

Abstract
C—H bond oxidation reactions underscore the existing paradigm wherein high reactivity and high selectivity are inversely correlated. The development of catalysts capable of oxidizing strong aliphatic C(sp3)—H bonds while displaying chemoselectivity (i.e. tolerance of more oxidizable functionality) remains an unsolved problem. Herein, we describe a catalyst, manganese tert-butylphthalocyanine [Mn(tBuPc)], that is an outlier to the reactivity-selectivity paradigm. It is unique in its capacity to functionalize all types of C(sp3)—H bonds intramolecularly, while displaying excellent chemoselectivity in the presence of π-functionality. Mechanistic studies indicate that [Mn(tBuPc)] transfers bound nitrenes to C(sp3)—H bonds via a pathway that lies between concerted C—H insertion, observed with reactive noble metals (e.g. rhodium), and stepwise radical C—H abstraction/rebound, observed with chemoselective base metals (e.g. iron). Rather than achieving a blending of effects, [Mn(tBuPc)] aminates even 1° aliphatic and propargylic C—H bonds, reactivity and selectivity unusual for previously known catalysts.
High-valent metal heteroatom species (i.e. oxos and nitrenes) oxidize inert C—H bonds with tunable site-selectivity and stereospecificity, but typically do not tolerate more readily oxidizable π-functionality1–6. Intramolecular metallonitrene-based C(sp3)—H amination of sulfamate esters, which installs medicinally important amino alcohol motifs, showcases the inverse correlation between reactivity and chemoselectivity for such catalysis7–10. Noble metal rhodium catalysts that stereospecifically functionalize robust aliphatic C—H bonds (secondary, 2° and tertiary, 3°) lack chemoselectivity due to competitive oxidation of π-bonds (Fig. 1a)1, 7–10. Conversely, base metal iron catalysts that chemoselectively aminate allylic C—H bonds over competing aziridination are only moderately reactive toward stronger aliphatic C—H bonds (Fig. 1a)11–15. Herein we describe the discovery of an outlier catalyst, manganese tert-butylphthalocyanine [Mn(tBuPc)] (3), that is the first to achieve both high reactivity and chemoselectivity for a C—H oxidation reaction. Catalyst [Mn(tBuPc)] preparatively aminates all C(sp3)—H bond types encompassed by rhodium and maintains the high chemoselectivity for allylic C—H amination observed with iron, all while demonstrating stereospecificity, site-selectivity, and high functional group tolerance (Fig. 1b). Additionally, [Mn(tBuPc)] aminates primary (1°) aliphatic and propargylic C—H bonds, reactivity and selectivity that is typically difficult to achieve with metallonitrene-based catalysis. Importantly, we show this reaction enables rapid and scalable late-stage diversification of bioactive molecules. The unique generality of [Mn(tBuPc)] is partially attributed to its mechanistically distinct pathway for nitrene transfer that lies between the stepwise mechanism of iron and the concerted mechanism of rhodium. Discovery of an Earth-abundant base metal catalyst that is capable of aminating all types of C(sp3)—H bonds, including those challenging to access with precious noble metals, underscores the potential benefits in the continued development of these inexpensive, underexplored metals as catalysts for important synthetic reactions5, 6, 11–14, 16–19.
Fig. 1. The C—H oxidation reactivity/selectivity paradigm.

a, Reactivity and chemoselectivity of existing C—H amination catalysts with sulfamate ester substrates. Rhodium catalysts aminate strong 2° C—H bonds in aliphatic substrates but aziridinate reactive π-functionality in allylic substrates. Iron and ruthenium catalysts aminate weak allylic bonds with high chemoselectivity, but demonstrate limited reactivity towards strong aliphatic C—H bonds. b, Novel [Mn(tBuPc)] catalyst demonstrates both high reactivity and chemoselectivity. It is capable of aminating strong aliphatic C—H bonds while tolerating reactive π-functionality in allylic substrates.
Noble metal rhodium catalysts functionalize strong aliphatic C—H bonds via a concerted asynchronous C—H insertion mechanism1. Conversely, base metal iron catalysts access mechanistically distinct single electron pathways for nitrene transfer, affording excellent chemoselectivity with diminished reactivity for stronger C—H bond types11–14. We hypothesized that a metal catalyst capable of transferring bound nitrenes to C(sp3)—H bonds via a stepwise mechanism with attenuated radical character of the metallonitrene oxidant relative to iron would achieve higher reactivity while maintaining chemoselectivity20. Low reactivity observed under iron catalysis with aliphatic C(sp3)—H bonds coupled with reports that sulfamate ester N-centered free radicals are unreactive towards intramolecular aminations of 3° C(sp3)—H bonds21 suggested to us that a metallonitrene with diminished radical character would be more reactive. Although nature rarely uses manganese metal species to mediate oxidations, early studies using synthetic metalloporphryins as models for cytochrome P450 demonstrated that manganese and iron oxos react via mechanistically analogous one electron pathways, with manganese exhibiting significantly higher C—H hydroxylation reactivity22. Importantly, the manganese catalysts were found to have smaller kinetic isotope effects (KIE) than their iron counterparts, suggestive of attenuated radical behavior23. Moreover, well-characterized nitridomanganese(V) porphyrin complexes have been shown to stoichiometrically transfer nitrenes when the nitrogen is rendered electron deficient, much like with iron24–26.
Results
Reaction development
We first compared a series of manganese complexes to their iron counterparts for the C—H amination of challenging 3° aliphatic substrate 4 (bond dissociation energy (BDE) ~96 kcal/mol)27. Improved yields of aminated product 5 were observed with the manganese complexes across all ligand classes (Table 1, entries 1–8). The previously reported phthalocyanine ligand was the most effective11, and so catalyst 2 was employed for further optimization. Notably, both iron and manganese porphyrin catalysts, among the first catalysts shown to be competent for metallonitrene based C—H amination28, exhibited significantly lower reactivity with this challenging substrate (entries 3 and 4). The enhanced reactivity for an electrophilic C—H amination reaction may be attributed to an electronic difference between these ligands, as evidence suggests that phthalocyanines are significantly better π-acceptor ligands and may lead to enhanced electrophilicity at the metal center29,30. Addition of molecular sieves significantly improved reactivity with both 10 mol% and 5 mol% catalyst 2, affording 60% and 58% of 5, respectively (entries 9, 10). Catalyst 3, in which tert-butyl groups were introduced into the periphery of the phthalocyanine ligand, further improved the yield to 75% (entry 11). This modification was not similarly beneficial for the corresponding iron complex (29% yield, entry 12). The enhanced productivity of 3 enables the catalyst loading to be reduced to 5 mol% (72%, entry 13) and in some cases to 2.5 mol% (71%, entry 14); additionally, the oxidant loading can be reduced to 1.2 equivalents while still maintaining good reactivity (68%, entry 15).
Table 1.
Reaction development with [Mn(tBuPc)] catalyst.
 | |||
|---|---|---|---|
| entry | catalyst | additive | % yield (% rsm) |
| 1 | [FePc]•SbF6 (1)* | - | 29 (32) |
| 2 | [MnPc]•SbF6 (2)* | - | 43 (27) |
| 3 | Fe(TPP)•SbF6* | - | 4 (85) |
| 4 | Mn(TPP)•SbF6* | - | 18 (62) |
| 5 | Fe(R,R-salen)•SbF6* | - | <1 (85) |
| 6 | Mn(R,R-salen)•SbF6* | - | 4 (78) |
| 7 | Fe(R,R-PDP)(SbF6)2 | - | <1 (91) |
| 8 | Mn(R,R-PDP)(SbF6)2 | - | 7 (82) |
| 9 | [MnPc]•SbF6 (2)* | 4Å MS | 60 (11) |
| 10 | [MnPc]•SbF6 (2)* | 4Å MS | 58 (20)† |
| 11 | [Mn(tBuPc)]•SbF6 (3)* | 4Å MS | 75 (<5) |
| 12 | [Fe(tBuPc)]•SbF6* | 4Å MS | 29 (34) |
| 13 | [Mn(tBuPc)]•SbF6 (3)* | 4Å MS | 72 (14)† |
| 14 | [Mn(tBuPc)]•SbF6 (3)* | 4Å MS | 71 (13)‡ |
| 15 | [Mn(tBuPc)]•SbF6 (3)* | 4Å MS | 68 (16)†,§ |
Isolated yields are average of three runs. Pc = phthalocyanine; TPP = tetraphenylporphyrin; salen = N,N'-bis(3,5-di-tert-butylsalicylidene)-1,2-cyclohexanediamine; PDP = [N,N'-Bis(2-pyridylmethyl)]-2,2'-bipyrrolidine.
Active catalyst formed via in situ metathesis using equimolar AgSbF6 to chloride pre-catalyst.
Used 5 mol% each Mn catalyst and AgSbF6.
Used 2.5 mol% each Mn catalyst and AgSbF6.
Used 1.2 equiv Phl(OPiv)2.
Reaction generality
This new catalytic method was examined with all other major sp3 C—H bond types (benzylic, allylic, 2° and 1° aliphatic) using unsubstituted linear sulfamate esters, among the most difficult substrate classes for intramolecular C—H amination (Table 2). In all cases, a significant improvement in yield was observed in switching from the iron to the manganese catalyst, with the benzylic and allylic substrates affording synthetically useful yields of aminated products 6 and 7 (entries 1 and 2). Catalyst 3 exhibited good reactivity across all bond types with 2° (8, BDE ~ 98 kcal/mol) and even 1° (9, BDE ~ 101 kcal/mol) 27 C—H bonds being readily intramolecularly aminated (entries 3 and 4). Significantly, 1° C—H bonds are at the lowest end of the reactivity spectrum under rhodium catalysis and amination of this bond type is rare (vide infra)31. Moreover, despite the high intramolecular reactivity of 3, excellent chemoselectivity (>20:1 ins./azir.) was maintained for allylic C—H amination, as compared to 1:1 ins./azir. observed for rhodium catalyst [Rh2(esp)2]31 (entry 2).
Table 2.
Manganese versus iron catalyst comparison with unsubstituted linear substrates.
 | ||||
|---|---|---|---|---|
| Entry | Product | Yield (based on catalyst) | ||
| [FePc]* (1) | [MnPc] (2) | [Mn(tBuPc)] (3) | ||
| 1 | 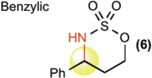 |
22% | 71% | 74% |
| 2 | 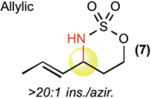 |
10% | 61 % | 61% |
| 3 | 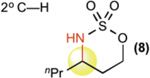 |
5%† | 39%† | 57%† |
| 4 | 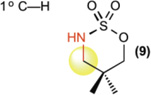 |
3%† | 37%† | 64%† |
Isolated yields are average of three runs.
Conditions: 5 mol% [FePc]Cl, 5 mol% AgSbF6, 2 equiv. Phl(OPiv)2, 4:1 PhMe/MeCN (0.5M), rt, 8h.
Used 10 mol% catalyst.
Catalyst 3 is capable of aminating 3°, 2°, and 1° aliphatic C—H bonds in a broad range of substrates with good functional group tolerance and site-selectivity (Table 3). Adjacent functionality, such as protected nitrogen (10, 79%) and silyl ethers (11, 71%), is well tolerated. In the presence of proximally equivalent C—H bonds, catalyst 3 discriminates according to BDE, functionalizing at the weaker β 3° C—H bond of an isopinocampheol derivative in the presence of a β 2° C—H bond (12, 63%; vide infra Fig. 2c). Amination of 3° C—H bonds is also effective for the formation of azaspirocycles (13, 52%) and fused bicycles (14, 86%). Additionally, 3 exhibits sensitivity to substrate electronics (i.e. inductive effects) in the amination of 2° C—H bonds. A remote electron-withdrawing ester moiety has limited impact on amination at the γ position (15, 57%), but will attenuate reactivity when made more proximal (Supplementary Table S3, S36). A distal tosylate group is tolerated under the reaction conditions to afford 16 in 54% yield; this functionality can be subsequently displaced intramolecularly to generate a heterobicycle in 80% yield (Supplementary Information, S37). In addition, 2° C—H amination in cyclohexanes can occur across the ring in a 1,3-fashion, functionalizing adjacent to a bulky tert-butyl group in a cis relationship (17, 90%), which would be challenging to accomplish through traditional synthetic methods.
Table 3.
Substrate scope of [Mn(tBuPc)] -catalyzed C—H amination.
 |
Isolated yields are average of three runs.
Conditions: 5 mol% [FePc]Cl, 5 mol% AgSbF6, 2 equiv. Phl(OPiv)2, 4:1 PhMe/MeCN (0.5M), rt, 8h.
Used 10 mol% catalyst.
ins./azir. rati
10% of 1° C—H animation also isolated.
Figure 2. Mechanistic studies of manganese and iron C—H amination catalysts.

a, Proposed stepwise mechanism for manganese and iron catalysis. b, Intramolecular Hammett analysis (σ+) reveals 3 is less sensitive to the electronics of the C—H bond than 1 but more so than reported for rhodium (ρ = −0.55). c, C—H bond reactivity trends for 3° aliphatic C—H bonds relative to other bond types show that 3 reacts according to relative C—H BDE but is less discriminating than 1, indicating attenuated radical character in C—H cleavage. d, KIE values for intramolecular competition experiments for catalysts 1, 2, 3 and Rh2(OAc)4 (quantitative 13C NMR) suggest manganese catalysis proceeds via a transition stucture where C—H bond breakage occurs to a greater extent than with rhodium but less than with iron. Intermolecular KIE studies with catalysts 2 and 3 suggest C—H cleavage contributes to the reaction rate but is not solely rate-determining. e, Partial isomerization of Z-olefin substrate 47 with catalysts 3 and 1 support a stepwise mechanism. f, Complete stereoretention in C—H amination of enantiometrically enriched (+)-48 with catalysts 3 and 1 supports a rapid radical rebound of the nitrogen from the base metal catalyst versus a free radical intermediate.
Under [Mn(tBuPc)] 3 catalysis, amination of γ C—H bonds to form six-membered oxathiazinanes is generally preferred over β C—H bonds to form five-membered oxathiazoles, regardless of relative bond strength. For example, in the functionalization of a borneol derivative, amination selectively occurs at the stronger γ 1° C—H bond of the adjacent methyl group to afford 18 in 53% yield. Competitive C—H amination at the β 2° C—H bond is geometrically possible with this substrate and is observed in 27% yield. This ring-size selectivity has been noted with rhodium catalysis and is based on the geometric constraints imposed by the favored N—S—O bond angles of the sulfamate tether1. However, the ability to aminate effectively at 1° methyl groups with metallonitrenes is rare, and the reported examples with iron12,14 and rhodium31,32 are low yielding. A limitation to this trend can be observed when amination to form a six-membered heterocycle requires functionalization of an exceptionally strong γ C—H bond over a much weaker β C—H bond. Amination of a cyclopropane-containing substrate occurs exclusively at the stereoelectronically activated β 2° C—H bond (BDE ~ 97 kcal/mol) to form five-membered sulfamidate 19 (51%) as opposed to the six-membered oxathiazinane, which would require abstraction of a much stronger 3° cyclopropane C—H bond (BDE ~ 106 kcal/mol)27.
Catalyst 3 successfully aminates at the allylic position in a variety of molecules, in all cases maintaining the excellent chemoselectivity previously observed with [FeIIIPc] for olefin-containing substrates (Table 3)11. For example, with a challenging linear terminal olefin substrate, catalyst 3 affords C—H amination product 20 in 50% yield and in 7:1 excess over the aziridine. Iron catalyst 1 exhibits similarly high chemoselectivity (7:1 ins./azir.) but poor reactivity (22%), whereas a noble metal ruthenium catalyst [Ru2(hp)4] designed to be highly chemoselective is less selective for insertion (2:1 ins./azir.)15 and Rh2(esp)2 favors π-functionalization (1:1.5 ins./azir.)31. Additionally, [Mn(tBuPc)] exhibits diminished sensitivity to electronics relative to its iron predecessor with weaker C(sp3)—H bond types (vide infra Fig. 2b). Catalyst 3 readily functionalizes C—H bonds proximal to an α,β-unsaturated ester (21, 77%), while other chemoselective catalysts 1 and [Ru2(hp)4] are less reactive (12% and 25%, respectively)11,15. This electronic insensitivity is further highlighted by the tolerance of electron-withdrawing nitrogen functionality (22, 73%) introduced via palladium-catalyzed intermolecular allylic C—H amination33. A cyclohexene derivative readily cyclizes to form bicycle 23 in 69% yield with excellent diastereoselectivity (>20:1 anti/syn). The high chemoselectivity for allylic C—H amination over aziridination with [Mn(tBuPc)] 3 is maintained even in cases where aziridination is geometrically accessible. Homoallylic sulfamate derivative of terpene (−)-nopol undergoes facile allylic C—H amination with catalyst 3 at the β-position to furnish the five-membered heterocycle (24, 60%) with no observed aziridine. This bias of 3 toward allylic C—H amination versus aziridination persists even in an acyclic styrenyl homoallylic sulfamate ester substrate (25, 62%, 7:1 ins./azir.). In contrast, both reactivity and chemoselectivity are lower with typically chemoselective chiral rhodium catalysts ([Rh2(S-nap)4], 48%, 2:1 ins./azir.), and formation of the aziridine product is strongly favored (1:20 ins./azir.) with standard Rh2(OAc)4.1,34
Although chemoselective propargylic C—H amination with carbamates to furnish 1,2-amino alcohols is precedented35, amination using sulfamate esters to afford the 1,3-amino alcohol motif is challenging as the alkyne typically undergoes alternate oxidation pathways36. Instead, two-step sequences have been developed that involve amination of activated ethereal C—H bonds to furnish N,O-acetals followed by Lewis acid-promoted alkylations to generate propargylic amines1. Further underscoring the high chemoselectivity achieved with manganese catalysis, a TMS-protected terminal alkyne sulfamate ester readily undergoes propargylic C—H amination (26, 64%). α-Substituted alkyne 27 functionalizes in moderate yields and serves as a viable intermediate for a streamlined synthesis of saxitoxin37. Alternatively, ethereal C—H amination can be performed in good yield and diastereoselectivity (64%; >20:1 d.r.) with catalyst 3 to furnish the sensitive oxathiazinane N,O-acetal 28, a known precursor to alkyne 27.
Catalyst 3 functionalizes benzylic C—H bonds in a variety of aromatic and heterocyclic compounds, further demonstrating the generality of this method (Table 3). Phenolic sulfamate esters cyclize with iron catalyst 1 in modest yields due to deleterious substrate decomposition (29, 43%), but these substrates are functionalized with manganese catalyst 3 in improved yields (29, 69% and 30, 69%). Catalyst 3 promotes benzylic C—H amination on substrates with varying degrees of electronic deactivation, such as para-Br- and para-CF3-substituted benzylic substrates (31, 68%; 32, 58%, respectively). In contrast, iron catalyst 1 is less reactive for electronically deactivated benzylic substrates (31, 30%). Both α- and β-branched sulfamates readily undergo benzylic C—H amination with excellent diastereoselectivity (>20:1) favoring the conformationally preferred syn and anti oxathiazinane, respectively (33, 64%; 34, 68%)1. Sterically encumbered benzylic substrates with quaternary centers adjacent to the site of functionalization still aminate in good yields (35, 63%). Given the prevalence of heterocycles in medicinally relevant compounds, we evaluated the tolerance of this method to aromatics with varying degrees of heteroatom incorporation. Pyrrole and indole substrates both afforded good yields and diastereoselectivities of the desired C—H amination products (36, 69%, 7:1 d.r. and 37, 71%, 12:1 d.r., respectively). N-aryloxazolidinones and oxadiazoles, which contain both nitrogen and oxygen heteroatoms, proceeded smoothly under the reaction conditions (38, 56% and 39, 63%, respectively). Exemplifying the potential application of this method to late-stage diversification of pharmaceuticals, an oxazole-based substrate derived from the commercial NSAID oxaprozin furnished oxathiazinane 40 in 63% yield.
Mechanistic studies
We sought to investigate our hypothesis that the unique generality of catalyst 3 can be attributed to the attenuated radical character of the manganese metallonitrene oxidant relative to iron. Intramolecular competition experiments were conducted to probe the C—H amination steps of the catalytic cycle independently from the reaction kinetics (Fig. 2a). The electronic nature of the transition state for C—H cleavage was assessed by way of Hammett analysis with a series of sulfamate ester substrates having two electronically dissimilar benzylic sites (Fig. 2b) (Supplementary Figure S1). Plotting log(kAr/kH) against substituent parameter σ+ furnished linear correlations, with manganese showing less sensitivity to the electronics of the C—H bond relative to iron (ρ = –0.88 for 3, –1.12 for 1), but significantly more than that reported for rhodium catalysts (ρ = –0.55)1,15. This data is consistent with the reactivity trends observed between manganese and iron with electronically deactivated substrates and suggests a transition structure in which C—H cleavage for manganese is less pronounced than for iron but more so than the related transition structure for the concerted rhodium-catalyzed process15. We also systematically compared the C—H bond reactivity trends for γ 3° aliphatic C—H bonds relative to other bond types (γ’) (41–44, Fig. 2c). The reactivity trends correlate with the homolytic C—H BDEs for both manganese and iron, but manganese shows less discrimination between the different bond types, consistent with diminished electrophilic radical character in the C—H cleavage transition state. Additionally, benzylic amination of monodeuterated substrate 45 provides an intramolecular KIE for manganese catalyst 3 (4.2 ± 0.1) that lies between iron catalyst 1 (4.8 ± 0.1) and Rh2(OAc)4 catalyst (3.8 ± 0.1) (Fig. 2d). Taken together, these data support our hypothesis that catalyst 3 proceeds through a stepwise mechanism with a transition structure in which C—H bond breakage occurs to a lesser extent than with iron15. Notably, catalyst 3 behaved comparably to its unsubstituted manganese analogue 2, with the exception that a slightly smaller intramolecular KIE was observed with 3 (4.2) relative to 2 (4.5), suggesting that this ligand modification results in further attenuation of radical character.
We next evaluated the recombination step for manganese catalyst 3. In the allylic C—H amination of Z-olefin substrate 47 (>20:1 Z/E), olefin isomerization was observed with manganese catalyst 3 to the same degree as with iron catalyst 1, affording a 10:1 Z/E mixture of 7 (Fig. 2e). Characteristic of a reaction proceeding via a concerted C—H amination mechanism, an analogous substrate has been reported to proceed with no isomerization under rhodium catalysis11. Importantly, C—H amination at a defined aliphatic stereocenter in 48 proceeds with complete stereoretention for manganese catalysts 2 and 3 as well as for iron catalyst 1 (49, 99% ee, Fig. 2f). These data further support a stepwise mechanism for manganese that proceeds through H–atom abstraction followed by rapid radical rebound from the base metal catalyst. In contrast to aminations that proceed via free radical intermediates21,38,39, this mechanistic feature of metallonitrene chemistry allows C—H amination reactions to proceed stereospecifically with high functional group tolerance, and enables the prospect of tuning the catalyst to control selectivity during functionalization6.
In order to gain further mechanistic insight, we investigated the influence of the manganese catalyst 3 on reaction kinetics. The reaction profile for a 2° aliphatic substrate suggested an overall reaction rate enhancement with 3 relative to both 1 and 2, resulting in significantly higher product yields with 3 (Supplementary Figure S2). Initial rate measurements for 2° and benzylic C—H amination with 3 quantitatively support catalyst involvement in the rate-determining step of the reaction, as changes in catalyst concentration (5 mol% to 10 mol%) result in proportional changes in the initial rate (Supplementary Figures S3–S5, S10). This is in contrast to rhodium catalysis where there is no rate dependence on catalyst concentration and iminoiodinane formation is hypothesized to be rate-determining1. Moreover, measuring initial rates on parallel reactions with benzylic substrate 46 and 46-d2 revealed a primary KIE of 1.7 for catalyst 3 and 1.9 for catalyst 2. Consistent with this, an intermolecular competition experiment with one equivalent of each 46 and 46-d2 gave a KIE of 1.6 for 3 and 1.9 for 2 from isolated product ratios (Fig. 2d). These two KIE experiments with manganese catalysts 2 and 3 provide values that are larger than would be expected for a rate-determining step that did not involve C—H cleavage (kH/kD ~1), but smaller than if C—H cleavage was solely rate-determining (kH/kD ~3.5–6)40. Ligand modification on the manganese catalyst results in small differences in KIE (Fig. 2d), providing support for a manganese bound nitrene species in the C—H cleavage step. Collectively, these data suggest that the C—H cleavage step contributes significantly to the overall reactivity and selectivity observed with catalyst 3, a mechanistic feature that enables tunable control over selectivity in oxidation.
Late-stage C—H amination
The reactivity, site- and chemoselectivity trends observed with catalyst 3 on relatively simple molecules are maintained in more topologically and functionally complex natural product settings (Fig. 3). A functionally dense picrotoxinin derivative, containing an unprotected 3° alcohol, reacted smoothly under standard reaction conditions at a 3° C—H bond to produce fused bicycle 50 in 57% yield (Fig. 3a). Allylic C—H amination of sulfamate ester derived steroids pregnenolone and stigmasterol furnished five-membered heterocycles 51 and 52 in 55% and 66% yields, respectively, as single diastereomers. A leelamine-derived phenolic sulfamate ester 53 gave 70% yield of the 3° benzylic C—H amination product 54. Isosteviol derivative 55 was functionalized at the equatorial γ 2° aliphatic C—H bond to furnish 56 in 92% yield as a single syn diastereomer. Importantly, this reaction is high yielding (75%) with reduced catalyst (2.5 mol% 3) and oxidant (1.5 equiv) loadings, and scales readily. The versatile oxathiazinane moiety can also be easily diversified41. For example, reaction of N-CBz-protected oxathiazinane 56 with NaN3 or KOAc affords 1,3-diamine (57, 56%) or amino alcohol (58, 76%) precursors.
Figure 3. Late-stage diversification of complex molecules via [Mn(tBuPc)]-catalyzed C—H amination.

Amination of native oxygen-containing complex molecules picrotoxinin, pregnenolone, stigmasterol, isosteviol, and betulinic acid, as well as those where hydroxyl functionality is readily installed, leelamine and dihydropleuromutilone. a, Predictably selective C—H amination occurs on allylic, benzylic, and 3° and 2° aliphatic C—H bonds in the presence of alternate reactive functionality. Isosteviol derivative (−)-55 undergoes C—H amination on a gram scale and the resulting oxathiazinane (−)-56 is derivatized to reveal 1,3 diamine and amino alcohol motifs. b, Site- and diastereoselective 1° aliphatic C—H amination of betulinic acid (+)-59 and dihydropleuromutilone (+)-61 sulfamate ester derivatives in the presence of alternate, accessible 2° and 3° C—H bonds to furnish geometrically favored six-membered oxathiazinanes.
Whereas C—H amination methods proceeding with noble metals (e.g. iridium) via organometallic intermediates have a steric preference for functionalizing 1° methyl groups42, electrophilic high valent metal-heteroatom species (i.e. oxos and nitrenes) typically display poor reactivity of with 1° methyl C—H bonds due to their high BDEs and low basicity. Based on our observation that γ 1° methyl C—H bonds can be selectively functionalized by catalyst 3 in preference to weaker β C—H bonds, we sought to explore this exciting new reactivity and selectivity in complex molecule settings (Fig. 3b). Betulinic acid is a readily available pentacyclic triterpenoid with demonstrated mitochondrial-targeted antitumor activity43. Betulinic acid-derived sulfamate ester 59 may undergo amination at either the γ 1° C—H bond of a C23 methyl group to furnish a 6-membered heterocycle or a β 2° C—H bond to generate a 5-membered heterocycle. Consistent with selectivities observed in simpler substrates (18, vide supra), catalyst 3 preferentially aminates at the γ 1° C—H bond of the equatorial C23 methyl group with high site- and diastereoselectivity to furnish oxathiazinane 60 in 76% yield. This provides a functional handle for a range of further modifications (vide supra) that may attenuate betulinic acid’s high lipophilicity that has thus far limited its bioavailability.
Dihydropleuromutilone sulfamate ester derivative 61, rapidly generated via Fe(PDP)-catalyzed C—H hydroxylation and subsequent sulfamoylation at C7, presented the opportunity for additional investigation of site-selectivity in a complex molecule setting44. For 61, C—H amination can feasibly occur at three sites: the γ 1° C—H bond (C16), the β 2° C—H bond (C8) or the β 3° C—H bond (C6). Notably, 3-catalyzed C—H amination again resulted in exclusive formation of 1° C—H amination oxathiazinane product 62 in 84% yield, a remarkably high level of reactivity for 1° aliphatic C—H bond amination under metallonitrene catalysis. These results establish that native alcohols present in a wide range of readily available natural products, as well as those that can be readily installed, serve as valuable handles to install nitrogen into a broad range of sp3 C—H bonds in predictable 1,3- or 1,2-relationships.
Conclusion
We have reported a novel manganese C—H amination catalyst, readily synthesized in one step from commercial materials (Supplementary Information), that is ten million times more abundant than its noble metal predecessor45. While further development of this reaction remains, [Mn(tBuPc)] 3 is unique in its capacity to intramolecularly functionalize all types of C(sp3)—H bonds, including 1° aliphatic and propargylic, while maintaining stereospecificity and broad functional group tolerance in complex molecule settings. Studies indicate that the mechanism of metallonitrene insertion into sp3 C—H bonds for [Mn(tBuPc)] lies between that of the concerted asynchronous C—H insertion observed with rhodium and the stepwise radical C—H abstraction/rebound observed for iron. Rather than demonstrating reactivity and selectivity in between the two, [Mn(tBuPc)] promotes intramolecular C—H amination with higher reactivity than rhodium while maintaining the high chemoselectivity observed with iron. This finding challenges the bounds of the reactivity/selectivity paradigm and informs an approach to discover other highly reactive and selective C—H oxidation reactions with catalysts that access tunable high valent metal-heteroatom species.
Supplementary Material
Acknowledgments
Financial support for this work was provided by the NIH/NIGMS (GM112492). S.M.P. is a NSF Graduate Research Fellow, J.R.G is a NSF Graduate Research Fellow and a Springborn Graduate Fellow, J.P.Z. is a R.C. Fuson graduate fellow, and S.M.M. is a UIUC Summer Undergraduate Research Fellow. We thank Bristol-Myers-Squibb for generous support in an unrestricted “Freedom to Discover” grant to M.C.W. and acknowledge Dr. J.M. Howell, Dr. J.R. Clark, and C.C. Patillo for checking our experimental procedure. The data reported in this paper are tabulated in the Supplementary Information. The University of Illinois has filed a patent application on the [Mn(tBuPc)] catalyst for general C—H functionalizations.
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/naturechemistry.
Author Contributions S.M.P., J.R.G., J.P.Z., A.L.P., S.M.M. conducted the experiments and analyzed the data. S.M.P., J.R.G., and M.C.W. conceived and designed the project, analyzed the data and prepared this manuscript.
References and Notes
- 1.Roizen JL, Harvey ME, Du Bois J. Metal-catalyzed nitrogen-atom transfer methods for the oxidation of aliphatic C—H bonds. Acc. Chem. Res. 2012;45:911–922. doi: 10.1021/ar200318q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.White MC. Adding aliphatic C—H bond oxidations to synthesis. Science. 2012;335:807–809. doi: 10.1126/science.1207661. [DOI] [PubMed] [Google Scholar]
- 3.Que L., Jr The road to non-heme oxoferryls and beyond. Acc. Chem. Res. 2007;40:493–500. doi: 10.1021/ar700024g. [DOI] [PubMed] [Google Scholar]
- 4.Nam W, Lee Y, Fukuzumi S. Tuning reactivity and mechanism in oxidation reactions by mononuclear nonheme iron(IV)-oxo complexes. Acc. Chem. Res. 2014;47:1146–1154. doi: 10.1021/ar400258p. [DOI] [PubMed] [Google Scholar]
- 5.Chen MS, White MCA. predictable selective aliphatic C—H oxidation reaction for complex molecule synthesis. Science. 2007;318:783–787. doi: 10.1126/science.1148597. [DOI] [PubMed] [Google Scholar]
- 6.Gormisky PE, White MC. Catalyst-controlled aliphatic C—H oxidations with a predictive model for site-selectivity. J. Am. Chem. Soc. 2013;135:14052–14055. doi: 10.1021/ja407388y. [DOI] [PubMed] [Google Scholar]
- 7.Jeffrey JL, Sarpong R. Intramolecular C(sp3)—H amination. Chem. Sci. 2013;4:4092–4106. [Google Scholar]
- 8.Lebel H. In: Catalyzed Carbon-Heteroatom Bond Formation. Yudin AK, editor. Weinheim: Wiley-VCH; 2011. pp. 137–155. [Google Scholar]
- 9.Collet F, Dodd RH, Dauban P. Catalytic C—H amination: recent progress and future directions. Chem. Commun. 2009:5061–5074. doi: 10.1039/b905820f. [DOI] [PubMed] [Google Scholar]
- 10.Che CM, Lo VKY, Zhou CY, Huang JS. Selective functionalization of saturated C—H bonds with metalloporphyrin catalysts. Chem. Soc. Rev. 2011;40:1950–1975. doi: 10.1039/c0cs00142b. [DOI] [PubMed] [Google Scholar]
- 11.Paradine SM, White MC. Iron-catalyzed intramolecular allylic C—H amination. J. Am. Chem. Soc. 2012;134:2036–2039. doi: 10.1021/ja211600g. [DOI] [PubMed] [Google Scholar]
- 12.Hennessy ET, Betley TA. Complex N-heterocycle synthesis via iron-catalyzed, direct C—H bond amination. Science. 2013;340:591–595. doi: 10.1126/science.1233701. [DOI] [PubMed] [Google Scholar]
- 13.Hennessy ET, Liu RY, Iovan DA, Duncan RA, Betley TA. Iron-mediated intermolecular N-group transfer chemistry with olefinic substrates. Chem. Sci. 2014;5:1526–1532. [Google Scholar]
- 14.Liu Y, et al. Nonheme iron-mediated amination of C(sp3)—H bonds. Quinquepyridine-supported iron-imide/nitrene intermediates by experimental studies and DFT calculations. J. Am. Chem. Soc. 2013;135:7194–7204. doi: 10.1021/ja3122526. [DOI] [PubMed] [Google Scholar]
- 15.Harvey ME, Musaev DG, Du Bois J. A diruthenium catalyst for selective, intramolecular allylic C—H amination: reaction development and mechanistic insight gained through experiment and theory. J. Am. Chem. Soc. 2011;133:17207–17216. doi: 10.1021/ja203576p. [DOI] [PubMed] [Google Scholar]
- 16.Suzuki K, Oldenburg PD, Que L., Jr Iron-catalyzed asymmetric olefin cis-dihydroxylation with 97% enantiomeric excess. Angew. Chem. Int. Ed. 2008;47:1887–1889. doi: 10.1002/anie.200705061. [DOI] [PubMed] [Google Scholar]
- 17.Friedfel MR, et al. Cobalt precursors for high-throughput discovery of base metal asymmetric alkene hydrogenation catalysts. Science. 2013;342:1076–1080. doi: 10.1126/science.1243550. [DOI] [PubMed] [Google Scholar]
- 18.Jagadeesh RV, et al. Nanoscale Fe2O3-based catalysts for selective hydrogenation of nitroarenes to anilines. Science. 2013;342:1073–1076. doi: 10.1126/science.1242005. [DOI] [PubMed] [Google Scholar]
- 19.Zuo W, Lough AJ, Young FL, Morris RH. Amine(imine)diphosphine iron catalysts for asymmetric transfer hydrogenation of ketones and imines. Science. 2013;342:1080–1083. doi: 10.1126/science.1244466. [DOI] [PubMed] [Google Scholar]
- 20.Nappa MJ, McKinney RJ. Selectivity control by axial ligand modification in manganese porphyrin catalyzed oxidations. Inorg. Chem. 1988;27:3740–3745. [Google Scholar]
- 21.Zalatan DN, Du Bois J. Oxidative cyclization of sulfamate esters using NaOCl – a metal-mediated Hoffman-Löffler-Freytag reaction. Synlett. 2009;1:143–146. doi: 10.1055/s-0028-1087392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Groves JT, Kruper WJ, Haushalter RC. Hydrocarbon oxidations with oxometalloporphinates. Isolation and reactions of a (porphinato)manganese(V) complex. J. Am. Chem. Soc. 1980;102:6375–6377. [Google Scholar]
- 23.Cook BR, Reinert TJ, Suslick KS. Shape-selective alkane hydroxylation by metalloporphyrin catalysts. J. Am. Chem. Soc. 1986;108:7281–7286. [Google Scholar]
- 24.Hill CJ, Hollander FJ. Structural characterization of a complex of manganese(V) nitrido[tetrakis(p-methoxyphenyl)porphinato]manganese(V) J. Am. Chem. Soc. 1982;104:7318–7319. [Google Scholar]
- 25.Mehn MP, Peters JC. Mid- to high-valent imido and nitrido complexes of iron. J. Inorg. Biochem. 2006;100:634–643. doi: 10.1016/j.jinorgbio.2006.01.023. [DOI] [PubMed] [Google Scholar]
- 26.Jensen MP, Mehn MP, Que L., Jr Intramolecular aromatic amination through iron-mediated nitrene transfer. Angew. Chem. Int. Ed. 2003;42:4357–4360. doi: 10.1002/anie.200351605. [DOI] [PubMed] [Google Scholar]
- 27.CRC Handbook of Chemistry & Physics. 94. Boca Raton, FL: CRC; 2013. pp. 9–65. [Google Scholar]
- 28.Breslow R, Gellman SH. Intramolecular nitrene C—H insertions mediated by transition-metal complexes as nitrogen analogues of cytochrome P-450 reactions. J. Am. Chem. Soc. 1983;105:6728–6729. [Google Scholar]
- 29.Lever ABP, Wilshire JP. Redox potentials of metal phthalocyanines in non-aqueous media. Can. J. Chem. 1976;54:2514–2516. [Google Scholar]
- 30.Löbbert G, editor. Ullmann’s Encyclopedia of Industrial Chemistry. Vol. 27. Weinheim: Wiley-VCH; 2012. pp. 181–213. [Google Scholar]
- 31.Fiori KW, Espino CG, Brodsky BH, Du Bois J. A Mechanistic analysis of the Rh-catalyzed intramolecular C—H amination reaction. Tetrahedron. 2009;65:3042–3051. [Google Scholar]
- 32.Huard K, Lebel H. Rhodium-catalyzed amination reaction from N-tosyloxycarbamates. Chem. Eur. J. 2008;14:6222–6230. doi: 10.1002/chem.200702027. [DOI] [PubMed] [Google Scholar]
- 33.Reed SA, Mazzotti AR, White MC. A catalytic, bronsted base strategy for intermolecular allylic C—H amination. J. Am. Chem. Soc. 2009;131:11701–11706. doi: 10.1021/ja903939k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liang C, et al. Toward a synthetically useful stereoselective C—H amination of hydrocarbons. J. Am. Chem. Soc. 2008;130:343–350. doi: 10.1021/ja076519d. [DOI] [PubMed] [Google Scholar]
- 35.Grigg DR, Rigoli JW, Pearce SD, Schomaker JM. Synthesis of propargylic and allenic carbamates via the C—H amination of alkynes. Org. Lett. 2012;14:280–283. doi: 10.1021/ol203055v. [DOI] [PubMed] [Google Scholar]
- 36.Thornton AR, Blakey SB. Catalytic metallonitrene/alkyne metathesis: A powerful cascade process for the synthesis of nitrogen-containing molecules. J. Am. Chem. Soc. 2008;130:5020–5021. doi: 10.1021/ja7111788. [DOI] [PubMed] [Google Scholar]
- 37.Fleming JJ, McReynolds MD, Du Bois J. (+)-Saxitoxin: A first and second generation stereoselective synthesis. J. Am. Chem. Soc. 2007;129:9964–9975. doi: 10.1021/ja071501o. [DOI] [PubMed] [Google Scholar]
- 38.Sharma A, Hartwig JH. Metal-catalysed azidation of tertiary C—H bonds suitable for late-stage functionalization. Nature. 2015;517:600–604. doi: 10.1038/nature14127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Michaudel Q, Thevenet D, Baran PS. Intermolecular Ritter-type C—H amination of unactivated sp3 carbons. J. Am. Chem. Soc. 2012;134:2547–2550. doi: 10.1021/ja212020b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhong HA, Labinger JA, Bercaw JE. C—H bond activation by cationic platinum(II) complexes: ligand electronic and steric effects. J. Am. Chem. Soc. 2002;124:1378–1399. doi: 10.1021/ja011189x. [DOI] [PubMed] [Google Scholar]
- 41.Meléndez RE, Lubell WD. Synthesis and reactivity of cyclic sulfamidites and sulfamidates. Tetrahedron. 2003;59:2581–2616. [Google Scholar]
- 42.Kang T, Kim Y, Lee D, Wang Z, Chang S. Iridium-catalyzed intermolecular amidation of sp3 C—H bonds: late-stage functionalization of an unactivated methyl group. J. Am. Chem. Soc. 2014;136:4141–4144. doi: 10.1021/ja501014b. [DOI] [PubMed] [Google Scholar]
- 43.Drag-Zalesinska M, et al. Esters of betulin and betulinic acid with amino acids have improved water solubility and are selectively cytotoxic toward cancer cells. Bioorg. Med. Chem. Lett. 2009;19:4814–4817. doi: 10.1016/j.bmcl.2009.06.046. [DOI] [PubMed] [Google Scholar]
- 44.Chen MS, White MC. Combined effects on selectivity in Fe-catalyzed methylene oxidation. Science. 2010;327:566–571. doi: 10.1126/science.1183602. [DOI] [PubMed] [Google Scholar]
- 45.Greenwood NN, Earnshaw A. Chemistry of the Elements. 2. Oxford: Elsevier; 1997. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.