

Summary
Hepatitis C virus (HCV) enters human hepatocytes through a multistep mechanism involving, among other host proteins, the virus receptor CD81. How CD81 governs HCV entry is poorly characterized, and CD81 protein interactions after virus binding remain elusive. We have developed a quantitative proteomics protocol to identify HCV-triggered CD81 interactions and found 26 dynamic binding partners. At least six of these proteins promote HCV infection, as indicated by RNAi. We further characterized serum response factor binding protein 1 (SRFBP1), which is recruited to CD81 during HCV uptake and supports HCV infection in hepatoma cells and primary human hepatocytes. SRFBP1 facilitates host cell penetration by all seven HCV genotypes, but not of vesicular stomatitis virus and human coronavirus. Thus, SRFBP1 is an HCV-specific, pan-genotypic host entry factor. These results demonstrate the use of quantitative proteomics to elucidate pathogen entry and underscore the importance of host protein-protein interactions during HCV invasion.
Graphical Abstract
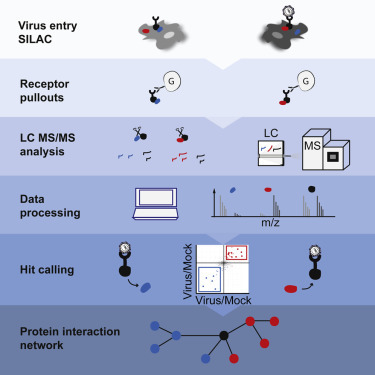
Highlights
-
•
Hepatitis C virus binding alters host protein interactions with the receptor CD81
-
•
Six out of 26 virus-dependent CD81-interacting proteins promote virus entry
-
•
SRFBP1 binds CD81 and aids infection of all HCV, but not VSV, genotypes
-
•
SRFBP1 is membrane-associated and required for HCV entry
Hepatitis C virus (HCV) enters human hepatocytes through a multistep mechanism. Gerold et al. apply quantitative proteomics to define the protein network responsible for HCV entry and identify SRFBP1 as a partner for the virus receptor CD81.
Introduction
Virus entry describes the process of delivering viral genomes in a replication-competent manner into a naive host cell. Successful penetration of cells involves receptor binding, virion uptake, membrane fusion or perturbation, transport of nucleocapsids to replication competent cellular compartments, and uncoating (Yamauchi and Helenius, 2013). Virus receptors are more than attachment factors, functionally supporting cell entry by several means: they mediate formation of receptor platforms, induce conformational changes in virus surface molecules, transmit signals within the cell, and induce virus translocation along the membrane and into the cell (Mercer et al., 2010). A number of virus receptors, however, lack signaling domains. Consequently, these receptors must initiate the virus uptake program through ligand-dependent interaction with additional host proteins.
In this study, we focus on the entry mechanism of hepatitis C virus (HCV), an enveloped RNA virus infecting 160 million individuals worldwide (Gravitz, 2011, Lavanchy, 2011). Hepatitis C is a slowly progressing disease, which can cause liver fibrosis, cirrhosis, and hepatocellular carcinoma 15–25 years after contraction (Seeff, 2002). To date, hepatitis C is the number one indication for liver transplantation in North America and Europe. Unfortunately, re-infection of the graft liver by virus residing in peripheral reservoirs is almost universal and leads to accelerated disease progression. For post-transplant patients, interfering with the entry of HCV into the engrafted hepatocytes would be a promising preventive treatment.
HCV penetration is a multistep process requiring the four entry factors scavenger receptor class B member 1 (SR-BI), CD81, claudin-1 (CLDN1), and occludin (OCLN) (Evans et al., 2007, Pileri et al., 1998, Ploss et al., 2009, Scarselli et al., 2002). CD81 is a central player in HCV entry as it directly binds the HCV E2 surface glycoprotein, renders it fusion competent (Pileri et al., 1998, Rajesh et al., 2012, Sharma et al., 2011), and activates the HCV entry cofactor epidermal growth factor receptor (EGFR) (Diao et al., 2012, Gerold and Rice, 2011, Lupberger et al., 2011). Moreover, CD81 is thought to laterally translocate with the virions to tight junctions, where CLDN1 and OCLN reside (Brazzoli et al., 2008). Finally, CD81 and CLDN1 co-internalize with the virus into endosomes (Farquhar et al., 2012). How CD81 orchestrates HCV uptake remained elusive. As a scaffolding protein, CD81 lacks intracellular signaling domains but coordinates protein-protein interactions in membrane microdomains termed tetraspanin webs (Charrin et al., 2003, Montpellier et al., 2011). We hypothesized that the binding of HCV to CD81 triggers protein interactions, which in turn coordinate HCV uptake.
Here, we determined changes in the protein interaction network coordinated by CD81 during uptake of HCV particles using quantitative proteomics (Meissner and Mann, 2014). We found 26 HCV-dependent CD81 interactions. Consistent with our hypothesis, a subset of the receptor-interacting proteins promoted HCV infectivity. In particular, we identified serum response factor binding protein 1 (SRFBP1) as an HCV host factor, which forms a complex with CD81 and coordinates host cell penetration. The method described here is applicable to various steps in the life cycle of viruses and other microbes. It holds the promise of revealing critical pathogen-induced changes in host protein-protein interactions, thus guiding development of anti-infective strategies.
Results
Quantitative Proteomics Identifies Virus Entry-Dependent Receptor Interactions
Quantitative proteomics allows the hypothesis-free characterization of protein-protein interactions between cellular states. Here, we use stable isotope labeling by amino acids in cell culture (SILAC) and quantitative interaction proteomics (Ong et al., 2002) to study host protein interactions with the HCV receptor CD81 upon HCV exposure. To this end, HCV permissive human hepatoma cells Huh-7 were labeled with heavy arginine ((15N4 13C6) Arg-10) and lysine ((15N2 13C6) Lys-8), achieving 95% incorporation of heavy amino acids into cellular proteins (heavy) or left unlabeled (light). As HCV induces clathrin-mediated endocytosis 15 min after binding (Coller et al., 2009), we incubated heavy Huh-7 cells for 15 min with HCV (J6/JFH-1 clone 2; MOI: 10) and light cells with non-infectious cell culture supernatants (forward label). To exclude isotope-specific effects, we swapped labels of the two conditions, so that light cells were HCV exposed and heavy cells mock treated (reverse label). Next, we affinity enriched CD81 and its interacting proteins (Figure S1A), combined proteins from HCV and uninfected samples, and digested proteins to peptides. Liquid chromatography (LC) coupled to mass spectrometry (MS) then identified, quantified, and distinguished peptides derived from HCV and uninfected conditions by their characteristic mass offset (Figures 1 A and S1B).
Figure 1.
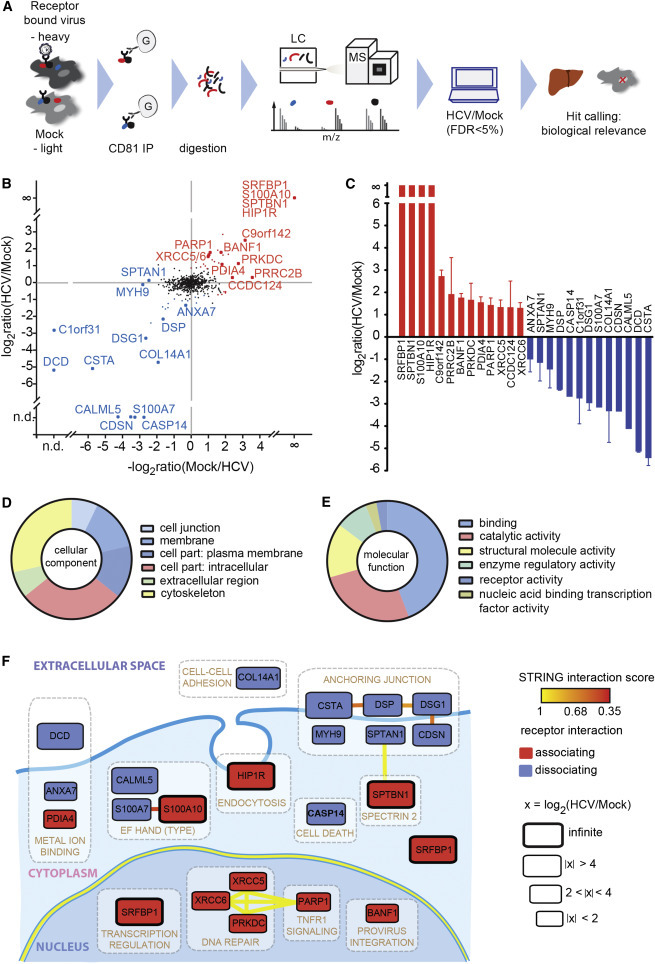
High-Resolution Quantitative MS Reveals Transient HCV Entry Factor Interactions
(A) Outline of the virus entry interaction proteomics procedure.
(B) CD81 interactome upon HCV exposure. Depicted are the mean log2 SILAC ratios of CD81-interacting proteins in HCV versus mock-treated samples from forward (y axis) and reverse experiments (x axis). Reverse label ratios are inverted, so that a positive correlation indicates reproducible interaction upon label swap. Significant (FDR < 5%) outliers are colored in red (CD81-associating proteins) and blue (CD81-dissociating proteins). Infinite ratio, interaction partners exclusively found in the presence of HCV. n.d., not quantified in either forward or reverse experiment.
(C) SILAC log2 ratios for each of the 13 CD81-associating and 13 CD81-dissociating factors. Shown are means ± SEM of four biological replicates with inverted reverse label ratios.
(D and E) Enrichment of Gene Ontology cellular component (GOCC) and molecular function (GOMF) annotations.
(F) Functional map of host factors transiently interacting with the HCV receptor CD81 during virus entry. Functional clusters (white boxes) and previously reported interactions (bold lines) of the here identified transient CD81-binding partners are depicted. We assigned individual proteins to the highest scoring DAVID cluster. Yellow lines between genes of different clusters indicate high-confidence (>0.9) STRING interactions. Within a functional annotation cluster, also lower confidence (>0.35) STRING interactions are shown. Proteins are placed in their predominant cellular location; SRFBP1 is shown twice as it localizes to nucleus and cytoplasm. The box size indicates the degree of CD81 association or dissociation upon HCV binding. Associating factors (red) and dissociating factors (blue) are shown. See also Figure S1 and Tables S1 and S2–S4.
LC-MS analysis revealed a total of 778 host proteins in CD81 co-immunoprecipitations (co-IPs). This high number is typical for affinity enrichment MS because it includes proteins, which non-specifically bind to the IP resin (Table S1) (Keilhauer et al., 2015). Subsequent data processing eliminates these background binders as described below. CD81 was detected in all co-IPs with high intensities independent of the presence of bound HCV (Figure S1C). To identify HCV-regulated protein-protein interactions, we next calculated the ratios of heavy over light protein abundances in cells with or without bound HCV. Protein quantification was reproducible as demonstrated by a median correlation of the SILAC ratios of r2 = 0.70 and 0.81 for forward and reverse label samples, respectively. Here, we focused on proteins, which differed between the two experimental conditions, thereby excluding background binders, which are equally abundant in HCV and mock samples. As hypothesized, we identified 55 protein interactors, which quantitatively differed between HCV and mock samples. Among these HCV-dependent transient interactors are proteins that associate with CD81 (Figure 1B, upper right quadrant) or dissociate from CD81 upon virus exposure (Figure 1B, lower left quadrant).
We stratified HCV-dependent interactors of CD81 based on statistical significance, interaction strength, and biological relevance. In particular, inclusion criteria were a minimum of 2.1-fold change in CD81 interaction strength upon HCV exposure, liver expression, and non-nuclear localization. Of note, we additionally excluded ribosomal proteins in this study, although they might have a potential role in virus uncoating. A total of 26 proteins fulfilled these inclusion criteria, half of which dissociated from and the other half associated with CD81 upon virus binding (labeled data points in Figures 1B and 1C and Table S2; IntAct: IM-24070). Proteins that were exclusively quantified in the presence of HCV were assigned an infinite ratio, because the strength of interaction could not be determined. Known steady-state interaction partners of CD81 were absent from this transient interactome. Instead, we identified huntingtin-interacting-protein-1-related protein (HIP1R), a previously reported HCV entry cofactor and a known component of clathrin-coated pits (Coller et al., 2009). Taken together, we confirmed our hypothesis that HCV binding to CD81 alters its protein interaction network and that some HCV entry cofactors are transient CD81 interactors.
Next, we investigated whether certain molecular functions and cellular compartments were enriched in our transient CD81 interactome. Most proteins were membrane associated (49%) or cytoskeletal components (31%), with one-third being plasma membrane associated (Figure 1D; Table S3). Molecular function analysis revealed a strong enrichment for proteins with binding function (44%), catalytic activity (27%), and structural molecule activity (15%; Figure 1E; Table S4). Taken together, our gene ontology (GO) enrichment analysis reflects the fact that the HCV-CD81 complex laterally translocates along the plasma membrane (Brazzoli et al., 2008, Coller et al., 2009) with a need for interaction with plasma membrane and cytoskeletal proteins.
To further reveal interconnected cellular structures and processes enriched in the HCV entry-dependent protein pool, we integrated a DAVID-based clustering analysis in a STRING-based protein interaction map (Figure 1F). Notably, we found a cluster of six cellular junction proteins not previously reported in HCV entry. These proteins, which include adherens junction, desmosomal, and cell envelope constituents, are interconnected by reported protein-protein interactions. Furthermore, we found cytoskeletal proteins (spectrins; myosin-9) and a clathrin-coated pit protein (HIP1R), which is in line with the reported clathrin-mediated endocytosis of HCV (Blanchard et al., 2006). Finally, we found clusters of calcium- and metal-binding proteins as well as nuclear proteins, with a secondary cytoplasmic localization. All identified proteins with extracellular or plasma membrane localization dissociated from CD81 upon HCV binding (Figure 1F, blue label), whereas proteins localizing to endosomes or intracellular compartments associated with CD81 (Figure 1F, red label). This confirms the notion that the HCV-CD81 complex needs to move out of plasma membrane microdomains to then get endocytosed. In summary, we identified 26 selected transient protein-receptor interactions during HCV entry including a known HCV entry cofactor and a cluster of junctional membrane proteins.
Virus-Dependent CD81-Binding Proteins Promote HCV Infectivity
We hypothesized that a subset of the 26 virus-dependent CD81-interacting proteins is required for productive virus entry. To test this, we silenced the 26 respective mRNAs and infected human hepatoma cells with a Renilla luciferase (RLuc) reporter strain of HCV (JcR2A; Figure 2 A). Eight of the 26 targets showed a significant reduction in HCV infection of Huh-7.5 cells upon RNAi. HIP1R, a previously reported HCV cofactor, also decreased HCV infectivity but did not meet our statistical significance criteria (Figure 2B; Table S5). CD81-targeting siRNAs served as positive control and reduced HCV infectivity more than 5-fold. None of the candidate targeting or scrambled siRNAs were cytotoxic or altered cell proliferation. Cystatin A (CSTA), a desmosomal regulator, dissociated almost completely from CD81 upon virus binding (Figures 1B and 1C), and its silencing led to a significant increase in HCV infectivity (Figure 2B).
Figure 2.
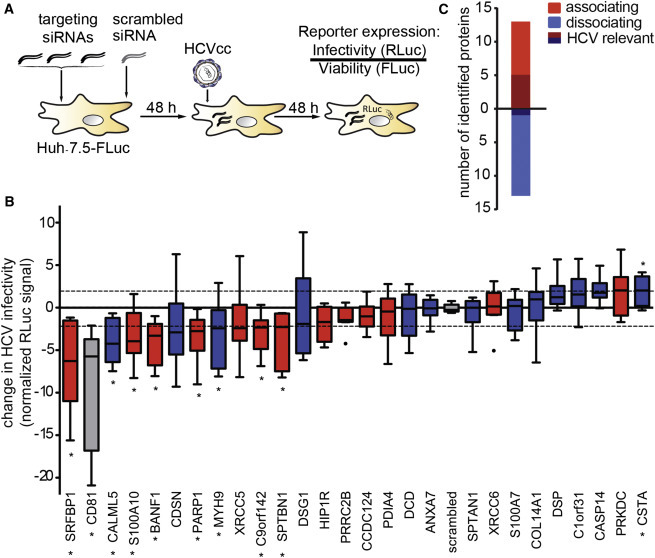
A Subset of CD81 Interaction Partners Is Required for HCV Infection
(A) Outline of the RNAi screen on transient CD81 interaction partners relevant for HCV infection.
(B) Functional RNAi follow-up screen on 26 selected transient CD81 interaction partners identifies nine putative host factors. We silenced the indicated transcript with a pool of three siRNAs in Huh-7.5 FLuc cells, infected 48 hr later with Renilla luciferase reporter HCV (JcR2A), and determined cell viability and HCV infectivity 48 hpi. Shown is the RLuc signal after normalization for cell viability and plate effects. Eight siRNA pools significantly decreased and one increased HCV infectivity (p ≤ 0.05; abs [z score] ≥ 2; ∗). Associating factors (red), dissociating factors (blue), CD81 and scrambled controls (gray) are shown. Box and whisker plot of nine biological replicates is shown.
(C) The combined SILAC co-IP RNAi strategy reveals a bias for CD81-associating factors to act as HCV host factors. Out of 26 HCV-dependent CD81-binding partners, six decreased HCV infectivity upon RNAi with a minimum transcript reduction of 75% (shaded color). See also Figure S2 and Tables S5 and S6.
Here, we chose to concentrate on the eight transient CD81 interaction partners, which reduced HCV infection when silenced (Figure 2B). Transcript levels of six of the eight targets were reduced to 25% or less of scrambled siRNA controls (Figure S2A). The six putative HCV host factors are SRFBP1, barrier-to-autointegration factor (BANF1), myosin-9 (MYH9), spectrin beta chain, non-erythrocytic 1 (SPTBN1), calpactin I light chain (S100A10), and poly [ADP-ribose] polymerase 1 (PARP1) (Table S6). Of all tested transient CD81 interactors, SRFBP1 showed the strongest reduction in HCV infectivity upon knockdown (z score [SRFBP1]: −6.3; z score [CD81]: −5.8). In summary, we found that at least six of the 26 tested transient CD81 interactors promote HCV infection with a clear bias for CD81 associating (five) over dissociating (one) factors (Figures 2C and S2B).
SRFBP1 Is Expressed in the Liver and Required for an Early Step in HCV Infection
SRFBP1 emerged as prime candidate for in-depth characterization as an HCV entry factor because the protein showed the strongest inhibition of HCV infection when silenced. We first quantified SRFBP1 mRNA in primary hepatocytes from resection specimens of five HCV-negative donors and observed an up to 6-fold higher SRFBP1 expression level than in Huh-7.5 cells (Figure 3 A). Importantly, SRFBP1 mRNA and protein levels correlated strongly (Figures S3A and S3B). The observed differences in SRFBP1 expression led us to examine whether endogenous SRFBP1 levels were limiting HCV infection in hepatoma cells. When overexpressing SRFBP1 in Huh-7.5 cells, we observed a dose-dependent 2- to 3-fold increase in HCV infectivity (Figure 3B). Conversely, when silencing SRFBP1 in primary hepatocytes, we observed a 4-fold decrease in HCV infectivity (Figure S3C). Collectively, this suggests that low SRFBP1 expression could contribute to the limited infectivity in current HCV cell culture models.
Figure 3.
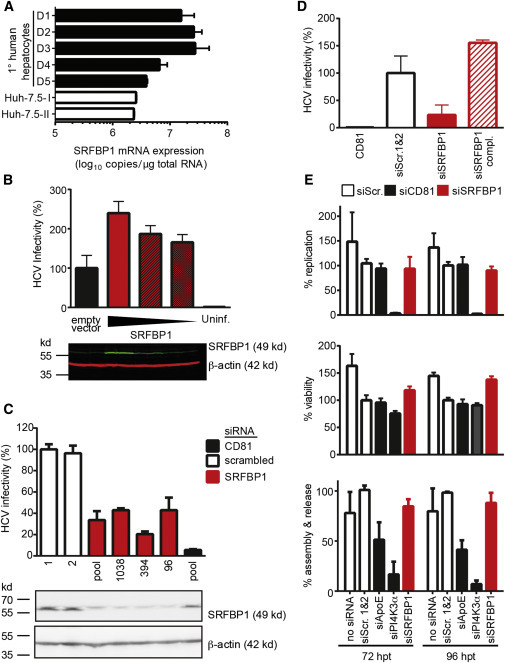
The CD81-Binding Partner SRFBP1 Is Expressed in Human Liver and Required for HCV Infection
(A) SRFBP1 transcript levels in primary human hepatocytes are up to 6-fold higher than in Huh-7.5 cells. Absolute transcript numbers of SRFBP1 in hepatocytes from five donors (D1–D5) and in two independent passages of human hepatoma cells (Huh-7.5) were determined in technical triplicates and displayed as mean + SD.
(B) HCV (JcR2A) infectivity increases in a dose-dependent manner in Huh-7.5 FLuc cells upon overexpression of full-length SRFBP1. Cells were transduced with lentiviruses encoding SRFBP1 or a blasticidin resistance gene (empty vector), 72 hr later infected with HCV, and infectivity measured 48 hpi by luciferase assay. Immunoblot analysis of lysates 72 post-transduction shows dose-dependent SRFBP1 overexpression (green). Actin served as loading control (red). The immunoblot is representative of three biological replicates.
(C) HCV (JcR2A) infectivity is reduced in Huh-7.5 FLuc cells 48 hp silencing of SRFBP1 or CD81. We used a pool of three siRNAs or individual siRNAs targeting the indicated ORF position and measured infectivity at 48 hpi by luciferase assay. Two scrambled siRNAs (1 and 2) served as controls. Immunoblot analysis confirms reduced SRFBP1 protein levels 48 hp RNAi. Mean + SD of three technical replicates are shown. Infectivity data and immunoblot are representative of three biological replicates.
(D) Lentiviral transduction with siRNA-resistant SRFBP1 rescues HCV infection in SRFBP1-silenced Huh-7.5 FLuc cells. Cells were transfected with siRNAs (SRFBP1: siRNA 394), 24 hr later transduced with blasticidin resistance gene encoding lentivirus (siSRFBP1) or siRNA-resistant SRFBP1 encoding lentivirus (siSRFBP1 compl.), and 24 hr later infected with HCV (JcR2A). Infectivity at 48 hpi measured by luciferase assay is shown.
(E) SRFBP1 is dispensable for HCV replication, assembly, and release. Huh-7.5 FLuc cells were transfected with genomic HCV RNA (JcR2A) and the indicated gene silenced 5 hr later (SRFBP1: siRNA 394). At 72 and 96 hp transfection (hpt), supernatants were harvested, cells lysed, and replication efficiency in lysates measured by luciferase assay (upper panel). Viability of HCV-replicating cells upon RNAi was determined using the cellular FLuc reporter at 72 or 96 hpt (middle panel). Supernatants from HCV-transfected and SRFBP1-silenced cells were titrated on naive Huh-7.5 cells to determine virus particle assembly and release rates (bottom panel). Values were normalized to a scrambled siRNA control. Unless stated otherwise, all experiments are displayed as mean + SD of three independent biological replicates each performed in technical triplicates. See also Figure S3.
Next, we asked which step in the HCV life cycle requires SRFBP1. First, we confirmed that infectivity of incoming virus was impaired in SRFBP1-silenced hepatoma cells. A pool of three SRFBP1-targeting siRNAs and three individual siRNAs (nt96, nt394, and nt1038) reduced SRFBP1 protein expression and resulted in an up to 4-fold reduction in HCV infectivity (Figure 3C). CD81-silenced cells showed 5-fold decreased CD81 surface expression and a 10-fold reduction in infectivity (Figures 3C and S4A). We next excluded off-target effects of SRFBP1 siRNA nt394 by complementing SRFBP1-silenced cells with a siRNA-resistant SRFBP1 variant (Figure 3D). Taken together, silencing of SRFBP1 led to a decrease in HCV susceptibility, and this phenotype could be rescued by SRFBP1 complementation.
The association of SRFBP1 with the HCV entry factor CD81 suggested a role in HCV entry or an early post-entry event. Our infectivity readout at 48 hr post-HCV inoculation determined accumulative effects of virus entry, translation, replication, and spread (Gerold and Pietschmann, 2014). Thus, we next sought to exclude that SRFBP1 would affect HCV translation or replication. When silencing SRFBP1 (nt394) in cells actively replicating HCV, we observed no impairment of replication and RLuc reporter translation at 48 or 72 hr post-RNAi. In stark contrast, silencing the known replication host factor phosphatidyl inositol 4 kinase 3 alpha (PI4KIIIalpha) reduced replication to background levels (Figure 3E, upper panel) (Berger et al., 2009, Reiss et al., 2011). Neither knockdown of SRFBP1 nor of PI4KIIIalpha affected cell viability or proliferation (Figure 3E, middle panel). To address a possible role of SRFBP1 in assembly, release, and spread of HCV, we collected supernatants from HCV-replicating cells at 48 or 72 hr post-siRNA transfection and infected naive Huh-7.5 cells. SRFBP1 silencing did not alter the released infectivity, whereas apolipoprotein E (APOE) silencing expectedly reduced the released infectivity to 40% (Figure 3E, lower panel) (Chang et al., 2007). Taken together, SRFBP1 silencing rendered cells less susceptible to HCV without altering replication or spread of the virus to naive cells.
SRFBP1 Colocalizes with CD81 without Affecting HCV Receptor Surface Expression
In light of our finding that SRFBP1 plays a role early during HCV infection, we investigated whether SRFBP1 colocalizes with established HCV entry factors in resting cells. Whereas SRFBP1 only weakly colocalized with CLDN1, OCLN, or SR-BI (Pearson’s coefficient < 0.2), a fraction of the protein colocalized with CD81 (Pearson’s coefficient 0.4). In particular, SRFBP1 and CD81 signals overlapped in perinuclear regions and in the cell periphery, where we observed a punctate, vesicular staining. We further observed a weak colocalization with the membrane marker wheat germ agglutinin (WGA) (Pearson’s coefficient 0.3; Figures 4A and 4B).
Figure 4.
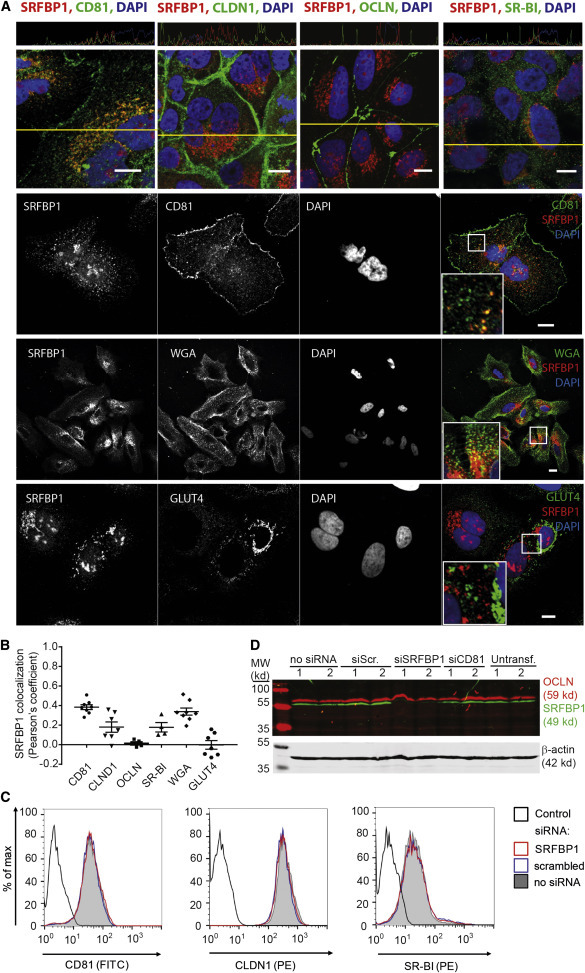
SRFBP1 Colocalizes with CD81 without Affecting Entry Factor Surface Expression
(A) SRFBP1 partially colocalizes with CD81 and the membrane marker WGA but only weakly with CLDN1, OCLN, SR-BI, and GLUT4. Huh-7.5 cells were stained with Alexa-conjugated membrane marker WGA (panel 3) for 1 min or left unstained (panels 1, 2, and 4), fixed, permeabilized, and stained for SRFBP1 and the indicated protein. Nuclei were stained with DAPI. Colocalization across a section (yellow line in panel 1) is depicted above the respective image. Representative confocal images; insert 2.2-fold magnification; scale bars 10 μm.
(B) Pearson’s correlation coefficient for SRFBP1 and the indicated cellular protein or the membrane marker WGA calculated by intensity correlation analysis. Each symbol represents an individual frame; horizontal lines indicate the mean ± SEM.
(C) SRFBP1-silenced cells (siRNA 394) express CD81, CLDN1, and SR-BI at the plasma membrane. Surface expression of CD81, CLDN1, and SR-BI on Huh-7.5 cells was analyzed 48 hpt with the indicated siRNAs. Cells were stained with antibodies against HCV entry factors followed by flow cytometric analysis of 10,000 cells per sample. For quantification and additional controls, see Figure S4. Control is directly conjugated isotype antibody (histogram 1) or secondary antibody only (histograms 2 and 3).
(D) OCLN expression levels are stable after SRFBP1 silencing (siRNA 394). Immunoblot analysis of OCLN (red) and SRFBP1 (green) after siRNA mediated silencing for 48 hr is shown. Actin served as loading control. Data are representative of at least three independent experiments. See also Figure S4.
Next, we sought to exclude that SRFBP1 acts as a chaperone for CD81, CLDN1, OCLN, and SR-BI. In adipocytes, SRFBP1 is required for expression of the insulin-responsive glucose transporter type 4 (GLUT4) and for shuttling of GLUT4 to the plasma membrane (Lisinski et al., 2006). In contrast, we did not observe SRFBP1 colocalization with GLUT4 in hepatoma cells (Figures 4A and 4B). In line with these observations, SRFBP1 silencing in hepatoma cells did not alter surface levels of CD81, CLDN1, or SR-BI, whereas silencing of CD81 and CLDN1 reduced surface expression of the respective protein (Figures 4C and S4A–S4C). Due to a lack of antibodies targeting the OCLN ectodomain, we addressed OCLN expression after knockdown by immunofluorescence and immunoblot. Total protein levels and plasma membrane expression of OCLN were similar in Huh-7.5 with or without SRFBP1 knockdown (Figure 4D; data not shown). Similarly, transcript levels of the HCV entry cofactors EGFR and Niemann-Pick C1-like protein 1 (NPC1L1) (Sainz et al., 2012) remained unaffected by SRFBP1 silencing (Figure S4D). Taken together, our data exclude that SRFBP1 acts as chaperone or transcriptional regulator of one of the previously characterized HCV entry factors.
SRFBP1 Partially Localizes to CD81-Positive Endosomes and Is Further Recruited to CD81 upon HCV Glycoprotein Exposure
To better visualize where and when during the HCV entry process SRFBP1 comes into play, we performed additional colocalization studies. We detected SRFBP1 in vesicular structures in the cell periphery, in perinuclear regions, and in heterochromatin regions of the nucleus. The nuclear localization of SRFBP1 is consistent with its transcription factor function described in cardiomyocytes (Zhang et al., 2004). Perinuclear SRFBP1 signals colocalized with the cytosolic trans-GOLGI marker p230 (Pearson’s correlation coefficient 0.4). The punctate, vesicular pattern of SRFBP1 in the cell periphery weakly stained positive for the endosomal markers EEA and LAMP1, as well as for F and G actin (Figures 5A and 5B). To achieve more-sensitive visualization of endosomal compartments, we transfected EGFP-Rab fusion proteins (Nielsen et al., 1999) into Huh-7.5 cells. We found overlapping signals for SRFBP1 and CD81 at Rab5-positive early and late endosomes (Figures 5C and 5D). In favor of a role for SRFBP1 early during HCV infection, SRFBP1 did not reside in HCV replication or assembly compartments as shown by co-staining with the p body marker DDX6, the stress granule marker ataxin-1, and the lipid droplet dye oil red O (Figures S5A and S5C).
Figure 5.
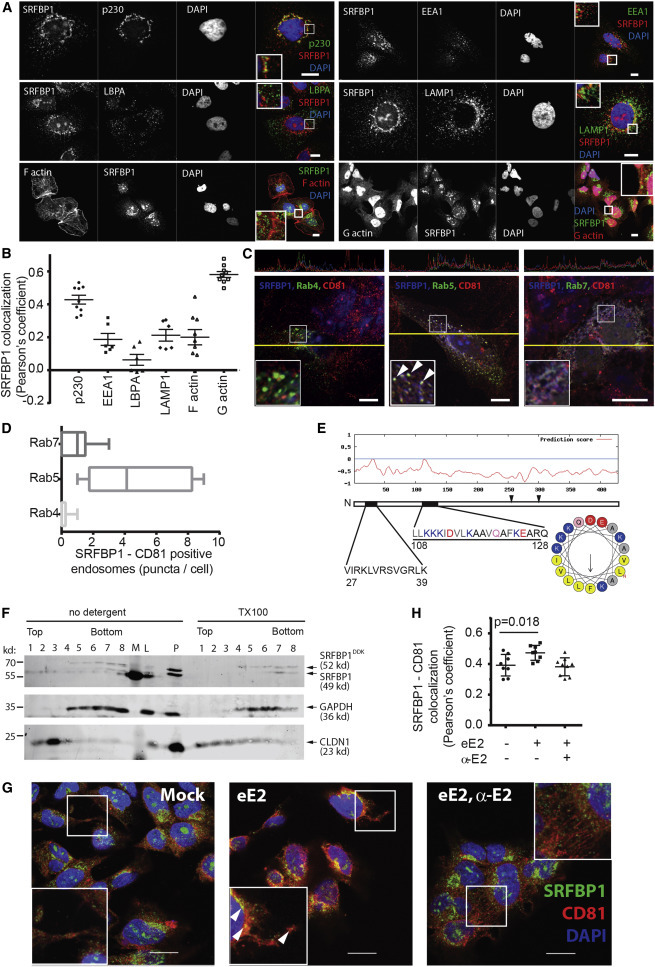
A Pool of SRFBP1 Localizes to CD81 on Endosomes and Is Recruited to CD81 upon HCV Glycoprotein Exposure
(A) SRFBP1 localizes to the trans-GOLGI, endosomes, and actin. Huh-7.5 cells were stained for SRFBP1; the trans-GOLGI marker p230; and the endosomal markers EEA1, LBPA, and LAMP1 as described in Figure 4A. F and G actin were stained with Alexa-conjugated phalloidin and DNase I, respectively.
(B) Pearson’s correlation coefficient for SRFBP1 and indicated cellular proteins calculated by intensity correlation analysis. Each symbol represents an individual frame; horizontal lines indicate the mean ± SEM.
(C) SRFBP1 localizes to CD81 on endosomes. Huh-7.5 cells were transfected with expression plasmids for EGFP-Rab4, -Rab5, and -Rab7 and stained for SRFBP1 and CD81. Colocalization of SRFBP1, CD81, and Rab proteins across a section (yellow line) is depicted in the upper panels. Arrowheads indicate colocalization.
(D) SRFBP1 and CD81 colocalize at early endosomes. Quantification of SRFBP1, CD81, and Rab triple-positive puncta is shown. Box and whisker plot showing median, minimum, and maximum values from six independent frames.
(E) Bioinformatics prediction of two weak amphipathic helices for SRFBP1 (black bars) with the second helix (aas 108–128) showing a small hydrophobic face of five amino acids (FLLVI). The hydrophobic face is highlighted in light gray in the primary sequence and in yellow in the helix model. Two cysteine residues (aa 254 and aa 300), which could serve as palmitoylation sites, are indicated by arrowheads.
(F) Membrane flotation assay suggests membrane association of SRFBP1. Huh-7.5 cells were transduced with mycDDK-tagged SRFBP1, 48 hr later lysed in hypotonic buffer, and analyzed by Nycodenz gradient ultracentrifugation followed by immunoblot analysis against SRFBP1, GAPDH, and CLDN1. TX-100-treated lysates served as solubilization control. L, precleared lysate; M, marker; P, pellet after lysate preclearing. One out of three independent experiments is shown.
(G) Exposure to soluble HCV glycoprotein (eE2) increases SRFBP1-CD81 colocalization in Huh-7.5 cells. Cells were incubated with eE2, with eE2 and an E2 blocking antibody (α-E2), or with PBS (mock) for 15 min; fixed; and stained for SRFBP1 and CD81 as described in Figure 4A. Arrowheads indicate colocalization.
(H) Pearson’s correlation coefficient for SRFBP1 and CD81 calculated by intensity correlation analysis. Each symbol represents an individual frame; horizontal lines indicate the mean ± SEM; p value is indicated.
Representative images; inserts show magnification; scale bars 10 μm (A and C) and 20 μm (G). See also Figure S5.
Next, we asked whether SRFBP1 could interact with intracellular membranes. SRFBP1 has no transmembrane domains, but we predicted two weak amphipathic helices at the N terminus of the protein (http://npsa-pbil.ibcp.fr/cgi-bin/npsa_automat.pl?page=/NPSA/npsa_amphipaseek.html; Sapay et al., 2006). In-depth analysis revealed that the putative helix at aa108 has a five-amino-acid hydrophobic side (FLLVI) flanked by lysine residues (http://heliquest.ipmc.cnrs.fr; Gautier et al., 2008). Moreover, two cysteine residues could serve as palmitoylation sites (Figure 5E). To confirm that a fraction of SRFBP1 is membrane associated, we performed membrane-flotation assays. In accordance with our prediction, a subfraction of endogenous SRFBP1 resided in the upper, membrane-associated gradient fractions (Figure 5F). As expected, CLDN1 and GAPDH resided in the upper and lower fractions, respectively. After solubilization of membranes using Triton X-100, we found endogenous SRFB1 exclusively in the soluble lower fractions and the CLDN1 control shifted partially to these fractions. Interestingly, a mycDDK-tagged overexpression construct of SRFBP1 resided in cytosolic fractions, suggesting that the soluble tag impaired membrane association of SRFBP1. Our analysis indicates that a fraction of SRFBP1 can associate with cellular membranes presumably through a weak amphipathic helix.
To test for relocalization of SRFBP1 during the HCV entry process, we exposed Huh-7.5 cells to purified ectodomain of the HCV E2 glycoprotein (eE2). This resulted in an increased colocalization of SRFBP1 and CD81 (Pearson’s correlation coefficient 0.5), which could be reverted when coadministering an E2 blocking antibody (Figures 5G and 5H). Notably, we observed a similar recruitment of SRFBP1 to CD81-positive compartments in primary hepatocytes after eE2 exposure (Figures S5B and S5D) and in hepatoma cells after HCV exposure (Figure S5E). In summary, our data suggest that SRFBP1 partially resides at intracellular membranes in human hepatoma cells and that HCV glycoprotein exposure promotes colocalization of SRFBP1 and CD81.
SRFBP1, a Pan-genotypic and HCV-Specific Host Entry Factor
Next, we tested whether SRFBP1 also supports infection with other enveloped viruses. In Huh-7.5 cells, SRFBP1 silencing neither reduced infectivity of vesicular stomatitis virus (VSV) or of human coronavirus 229E (Figures 6A and 6B), hinting that SRFBP1 is an HCV-specific host factor.
Figure 6.
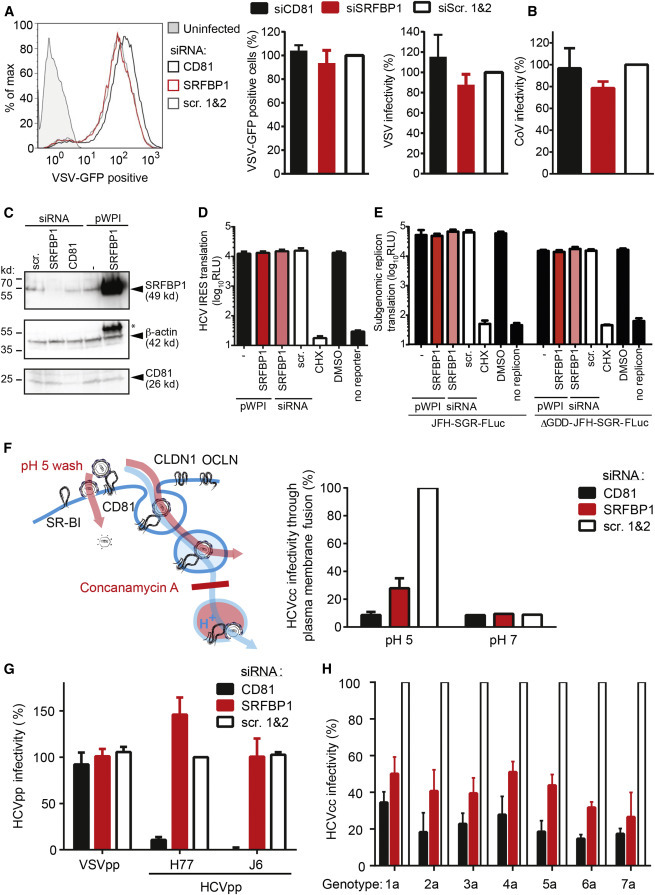
SRFBP1 Is a Pan-genotypic and HCV-Specific Host Entry Factor
(A) SRFBP1 is dispensable for VSV infection. SRFBP1-silenced Huh-7.5 cells were infected with VSV∗MQ (MOI 0.1) and analyzed for GFP expression by flow cytometry 20 hpi. Histogram is representative of biological triplicates (left panel). Quantification of VSV∗MQ infectivity 20 hpi in SRFBP1-silenced cells is determined as percentage of GFP-positive cells (middle panel) or by normalization of the mean fluorescence intensity (MFI) of VSV-infected SRFBP1- or CD81-silenced cells to MFI of scrambled siRNA-transfected cells (right panel).
(B) SRFBP1 is dispensable for coronavirus infection. SRFBP1-silenced cells were infected with HCoV229E-luc (MOI 0.1) and RLuc activity in cell lysates measured 24 hpi. Infectivity relative to a scrambled siRNA control is shown.
(C) Immunoblot analysis of SRFBP1 and CD81 48 hp siRNA transfection. Huh-7.5 cells were transfected with siRNA or transduced with the indicated pWPI expression construct as in (A)–(H), 48 hr later lysed, and analyzed by immunoblot. Actin served as loading control. ∗, residual SRFBP1 signal.
(D) Bicistronic translational reporter assay with HCV IRES-driven RLuc and cap-dependent FLuc (see also Figures S6A and S6B). SRFBP1 silencing and overexpression was performed as in (C), and 48 hr later, cells were transfected with translational reporter RNA. Eight hours after reporter transfection, luciferase activity in lysates was monitored.
(E) Early replication reporter assay using a subgenomic HCV genome expressing FLuc. SRFBP1 silencing and overexpression was performed as in (C), and 48 hr later, JFH-SGR-FLuc RNA was transfected into cells; cells lysed after 8 hr; and luciferase activity monitored. A polymerase mutant JFH-SGR-FLuc replicon (ΔGDD) was used to assess translation of HCV genomes independent of de novo replication. See also Figures S6C and S6D for additional controls.
(F) SRFBP1 is required in a plasma membrane fusion assay of HCV infection. Huh-7.5 cells silenced for SRFBP1 were pretreated with concanamycin A (5 nM; 1 hr) to block vacuolar type H+-ATPases, incubated with HCV (JcR2A) for 2 hr at 4°C in the presence of concanamycin A, shifted to 37°C, and washed with a pH 5 or pH 7 buffer for 5 min. After incubation with concanamycin A for 4 hr, medium was changed and endosomal acidification independent infectivity measured at 48 hpi. See also Figures S6E and S6F for additional controls.
(G) Lentiviral pseudotypes infect Huh-7.5 cells independently of SRFBP1. Cells in which SRFBP1 had been silenced (48 hr) were infected with HIV-1 pseudotypes encoding FLuc and displaying glycoproteins from HCV genotype 1 (H77), HCV genotype 2 (J6), VSV, or no glycoprotein. At 72 hpi, cells were lysed and FLuc activity measured. Infectivity was calculated by subtraction of background read for glycoprotein-free particles and relative to VSVG particles.
(H) Silencing of SRFBP1 reduces infectivity of chimeric HCV viruses with glycoproteins from all seven genotypes. Huh-7.5 FLuc cells were subjected to siRNA-mediated silencing followed by infection with intergenotypic HCV chimeras (MOI 0.1) expressing RLuc. Forty-eight hours post-infection, infectivity was determined by RLuc activity measurement. Cells treated with CD81 targeting or scrambled siRNAs served as controls. SRFBP1-targeting siRNA 394 was used in all experiments. Data from three to five biological replicates are displayed as mean + SD. See also Figure S6.
To further elucidate how SRFBP1 aids early HCV infection, we experimentally addressed HCV translation, replication complex formation, and membrane fusion. Using a bicistronic HCV IRES-driven translation reporter, we excluded a role for SRFBP1 in viral genome translation (Figures 6C, 6D, S6A, and S6B). Replication complex formation of subgenomic and full-length HCV replicons similarly remained unaltered upon SRFBP1 silencing (Figures 6E, S6C, and S6D). In line with this observation, SRFBP1 did not colocalize with the HCV protein NS5A (Pearson’s coefficient < 0.2; Figures S6E and S6F). We next induced HCV fusion at the plasma membrane by a low-pH wash and by concomitantly blocking endosomal acidification. In this assay, HCV enters cells by fusion at the plasma membrane or at the limiting endosomal membrane before acidification (Figure 6F). As expected, CD81 silencing led to a 5-fold reduced HCV infectivity, as CD81 interactions prime the HCV glycoproteins for membrane fusion. Notably, silencing of SRFBP1 reduced HCV infection by 3-fold, indicating that SRFBP1 functions in cell entry steps other than the acidification of endosomes. In confirmation of our assay setup, human coronavirus fusion at the plasma membrane was independent of pH, whereas VSV required a low-pH wash. Both coronavirus and VSV infectivity remained unaffected by SRFBP1 or CD81 silencing in this bypass assay (Figures S6G and S6H). Taken together, our data show that SRFBP1 is aiding an early step in HCV infection even when viral envelope fusion is artificially induced at the plasma membrane.
To pinpoint the requirements of the virus particle toward SRFBP1 usage, we tested whether lentiviral pseudoparticles decorated with HCV glycoproteins depend on SRFBP1. Interestingly, lentiviral pseudotypes for HCV genotype 1 (H77) and 2 (J6; Hsu et al., 2003) transduced SRFBP1-silenced cells efficiently. CD81 silencing reduced HCV pseudoparticle entry 10-fold, whereas none of the tested conditions affected control pseudoparticles carrying VSV glycoproteins (Figure 6G). Thus, SRFBP1 does not affect receptor binding but instead supports an infection step not reflected by HCV pseudoparticles. Furthermore, SRFBP1 did not influence lentiviral transduction. Collectively, our results show that SRFBP1 is dispensable for lentiviral pseudotype, VSV, and coronavirus infection but required to render cells fully susceptible to HCV.
Lastly, we addressed whether, in addition to cell culture HCV of genotype 2a, other clinically relevant HCV genotypes require SRFBP1 for efficient penetration. SRFBP1 interference reduced infectivity of chimeric viruses displaying the glycoproteins of either one of the seven HCV genotypes to a similar degree (Figure 6H). Thus, through quantitative interaction proteomics, we could identify six putative HCV host factors and, in particular, SRFBP1 as a pan-genotypic entry factor for HCV.
Discussion
Our results demonstrate that quantitative interaction proteomics combined with RNAi provides a valuable approach to study host-pathogen interactions. Quantitative MS provides direct information on protein-protein interactions and interaction strength upon perturbation of a cellular system. Here, we developed a SILAC co-IP strategy to identify host factors, which transiently interact with the HCV receptor CD81. The data set allowed generation of a weighted virus entry network and identification of cellular processes during entry.
Among the identified 26 transient interaction partners of CD81, four (DSG1, CSTA, DSP, and CDSN) are integral parts of cellular junctions, in particular desmosomes. These proteins dissociated from CD81 during virus entry, suggesting that the virus receptor complex leaves desmosomal membrane compartments during uptake. The second enriched cluster of Ca2+-binding proteins (CALML5, S100A7, and S100A10) could contribute to the reported Ca2+-dependent ER stress and deregulation of Ca2+ homeostasis induced by HCV (Benali-Furet et al., 2005, Piccoli et al., 2009). Here, we focused on CD81 interaction partners, which support HCV infection, and found that these comprise at least 23% of the 26 interactors. The putative HCV host factors are SRFBP1, S100A10, BANF1, PARP1, MYH9, and SPTBN1 (Table S6). The latter two guide cytoskeleton movement at the plasma membrane, which is in line with the reported membrane “surfing” of HCV (Brazzoli et al., 2008). S100A10 is a component of the annexin 2 heterotetramer and regulates membrane organization and endocytosis. We conclude that quantitative proteomics can identify functional virus-host cell interactions.
Several HCV host factors described in this study play a role in the life cycle of other enveloped viruses. MYH9 (also known as myosin IIA) regulates Kaposi’s sarcoma-associated herpesvirus macropinocytosis (Valiya Veettil et al., 2010). S100A10 is phosphorylated by the HCV host factor EGFR (Lupberger et al., 2011) and promotes uptake of papillomaviruses upon EGFR-driven phosphorylation (Dziduszko and Ozbun, 2013, Woodham et al., 2012). The protein is also thought to be a cofactor for entry of HIV-1, cytomegalovirus, and respiratory syncytial virus (Ma et al., 2004, Malhotra et al., 2003, Raynor et al., 1999). BANF1 is involved in nuclear DNA repair and HIV-1 genome integration (Chen and Engelman, 1998) and senses vaccinia virus genomes in the cytoplasm (Ibrahim et al., 2011). PARP1 has poly(ADP-ribosyl)ation activity and is sequestered to the cytoplasm during HIV-1 and Sindbis virus infection (Muthumani et al., 2006, Park and Griffin, 2009). Hence, we identified six putative HCV host factors, which are linked to known HCV entry machineries, e.g., clathrin-mediated endocytosis and EGFR signaling, or have a reported role in infection or sensing of other enveloped viruses.
SRFBP1 emerged as prime candidate for follow up and proof of principle analysis as the protein had the strongest RNAi phenotype in our screen. The protein promotes plasma membrane expression of GLUT4 in adipocytes (Lisinski et al., 2006), associates with actin, and has transcription factor activity in cardiomyocytes (Zhang et al., 2004, Zhang et al., 2014). In this study, we identify SRFBP1 as transient binding partner of the HCV receptor CD81 in human hepatoma cells. Our results demonstrate that SRFBP1 is required for productive uptake of HCV without affecting expression or membrane localization of known HCV entry factors. We further show that primary hepatocytes express up to 6-fold higher transcript levels of SRFBP1 than hepatoma cells, in which SRFBP1 expression seems to limit HCV infectivity. Thus, SRFBP1 might play a critical role in HCV infection in patients.
Collectively, our data suggest that SRFBP1 is a bona fide entry factor for HCV (Figure 7 ). During the HCV entry pathway, SRFBP1 supports a step independent of receptor binding, clathrin-mediated endocytosis, and endosomal acidification. The latter three steps are reliably mimicked by lentiviral HCV pseudotypes, which enter cells independent of SRFBP1. The observed discrepancy between cell culture HCV and HCV pseudoparticles confirms other studies suggesting that HCV pseudoparticles cannot fully mimic the entry pathway of HCV (Sainz et al., 2012). This can be attributed either to the lower avidity of HCV pseudoparticles, which display a lower density of glycoproteins, to the lack of serum lipoprotein association of HCV pseudoparticles, or to the different nucleocapsid.
Figure 7.
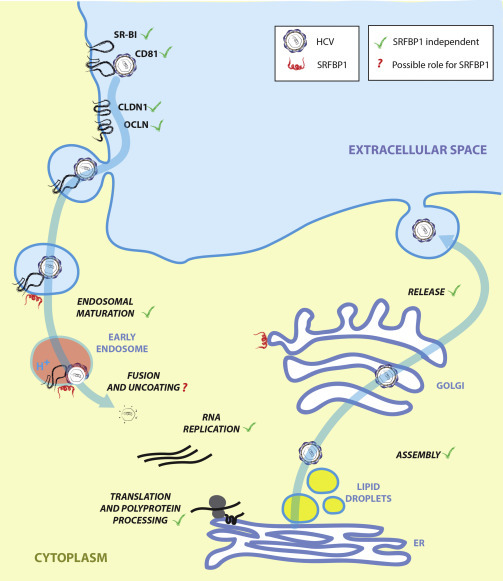
HCV Life Cycle and Possible Role for SRFBP1 during HCV Entry
Our data point toward a role for SRFBP1 in the last step of entry; i.e., nucleocapsid uncoating.
Here, we propose two possible modes of action of SRFBP1 during HCV entry. First, SRFBP1 could be involved in actin retrograde transport of HCV particles as observed by Coller et al. (2009). Such actin cortex remodeling is not induced by HCV pseudoparticles (Harris et al., 2013). Second, SRFBP1 might assist HCV uncoating or nucleocapsid transport, both of which are not reflected by the pseudoparticle system. Generally, SRFBP1 could be the missing link between HCV receptor binding and actin-dependent movement during HCV invasion. Indeed, we observed an interaction of SRFBP1 with G actin. Moreover, EGFR, Arp2/3, Rho GEFs, and Rho GTPases are reported upstream activators of the SRFBP1 protein family and at the same time support HCV entry (Brazzoli et al., 2008, Lupberger et al., 2011, Zona et al., 2013). Future studies including high-resolution imaging will be necessary to experimentally validate either model.
The endogenous function of cytoplasmic SRFBP1 is currently elusive. In rat cardiomyocytes, SRFBP1 localizes to actin fibers close to their attachment site to the cell cortex and SRFBP1 overexpression leads to actin depolymerization (Zhang et al., 2014). This hints that SRFBP1 belongs to the class of myocardin-related transcription factor (MRTF) cofactors (Olson and Nordheim, 2010), which regulate actin polymerization by cycling between a G-actin-bound cytoplasmic state and a nuclear state. Concordantly, we found SRFBP1 in the nucleus and at cytoplasmic G actin in human hepatoma cells. Like other MRTF cofactors, SRFBP1 could regulate actin dynamics downstream of plasma membrane receptor signaling. In line with this, CD81 engagement by antibodies was recently reported to promote actin-dependent hepatoma spread (Brimacombe et al., 2014). Thus, HCV might exploit endogenous mechanisms of physical force generation to traffic during its entry.
Taken together, we established a combination of high-resolution MS, computational proteomics, and RNAi to elucidate receptor complex rearrangements during HCV entry. We believe that quantitative interaction proteomics is an attractive strategy to identify host factors of infectious agents. A particular strength lies in the unbiased identification of yet uncharacterized proteins as we demonstrate for SRFBP1, a protein with previously unknown function in hepatocytes. Moreover, interaction proteomics allows the identification of host factors, which are ubiquitously expressed, and are thus not accessible by genetic complementation screens. Similarly, host factors with essential endogenous function are poorly suited for stable knockdown or knockout screens and can readily be found by quantitative proteomics. Thus, the minimal system perturbation during the above-described workflow is a clear benefit over classical genetic screening methods. On the other hand, functional information on the identified host factors is limited to the interaction with a given virus receptor. A functional follow-up screen, as we describe here, is therefore critical to evaluate the protein interaction data. Consequently, interaction proteomics is a complementary method in the thus far genomics-oriented toolbox for systems virology (Law et al., 2013). As for other systems biology methods, interaction proteomics is not error free. Although detection limits for MS fingerprinting have increased tremendously in the past years (Cox and Mann, 2012), false negatives might arise depending on the affinity enrichment method used. False positive interactions could obviously arise after cell lysis and disruption of cellular compartments, and thus, careful follow-up analysis is critical for the here-described methodology. Clearly, receptor interaction proteomics will not only allow the follow-up search for entry factors but can also reveal host factors involved in innate immune recognition or cellular perturbations triggered by the virus like, e.g., apoptosis (Figure S7). Given that co-IP proteomics reveals interconnectivity of pathogen receptors and host cofactors, we envision that the technique can spur the development of peptidomimetics or small molecules for therapeutic intervention of pathogen invasion (de Chassey et al., 2012).
Experimental Procedures
SILAC Labeling and HCV Inoculation
Huh-7 cells were passaged eight times in light or heavy label media, i.e., Arg- and Lys-free DMEM (PAA Laboratories) supplemented with 2 mM L-glutamine, 1 mM pyruvate, 10% dialyzed FBS, 0.375% sodium bicarbonate, 48 μg/ml Arg (Arg-0, Arg-6, and Arg-10, respectively), and 73 μg/ml Lys (Lys-0 and Lys-8, respectively; Cambridge Isotope Labs). Confluent P150 cultures were incubated with J6/JFH clone 2 (MOI 10 after 1:5 dilution in serum-free label media) for 15 min at 37°C or treated in a similar manner with virus-free conditioned cell culture media processed in the same way as the HCV preparation (mock electroporation). One-step immunoprecipitations of membrane proteins were performed as detailed in the Supplemental Experimental Procedures. Experiments were conducted in four replicates (two heavy- and two light-labeled cultures per experimental condition).
MS Bioinformatics, Hierarchical Clustering, and Hit Scoring
Mass spectra were acquired and analyzed as described in the Supplemental Experimental Procedures. For each HCV and mock-treated SILAC pair with a given label combination, normalized ratios were calculated from the individual heavy and light peptide intensities as described in Cox and Mann (2008).
Transient protein interactions (i.e., regulated CD81 binding during HCV entry) were defined by differing significantly from the main distribution of steady-state interactors using significance B with a false discovery rate of 5% as described in Cox and Mann (2008). By this analysis, 55 proteins from a total of 778 proteins were identified, which grouped into 29 associating and 26 dissociating proteins (Table S1). Proteins were required to be significantly regulated in at least one experiment to be included for further analysis. To quantify proteins, which were only detected in one experimental condition (HCV or mock), we analyzed the total ion intensities for heavy and light peptides separately. We detected five proteins exclusively in HCV samples, four of which were liver expressed. No proteins were exclusively detected in mock samples.
Proteins significantly regulated upon HCV incubation were ranked by their fold change and only those with >2.1-fold change considered for further analysis. Forty-seven interaction partners of CD81 fulfilled this criterion. For functional follow up of CD81-binding proteins, we additionally filtered for expression in human liver and for subcellular localization. Proteins with lacking liver expression or strict ribosomal and nuclear localization were excluded from downstream analysis, resulting in 26 selected CD81-binding partners to be tested for their role in HCV infection.
The protein interactions from this publication have been submitted to the IMEx (http://www.imexconsortium.org) consortium through IntAct and assigned the identifier IM-24070 (Orchard et al., 2014).
RNAi Screen for HCV Host Factors and Bioinformatic Analysis
Huh-7.5 cells stably expressing Firefly luciferase (Huh-7.5 FLuc) were transfected with pools of three siRNAs against the 26 selected transient CD81 interaction partners (Ambion Silencer Select) and infected with the Renilla luciferase reporter virus JcR2A (MOI 0.1) as detailed in the Supplemental Experimental Procedures. The screen was performed nine times on Huh-7.5 FLuc cells of three independent passages. Normalization and statistical analysis was performed on a set of 34 targets total in R using the Bioconductor package RNAither (Rieber et al., 2009).
Cell lines, viruses, used reagents, and detailed methods are described in the Supplemental Experimental Procedures. Human samples were handled under oversight of the ethics committee of the Hannover Medical School.
Author Contributions
G.G. designed experiments, performed experiments, evaluated and interpreted the data, and wrote the manuscript. F.M. designed and performed the mass spec analysis and assisted with writing the manuscript. J.B. and K.W. performed experiments. P.M.P. performed fusion assays. T.F.B. generated and provided monoclonal anti-SR-BI antibody. A.G.K. and J.M. provided purified eE2. F.W.V. isolated and provided primary hepatocytes. L.K. performed RNAi statistics. M.M. advised on quantitative mass spec analysis. C.M.R. and T.P. advised on experimental approaches, evaluated and interpreted the data, and assisted with writing the manuscript.
Acknowledgments
We thank Takaji Wakita for the JFH1 isolate; Jens Bukh for intergenotypic chimeric HCV strains; Marino Zerial for Rab-GFP constructs; Dirk Lindemann for the pc.Z.VSV-G plasmid; Gert Zimmer for VSV∗MQ; Volker Thiel for coronavirus 229E-Luc; Didier Trono for pWPI and pCMVdeltaR8.74 constructs; and Annie Cahour, Jean Dubuisson, and Yves Rouille for the pIRF construct. This work was supported by fellowships from the Human Frontier Science Program (LT-000048-2009), the German Academy of Science Leopoldina (LPDS 2009-9), the German Research Foundation (DFG; GE 2145/3-1), and the German Liver Foundation (S163/10073/2011; to G.G.). Grants from the Deutsche Forschungsgemeinschaft (SFB 900; project A6) and the Helmholtz Association SO-024 funded T.P. The work was also supported by US Public Health Service NIH grant R01 AI072613 (to C.M.R.), the Greenberg Medical Research Institute, and the Starr Foundation. C.M.R. has equity in Apath, LLC, which holds commercial licenses for the Huh-7.5 cell line and HCV cell culture system.
Published: July 23, 2015
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and six tables and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2015.06.063.
Contributor Information
Gisa Gerold, Email: gisa.gerold@twincore.de.
Thomas Pietschmann, Email: thomas.pietschmann@twincore.de.
Supplemental Information
SILAC ratios, heavy and light intensities for each label direction, significance analysis, and protein annotations are listed.
References
- Benali-Furet N.L., Chami M., Houel L., De Giorgi F., Vernejoul F., Lagorce D., Buscail L., Bartenschlager R., Ichas F., Rizzuto R., Paterlini-Bréchot P. Hepatitis C virus core triggers apoptosis in liver cells by inducing ER stress and ER calcium depletion. Oncogene. 2005;24:4921–4933. doi: 10.1038/sj.onc.1208673. [DOI] [PubMed] [Google Scholar]
- Berger K.L., Cooper J.D., Heaton N.S., Yoon R., Oakland T.E., Jordan T.X., Mateu G., Grakoui A., Randall G. Roles for endocytic trafficking and phosphatidylinositol 4-kinase III alpha in hepatitis C virus replication. Proc. Natl. Acad. Sci. USA. 2009;106:7577–7582. doi: 10.1073/pnas.0902693106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchard E., Belouzard S., Goueslain L., Wakita T., Dubuisson J., Wychowski C., Rouillé Y. Hepatitis C virus entry depends on clathrin-mediated endocytosis. J. Virol. 2006;80:6964–6972. doi: 10.1128/JVI.00024-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brazzoli M., Bianchi A., Filippini S., Weiner A., Zhu Q., Pizza M., Crotta S. CD81 is a central regulator of cellular events required for hepatitis C virus infection of human hepatocytes. J. Virol. 2008;82:8316–8329. doi: 10.1128/JVI.00665-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brimacombe C.L., Wilson G.K., Hübscher S.G., McKeating J.A., Farquhar M.J. A role for CD81 and hepatitis C virus in hepatoma mobility. Viruses. 2014;6:1454–1472. doi: 10.3390/v6031454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang K.S., Jiang J., Cai Z., Luo G. Human apolipoprotein e is required for infectivity and production of hepatitis C virus in cell culture. J. Virol. 2007;81:13783–13793. doi: 10.1128/JVI.01091-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charrin S., Le Naour F., Labas V., Billard M., Le Caer J.P., Emile J.F., Petit M.A., Boucheix C., Rubinstein E. EWI-2 is a new component of the tetraspanin web in hepatocytes and lymphoid cells. Biochem. J. 2003;373:409–421. doi: 10.1042/BJ20030343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H., Engelman A. The barrier-to-autointegration protein is a host factor for HIV type 1 integration. Proc. Natl. Acad. Sci. USA. 1998;95:15270–15274. doi: 10.1073/pnas.95.26.15270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coller K.E., Berger K.L., Heaton N.S., Cooper J.D., Yoon R., Randall G. RNA interference and single particle tracking analysis of hepatitis C virus endocytosis. PLoS Pathog. 2009;5:e1000702. doi: 10.1371/journal.ppat.1000702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J., Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- Cox J., Mann M. 1D and 2D annotation enrichment: a statistical method integrating quantitative proteomics with complementary high-throughput data. BMC Bioinformatics. 2012;13(16):S12. doi: 10.1186/1471-2105-13-S16-S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Chassey B., Meyniel-Schicklin L., Aublin-Gex A., André P., Lotteau V. New horizons for antiviral drug discovery from virus-host protein interaction networks. Curr Opin Virol. 2012;2:606–613. doi: 10.1016/j.coviro.2012.09.001. [DOI] [PubMed] [Google Scholar]
- Diao J., Pantua H., Ngu H., Komuves L., Diehl L., Schaefer G., Kapadia S.B. Hepatitis C virus induces epidermal growth factor receptor activation via CD81 binding for viral internalization and entry. J. Virol. 2012;86:10935–10949. doi: 10.1128/JVI.00750-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dziduszko A., Ozbun M.A. Annexin A2 and S100A10 regulate human papillomavirus type 16 entry and intracellular trafficking in human keratinocytes. J. Virol. 2013;87:7502–7515. doi: 10.1128/JVI.00519-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans M.J., von Hahn T., Tscherne D.M., Syder A.J., Panis M., Wölk B., Hatziioannou T., McKeating J.A., Bieniasz P.D., Rice C.M. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature. 2007;446:801–805. doi: 10.1038/nature05654. [DOI] [PubMed] [Google Scholar]
- Farquhar M.J., Hu K., Harris H.J., Davis C., Brimacombe C.L., Fletcher S.J., Baumert T.F., Rappoport J.Z., Balfe P., McKeating J.A. Hepatitis C virus induces CD81 and claudin-1 endocytosis. J. Virol. 2012;86:4305–4316. doi: 10.1128/JVI.06996-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier R., Douguet D., Antonny B., Drin G. HELIQUEST: a web server to screen sequences with specific α-helical properties. Bioinformatics. 2008;24:2101–2102. doi: 10.1093/bioinformatics/btn392. [DOI] [PubMed] [Google Scholar]
- Gerold G., Pietschmann T. The HCV life cycle: in vitro tissue culture systems and therapeutic targets. Dig. Dis. 2014;32:525–537. doi: 10.1159/000360830. [DOI] [PubMed] [Google Scholar]
- Gerold G., Rice C.M. Locking out hepatitis C. Nat. Med. 2011;17:542–544. doi: 10.1038/nm0511-542. [DOI] [PubMed] [Google Scholar]
- Gravitz L. Introduction: a smouldering public-health crisis. Nature. 2011;474:S2–S4. doi: 10.1038/474S2a. [DOI] [PubMed] [Google Scholar]
- Harris H.J., Clerte C., Farquhar M.J., Goodall M., Hu K., Rassam P., Dosset P., Wilson G.K., Balfe P., Ijzendoorn S.C. Hepatoma polarization limits CD81 and hepatitis C virus dynamics. Cell. Microbiol. 2013;15:430–445. doi: 10.1111/cmi.12047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu M., Zhang J., Flint M., Logvinoff C., Cheng-Mayer C., Rice C.M., McKeating J.A. Hepatitis C virus glycoproteins mediate pH-dependent cell entry of pseudotyped retroviral particles. Proc. Natl. Acad. Sci. USA. 2003;100:7271–7276. doi: 10.1073/pnas.0832180100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim N., Wicklund A., Wiebe M.S. Molecular characterization of the host defense activity of the barrier to autointegration factor against vaccinia virus. J. Virol. 2011;85:11588–11600. doi: 10.1128/JVI.00641-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keilhauer E.C., Hein M.Y., Mann M. Accurate protein complex retrieval by affinity enrichment mass spectrometry (AE-MS) rather than affinity purification mass spectrometry (AP-MS) Mol. Cell. Proteomics. 2015;14:120–135. doi: 10.1074/mcp.M114.041012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavanchy D. Evolving epidemiology of hepatitis C virus. Clin. Microbiol. Infect. 2011;17:107–115. doi: 10.1111/j.1469-0691.2010.03432.x. [DOI] [PubMed] [Google Scholar]
- Law G.L., Korth M.J., Benecke A.G., Katze M.G. Systems virology: host-directed approaches to viral pathogenesis and drug targeting. Nat. Rev. Microbiol. 2013;11:455–466. doi: 10.1038/nrmicro3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisinski I., Matsumoto H., Yver D.R., Schürmann A., Cushman S.W., Al-Hasani H. Identification and characterization of p49/STRAP as a novel GLUT4-binding protein. Biochem. Biophys. Res. Commun. 2006;344:1179–1185. doi: 10.1016/j.bbrc.2006.04.017. [DOI] [PubMed] [Google Scholar]
- Lupberger J., Zeisel M.B., Xiao F., Thumann C., Fofana I., Zona L., Davis C., Mee C.J., Turek M., Gorke S. EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat. Med. 2011;17:589–595. doi: 10.1038/nm.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma G., Greenwell-Wild T., Lei K., Jin W., Swisher J., Hardegen N., Wild C.T., Wahl S.M. Secretory leukocyte protease inhibitor binds to annexin II, a cofactor for macrophage HIV-1 infection. J. Exp. Med. 2004;200:1337–1346. doi: 10.1084/jem.20041115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra R., Ward M., Bright H., Priest R., Foster M.R., Hurle M., Blair E., Bird M. Isolation and characterisation of potential respiratory syncytial virus receptor(s) on epithelial cells. Microbes Infect. 2003;5:123–133. doi: 10.1016/s1286-4579(02)00079-5. [DOI] [PubMed] [Google Scholar]
- Meissner F., Mann M. Quantitative shotgun proteomics: considerations for a high-quality workflow in immunology. Nat. Immunol. 2014;15:112–117. doi: 10.1038/ni.2781. [DOI] [PubMed] [Google Scholar]
- Mercer J., Schelhaas M., Helenius A. Virus entry by endocytosis. Annu. Rev. Biochem. 2010;79:803–833. doi: 10.1146/annurev-biochem-060208-104626. [DOI] [PubMed] [Google Scholar]
- Montpellier C., Tews B.A., Poitrimole J., Rocha-Perugini V., D’Arienzo V., Potel J., Zhang X.A., Rubinstein E., Dubuisson J., Cocquerel L. Interacting regions of CD81 and two of its partners, EWI-2 and EWI-2wint, and their effect on hepatitis C virus infection. J. Biol. Chem. 2011;286:13954–13965. doi: 10.1074/jbc.M111.220103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthumani K., Choo A.Y., Zong W.X., Madesh M., Hwang D.S., Premkumar A., Thieu K.P., Emmanuel J., Kumar S., Thompson C.B., Weiner D.B. The HIV-1 Vpr and glucocorticoid receptor complex is a gain-of-function interaction that prevents the nuclear localization of PARP-1. Nat. Cell Biol. 2006;8:170–179. doi: 10.1038/ncb1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen E., Severin F., Backer J.M., Hyman A.A., Zerial M. Rab5 regulates motility of early endosomes on microtubules. Nat. Cell Biol. 1999;1:376–382. doi: 10.1038/14075. [DOI] [PubMed] [Google Scholar]
- Olson E.N., Nordheim A. Linking actin dynamics and gene transcription to drive cellular motile functions. Nat. Rev. Mol. Cell Biol. 2010;11:353–365. doi: 10.1038/nrm2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong S.E., Blagoev B., Kratchmarova I., Kristensen D.B., Steen H., Pandey A., Mann M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics. 2002;1:376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- Orchard S., Ammari M., Aranda B., Breuza L., Briganti L., Broackes-Carter F., Campbell N.H., Chavali G., Chen C., del-Toro N. The MIntAct project—IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 2014;42:D358–D363. doi: 10.1093/nar/gkt1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park E., Griffin D.E. Interaction of Sindbis virus non-structural protein 3 with poly(ADP-ribose) polymerase 1 in neuronal cells. J. Gen. Virol. 2009;90:2073–2080. doi: 10.1099/vir.0.012682-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccoli C., Quarato G., Ripoli M., D’Aprile A., Scrima R., Cela O., Boffoli D., Moradpour D., Capitanio N. HCV infection induces mitochondrial bioenergetic unbalance: causes and effects. Biochim. Biophys. Acta. 2009;1787:539–546. doi: 10.1016/j.bbabio.2008.11.008. [DOI] [PubMed] [Google Scholar]
- Pileri P., Uematsu Y., Campagnoli S., Galli G., Falugi F., Petracca R., Weiner A.J., Houghton M., Rosa D., Grandi G., Abrignani S. Binding of hepatitis C virus to CD81. Science. 1998;282:938–941. doi: 10.1126/science.282.5390.938. [DOI] [PubMed] [Google Scholar]
- Ploss A., Evans M.J., Gaysinskaya V.A., Panis M., You H., de Jong Y.P., Rice C.M. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature. 2009;457:882–886. doi: 10.1038/nature07684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajesh S., Sridhar P., Tews B.A., Fénéant L., Cocquerel L., Ward D.G., Berditchevski F., Overduin M. Structural basis of ligand interactions of the large extracellular domain of tetraspanin CD81. J. Virol. 2012;86:9606–9616. doi: 10.1128/JVI.00559-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raynor C.M., Wright J.F., Waisman D.M., Pryzdial E.L. Annexin II enhances cytomegalovirus binding and fusion to phospholipid membranes. Biochemistry. 1999;38:5089–5095. doi: 10.1021/bi982095b. [DOI] [PubMed] [Google Scholar]
- Reiss S., Rebhan I., Backes P., Romero-Brey I., Erfle H., Matula P., Kaderali L., Poenisch M., Blankenburg H., Hiet M.S. Recruitment and activation of a lipid kinase by hepatitis C virus NS5A is essential for integrity of the membranous replication compartment. Cell Host Microbe. 2011;9:32–45. doi: 10.1016/j.chom.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieber N., Knapp B., Eils R., Kaderali L. RNAither, an automated pipeline for the statistical analysis of high-throughput RNAi screens. Bioinformatics. 2009;25:678–679. doi: 10.1093/bioinformatics/btp014. [DOI] [PubMed] [Google Scholar]
- Sainz B., Jr., Barretto N., Martin D.N., Hiraga N., Imamura M., Hussain S., Marsh K.A., Yu X., Chayama K., Alrefai W.A., Uprichard S.L. Identification of the Niemann-Pick C1-like 1 cholesterol absorption receptor as a new hepatitis C virus entry factor. Nat. Med. 2012;18:281–285. doi: 10.1038/nm.2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapay N., Guermeur Y., Deléage G. Prediction of amphipathic in-plane membrane anchors in monotopic proteins using a SVM classifier. BMC Bioinformatics. 2006;7:255. doi: 10.1186/1471-2105-7-255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarselli E., Ansuini H., Cerino R., Roccasecca R.M., Acali S., Filocamo G., Traboni C., Nicosia A., Cortese R., Vitelli A. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J. 2002;21:5017–5025. doi: 10.1093/emboj/cdf529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeff L.B. Natural history of chronic hepatitis C. Hepatology. 2002;36(1):S35–S46. doi: 10.1053/jhep.2002.36806. [DOI] [PubMed] [Google Scholar]
- Sharma N.R., Mateu G., Dreux M., Grakoui A., Cosset F.L., Melikyan G.B. Hepatitis C virus is primed by CD81 protein for low pH-dependent fusion. J. Biol. Chem. 2011;286:30361–30376. doi: 10.1074/jbc.M111.263350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valiya Veettil M., Sadagopan S., Kerur N., Chakraborty S., Chandran B. Interaction of c-Cbl with myosin IIA regulates Bleb associated macropinocytosis of Kaposi’s sarcoma-associated herpesvirus. PLoS Pathog. 2010;6:e1001238. doi: 10.1371/journal.ppat.1001238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodham A.W., Da Silva D.M., Skeate J.G., Raff A.B., Ambroso M.R., Brand H.E., Isas J.M., Langen R., Kast W.M. The S100A10 subunit of the annexin A2 heterotetramer facilitates L2-mediated human papillomavirus infection. PLoS ONE. 2012;7:e43519. doi: 10.1371/journal.pone.0043519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamauchi Y., Helenius A. Virus entry at a glance. J. Cell Sci. 2013;126:1289–1295. doi: 10.1242/jcs.119685. [DOI] [PubMed] [Google Scholar]
- Zhang X., Azhar G., Zhong Y., Wei J.Y. Identification of a novel serum response factor cofactor in cardiac gene regulation. J. Biol. Chem. 2004;279:55626–55632. doi: 10.1074/jbc.M405945200. [DOI] [PubMed] [Google Scholar]
- Zhang X., Azhar G., Rogers S.C., Foster S.R., Luo S., Wei J.Y. Overexpression of p49/STRAP alters cellular cytoskeletal structure and gross anatomy in mice. BMC Cell Biol. 2014;15:32. doi: 10.1186/1471-2121-15-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zona L., Lupberger J., Sidahmed-Adrar N., Thumann C., Harris H.J., Barnes A., Florentin J., Tawar R.G., Xiao F., Turek M. HRas signal transduction promotes hepatitis C virus cell entry by triggering assembly of the host tetraspanin receptor complex. Cell Host Microbe. 2013;13:302–313. doi: 10.1016/j.chom.2013.02.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SILAC ratios, heavy and light intensities for each label direction, significance analysis, and protein annotations are listed.