

Hypercapnic respiratory acidosis (HRA) causes cerebral vasodilation via perivascular extracellular pH changes.1 Its ability to precipitate global cerebral edema (GCE) in the absence of acute brain injury is rare. Treatment in these reports involves mechanical ventilation.2 We describe a rare case of HRA-induced GCE in a patient without brain injury and the efficacy of hyperosmolar treatment after mechanical ventilation failed.
This case provides Class IV evidence. This is a single observational study without controls.
Case report.
A 32-year-old quadriparetic man with cervico-thoracic syringomyelia and ventriculoperitoneal shunt (VPS) presented with recurrent dyspnea. Prior episodes were treated with noninvasive positive pressure ventilation (NIPPV). However, he presented 6 hours after symptom onset, much later than prior episodes. He denied new sensorimotor or constitutional symptoms. He was afebrile, tachycardic, tachypneic, and without evidence of hypoxia or dysautonomia. General examination revealed rapid, shallow breathing. Mental status and cranial nerves were intact with baseline spastic quadriparesis and hyperreflexia.
Arterial blood gas (ABG) on room air revealed HRA (pH: 7.18, PaCO2: >96 mm Hg, PaO2: 128 mm Hg, HCO3: 42 mEq/L). Routine laboratory studies, cardiopulmonary, and infectious workup were unrevealing. NIPPV failed to improve HRA. A previous do not intubate order was reversed after discussion with the patient. After 65 hours of HRA, the patient progressed to coma and was intubated. CT brain (figure, A) revealed GCE and he was transferred to the neurointensive care unit.
Figure. Hypercapnic cerebral edema and resolution after hyperosmolar therapy.
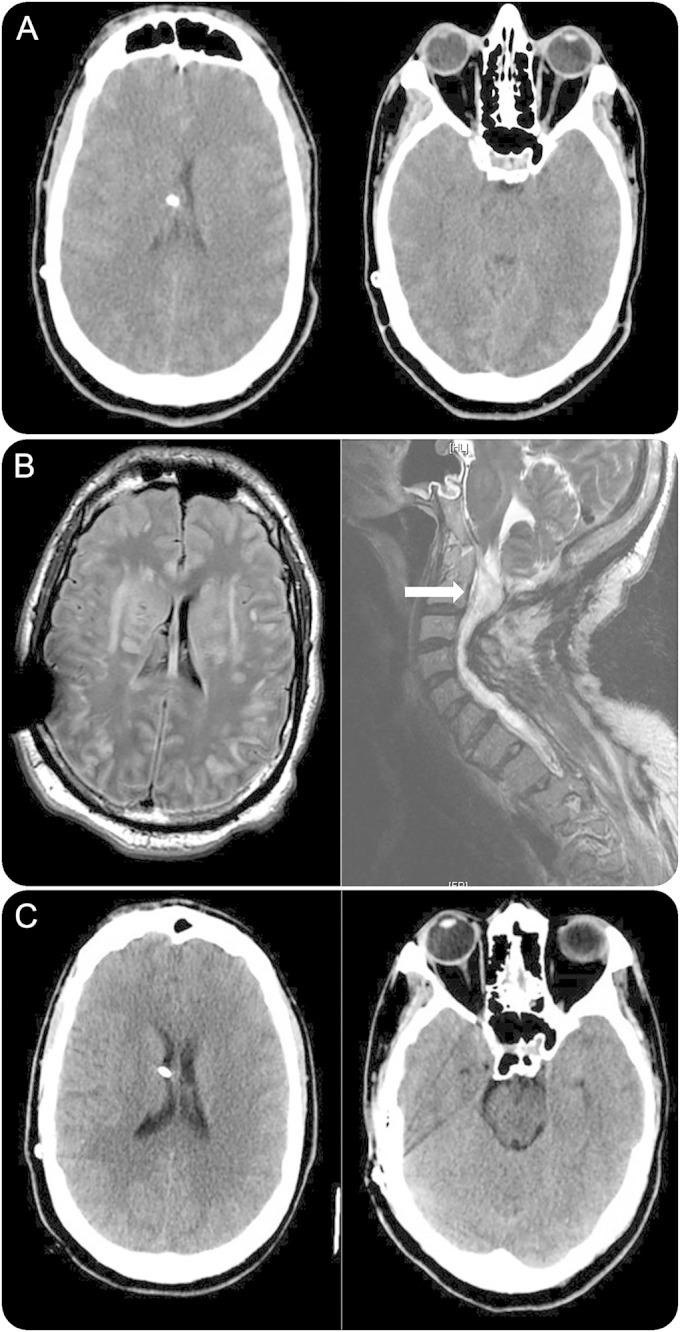
(A) CT (65 hours): global cerebral edema (GCE) (functioning ventriculoperitoneal shunt). (B) MRI (96 hours): diffuse subcortical hyperintensities despite 30 hours of acidosis/CO2 correction; severe/stable cervico-thoracic syringomyelia. (C) CT (2-month follow-up): resolved GCE.
Despite HRA resolution after intubation (ABG: pH: 7.58, PaCO2: 42 mm Hg), the patient had a Glasgow Coma Scale (GCS) score of 3T. A total of 30 mL of 23% NaCl on arrival provided transient improvement of GCS to 7T (motor localization) before returning to GCS 3T. Metabolic workup and EEG were unrevealing. MRI brain at 96 hours revealed diffuse subcortical hyperintensities despite HRA resolution for over 30 hours (figure, B). There was no evidence of hypoxic injury, thrombosis/infarct, infection, or hydrocephalus. VPS was functional via shunt series examination. MRI spine revealed stable syringomyelia. Persistent coma and prior response to hyperosmolar therapy prompted scheduled hyperosmolar therapy. Thirty milliliters of 23% NaCl at 103 hours improved the GCS from 3T to 10T within an hour (following commands). The GCS worsened from 10T to 7T at 105 hours and mannitol 80 g (1g/kg) was given, improving the GCS back to 10T within 3 hours.
Alternating scheduled 23% NaCl and mannitol were continued every 6 hours with titration to clinical response. The patient returned to his neurologic baseline in 24 hours but required a tracheostomy and phrenic nerve pacer placement for ventilator support. A 2-month follow-up CT scan revealed resolution of GCE (figure, C).
Discussion.
Etiologies for edema such as hydrocephalus, cervical dysautonomia, infection, stroke, toxins, and hepatic failure3 were excluded in our patient. Without other provoking factors, his delayed presentation and subtle extension of the syringomyelia (not appreciated on imaging) resulted in severe HRA. Central respiratory drive was intact, but was limited by neuromuscular weakness resulting in GCE.
PaCO2 >80 mm Hg increases cerebral blood flow up to 6 times its baseline4 via pH-induced chemoregulatory cerebrovasodilation. Hyperemia in conjunction with increased vascular permeability from severe vasodilation allowed for resultant edema formation and subsequent elevated intracranial pressure. The length of time required to precipitate GCE from HRA is unknown. Our patient had severe HRA for more than 60 hours prior to deterioration. Furthermore, he remained comatose for approximately 30 hours following HRA correction until hyperosmolar therapy was initiated. Although documented as >96 mm Hg, our patient's PaCO2 level was most likely much higher than what was reported (due to laboratory limitations) and continued to rise until mechanical ventilation was implemented. This prolonged HRA led to sustained cerebral acidosis unamenable to compensatory measures inducing a vasoplegia in which correction of arterial pH no longer mitigated a vasoconstrictive response.
While critical in other types of brain injury, the role of hyperosmolar therapy in GCE incited by HRA has not been described previously.5 The use of hyperosmolar therapy may have had a 2-fold effect in returning the patient back to baseline: (1) water extraction through the creation of an intravascular osmotic gradient and (2) rheologic and cardiac output augmentation resulting in mechanoregulatory vasoconstriction and a subsequent decrease in intracranial pressure.6 The role of hyperosmolar therapy in treating GCE in this case is further supported by the temporal synchronization of our patient's clinical fluctuations reflecting the pharmacokinetic profile of mannitol/hypertonic saline: onset 30 minutes and duration of action 2–12 hours.7 Our patient's extreme hypercapnia in the absence of hypoxemia did not alter the cerebral energy state and subsequently did not result in irreversible cell damage. Consequently, our patient's case illustrates that hyperosmolar therapy in patients with GCE secondary to HRA should be considered should mechanical ventilation fail.
Supplementary Material
Footnotes
Author contributions: Dr. Roh: acquisition of data, analysis and interpretation, draft of the manuscript, critical revision of the manuscript for important intellectual content. Dr. Merkler: acquisition of data, critical revision of the manuscript for important intellectual content. Dr. Morris: acquisition of data, critical revision of the manuscript for important intellectual content. Dr. Al-Mufti: acquisition of data, critical revision of the manuscript for important intellectual content. Dr. Agarwal: acquisition of data, critical revision of the manuscript for important intellectual content. Dr. Claassen: acquisition of data, critical revision of the manuscript for important intellectual content. Dr. Park: critical revision of the manuscript for important intellectual content, study supervision.
Study funding: No targeted funding reported.
Disclosure: The authors report no disclosures relevant to the manuscript. Dr. Roh takes full responsibility for the data, analyses, and interpretation. This author has full access to all of the data and has the right to publish any and all data separate and apart from any sponsor. The author has received consent from the patient whose case is depicted. Go to Neurology.org for full disclosures.
References
- 1.Meng L, Gelb AW. Regulation of cerebral autoregulation by carbon dioxide. Anesthesiology 2015;122:196–205. [DOI] [PubMed] [Google Scholar]
- 2.Joyce RR, McGee WT. Hypercapnic cerebral edema presenting in a woman with asthma: a case report. J Med Case Rep 2011;5:192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baethmann A. Pathophysiological and pathochemical aspects of cerebral edema. Neurosurg Rev 1978;1:85–100. [Google Scholar]
- 4.Curley G, Laffey JG, Kavanagh BP. Bench-to-bedside review: carbon dioxide. Crit Care Lond Engl 2010;14:220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brain Trauma Foundation, American Association of Neurological Surgeons & Congress of Neurological Surgeons. Guidelines for the management of severe traumatic brain injury. J Neurotrauma 2007;24(suppl 1):S1–S106. [DOI] [PubMed] [Google Scholar]
- 6.Diringer MN, Zazulia AR. Osmotic therapy: fact and fiction. Neurocrit Care 2004;1:219–233. [DOI] [PubMed] [Google Scholar]
- 7.Bhardwaj A, Ulatowski JA. Cerebral edema: hypertonic saline solutions. Curr Treat Options Neurol 1999;1:179–188. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.