humans. J Am Coll Cardiol. 2013;61:723–727. doi: 10.1016/j.jacc.2012.11.032. [DOI] [PubMed] [Google Scholar]
- 34.Alexopoulos D, Moulias A, Koutsogiannis N, Xanthopoulou I, Kakkavas A, Mavronasiou E, Davlouros P, Hahalis G. Differential effect of ticagrelor versus prasugrel on coronary blood flow velocity in patients with non-ST-elevation acute coronary syndrome undergoing percutaneous coronary intervention: an exploratory study. Circ Cardiovasc Interv. 2013;6:277–283. doi: 10.1161/CIRCINTERVENTIONS.113.000293. [DOI] [PubMed] [Google Scholar]
- 35.Nanhwan MK, Ling S, Kodakandla M, Nylander S, Ye Y, Birnbaum Y. Chronic treatment with ticagrelor limits myocardial infarct size: an adenosine and cyclooxygenase-2-dependent effect. Arterioscler Thromb Vasc Biol. 2014;34:2078–2085. doi: 10.1161/ATVBAHA.114.304002. [DOI] [PubMed] [Google Scholar]
- 36.Wang K, Zhou X, Huang Y, Khalil M, Wiktor D, van Giezen JJ, Penn MS. Adjunctive treatment with ticagrelor, but not clopidogrel, added to tPA enables sustained coronary artery recanalisation with recovery of myocardium perfusion in a canine coronary thrombosis model. Thromb Haemost. 2010;104:609–617. doi: 10.1160/TH09-12-0823. [DOI] [PubMed] [Google Scholar]
- 37.Mahaffey KW, Wojdyla DM, Carroll K, Becker RC, Storey RF, Angiolillo DJ, Held C, Cannon CP, James S, Pieper KS, Horrow J, Harrington RA, Wallentin L, PLATO Investigators Ticagrelor compared with clopidogrel by geographic region in the Platelet Inhibition and Patient Outcomes (PLATO) trial. Circulation. 2011;124:544–554. doi: 10.1161/CIRCULATIONAHA.111.047498. [DOI] [PubMed] [Google Scholar]
- 38.Gurbel PA, Bliden KP, Butler K, et al. Randomized double-blind assessment of the ONSET and OFFSET of the antiplatelet effects of ticagrelor versus clopidogrel in patients with stable coronary artery disease: the ONSET/OFFSET study. Circulation. 2009;120:2577–2585. doi: 10.1161/CIRCULATIONAHA.109.912550. [DOI] [PubMed] [Google Scholar]
- 39.Angiolillo DJ, Curzen N, Gurbel P, Vaitkus P, Lipkin F, Li W, Jakubowski JA, Zettler M, Effron MB, Trenk D. Pharmacodynamic evaluation of switching from ticagrelor to prasugrel in patients with stable coronary artery disease: Results of the SWAP-2 Study (Switching Anti Platelet-2) J Am Coll Cardiol. 2014;63:1500–1509. doi: 10.1016/j.jacc.2013.11.032. [DOI] [PubMed] [Google Scholar]
- 40.Bhavaraju K, Georgakis A, Jin J, Gartner TK, Tomiyama Y, Nurden A, Nurden P, Kunapuli SP. Antagonism of P2Y12 reduces physiological thromboxane levels. Platelets. 2010;21:604–609. doi: 10.3109/09537104.2010.511684. [DOI] [PubMed] [Google Scholar]
- 41.Paul BZ, Jin J, Kunapuli SP. Molecular mechanism of thromboxane A(2)-induced platelet aggregation. Essential role for p2t(ac) and alpha(2a) receptors. J Biol Chem. 1999;274:29108–29114. doi: 10.1074/jbc.274.41.29108. [DOI] [PubMed] [Google Scholar]
- 42.Kirkby NS, Leadbeater PD, Chan MV, Nylander S, Mitchell JA, Warner TD. Antiplatelet effects of aspirin vary with level of P2Y12 receptor blockade supplied by either ticagrelor or prasugrel. J Thromb Haemost. 2011;9:2103–2105. doi: 10.1111/j.1538-7836.2011.04453.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gurbel PA, Bliden KP, DiChiara J, Newcomer J, Weng W, Neerchal NK, Gesheff T, Chaganti SK, Etherington A, Tantry US. Evaluation of dose-related effects of aspirin on platelet function: results from the Aspirin-Induced Platelet Effect (ASPECT) study. Circulation. 2007;115:3156–3164. doi: 10.1161/CIRCULATIONAHA.106.675587. [DOI] [PubMed] [Google Scholar]
- 44.Warner TD, Nylander S, Whatling C. Anti-platelet therapy: cyclooxygenase inhibition and the use of aspirin with particular regard to dual anti-platelet therapy. Br J Clin Pharmacol. 2011;72:619–633. doi: 10.1111/j.1365-2125.2011.03943.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grześk G, Kozinski M, Tantry US, Wicinski M, Fabiszak T, Navarese EP, Grzesk E, Jeong YH, Gurbel PA, Kubica J. High-dose, but not low-dose, aspirin impairs anticontractile effect of ticagrelor following ADP stimulation in rat tail artery smooth muscle cells. Biomed Res Int. 2013;2013:928271. doi: 10.1155/2013/928271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ingall AH, Dixon J, Bailey A, Coombs ME, Cox D, McInally JI, Hunt SF, Kindon ND, Teobald BJ, Willis PA, Humphries RG, Leff P, Clegg JA, Smith JA, Tomlinson W. Antagonists of the platelet P2T receptor: a novel approach to antithrombotic therapy. J Med Chem. 1999;42:213–220. doi: 10.1021/jm981072s. [DOI] [PubMed] [Google Scholar]
- 47.Huang J, Driscoll EM, Gonzales ML, Park AM, Lucchesi BR. Prevention of arterial thrombosis by intravenously administered platelet P2T receptor antagonist AR-C69931MX in a canine model. J Pharmacol Exp Ther. 2000;295:492–499. [PubMed] [Google Scholar]
- 48.Faxon DP. Antiplatelet therapy: cangrelor for ACS-lessons from the CHAMPION trials. Nat Rev Cardiol. 2010;7:124–125. doi: 10.1038/nrcardio.2009.247. [DOI] [PubMed] [Google Scholar]
- 49.Bhatt DL, Stone GW, Mahaffey KW, et al. CHAMPION PHOENIX Investigators. Effect of platelet inhibition with cangrelor during PCI on ischemic events. N Engl J Med. 2013;368:1303–1313. doi: 10.1056/NEJMoa1300815. [DOI] [PubMed] [Google Scholar]
- 50.Angiolillo DJ, Firstenberg MS, Price MJ, et al. BRIDGE Investigators. Bridging antiplatelet therapy with cangrelor in patients undergoing cardiac surgery: a randomized controlled trial. JAMA. 2012;307:265–274. doi: 10.1001/jama.2011.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Steinhubl SR, Oh JJ, Oestreich JH, Ferraris S, Charnigo R, Akers WS. Transitioning patients from cangrelor to clopidogrel: pharmacodynamic evidence of a competitive effect. Thromb Res. 2008;121:527–534. doi: 10.1016/j.thromres.2007.05.020. [DOI] [PubMed] [Google Scholar]
- 52.Dovlatova NL, Jakubowski JA, Sugidachi A, Heptinstall S. The reversible P2Y antagonist cangrelor influences the ability of the active metabolites of clopidogrel and prasugrel to produce irreversible inhibition of platelet function. J Thromb Haemost. 2008;6:1153–1159. doi: 10.1111/j.1538-7836.2008.03020.x. [DOI] [PubMed] [Google Scholar]
- 53.Ravnefjord A, Weilitz J, Emanuelsson BM, van Giezen JJ. Evaluation of ticagrelor pharmacodynamic interactions with reversibly binding or non-reversibly binding P2Y(12) antagonists in an ex-vivo canine model. Thromb Res. 2012;130:622–628. doi: 10.1016/j.thromres.2012.07.021. [DOI] [PubMed] [Google Scholar]
- 54.Schneider DJ, Agarwal Z, Seecheran N, Keating FK, Gogo P. Pharmacodynamic effects during the transition between cangrelor and ticagrelor. JACC Cardiovasc Interv. 2014;7:435–442. doi: 10.1016/j.jcin.2013.08.017. [DOI] [PubMed] [Google Scholar]
- 55.Angiolillo DJ, Saucedo JF, Deraad R, Frelinger AL, Gurbel PA, Costigan TM, Jakubowski JA, Ojeh CK, Effron MB, SWAP Investigators Increased platelet inhibition after switching from maintenance clopidogrel to prasugrel in patients with acute coronary syndromes: results of the SWAP (SWitching Anti Platelet) study. J Am Coll Cardiol. 2010;56:1017–1023. doi: 10.1016/j.jacc.2010.02.072. [DOI] [PubMed] [Google Scholar]
- 56.Bach P, Antonsson T, Bylund R, Björkman JA, Österlund K, Giordanetto F, van Giezen JJ, Andersen SM, Zachrisson H, Zetterberg F. Lead optimization of ethyl 6-aminonicotinate acyl sulfonamides as antagonists of the P2Y12 receptor. separation of the antithrombotic effect and bleeding for candidate drug AZD1283. J Med Chem. 2013;56:7015–7024. doi: 10.1021/jm400820m. [DOI] [PubMed] [Google Scholar]
- 57.Chang H, Yanachkov IB, Dix EJ, Yanachkova M, Li Y, Barnard MR, Wright GE, Michelson AD, Frelinger AL., III Antiplatelet activity, P2Y1 and P2Y12 inhibition, and metabolism in plasma of stereoisomers of diadenosine 5′,5′″-P1,P4-dithio-P2,P3-chloromethylenetetraphosphate. PLoS One. 2014;9:e94780. doi: 10.1371/journal.pone.0094780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gurbel PA, Jeong YH, Tantry US. Vorapaxar: a novel protease-activated receptor-1 inhibitor. Expert Opin Investig Drugs. 2011;20:1445–1453. doi: 10.1517/13543784.2011.606809. [DOI] [PubMed] [Google Scholar]
- 59.Brass LF, Vassallo RR, Jr, Belmonte E, Ahuja M, Cichowski K, Hoxie JA. Structure and function of the human platelet thrombin receptor. Studies using monoclonal antibodies directed against a defined domain within the receptor N terminus. J Biol Chem. 1992;267:13795–13798. [PubMed] [Google Scholar]
- 60.Leger AJ, Jacques SL, Badar J, Kaneider NC, Derian CK, Andrade-Gordon P, Covic L, Kuliopulos A. Blocking the protease-activated receptor 1–4 heterodimer in platelet-mediated thrombosis. Circulation. 2006;113:1244–1254. doi: 10.1161/CIRCULATIONAHA.105.587758. [DOI] [PubMed] [Google Scholar]
- 61.Austin KM, Covic L, Kuliopulos A. Matrix metalloproteases and PAR1 activation. Blood. 2013;121:431–439. doi: 10.1182/blood-2012-09-355958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Duvernay M, Young S, Gailani D, Schoenecker J, Hamm HE, Hamm H. Protease-activated receptor (PAR) 1 and PAR4 differentially regulate factor V expression from human platelets. Mol Pharmacol. 2013;83:781–792. doi: 10.1124/mol.112.083477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Andersen H, Greenberg DL, Fujikawa K, Xu W, Chung DW, Davie EW. Protease-activated receptor 1 is the primary mediator of thrombin-stimulated platelet procoagulant activity. Proc Natl Acad Sci U S A. 1999;96:11189–11193. doi: 10.1073/pnas.96.20.11189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim S, Foster C, Lecchi A, Quinton TM, Prosser DM, Jin J, Cattaneo M, Kunapuli SP. Protease-activated receptors 1 and 4 do not stimulate G(i) signaling pathways in the absence of secreted ADP and cause human platelet aggregation independently of G(i) signaling. Blood. 2002;99:3629–3636. doi: 10.1182/blood.v99.10.3629. [DOI] [PubMed] [Google Scholar]
- 65.Chackalamannil S, Wang Y, Greenlee WJ, Hu Z, Xia Y, Ahn HS, Boykow G, Hsieh Y, Palamanda J, Agans-Fantuzzi J, Kurowski S, Graziano M, Chintala M. Discovery of a novel, orally active himbacine-based thrombin receptor antagonist (SCH 530348) with potent antiplatelet activity. J Med Chem. 2008;51:3061–3064. doi: 10.1021/jm800180e. [DOI] [PubMed] [Google Scholar]
- 66.Gurbel PA, Bliden KP, Saucedo JF, Suarez TA, DiChiara J, Antonino MJ, Mahla E, Singla A, Herzog WR, Bassi AK, Hennebry TA, Gesheff TB, Tantry US. Bivalirudin and clopidogrel with and without eptifibatide for elective stenting: effects on platelet function, thrombelastographic indexes, and their relation to periprocedural infarction results of the CLEAR PLATELETS-2 (Clopidogrel with Eptifibatide to Arrest the Reactivity of Platelets) study. J Am Coll Cardiol. 2009;53:648–657. doi: 10.1016/j.jacc.2008.10.045. [DOI] [PubMed] [Google Scholar]
- 67.Zhang P, Covic L, Kuliopulos A. Platelet receptors and drug targets: PAR1, collagen, vWF, thromboxane, and other novel targets. In: Waksman R, Gurbel PA, Giglia MA, editors. Antiplatelet therapy in Cardiovascular Disease. West Susses, United Kingdom: John Wiley and Sons, Ltd; 2014. [Google Scholar]
- 68.Wu CC, Hwang TL, Liao CH, Kuo SC, Lee FY, Lee CY, Teng CM. Selective inhibition of protease-activated receptor 4-dependent platelet activation by YD-3. Thromb Haemost. 2002;87:1026–1033. [PubMed] [Google Scholar]
- 69.Wu CC, Teng CM. Comparison of the effects of PAR1 antagonists, PAR4 antagonists, and their combinations on thrombin-induced human platelet activation. Eur J Pharmacol. 2006;546:142–147. doi: 10.1016/j.ejphar.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 70.Coughlin SR. Protease-activated receptors in hemostasis, thrombosis and vascular biology. J Thromb Haemost. 2005;3:1800–1814. doi: 10.1111/j.1538-7836.2005.01377.x. [DOI] [PubMed] [Google Scholar]
- 71.Zhang C, Srinivasan Y, Arlow DH, Fung JJ, Palmer D, Zheng Y, Green HF, Pandey A, Dror RO, Shaw DE, Weis WI, Coughlin SR, Kobilka BK. High-resolution crystal structure of human protease-activated receptor 1. Nature. 2012;492:387–392. doi: 10.1038/nature11701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.FDA approves Zontivity to reduce the risk of heart attacks and stroke in high-risk patients. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm396585.htm. Accessed August 7, 2014.
- 73.Kimmelstiel C, Zhang P, Kapur NK, Weintraub A, Krishnamurthy B, Castaneda V, Covic L, Kuliopulos A. Bivalirudin is a dual inhibitor of thrombin and collagen-dependent platelet activation in patients undergoing percutaneous coronary intervention. Circ Cardiovasc Interv. 2011;4:171–179. doi: 10.1161/CIRCINTERVENTIONS.110.959098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Covic L, Singh C, Smith H, Kuliopulos A. Role of the PAR4 thrombin receptor in stabilizing platelet-platelet aggregates as revealed by a patient with Hermansky-Pudlak syndrome. Thromb Haemost. 2002;87:722–727. [PubMed] [Google Scholar]
- 75.Dimond P, Carlson K, Bouvier M, Gerard C, Xu L, Covic L, Agarwal A, Ernst OP, Janz JM, Schwartz TW, Gardella TJ, Milligan G, Kuliopulos A, Sakmar TP, Hunt SW., III G protein-coupled receptor modulation with pepducins: moving closer to the clinic. Ann N Y Acad Sci. 2011;1226:34–49. doi: 10.1111/j.1749-6632.2011.06039.x. [DOI] [PubMed] [Google Scholar]
- 76.Covic L, Misra M, Badar J, Singh C, Kuliopulos A. Pepducin-based intervention of thrombin-receptor signaling and systemic platelet activation. Nat Med. 2002;8:1161–1165. doi: 10.1038/nm760. [DOI] [PubMed] [Google Scholar]
- 77.Zhang P, Gruber A, Kasuda S, Kimmelstiel C, O’Callaghan K, Cox DH, Bohm A, Baleja JD, Covic L, Kuliopulos A. Suppression of arterial thrombosis without affecting hemostatic parameters with a cell-penetrating PAR1 pepducin. Circulation. 2012;126:83–91. doi: 10.1161/CIRCULATIONAHA.112.091918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fiessinger JN, Bounameaux H, Cairols MA, Clement DL, Coccheri S, Fletcher JP, Hoffmann U, Turpie AG, TAIPAD Investigators Thromboxane antagonism with terutroban in peripheral arterial disease: the TAIPAD study. J Thromb Haemost. 2010;8:2369–2376. doi: 10.1111/j.1538-7836.2010.04020.x. [DOI] [PubMed] [Google Scholar]
- 79.Bal Dit, Sollier C, Crassard I, Simoneau G, Bergmann JF, Bousser MG, Drouet L. Effect of the thromboxane prostaglandin receptor antagonist terutroban on arterial thrombogenesis after repeated administration in patients treated for the prevention of ischemic stroke. Cerebrovasc Dis. 2009;28:505–513. doi: 10.1159/000236915. [DOI] [PubMed] [Google Scholar]
- 80.Bousser MG, Amarenco P, Chamorro A, Fisher M, Ford I, Fox KM, Hennerici MG, Mattle HP, Rothwell PM, de Cordoüe A, Fratacci MD, PERFORM Study Investigators Terutroban versus aspirin in patients with cerebral ischaemic events (PERFORM): a randomised, double-blind, parallel-group trial. Lancet. 2011;377:2013–2022. doi: 10.1016/S0140-6736(11)60600-4. [DOI] [PubMed] [Google Scholar]
- 81.Randomized trial of ridogrel, a combined thromboxane A2 synthase inhibitor and thromboxane A2/prostaglandin endoperoxide receptor antagonist, versus aspirin as adjunct to thrombolysis in patients with acute myocardial infarction. The Ridogrel Versus Aspirin Patency Trial (RAPT) Circulation. 1994;89:588–595. doi: 10.1161/01.cir.89.2.588. [DOI] [PubMed] [Google Scholar]
- 82.Guth BD, Narjes H, Schubert HD, Tanswell P, Riedel A, Nehmiz G. Pharmacokinetics and pharmacodynamics of terbogrel, a combined thromboxane A2 receptor and synthase inhibitor, in healthy subjects. Br J Clin Pharmacol. 2004;58:40–51. doi: 10.1111/j.1365-2125.2004.02083.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jackson SP, Schoenwaelder SM, Goncalves I, et al. PI 3-kinase p110beta: a new target for antithrombotic therapy. Nat Med. 2005;11:507–514. doi: 10.1038/nm1232. [DOI] [PubMed] [Google Scholar]
- 84.Martin V, Guillermet-Guibert J, Chicanne G, Cabou C, Jandrot-Perrus M, Plantavid M, Vanhaesebroeck B, Payrastre B, Gratacap MP. Deletion of the p110beta isoform of phosphoinositide 3-kinase in platelets reveals its central role in Akt activation and thrombus formation in vitro and in vivo. Blood. 2010;115:2008–2013. doi: 10.1182/blood-2009-04-217224. [DOI] [PubMed] [Google Scholar]
- 85.Jackson SP, Schoenwaelder SM. Antithrombotic phosphoinositide 3-kinase β inhibitors in humans: a ‘shear’ delight! J Thromb Haemost. 2012;10:2123–2126. doi: 10.1111/j.1538-7836.2012.04912.x. [DOI] [PubMed] [Google Scholar]
- 86.Uemura T, Kawasaki T, Taniguchi M, Moritani Y, Hayashi K, Saito T, Takasaki J, Uchida W, Miyata K. Biological properties of a specific Galpha q/11 inhibitor, YM-254890, on platelet functions and thrombus formation under high-shear stress. Br J Pharmacol. 2006;148:61–69. doi: 10.1038/sj.bjp.0706711. [DOI] [PMC free article] [PubMed] [Google Scholar]