

Abstract
Objective:
To determine the safety and tolerability of mexiletine in a phase II double-blind randomized controlled trial of sporadic amyotrophic lateral sclerosis (SALS).
Methods:
Sixty participants with SALS from 10 centers were randomized 1:1:1 to placebo, mexiletine 300 mg/d, or mexiletine 900 mg/d and followed for 12 weeks. The primary endpoints were safety and tolerability. Secondary endpoints were pharmacokinetic study from plasma and CSF, ALS Functional Rating Scale–Revised (ALSFRS-R) score, slow vital capacity (SVC), and muscle cramp frequency and severity.
Results:
The only serious adverse event among active arm participants was one episode of imbalance. Thirty-two percent of participants receiving 900 mg of mexiletine discontinued study drug vs 5% on placebo (p = 0.026). Pharmacokinetic study demonstrated a peak plasma concentration 2 hours postdose and strong correlation between plasma and CSF (p < 0.001). Rates of decline of ALSFRS-R and SVC did not differ from placebo. Analysis of all randomized patients demonstrated significant reductions of muscle cramp frequency (300 mg: rate = 31% of placebo, p = 0.047; 900 mg: 16% of placebo, p = 0.002) and cramp intensity (300 mg: mean = 45% of placebo, p = 0.08; 900 mg: 25% of placebo, p = 0.005).
Conclusions:
Mexiletine was safe at both doses and well-tolerated at 300 mg/d but adverse effects at 900 mg/d led to a high rate of discontinuation. Mexiletine treatment resulted in large dose-dependent reductions in muscle cramp frequency and severity. No effect on rate of progression was detected, but clinically important differences could not be excluded in this small and short-duration study.
Classification of evidence:
This study provides Class I evidence that mexiletine is safe when given daily to patients with amyotrophic lateral sclerosis at 300 and 900 mg and well-tolerated at the lower dose.
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disorder characterized by death of cortical, brainstem, and spinal motor neurons. Riluzole, the sole Food and Drug Administration–approved therapy for ALS, has only a limited effect on slowing progression.1 Recent in vivo2–5 and in vitro6–12 studies support a role for hyperexcitability of both peripheral motor nerve axons and cortical motor neurons as a possible pathogenic mechanism of ALS. Additionally, cortical motor neuronal hyperexcitability has been demonstrated in patients with both familial and sporadic ALS (SALS) using transcranial magnetic stimulation.8–12 Threshold tracking studies have demonstrated hyperexcitability of peripheral motor nerve axons, which could also play a role in the generation of fasciculations and muscle cramps, problematic symptoms in ALS that lack effective treatment.6,7 Muscle cramps occur in most patients with ALS during the course of the disease13 and frequently cause distress.14
Mexiletine, a cardiac antiarrhythmic agent and use-dependent sodium channel blocker, has recently been demonstrated to inhibit neuronal hyperexcitability and prevent cell death in a motor neuron cell line exposed to cultured media from astrocytes engineered to express the human SOD1 gene15,16 and could potentially benefit patients with ALS. Additionally, mexiletine reduced muscle cramping in a small open-label study in Machado-Joseph disease, a hereditary spinocerebellar neurodegenerative disorder that also leads to degeneration of anterior horn cells.17 Based on these studies, we performed a randomized placebo-controlled multicenter study to determine the safety and tolerability of mexiletine in patients with SALS. Effects of the medication on markers of disease progression and muscle cramps were also determined.
METHODS
Standard protocol approvals, registrations, and patient consents.
The study was performed at 10 member sites of the Northeast ALS clinical trials consortium from July 2013 to September 2014. The institutional review boards of each participating site approved the study protocol and all amendments. Written informed consent was obtained at the screening visit. The study is registered on clinicaltrials.gov (NCT01849770).
General study design.
A randomized, double-blind, placebo-controlled, 3-arm trial design was used. Sixty participants with SALS were randomized in a 1:1:1 fashion to receive for 12 weeks 900 mg/d of mexiletine, 300 mg/d of mexiletine, or placebo divided into 2 dosages daily. We decided upon 2 dosages of the medication, 300 and 900 mg/d, suggested by the known safety profile of this drug for the treatment of cardiac arrhythmias. The study drug was manufactured by the University of Iowa research pharmacy and distributed by the Clinical Material Services Unit of the University of Rochester. Participants were assigned to treatment groups using a computer-generated permuted-block randomization schedule, stratified by site, by a coordination center (Massachusetts General Hospital). At the time of treatment assignment, participants were also randomly assigned by a computer-generated permuted-block randomization schedule, stratified by treatment assignment, to 1 of 5 lumbar puncture times for collection of CSF at their week 6 visit. All investigators, coordinators, and evaluators and coordination center staff were blind to treatment assignment. An independent data safety monitoring board (DSMB) reviewed safety data every 4 months throughout the study. The DSMB had the ability to recommend modifying or terminating the study if it believed participant safety was compromised.
Participant selection criteria.
Eligible participants were 18 years old or older with possible, laboratory-supported probable, probable, or definite ALS as defined by revised El Escorial criteria,18 slow vital capacity (SVC) of ≥50% of predicted value, and disease duration ≤3 years from symptom onset. Participants were excluded if they had a history of myocardial infarction, cardiomyopathy, or cardiac arrhythmia, previous use of mexiletine, implantation of a diaphragmatic pacer ≤60 days prior to the baseline study visit, or use of another investigative medication ≤30 days prior to the baseline study visit. They had to be on a stable dosage of riluzole and any medications used to treat muscle cramps for ≥60 days or have been off these medications ≥30 days prior to randomization.
Study procedures.
Screening procedures included assessment of eligibility requirements, a medical history, a physical and neurologic examination, vital signs, ECG, laboratory safety panel (blood chemistry, complete blood count, serum pregnancy test for women of childbearing potential, and urinalysis), administration of the revised ALS Functional Rating Scale (ALSFRS-R) questionnaire, SVC determination, and medication review. At the baseline visit performed within 21 days of the screening visit for eligible participants, procedures included recording of vital signs, ECG, assessment of adverse events, and distribution of a muscle cramp diary. A muscle cramp was defined as a sustained painful muscle contraction lasting seconds to minutes, as distinguished from a fasciculation, which was defined as a transient painless muscle twitch typically lasting a few seconds, though frequently recurrent. Participants were instructed to record the number of muscle cramps they experienced daily and the intensity of the cramps using a visual analogue scale for pain from 0 (no pain) to 10 (maximal pain). Additionally, participants were asked to estimate the frequency of muscle cramps they had experienced in the previous 24 hours and 30 days.
Participants were assessed in person at weeks 2, 6, and 12 and contacted by telephone at weeks 1, 10, and 16 from the baseline visit. Study drug was discontinued at week 12. Study procedures at each in-person visit included physical examination, vital signs, ALSFRS-R, collection of the muscle cramp diary, assessment of drug compliance and collection of a dosing diary, reports of concomitant medications and adverse events, ECG, and laboratory safety panel (blood chemistry, complete blood count, serum pregnancy test for women, urinalysis). SVC was recorded at the week 6 and 12 visit only. Plasma and CSF samples were collected for pharmacokinetic analysis at week 6. Blood draws were performed immediately prior to the first dose of the day, and at 1, 2, 4, and 6 hours postdose. CSF was collected by lumbar puncture at a single, randomly assigned time point only. At the telephone visits, drug compliance was assessed if still on study drug, concomitant medications and adverse events were reported and documented, and the ALSFRS-R was administered at the week 16 visit.
Outcomes.
The primary outcomes were safety and tolerability (Class I). Secondary outcomes were pharmacokinetic study from plasma and CSF and differences in the change from baseline of the ALSFRS-R score, SVC, and muscle cramp frequency and severity.
Statistical analysis.
Sample size.
A target sample size of 60 volunteers, 20 per group, was selected to have 80% power to demonstrate that a given dosage was tolerable, defining tolerance as a proportion of treatment failures of less than 40% with 80% confidence, one-sided, and assuming that the true treatment failure rate was no more than 20%. Power for detecting treatment differences in rate of disease progression as measured by slope in ALSFRS-R total score was low. Based on observed variance component estimates from the study, the effective standard deviation after adjustment was 1.3 units/month, yielding 7% power for a 30% difference in rate of progression between either active group and placebo, assuming 2-tailed testing at α = 0.027 by the Dunnett method for comparison of multiple doses to placebo.
Analysis.
All participants who initiated study drug were included in all analyses, following a modified intention-to-treat principle. Baseline characteristics were compared across treatment groups by Fisher exact test and 1-way analysis of variance for categorical and continuous variables, respectively. Safety was assessed with respect to adverse events (AEs) and serious AEs (SAEs) classified according to MedDRA system organ class and preferred term, abnormal clinical laboratory data, vital signs, physical examination findings, and changes in ECG data. The proportion of participants experiencing a given AE or abnormality was compared by Fisher exact test between each pair of treatments. ALSFRS-R and SVC data were analyzed in separate shared-baseline random-slope linear mixed effect models with fixed effects of time from treatment initiation and treatment × time interaction. Fixed effect of bulbar onset, symptom onset to baseline time, and baseline riluzole use and their interactions with time were included to avoid confounding by chance differences in those characteristics across treatment groups. Random participant-specific intercepts and slopes were modeled with unstructured covariance to account for correlation among repeated measurements over time. Models for muscle cramp frequency and intensity were similar except that weekly cramp frequencies and mean maximum intensity were modeled as negative binomial distributed data, data from the first 2 weeks of follow-up during dose titration were excluded, a single treatment main effect rather than time and treatment × time interactions was included as fixed effect, and an additional adjustment for cramp frequency or maximum cramp pain intensity over the 30 days before baseline were included, respectively. Treatment slopes or means were compared by linear contrast. Plasma and CSF mexiletine concentrations were determined by high-pressure liquid chromatography. Pharmacokinetic analysis of plasma mexiletine levels assumed a noncompartmental model and was summarized by area under the concentration curve (AUC) from time 0 through 6 hours after dosage. The ratio of CSF to plasma mexiletine concentration was estimated by linear regression of time-coincident samples. Except where specified, all tests and confidence intervals are 2-sided with a nominal 5% type I error rate. No corrections for multiple comparisons were made. This is conservative with respect to detection of differences in safety outcomes and liberal with respect to efficacy outcomes. All analyses were performed using SAS (version 9.4, SAS Institute, Cary, NC).
RESULTS
Participants.
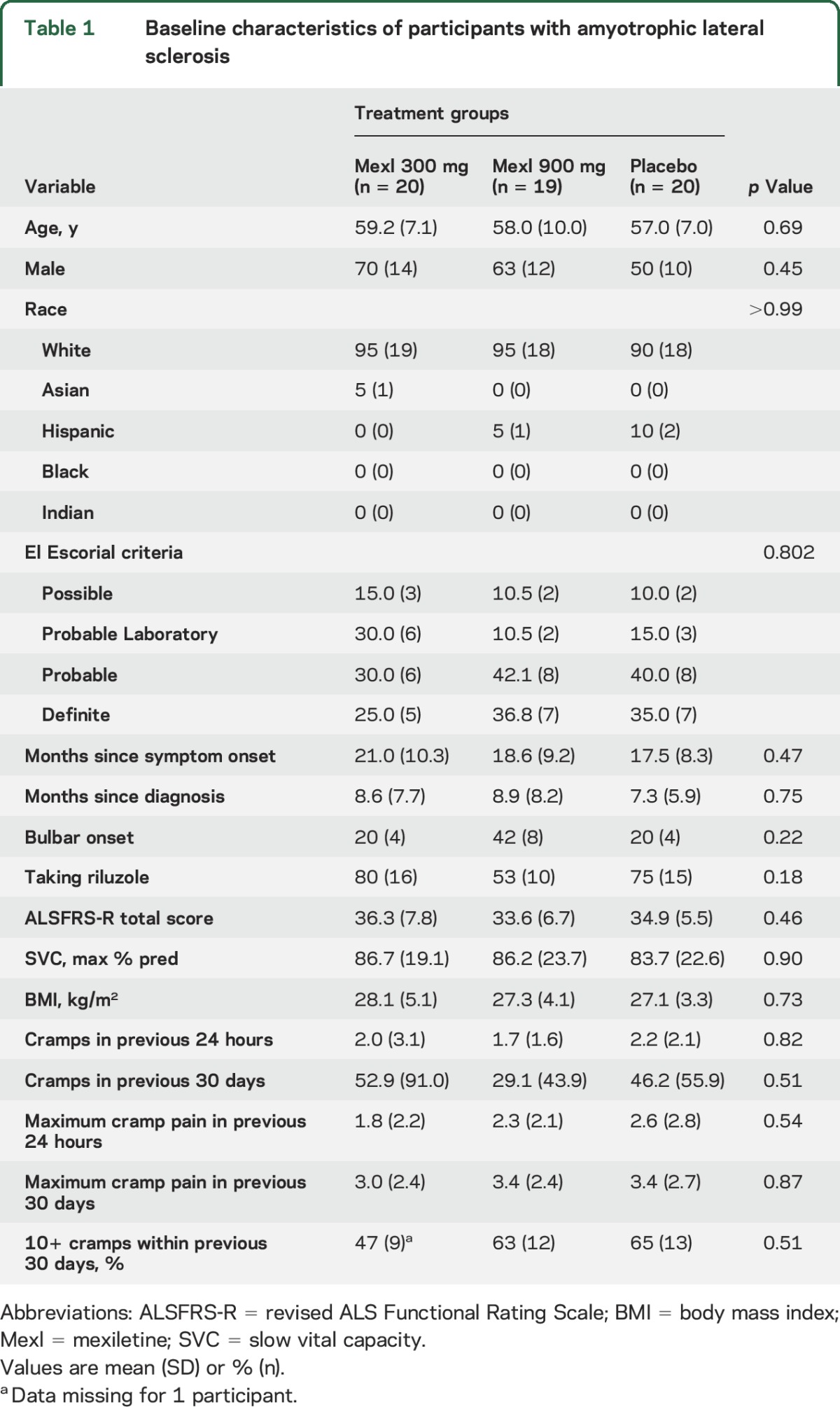
The flow of participants is shown in figure 1. Between July 2013 and May 2014, 75 patients were consented and 60 were enrolled from 10 centers (1–12 enrolled per site), with follow-up completed in August 2014. Of the 60 participants randomized, 59 received the study drug (1 participant assigned to placebo was unable to swallow the study drug capsule). Baseline characteristics did not differ significantly among treatment groups (table 1). By chance, bulbar onset was more common and use of riluzole less common among those randomized to 900 mg mexiletine and time since symptom onset was longer among those randomized to 300 mg mexiletine, but these comparisons did not reach significance. About 16% of all participants reported no muscle cramps and 60% at least 10 muscle cramps in the 30-day period prior to baseline.
Figure 1. Consolidated Standards of Reporting Trials diagram.

AE = adverse event; Mexl = mexiletine.
Table 1.
Baseline characteristics of participants with amyotrophic lateral sclerosis
Safety.
AEs reported in more than 5% of either mexiletine group are shown in table e-1 on the Neurology® Web site at Neurology.org, excluding AEs directly related to lumbar puncture. More than 25% of participants taking 900 mg of mexiletine a day reported dizziness, falls, tremor, or nausea. Tremor occurred in 26% of participants on 900 mg of mexiletine compared to 5% of participants on placebo (p = 0.091) and 0% of participants on 300 mg of mexiletine (p = 0.020). Forty-two percent of participants on 900 mg of mexiletine reported nausea compared to 10% on placebo (p = 0.031) and 5% on 300 mg of mexiletine (p = 0.008). Compared to placebo, no differences were noted in laboratory safety studies, ECG, or vital signs. There were 3 SAEs after treatment initiation, 1 death due to respiratory distress in a participant on placebo, 1 hospital admission for a fractured tibia and fibula following a fall in a participant on placebo, and 1 hospital admission for loss of balance leading to a dose suspension in a participant on 900 mg of mexiletine.
Tolerability.
Of the 59 participants who initiated study drug, 5 did not complete the study (4 participants on 900 mg of mexiletine and 1 participant on placebo who died) and 3 additional participants permanently discontinued study drug but remained on study (2 participants on 900 mg of mexiletine and 1 participant on 300 mg mexiletine). Overall, 32% (6/19) of participants receiving 900 mg of mexiletine discontinued study drug vs 5% (1/20) receiving 300 mg of mexiletine and 5% (1/20) on placebo (p = 0.026; Fisher exact test). The upper exact 1-sided 80% confidence bound for the proportion intolerant to 300 mg of mexiletine was 14% (the 2-sided 95% confidence interval was 0.1%–25%), well below the 40% threshold prespecified to declare a dose tolerable.
Pharmacokinetic studies.
For participants reaching the week 6 visit (n = 55), the mean peak mexiletine levels at 2 hours postdose were 0.41 μg/mL for the 300 mg group and 1.27 μg/mL for the 900 mg group. AUC was proportional to dose. For a variety of reasons related to consent and timing, CSF was collected from only 43 participants. The CSF:plasma ratio averaged 0.38 among participants receiving 300 mg of mexiletine and 0.46 among those receiving 900 mg, with a strong correlation between CSF and plasma levels by linear regression analysis (R2 = 0.85, p < 0.001).
Outcome measures.
The change in ALSFRS-R and SVC are shown in figure 2 and summarized in table 2. Rates of decline of ALSFRS-R and SVC did not differ from placebo. The change in muscle cramp frequency and pain intensity is noted in figure 3 and summarized in table 2. Among all participants who initiated treatment (figure 3, A and B), mexiletine treatment resulted in significant reductions of muscle cramp frequency (300 mg mexiletine: rate = 31% of placebo, p = 0.047; 900 mg mexiletine: rate = 16% of placebo, p = 0.002) and intensity (300 mg mexiletine: mean = 45% of placebo, p = 0.08; 900 mg mexiletine: mean = 25% of placebo, p = 0.005). The analysis of the subset with 10 or more muscle cramps in the 30 days prior to baseline (figure 3, C and D) revealed even larger dose-dependent reductions in cramp frequency (300 mg mexiletine: 22% of placebo, p = 0.005; 900 mg mexiletine: 7% of placebo, p = 0.002) and pain intensity (300 mg mexiletine: 37% of placebo, p = 0.058; 900 mg mexiletine: 16% of placebo, p = 0.025).
Figure 2. Changes in mean ALS Functional Rating Score–revised (ALSFRS-R) and slow vital capacity (SVC) from baseline.
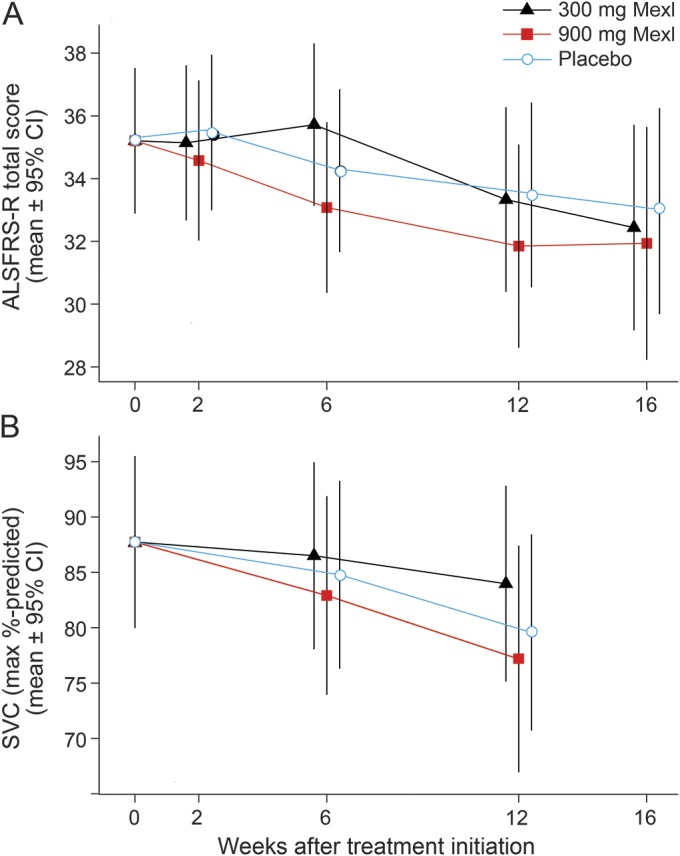
No apparent effects were noted on the ALSFRS-R (A) or SVC (B) at either dose of mexiletine (Mexl) compared to baseline. Bars indicate 95% confidence intervals (CI).
Table 2.
Outcomes of mexiletine (Mexl) treatment vs placebo
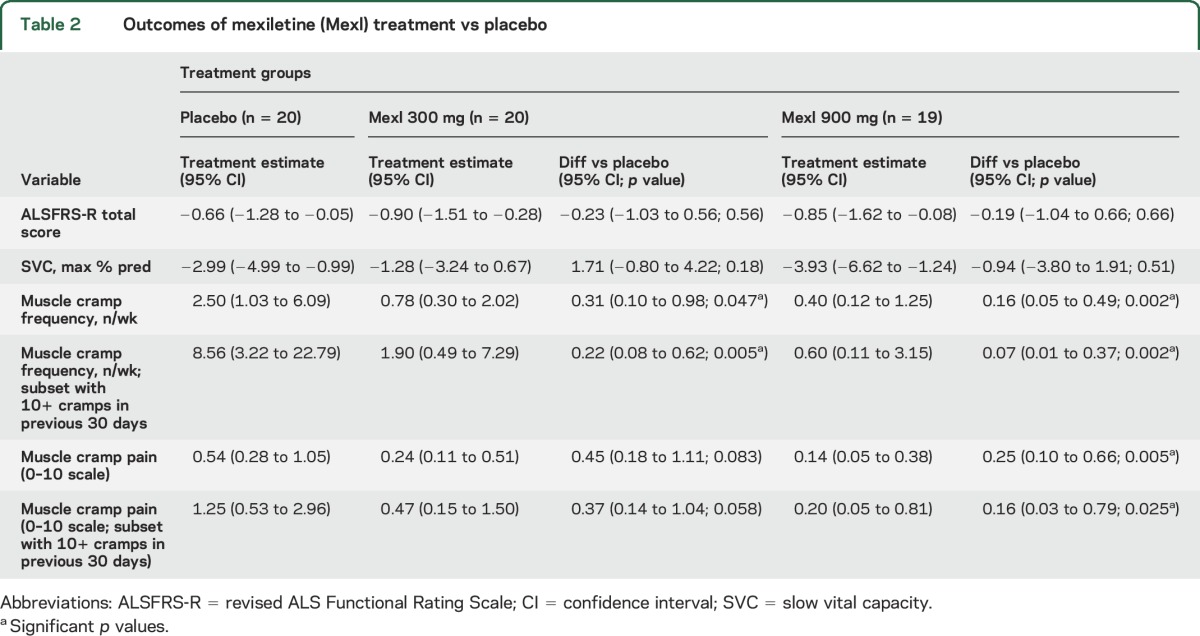
Figure 3. Reduction in mean muscle cramp frequency and pain intensity with mexiletine (Mexl) compared with placebo.

Mexiletine treatment demonstrated dose-dependent reductions of both mean cramp frequency/week (A) and intensity (B) for the entire cohort, but more so for the subset of participants with at least 10 cramps per month at baseline (C, D). Bars indicate 95% confidence intervals (CI).
DISCUSSION
Mexiletine at 300 and 900 mg per day was found to be safe in patients with SALS treated over 12 weeks; however, only 300 mg per day of mexiletine was tolerable, with only 1 in 20 discontinuing study drug at that dosage. In contrast, nearly a third of those taking the higher dosage discontinued study drug due to AEs. The reported AEs, including nausea and tremor, are known side effects of mexiletine. Pharmacokinetic study, performed to determine adequate entry of the study medication into the CNS, the relationship between plasma and CSF levels, and peak plasma and CSF concentrations, demonstrated good penetration of mexiletine into CSF and strong correlation with plasma concentrations. The 2-hour peak concentration of mexiletine in plasma is similar to studies in patients without ALS. The results of pharmacokinetic study of mexiletine from CSF were similar to those described in a small unblinded study of participants with spasmodic torticollis.19 Rates of decline of ALSFRS-R and SVC did not differ from placebo. However, as is typical of small therapeutic ALS trials, the intention was not to determine effects of mexiletine on progression of disease but rather on safety and tolerability and whether the medication penetrates the CNS in a sufficient fashion to target diseased motor neurons.
Mexiletine treatment resulted in robust dose-dependent reductions in average muscle cramp frequency/week and severity among all participants and more substantially in the subset that reported 10 or more muscle cramps in the 30 days before baseline. The dose-dependent reduction in cramp frequency and intensity, especially for those participants with more frequent cramps at baseline, was dramatic, suggesting that mexiletine could become an important therapy for this symptom. A possible dosage escalation strategy for the use of mexiletine as a treatment for muscle cramps in ALS could be to initiate the medication at 300 mg a day with gradual increments up to 900 mg a day as needed and as tolerated.
Muscle cramps are common in ALS, poorly responsive to treatment, and often debilitating.20 Typically, they are more prevalent in the first year of symptoms of ALS and lessen over time, occurring in over 75% of patients.13 Similar to a recent report,13 we found considerable variability in the frequency and severity of muscle cramps, as a minority of participants had no cramps and others hundreds in a month. There have been a number of randomized treatment trials addressing this often disabling symptom but none has been successful to date.21 These have included the use of gabapentin,22 vitamin E,23 l-threonine,24 memantine,25 xaliproden,26 indinavir,27 and baclofen.28 Though riluzole, like mexiletine, is a use-dependent sodium channel blocker, it has not been shown to have any significant effects on muscle cramps.29–31 Most of the studies investigated muscle cramps as a secondary outcome measure using primarily changes on a visual analogue scale of severity. A minority also employed the Short Form–36 Health Survey (SF-36) as a quality of life measure. The only randomized study to date to explore effects on muscle cramps as a primary endpoint was a tetrahydrocannabinol study, which evaluated 44 participants in a crossover design over 4 weeks with mean change of muscle cramp intensity on a visual analogue scale used as the primary endpoint.32
There were several limitations to this trial. As this study recruited participants from large referral centers, employing strict inclusion and exclusion criteria, the participants may not be representative of the ALS population as a whole. Additionally, no quality of life measure such as the SF-36 was utilized to determine the effects on participants of the reduction of muscle cramp frequency and severity. Finally, given the small number of patients, the study was not sufficiently powered to detect a slowing of progression and was also of short duration. As such, potential benefit could not be ruled out and additional study of the drug in larger number of participants and of longer duration may still be warranted.
Mexiletine is the only medication to date to demonstrate significant benefit in reducing the frequency and severity of muscle cramps in patients with ALS in a randomized controlled study. While it remains of uncertain benefit in regard to effects on disease progression, mexiletine could potentially become a first-line therapy for this frequently debilitating complication.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the patients who participated in this study; Anthony Amato, MD, the medical monitor, and the members of the Data Safety Monitoring Board, John Ravits, MD (University of California, San Diego), April Stempien-Otero, MD (University of Washington Medical Center), and Maciej Mrugala, MD, PhD (University of Washington Medical Center); and William David, MD, PhD (Massachusetts General Hospital), for his suggestion about including an assessment of muscle cramp frequency and severity as an exploratory outcome measure in this study.
GLOSSARY
- AE
adverse event
- ALS
amyotrophic lateral sclerosis
- ALSFRS-R
revised ALS Functional Rating Scale
- AUC
area under the concentration curve
- DSMB
data safety monitoring board
- SAE
serious adverse event
- SALS
sporadic amyotrophic lateral sclerosis
- SF-36
Short Form–36 Health Survey
- SVC
slow vital capacity
Footnotes
Editorial, page 1468
Supplemental data at Neurology.org
Contributor Information
Collaborators: Mexiletine ALS Study Group, Hong Yu, Amanda Nichols, Jason Walker, Igor Katsovskiy, Ervin Sinani, Alexander Sherman, Annette De Mattos, Daniela Grasso, Katelyn Gilardi, Meghan Hall, Mary Lou Watson, Sharon Downing, Divpreet Kaur, Max Lowden, Divisha Raheja, Helen Stephens, Travis Haines, Martina Wiedau-Pazos, Thomas Hintz, Rebecca Alvarez, Diane McKenna-Yasek, Sharon Nations, Lauren Phillips, Lydia Sharp, Nina Gorham, Mazen M. Dimachkie, April L. Mcvey, Jeffrey Statland, Mamatha Pasnoor, Omar Jawdat, Maureen Walsh, Julaine M. Florence, Jeri Sieren, and Heena Olalde
AUTHOR CONTRIBUTIONS
M. Weiss: design and conceptualization of study, acquisition, analysis, and interpretation of data, drafting and revising the manuscript. E. Macklin: design and conceptualization of study, acquisition, analysis, and interpretation of data, revising the manuscript. Z. Simmons: acquisition, analysis, and interpretation of data, revising the manuscript. A. Knox: design and conceptualization of study. D. Greenblatt: design and conceptualization of study, acquisition, analysis, and interpretation of data. N. Attasi: acquisition, analysis, and interpretation of data, revising the manuscript. M. Graves: acquisition, analysis, and interpretation of data. N. Parziale: acquisition, analysis, and interpretation of data. J. Salameh: acquisition, analysis, and interpretation of data. C. Quinn: acquisition, analysis, and interpretation of data. R. Brown Jr.: design and conceptualization of study. J. Distad: acquisition, analysis, and interpretation of data. J. Trivedi: acquisition, analysis, and interpretation of data. J. Sheffner: acquisition, analysis, and interpretation of data. R. Barohn: acquisition, analysis, and interpretation of data. A. Pestronk: acquisition, analysis, and interpretation of data, revising the manuscript. A. Swenson: acquisition, analysis, and interpretation of data. M. Cudkowicz: design and conceptualization of study, acquisition, analysis, and interpretation of data, revising the manuscript.
STUDY FUNDING
Supported by funding from the ALS Therapy Alliance and Neurology Clinical Research Institute/Northeast ALS Consortium.
DISCLOSURE
M. Weiss received personal compensation for speaking for Walgreens and NuFactor and research support from ALS therapy Alliance and the Northeast ALS Consortium. E. Macklin served as a DSMB member for trials by Lantheus Medical Imaging, Shire Human Genetic Therapies, and Acorda Therapeutics; was an unpaid consultant to Knopp Biosciences; and received research support via grants awarded to Massachusetts General Hospital from ALS Association, ALS Therapy Alliance, ALS Therapy Development Institute, Muscular Dystrophy Association, Michael J. Fox Foundation, Autism Speaks, Adolph Coors Foundation, NIH, and HRSA. Z. Simmons served as a consultant for Neuralstem, Inc. and for Cytokinetics, Inc. A. Knox and D. Greenblatt report no disclosures relevant to the manuscript. N. Attasi served as a consultant for Biogen-Idec. M. Graves received research support from Avanir and Knopp Pharmaceuticals. N. Parziale, J. Salameh, C. Quinn, R. Brown, B. Distad, and J. Trivedi report no disclosures relevant to the manuscript. J. Shefner received personal compensation from Cytokinetics, Inc., from Biogen Idec, and from Isis Pharmaceuticals for consultation related to clinical development of compounds for motor neuron disease. He has received research funding from ALS Therapy Alliance, ALSA, MDA, NINDS, Michael J. Fox Foundation, ALSTDI, GSK, and Cytokinetics. R. Barohn received personal compensation for speaking for Walgreens, Grifols Therapeutics, and NuFactor. A. Pestronk received speaker honoraria from the Myositis Association and Athena Diagnostics, revenue related to antibody patient licenses, owns stock in Johnson and Johnson, directs the Washington University Neuromuscular Clinical Laboratory, which performs antibody testing, and received research support from the NIH, Muscular Dystrophy Association, Neuromuscular Research Fund, Insmed, Knopp, Cytokinetics, Biogen-Idec, ISIS, Genzyme, GSK, Sanofi, and Ultragenyx. A. Swenson reports no disclosures relevant to the manuscript. M. Cudkowicz received compensation for consulting from Cytokinetics, Neuraltis, Genetech, Clintrex, and Biogen-Idec. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Miller RG, Jackson CE, Kasarskis EJ, et al. Practice parameter update: the care of the patient with amyotrophic lateral sclerosis: drug, nutritional, and respiratory therapies (an evidence-based review). Neurology 2009;73:1218–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kuo JJ, Schonewille M, Siddique T, et al. Hyperexcitability of cultured spinal motoneurons from presymptomatic ALS mice. Neurophysiol 2004;91:571–575. [DOI] [PubMed] [Google Scholar]
- 3.Kuo JJ, Siddique T, Fu R, et al. Increased persistent Na(+) current and its effect on excitability in motoneurones cultured from mutant SOD1 mice. J Physiol 2005;563:843–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pieri M, Carunchi I, Curcio L, et al. Increased persistent sodium current determines cortical hyperexcitability in a genetic model of amyotrophic lateral sclerosis. Exp Neurol 2009;215:368–379. [DOI] [PubMed] [Google Scholar]
- 5.Wainger BJ, Kiskinis E, Mellin C, et al. Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. Cell Rep 2014;7:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vucic S, Kiernan MC. Distal excitability changes in motor axons in amyotrophic lateral sclerosis. Clin Neurophysiol 2006;117:1444–1448. [DOI] [PubMed] [Google Scholar]
- 7.Bostock H, Sharief MK, Reid G, Murray NM. Axonal ion channel dysfunction in amyotrophic lateral sclerosis. Brain 1995;118:217–225. [DOI] [PubMed] [Google Scholar]
- 8.Vucic S, Nicholson GA, Kiernan MC. Cortical hyperexcitability may precede the onset of familial amyotrophic lateral sclerosis. Brain 2008;131:1540–1550. [DOI] [PubMed] [Google Scholar]
- 9.Mills K, Nithi K. Corticomotor threshold is reduced in early sporadic amyotrophic lateral sclerosis. Muscle Nerve 1997;20:1137–1141. [DOI] [PubMed] [Google Scholar]
- 10.Ziemann U, Winter M, Reimers CD, Reimers K, Tergau F, Paulus W. Impaired motor cortex inhibition in patients with amyotrophic lateral sclerosis: evidence from paired transcranial magnetic stimulation. Neurology 1997;49:1292–1298. [DOI] [PubMed] [Google Scholar]
- 11.de Carvalho M, Evangelista T, Sales-Luís ML. The corticomotor threshold is not dependent on disease duration in amyotrophic lateral sclerosis (ALS). Amyotroph Lateral Scler Other Motor Neuron Disord 2002;3:39–42. [DOI] [PubMed] [Google Scholar]
- 12.Bae JS, Simon NG, Menon P, Vucic S, Kiernan MC. The puzzling case of hyperexcitability in ALS. J Clin Neurol 2013;9:65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sullivan E, Blankenship S, Griffin L, et al. The natural history of muscle cramps in ALS. Neurology 2013;80:P06.13. [Google Scholar]
- 14.Ganzini L, Johnston WS, Hoffman WF. Correlates of suffering in amyotrophic lateral sclerosis. Neurology 1999;52:1434–1440. [DOI] [PubMed] [Google Scholar]
- 15.Fritz E, Izaurieta P, Weiss A, et al. Mutant SOD1-expressing astrocytes release toxic factors that trigger motoneuron death by inducing hyperexcitability. J Neurophysiol 2013;109:2803–2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rojas F, Cortes N, Abarzua S, et al. Astrocytes expressing mutant SOD1 and TDP43 trigger motoneuron death that is mediated via sodium channels and nitroxidative stress. Front Cell Neurosci 2014;8:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kanai K, Kuwabara S, Arai K, Sung JY, Ogawara K, Hattori T. Muscle cramp in Machado-Joseph disease: altered motor axonal excitability properties and mexiletine treatment. Brain 2003;126:965–973. [DOI] [PubMed] [Google Scholar]
- 18.Brooks BR, Miller RG, Swash M, Munsat TL, World Federation of Neurology Research Group on Motor Neuron Diseases. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord 2000;1:293–299. [DOI] [PubMed] [Google Scholar]
- 19.Ohara S, Hayashi R, Momoi H, Miki J, Yanagisawa N. Mexiletine in the treatment of spasmodic torticollis. Mov Disord 1998;13:934–940. [DOI] [PubMed] [Google Scholar]
- 20.Miller RG, Jackson CE, Kasarskis EJ, et al. Practice parameter update: the care of the patient with amyotrophic lateral sclerosis: multidisciplinary care, symptom management, and cognitive/behavioral impairment (an evidence-based review). Neurology 2009;73:1227–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baldinger R, Katzberg HD, Weber M. Treatment for cramps in amyotrophic lateral sclerosis/motor neuron disease. Cochrane Database Syst Rev 2012;4:CD004157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miller R, Moore D, Young L, et al. Placebo-controlled trial of gabapentin in patients with amyotrophic lateral sclerosis. Neurology 1996;47:1383–1388. [DOI] [PubMed] [Google Scholar]
- 23.Desnuelle C, Dib M, Garrel C, Favier A. A double-blind placebo-controlled randomized clinical trial of alpha-tocopherol (vitamin E) in the treatment of amyotrophic lateral sclerosis: ALS Riluzole-Tocopherol Study Group. Amyotroph Lateral Scler Other Motor Neuron Disord 2001;2:9–18. [DOI] [PubMed] [Google Scholar]
- 24.Blin O, Pouget J, Aubrespy G, Guelton C, Crevat A, Serratrice G. A double-blind placebo-controlled trial of L-threonine in amyotrophic lateral sclerosis. J Neurol 1992;239:79–81. [DOI] [PubMed] [Google Scholar]
- 25.De Carvalho M, Pinto S, Costa J, Evangelista T, Ohana B, Pinto A. A randomized, placebo- controlled trial of memantine for functional disability in amyotrophic lateral sclerosis. Amyotroph Lateral Scler 2010;5:456–460. [DOI] [PubMed] [Google Scholar]
- 26.Meininger V, Bensimon G, Bradley WG, et al. Efficacy and safety of xaliproden in amyotrophic lateral sclerosis: results of two phase III trials. Amyotroph Lateral Scler Other Motor Neuron Disord 2004;5:107–117. [DOI] [PubMed] [Google Scholar]
- 27.Scelsa SN, MacGowan DJL, Mitsumoto H, et al. A pilot, double-blind, placebo controlled trial of indinavir in patients with ALS. Neurology 2005;64:1298–1300. [DOI] [PubMed] [Google Scholar]
- 28.Norris FH, Jr, Sang UK, Sachais B, Carey M. Trial of baclofen in amyotrophic lateral sclerosis. Arch Neurol 1979;36:715–716.29. [DOI] [PubMed] [Google Scholar]
- 29.Bensimon G, Lacomblez L, Meininger V. A controlled trial of riluzole in amyotrophic lateral sclerosis. New Engl J Med 1994;330:585–591. [DOI] [PubMed] [Google Scholar]
- 30.Bensimon G, Lacomblez L, Delumeau JC, Bejuit R, Truffinet P, Meininger V. A study of riluzole in the treatment of advanced stage or elderly patients with amyotrophic lateral sclerosis. J Neurol 2002;249:609–615. [DOI] [PubMed] [Google Scholar]
- 31.Lacomblez L, Bensimon G, Leigh PN, Guillet P, Meininger V. Dose-ranging study of riluzole in amyotrophic lateral sclerosis. Lancet 1996;347:1425–1431. [DOI] [PubMed] [Google Scholar]
- 32.Weber M, Goldman B, Truniger S. Tetrahydrocannabinol (THC) for cramps in amyotrophic lateral sclerosis: a randomized, double-blind crossover trial. J Neurol Neurosurg Psychiatry 2010;81:1135–1140. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.