

Abstract
Clinical classification of early dementia and mild cognitive impairment is imprecise. We reported previously that molecular imaging classification of early dementia and mild cognitive impairment with dual amyloid and dopamine terminal positron emission tomography differs significantly from expert clinical classification. We now report pathologic diagnoses in a substantial subset of our previously imaged subjects. Among 36 subjects coming to autopsy, imaging classifications and pathologic diagnosis were concordant in 33 cases (κ=0.85). This approach enhanced specificity of Alzheimer disease diagnosis. The strong concordance of imaging based classifications and pathologic diagnoses suggests that this imaging approach will be useful in establishing more accurate and convenient classification biomarkers for dementia research.
Introduction
Clinical classification of dementias, particularly in early disease phases, is imprecise.1 There are 3 common neurodegenerative dementias; Alzheimer disease (AD), Lewy body dementia (LBD), and Frontotemporal dementias (FTDs). Even expert clinical characterization does relatively poorly in differentiating AD from FTDs.2 Clinical criteria for LBD possess good specificity but relatively poor sensitivity.3 Mild Cognitive Impairment (MCI), a common precursor of dementia, is a heterogeneous category associated with all major neurodegenerative pathologies and vascular etiologies. Imprecise classification of MCI and early dementia subjects is an obstacle to clinical research as heterogeneous study populations dilute power to detect effects of trial interventions or associations with potential biomarkers. The emergence of PET ligands identifying specific pathologic features of neurodegenerative disorders raises the possibility of minimally invasive characterization of MCI and early dementia subjects. We previously reported results of combined amyloid ([11C]PIB) and dopamine terminal ([11C]DTBZ) PET imaging in 102 MCI and early dementia subjects, demonstrating only moderate concordance (κ=0.41) between imaging based and expert clinical consensus classifications.4,5 Our prior results raise the possibility that this imaging based approach to classification more faithfully reflects underlying pathologies than clinical characterization. We now report neuropathologic follow-up of a substantial fraction of our study subjects.
Methods
Study participants were individuals with MCI or relatively mild dementia (MMSE > 17) as described previously and enrolled in our prior imaging study from 2005 to 2009.4,5 The purpose of the prior study was to compare amyloid-dopamine terminal PET based classification of early cognitive impairment subjects with expert clinical classification. Subjects with primary features of cognitive impairment were recruited from the University of Michigan Cognitive Disorders Clinic. Patients with primary neurological presentations involving non-cognitive domains (ataxia, parkinsonism, etc.) were excluded. Inclusion-exclusion criteria are described in prior publications; patients with possible vascular dementia (modified Hachinski score >4 or meeting NINDS-AIREN criteria or large infarcts on structural imaging) were excluded.4 Clinical classifications were established via expert consensus conference based on clinical and neuropsychological data accumulated at the time of visits for imaging as described previously.4 Enrollees agreed to follow-up autopsy. To date, 41 study participants died and autopsies were completed on 36. Autopsy results of one subject were reported previously.6 All autopsies were performed at the University of Michigan Health System. Neuropathology was assessed by standard methods and using standard diagnostic criteria.7–11 The examining neuropathologists (AF-H, APL, SC-P) were blind to results of imaging studies. Thal scores of amyloid plaque density were compiled for 3 neocortical regions; mid-frontal (Brodmann’s areas [BA] 10 & 46), parietal (BA 7 & 39), and primary occipital (BA 17). Plaques were identified with Aβ immunohistochemistry (6F/3D; Leica Biosystems, 1:50). Thal scoring was available for all subjects. Regional [11C]PiB binding was quantified as distribution volume ratios (DVRs) with the cerebellar gray matter as the reference region. Image based classifications established in our prior studies were used for categorical comparison with pathologic diagnoses.4,5 Standardized DVR image datasets were classified qualitatively by an expert interpreter (KAF) familiar with the normal and pathologic distributions of these tracers and blind to all clinical and routine structural imaging data as described previously.4 In our prior study, use of parametric regional DVR thresholds for classification did not alter results.4 The unweighted Cohen’s Kappa statistic was used to estimate concordance between imaging based and pathologic classifications. Spearman’s rank-order correlation was used to compare amyloid burden assessed pathologically with the [11C]PiB DVR estimates of regional amyloid burden. Sixteen subjects also underwent [18F]fluoro-deoxyglucose PET (FDG-PET) at the same time they underwent DTBZ-PiB imaging. These studies were interpreted by the same expert interpreter (KAF) blind to the clinical histories, and structural and PET imaging data.
Results
There was overall excellent concordance of imaging based classifications with neuropathologic diagnoses; κ=0.85 (95% CI = 0.69–1.0; Table 1; details of pathologic results in Supplementary Table). Regional amyloid DVRs correlated well with neuropathologic scoring of amyloid burden in the selected neocortical regions (Figure). For mid-frontal cortex, rho = 0.72; for parietal cortex, rho = 0.79; for primary occipital cortex, rho = 0.64 (all p<0.05). There were 3 cases with discordant imaging-pathologic classifications. One subject had a clinical diagnosis and imaging classification of LBD but a pathologic diagnosis of AD. α–Synuclein immunoreactive Lewy bodies were found in midbrain neurons in this subject, suggesting the presence of mixed AD-LBD pathology. The second discordant subject had marked frontal and temporal atrophy secondary to multiple small infarctions and imaging classification as FTD. The final discordant case was classified by imaging as LBD but remarkable only for the presence of TDP-43-immunoreactive neurites in the frontal cortex and hippocampal formation. This was an unusual case in that there was marked unilateral striatal loss of [11C]DTBZ binding. Three cases were assessed pathologically as meeting criteria for both AD and LBD. These individuals had imaging classifications as LBD with amyloid deposition and are assessed as concordant classifications. There was excellent concordance between imaging assessments of increased amyloid burden and pathologic results; all subjects found to have moderate to high amyloid plaque burden at autopsy were classified as amyloid positive in imaging classifications.
Table 1.
Expert consensus clinical classifications, clinical data, imaging classifications, and neuropathologic diagnoses.
| Clinical Consensus Classification | Age at Initial Evaluation | Age at Imaging Classification | Age at Death | Disease Duration | PiB-DTBZ Imaging Classification | Pathologic Diagnosis | Comment |
|---|---|---|---|---|---|---|---|
| LBD | 75 | 76 | 80 | 5 | LBD | AD | Midbrain Lewy Bodies |
| AD | 59 | 60 | 66 | 7 | AD | AD | |
| AD | 66 | 67 | 70 | 4 | AD | AD | |
| LBD | 68 | 70 | 71 | 3 | LBD | LBD | |
| LBD | 74 | 74 | 78 | 4 | LBD | LBD | Subacute Right Basal Ganglia Infarct |
| AD | 76 | 78 | 80 | 4 | AD | AD | Subacute Left Parahippocampal Infarct |
| mdMCI | 78 | 78 | 83 | 5 | AD | AD | |
| FTD | 71 | 72 | 75 | 4 | FTD | Multiple Infarcts | Frontal and Temporal Atrophy, Multiple Small Infarctions, Mild Loss of Nigral Neurons |
| AD | 64 | 65 | 67 | 3 | AD | AD | |
| AD | 69 | 71 | 73 | 4 | AD | AD | |
| naMCI | 66 | 67 | 71 | 5 | FTD | FTD | C9ORF73 Mutation |
| LBD | 65 | 65 | 67 | 2 | LBD | LBD + AD | |
| LBD | 75 | 79 | 86 | 11 | LBD | LBD | |
| FTD | 71 | 72 | 76 | 5 | AD | AD | |
| LBD | 74 | 74 | 79 | 5 | LBD | LBD | |
| LBD | 65 | 66 | 73 | 8 | LBD | LBD | |
| mdMCI | 65 | 65 | 66 | 1 | AD | AD | |
| FTD | 78 | 80 | 82 | 4 | LBD | TDP-43+ | TDP-43 Immunoreactive Neurites in Frontal Cortex and Hippocampal Formation |
| aMCI | 77 | 80 | 83 | 5 | AD | AD | |
| FTD | 62 | 62 | 63 | 1 | AD | AD | Sparse TDP-43 Immunoreactive Inclusions in Frontal Cortex and Dentate Gyrus |
| AD | 76 | 76 | 80 | 4 | AD | AD + LBD | |
| AD | 64 | 65 | 69 | 5 | AD | AD | |
| naMCI | 62 | 64 | 67 | 5 | LBD | AD + LBD | |
| FTD | 67 | 67 | 71 | 4 | FTD | PSP | |
| AD | 76 | 81 | 82 | 6 | AD | AD | |
| AD | 79 | 79 | 82 | 3 | AD | AD | |
| AD | 85 | 85 | 89 | 4 | AD | AD | |
| naMCI | 78 | 79 | 82 | 4 | FTD | PSP | Remote Cerebellar Infarct |
| AD | 66 | 67 | 72 | 6 | LBD | LBD | |
| aMCI | 79 | 80 | 84 | 5 | AD | AD | |
| CBS | 62 | 66 | 71 | 9 | AD | AD | |
| AD | 55 | 55 | 59 | 4 | AD | AD | |
| AD | 65 | 68 | 72 | 7 | LBD | LBD + AD | |
| LBD | 76 | 77 | 83 | 7 | AD | AD | |
| AD | 56 | 59 | 63 | 7 | AD | AD | |
| AD | 85 | 88 | 92 | 7 | AD | AD | Remote Midbrain Infarct |
| Mean = 70.3 yrs | Mean = 71.6 yrs | Mean = 75.2 yrs | Mean = 4.9 yrs |
Disease duration = interval from age at initial evaluation to age at death. AD = Alzheimer disease, LBD = Lewy body dementia, FTD = Frontotemporal dementia, MCI = Mild Cognitive Impairment, aMCI = Amnestic MCI, naMCI = Non-Amnestic MCI, mdMCI = Multidomain MCI, CBS = Corticobasal syndrome, PSP = Progressive Supranuclear Palsy, PiB-DTBZ = [11C]Pittsburgh B & [11C]dihydrotetrabenazine Positron Emission Tomography, TDP-43 = Transactive response DNA binding protein 43 kDa.
Figure.
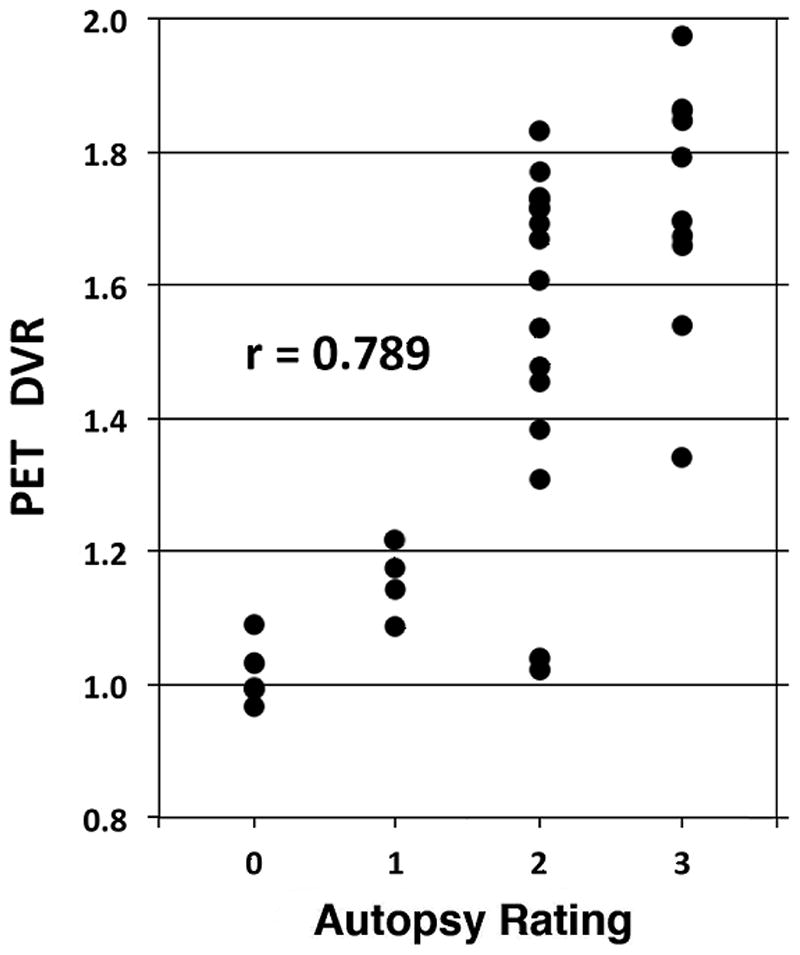
Scatter plot of parietal cortex Thal scores (Autopsy Rating) versus [11C]PiB DVRs. r – Spearman’s rho.
We performed a more limited comparison of combined amyloid and dopamine terminal imaging classifications, neuropathologic diagnoses, and FDG-PET classifications. Approximately thirty percent of the FDG-PET classifications differed from final neuropathologic diagnoses (Table 2). There were 3 cases where the FDG-PET classification was FTD with pathologic diagnoses of AD and 2 cases where the FDG-PET classification was AD with pathologic diagnoses of LDB. In all cases with discrepant FDG-PET classifications and neuropathologic diagnoses, combined amyloid and dopamine terminal PET imaging correctly identified the pathologic diagnosis (Table 2).
Table 2.
Expert consensus clinical classifications, PiB-DTBZ imaging classifications, neuropathologic diagnoses, and FDG-PET classifications.
| Clinical Consensus Classification | Age at Initial Evaluation | Age at Imaging Classification | Age at Death | Disease Duration | PiB-DTBZ Imaging Classification | Pathologic Diagnosis | FDG-PET Classification |
|---|---|---|---|---|---|---|---|
| AD | 59 | 60 | 66 | 7 | AD | AD | AD |
| LBD | 68 | 70 | 71 | 3 | LBD | LBD | AD |
| AD | 64 | 65 | 67 | 3 | AD | AD | FTD |
| AD | 69 | 71 | 73 | 4 | AD | AD | AD |
| LBD | 75 | 79 | 86 | 11 | LBD | LBD | LBD |
| LBD | 74 | 74 | 79 | 5 | LBD | LBD | LBD |
| LBD | 65 | 66 | 73 | 8 | LBD | LBD | AD/LBD |
| mdMCI | 65 | 65 | 66 | 1 | AD | AD | AD |
| aMCI | 77 | 80 | 83 | 5 | AD | AD | FTD |
| AD | 76 | 81 | 82 | 6 | AD | AD | AD |
| AD | 85 | 85 | 89 | 4 | AD | AD | FTD |
| AD | 66 | 67 | 72 | 6 | LBD | LBD | AD |
| aMCI | 79 | 80 | 84 | 5 | AD | AD | AD |
| AD | 65 | 68 | 72 | 7 | LBD | LBD + AD | LBD |
| LBD | 76 | 77 | 83 | 7 | AD | AD | AD |
| AD | 56 | 59 | 63 | 7 | AD | AD | AD |
Disease duration = interval from age at initial evaluation to age at death. AD = Alzheimer disease, LBD = Lewy body dementia, FTD = Frontotemporal dementia, MCI = Mild Cognitive Impairment, aMCI = Amnestic MCI, mdMCI = Multidomain MCI, PiB-DTBZ = [11C]Pittsburgh B & [11C]dihydrotetrabenazine Positron Emission Tomography, FDG-PET = [18F]fluorodeoxyglucose-Positron Emission Tomography. FDG-PET classification criteria: AD – temporoparietal and posterior cingulate hypometabolism; LDB - temporoparietal and posterior cingulate hypometabolism plus occipital hypometabolism; FTD – frontal, anterior temporal, and anterior cingulate hypometabolism.
Discussion
Our results indicate that classifications based on combined amyloid and dopamine terminal PET imaging correlate well with neuropathologic diagnostic classifications. Of 36 subjects studied, there were 3 cases (8.3%) where imaging based and pathologic classifications differed. In our prior studies, in contrast, ~35% of participants had discordant expert clinical consensus and imaging diagnostic classifications.4,5 One discordant case was classified as LBD on the basis of significantly reduced striatal [11C]DTBZ binding. Though not meeting pathologic criteria for LBD, this subject had nigral Lewy bodies, suggesting mixed pathology.
Our results are consistent with other recent studies. In trials of anti-amyloid therapy of clinically classified early AD subjects where participants underwent amyloid imaging, ~15% of enrolled subjects had negative amyloid imaging, excluding AD.12,13,14 These results likely underestimate diagnostic misclassifications as ~50% of LBD cases exhibit significant amyloid burden, likely leading to misclassification of some LBD subjects as AD.15 A clinicopathologic study using the National Alzheimer’s Coordinating Center (NACC) dataset found that ~15% of classified clinically AD subjects failed pathologic criteria for AD.16 In our prior studies, a major cause of discrepant clinical and imaging classifications were subjects classified clinically as FTD but with positive amyloid imaging results suggesting AD.4 Our results comparing amyloid/dopamine terminal imaging and clinical diagnostic classifications are similar to those reported by Beach et al. using the NACC dataset to compare clinical and neuropathologic diagnoses.17
Our limited evaluation of FDG-PET classifications suggests that this method is less precise than combined amyloid and dopamine terminal PET imaging. These types of FDG-PET misclassifications are well described in prior literature. Disproportionate frontal amyloid deposition may give rise to frontal predominant hypometabolism.18 The canonical pattern of cerebral metabolic deficits in LDB is the pattern of temporoparietal and frontal deficits found in AD plus occipital hypometabolism, but the distinguishing occipital metabolic deficits are absent is a significant fraction of patients.19
Amyloid imaging is accepted as a useful biomarker of fibrillar amyloid deposition. The high prevalence of amyloidopathy in LBD, however, indicates that increased amyloid burden is not a unique AD biomarker. Combining amyloid imaging with a dopamine terminal marker enhances accuracy. Our study, and this approach in general, have some limitations. Our number of autopsied subjects is relatively small. Because amyloid imaging is relatively sensitive for detecting AD, and dopamine terminal imaging allows exclusion of LBD, this method is arguably best at improving identification of AD. Two of the imaging misclassifications assessed subjects as LBD were found at autopsy to have another diagnosis. This result and the existence of nigrostriatal pathology in FTD and related syndromes indicates that dopamine terminal imaging possesses good sensitivity but less specificity for detection of LBD. In amyloid negative individuals, substantial nigrostriatal terminal loss could indicate either LBD or FTD, as some FTD patients develop parkinsonism with nigrostriatal degeneration, particularly those with MAPT or GRN mutations, decreasing the specificity of this approach of accurate classification of LBD.20 Identification of FTD is most problematic as our classification of FTD is based on negative imaging results – the absence of pathologic amyloid or dopamine terminal imaging changes. This may be misleading as there are multiple potential causes of cognitive impairment without amyloid or nigrostriatal pathology, for example, our case where the neuropathologic evaluation revealed multiple small infarcts instead of neurodegeneration. Positive imaging markers for tau deposition and other FTD associated pathologies would be useful additions to this imaging approach.21
Our results point to another problem secondary to use of the trinary classification scheme. This conventional approach is artificial in that mixed pathologies are common, though the presence of other pathologies does not confound amyloid ligand binding.22 Mixed pathologies are seen in our dataset with 3 subjects with both AD and LBD, and the discordant subject who met neuropathologic criteria for AD and had midbrain Lewy bodies. Identification of individuals with both AD and LBD is particularly difficult, both with our dual tracer approach and with conventional clinical classifications. This is an area where addition of a tau tracer may be valuable.
Our study has significant advantages. Our subjects were enrolled during relatively early disease stages, either MCI status or relatively mild dementia (MMSE >17). Prior studies correlating amyloid imaging results with neuropathology enrolled individuals with advanced dementia.23–26 Our study population is more typical of clinical research studies and offers reassurance that prior imaging-pathologic correlation studies of amyloid imaging are relevant to earlier phases of neurodegeneration. Our study design may underestimate the utility of this multi-tracer approach. Imaging classifications were made in the absence of clinical and structural (CT or MRI) imaging information. Conversely, our clinical classifications were made without the PET results. Use of this multi-tracer PET approach in conjunction with clinical and structural imaging data would likely enhance accuracy of classifications. These methods may provide additional useful data. We showed previously that regional cerebral blood flow data derived from [11C]DTBZ PET closely mimics the patterns of regional cerebral metabolism visualized with [18F]FDG-PET imaging.27,28 Analysis of [11C]DTBZ-based regional perfusion data would add a functional dimension to analysis and might further enhance classifications. Individuals, for example, with abnormal striatal [11C]DTBZ ligand binding could have either LBD or FTD. These syndromes exhibit distinctive regional cerebral metabolic-perfusion deficit patterns which could be helpful in classifying LBD and FTD subjects more accurately.
We do not suggest that this approach to classification would be broadly useful in clinical practice. It is more plausible that this approach, or approaches using similar tracers or incorporating additional tracers such as a tau ligand, will be useful in clinical research. These methods may allow purer subject samples or better subject stratification, particularly for selection of AD subjects, for clinical research studies. This approach may be useful in establishing the utility of more accessible classification biomarkers. Rather than waiting years for autopsy results, this dual tracer approach or similar methods could be used as surrogates to validate more convenient classification biomarkers.
Supplementary Material
Acknowledgments
We thank the families of our research participants for their commitment to research. We thank the 2 anonymous reviewers for helpful criticisms. Supported by AG08671, NS15655, R56 NS082941, P50 NS091856, and a gift from an anonymous donor.
Footnotes
Disclosures:
Drs. Albin, Burke, Koeppe, Giordani, Fisher-Hubbard, Camelo-Piragua, Lieberman, Frey, and Ms. Shanmugasundaram do not have any financial relationships that could be perceived as a conflict of interest.
Author Contributions:
Study Concept and Design: RLA, KAF, JFB
Data Acquisition and Analysis: RLA, KAF, RAK, JFB, BG, AF-H, APL, SC-P, KS.
Drafting Text and Figures: RLA, KAF, RAK.
References
- 1.Knopman DS, DeKosky ST, Cummings JL, et al. Practice parameter: diagnosis of dementia (an evidence-based review). Report of the quality standards subcommittee of the American Academy of Neurology. Neurology. 2001;56:1143–1153. doi: 10.1212/wnl.56.9.1143. [DOI] [PubMed] [Google Scholar]
- 2.Mendez MF, Shapira JS, McMurtray A, Licht E, Miller BL. Accuracy of the clinical evaluation for frontotemporal dementia. Arch Neurol. 2007;64:830–835. doi: 10.1001/archneur.64.6.830. [DOI] [PubMed] [Google Scholar]
- 3.McKeith IG, Ballard CG, Perry RH, et al. Prospective validation of consensus criteria for the diagnosis of dementia with Lewy bodies. Neurology. 2000;54:1050–1058. doi: 10.1212/wnl.54.5.1050. [DOI] [PubMed] [Google Scholar]
- 4.Burke JF, Albin RL, Koeppe RA, Giordani B, Kilbourn MR, Gilman S, Frey KA. Assessment of mild dementia with amyloid and dopamine terminal positron emission tomography. Brain. 2011;134:1647–1657. doi: 10.1093/brain/awr089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Albin RL, Burke JF, Koeppe RA, Giordani B, Gilman S, Frey KA. Assessing mild cognitive impairment with amyloid and dopamine terminal molecular imaging. J Nucl Med. 2013;54:887–893. doi: 10.2967/jnumed.112.112599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meisler MH, Grant AE, Jones JM, Lenk GM, He F, Todd PK, Kamali M, Albin RL, Lieberman AP, Langenecker SA, McInnis MG. C9ORF72 expansion in a family with bipolar disorder. Bipolar Disord. 2013;15:326–332. doi: 10.1111/bdi.12063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hyman BT, Phelps CH, Beach TG, et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement. 2012;58:1–13. doi: 10.1016/j.jalz.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mirra SS, Heyman AT, Mckeel D, et al. The consortium to establish a registry for Alzheimer disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology. 1991;41:479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 9.Thal DR, Rub U, Braak H. Phases of Abeta deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58:1791–1800. doi: 10.1212/wnl.58.12.1791. [DOI] [PubMed] [Google Scholar]
- 10.Mackenzie IRA, Neumann M, Bigio EH, Cairns NJ, Alafuzoff I, et al. Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol. 2010;119:1–4. doi: 10.1007/s00401-009-0612-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McKeith IG. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the Consortium on DLB International Workshop. J Alzheimers Dis. 2006;9(3 Suppl):417–23. doi: 10.3233/jad-2006-9s347. [DOI] [PubMed] [Google Scholar]
- 12.Rinne JO, Brooks DJ, Rossor MN, et al. 11C-PiB PET assessment of change in fibrillar amyloid-beta load in patients with Alzheimer’s disease treated with bapineuzumab: a phase 2, double-blind, placebo-controlled, ascending-dose study. Lancet Neurol. 2010;9:363–372. doi: 10.1016/S1474-4422(10)70043-0. [DOI] [PubMed] [Google Scholar]
- 13.Salloway S, Sperling R, Fox NC, et al. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N Engl J Med. 2014;370:322–333. doi: 10.1056/NEJMoa1304839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Doody RS, Thomas RG, Farlow M, et al. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N Engl J Med. 2014;370:311–32. doi: 10.1056/NEJMoa1312889. [DOI] [PubMed] [Google Scholar]
- 15.Petrou M, Dwamena B, Foerster B, et al. Amyloid deposition in Parkinson disease and cognitive impairment: a systematic review. Mov Dis. 2015 doi: 10.1002/mds.26191. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Serrano-Pozo A, Qian J, Monsell SE, et al. Mild to moderate Alzheimer dementia with insufficient neuropathological changes. Ann Neurol. 2014;75:597–601. doi: 10.1002/ana.24125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beach TG, Monsell SE, Phillips LE, Kukull W. Accuracy of the clinical diagnosis of Alzheimer disease at National Institute on Aging Alzheimer Disease Centers, 2005–2010. J Neuropathol Exp Neurol. 2012;71:266–273. doi: 10.1097/NEN.0b013e31824b211b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rabinovici GD, Jagust WJ, Furst AJ, et al. Abeta amyloid and glucose metabolism in three variants of primary progressive aphasia. Ann Neurol. 2008;64:388–401. doi: 10.1002/ana.21451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kantarci K, Lowe VJ, Boeve BF, et al. Multimodality imaging characteristics of dementia with Lewy bodies. Neurobiol Aging. 2012;33:2091–105. doi: 10.1016/j.neurobiolaging.2011.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karageorgiou E, Miller BL. Frontotemporal lobar dementia: a clinical approach. Sem Neurol. 2014;34:189–201. doi: 10.1055/s-0034-1381735. [DOI] [PubMed] [Google Scholar]
- 21.Maruyama M, Shimada H, Suhara T, et al. Imaging of tau pathology in a tauopathy mouse model and in Alzheimer patients compared to normal controls. Neuron. 2013;79:1094–1108. doi: 10.1016/j.neuron.2013.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dugger BN, Clark CM, Serrano G, et al. Neuropathologic heterogeneity does not impair florbetapir-positron emission tomography postmortem correlates. J Neuropathol Exp Neurol. 2014;73:72–80. doi: 10.1097/NEN.0000000000000028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clark CM, Pontecorvo MJ, Beach TG, et al. Cerebral PET with florbetapir compared with neuropathology at autopsy for detection of neuritic amyloid-β plaques: a prospective cohort study. Lancet Neurol. 2012;11:669–678. doi: 10.1016/S1474-4422(12)70142-4. [DOI] [PubMed] [Google Scholar]
- 24.Clark CM, Schneider JA, Bedell BJ, et al. Use of florbetapir-PET for imaging beta-amyloid pathology. JAMA. 2011;305:275–283. doi: 10.1001/jama.2010.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Curtis C, Gamez JE, Singh U, et al. Phase 3 trial of flutemetamol labeled with radioactive fluorine 18 imaging and neuritic plaque density. JAMA Neurol. 2015;72:287–294. doi: 10.1001/jamaneurol.2014.4144. [DOI] [PubMed] [Google Scholar]
- 26.Murray ME, Lowe VJ, Graff-Radford NR, et al. Clinicopathologic and 11C-pittsburgh compound B implications of Thal amyloid phase across the Alzheimer’s disease spectrum. Brain. 2015;138:1370–1381. doi: 10.1093/brain/awv050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Albin RL, Koeppe RA, Burke JF, Giordani B, Kilbourn MR, Gilman S, Frey KA. Comparing fludeoxyglucose F18-PET assessment of regional cerebral glucose metabolism and [11C]dihydrotetrabenazine-PET in evaluation of early dementia and mild cognitive impairment. Arch Neurol. 2010;67:440–446. doi: 10.1001/archneurol.2010.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koeppe RA, Gilman S, Joshi A, Liu S, Little R, Junck L, Heumann M, Frey KA, Albin RL. 11C-DTBZ and 18F-FDG PET measures in differentiating dementias. J Nucl Med. 2005;46:936–944. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.